

Abstract
Cell‐penetrating peptides (CPPs) are still an interesting and viable alternative for drug delivery applications. CPPs contain considerably high amounts of positively charged amino acids, imparting them with cationic character. Tumor cells are characterized by an enhanced anionic nature of their membrane surface, a property that could be used by CPPs to target these cells. We recently identified a branched CPP that displays a high internalization capacity while exhibiting selectivity for certain tumor cell types. In this study we elucidated this observation in greater detail by investigating the underlying mechanism behind the cellular uptake of this peptide. An additional cytotoxicity screen against several cancer cell lines indeed demonstrates high cytotoxic activity against cancer cells over normal fibroblasts. Furthermore, we show that this feature can be used for delivering the anticancer drug actinomycin D with high efficiency in the MCF‐7 cancer cell line.
Keywords: anticancer activity, cell-penetrating peptides, lytic peptides, tumor cells, tumor targeting
Introduction
Recently, the use of anticancer peptides (ACPs) has been described as promising new tool to target cancer cells.1 This strategy makes advantage of the difference in terms of membrane composition of healthy cells versus neoplastic cells. Plasma membranes of mammalian cells mainly consist of glycerol‐based phospholipids (PLs), such as phosphatidylcholine (PC), phosphatidylserine (PS), phosphatidylinositol (PI), phosphatidylethanolamine (PE), sterols (mainly cholesterol), and sphingomyelins (SM).2 The outer leaflet of tumor cells is increased in the amount of anionic PLs, such as PS, resulting in a slightly more negatively charged membrane.3 Thus, electrostatic interactions with positively charged peptides constitute the major factor leading to a preferential targeting of cancer cells. This group of anticancer peptides is mostly of cationic nature and normally not attracted to normal vertebrate cells that exhibit zwitterionic outer membranes. Beside an increased PS level, also other factors contribute to the selectivity of ACPs like increased levels of sialic acid in membrane bound glycoconjugates and the high negative transmembrane potential associated with cancer cell membranes.1 One group of anticancer peptides comprises host defense peptides that play a crucial role as antimicrobial peptides in the innate immune system of various organisms. Mostly, these peptides act by direct attack of the microbial membrane itself, whereas also intracellular targets might be of relevance.4 Interestingly, they have been shown to interact strongly with membranes of cancer cells too, leading to invasion of the plasma membrane and membrane lysis. Recently, also cell‐penetrating peptides (CPPs) have been described as molecules with a tumor‐homing potential.5 CPPs are short cationic peptide sequences with the ability to overcome cell membranes and to deliver attached cargos inside cells.
We recently reported the development of the CPP sC18 that is derived from the C‐terminal domain of the cationic antimicrobial peptide CAP18.6 This peptide was highlighted by us as an effective delivery vector for metal‐containing cytostatic drugs7 and imaging probes.8 The antimicrobial activity of sC18 was only moderate, but could be enhanced by the conjugation to imidazolium salts.9 Furthermore, we could show that dimerization of sC18, providing the novel peptide (sC18)2, leads to increased cellular uptake and interestingly, to a certain tumor cell selectivity.10 Driven by this latter observation, the present work aims to elucidate in more detail the underlying mechanistic features of (sC18)2 and its activity profile in presence of different cancer and non‐cancer cell lines.
Results and Discussion
Peptide design and synthesis
We previously showed that dimerization of sC18 leads to an enhanced cellular uptake and also increases cytotoxicity depending on the cell type used.10 The importance of branching for activity was formerly delineated by us when investigating drug delivery properties of another cell‐penetrating peptide.11 However, we also investigated the linear dimer of the (sC18)2 version. Although acting relatively similar, microscopic images and cytotoxicity assays revealed a trend to a slightly higher toxic profile of this peptide. Indeed, without the branching, the peptide forms nearly a perfect α‐helix leading to an intense attack of membranous structures (Supporting Information, Figures S1 and S2). In our opinion, the branching helps to decrease this relatively unselective toxic property, thus increasing potential selectivity. Therefore, we stayed with the branched dimer for the following studies.
All peptides were synthesized using standard Fmoc/tBu solid‐phase peptide synthesis protocols. For subsequent studies the peptides were labeled either with the fluorophore 5,6‐carboxyfluorescein (CF), or radiolabeled with the radioisotope 68Ga after introduction of the chelator NODA‐Ga(tBu)3 (NODAGA). Identification was realized by using LC–ESI mass spectrometry, and all peptides were purified prior use by preparative HPLC (purity >95 %).
Internalization pattern and intracellular fate
To get a deeper understanding of our previous work, we first established and compared the uptake profiles of the monomer and dimer in two different cell lines (see Table 1 and Table S1 for peptide names, molecular weights and sequences used in this study). Human breast cancer MCF‐7 cells were taken as representative cancer cell line, and human epithelial kidney HEK‐293 cells as non‐cancer cells. To investigate the time‐dependence of peptide uptake, the cells were incubated with fluorescently labeled sC18 and (sC18)2 for the time lengths of 30, 60, 90 and 120 min. From this it became clear that the uptake occurs in a time‐dependent manner and the amount of internalized peptide was higher for the dimeric than for the monomeric CPP at all time points evaluated (Figure 1). This effect was quite more intense for MCF‐7 cells than for HEK‐293 cells. Notably, both peptides seem to behave differently in their uptake mechanisms and intracellular distribution depending on the cell line used. In HEK‐293 cells, the peptides are presumably internalized by endocytosis, accumulate in vesicles and are not present in the nucleus. In contrast, for MCF‐7 cells, it was visible that the uptake probably occurs via direct penetration and endocytosis with followed endosomal release, as the CPPs accumulated within the cytoplasm as well as within the nucleus (Figure 1). This effect was much more pronounced for the dimeric peptide, considering also the much lower concentration used when incubating the peptide with MCF‐7 cells. Clearly the increase in positive charge density within the structure of (sC18)2 is one reason for this observation. Recently, also for other CPPs nuclear targeting within cancer cells was observed.12
Table 1.
Peptides used in this study, their molecular weights and net charges.
| Peptide[a] | MWcalcd [Da] | Net charge |
|---|---|---|
| sC18 | 2069.6 | +9 |
| (sC18)2 | 4122.2 | +17 |
| CF‐sC18 | 2427.9 | +8 |
| CF‐(sC18)2 | 4480.5 | +16 |
| NODAGA‐sC18 | 2426.9 | +8 |
| NODAGA‐(sC18)2 | 4479.5 | +16 |
[a] Details about peptide sequences can be found in the Supporting Information. CF: 5,6‐carboxyfluorescein; NODAGA: NODA‐Ga(tBu)3.
Figure 1.
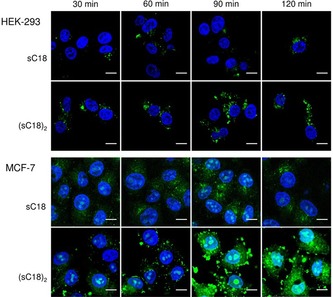
CLSM images of HEK‐293 and MCF‐7 cells after incubation with various concentrations of CF‐sC18 (10 μm for HEK‐293, 5 μm for MCF‐7) and CF‐(sC18)2 (10 μm for HEK‐293, 1 μm for MCF‐7), for different time points. Blue: Hoechst nuclear stain, green: CF‐labeled peptides. Scale bar: 10 μm.
To explore the mode of uptake in more detail, we tested the influence of incubation temperature. Therefore, we let the cells incubate by either 37 °C or 4 °C, respectively, whereby energy‐dependent endocytosis is inhibited in the latter case. Flow cytometry studies approved the previous CLSM results, showing that internalization of the dimeric (sC18)2 is increased relative to the parent monomer sC18 in the two cell lines. Furthermore, it can be clearly observed that the dimeric peptide is taken up to a higher extent in MCF‐7 cancer cells than in HEK‐293 cells (Figure 2 A). Moreover, when incubating the MCF‐7 cells at 4 °C with (sC18)2, no significant decrease in peptide accumulation can be observed, which confirms the theory that the CPPs are entering the cancer cells mainly by direct penetration. In contrast to this, there is a clear difference in the amount of internalized (sC18)2 at 37 °C and 4 °C in HEK‐293 cells (***p<0.001) approving the idea that the peptides enter the non‐cancer cells mainly by endocytosis (Figure 2 A). This is in agreement to the microscopy images (Figure 2 B) demonstrating more or less the same phenotype in MCF‐7 cells, whereas in HEK‐293 cells, at 4 °C, also nuclear accumulation is observed. May be in this case, some of the (sC18)2 peptides are able to directly enter the cells and target the nuclei, whereas at 37 °C mainly endocytosis takes place.
Figure 2.
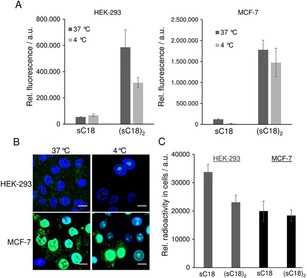
A) Flow cytometric analysis of cellular uptake of 10 μm CF‐labeled peptides in HEK‐293 and MCF‐7 cells at 4 °C and 37 °C incubated for 30 min. B) CLSM images of MCF‐7 and HEK‐293 cells after 30 min with 10 μm CF‐(sC18)2 at 4 °C or 37 °C, respectively. Blue: Hoechst nuclear stain, green: CF‐labeled peptides. Scale bar: 10 μm. C) Cellular uptake of 68Ga‐NODAGA‐modified peptides measured in a gamma spectroscopy system. Cells were incubated with 0.2 μm 68Ga‐NODAGA peptide.
In another set of experiments we further determined the peptide uptake at really low peptide concentrations. Because fluorescence techniques are not very suitable to track such low amounts, we used radiolabeling in this case. Cells were treated by a 0.2 μm Ga68‐NODAGA‐peptide solution, but after measuring the relative radioactivity, the uptake in both cell lines for both peptides was not significantly different (Figure 2 C). It is known that the uptake mechanism of cell‐penetrating peptides is highly depending on various factors, for example, the CPP concentration.13 However, at these low concentrations used in our experiment, the internalization rates in MCF‐7 and HEK‐293 cells were not statistically different. Moreover, we cannot ascertain if endocytotic processes still play a role for cell entry.
Next, we elucidated the intracellular fate of the CPPs after a longer time period of incubation in these both cell‐lines. Therefore, we removed the peptide solutions after 30 min of incubation with the cells and let them further incubated for 6 h in the appropriate medium. As can be seen in Figure 3, the peptides differ in their internalization pattern depending on the investigated cell lines. In HEK‐293 cells, the amount of peptide, as well as its destination inside the cells (e.g., vesicles) stayed more or less equal. These results are in line with the theory that the uptake in these cells occurs mainly via endocytosis. Thereby, the peptides remained encapsulated in the vesicles and were not released into the cytosol. The endosomes are then undergoing the degradative pathway and will be developed to endolysosomes.14 These lysosomes are visually not distinguishable from the endosomes. In MCF‐7 cells, a difference between the pictures at time points 0 and 6 h was clear. The amount of sC18 is lower after 6 h and just appeared in fluorescent dots, presenting the vesicles containing the peptide. Probably, most of the peptides that entered the cells by endocytosis is released from the vesicles and was then degraded in the cytosol. Thus, after 6 h, there was just a few amount of peptide left in the lysosomal pathway. After endosomal release over time, the free CPP within the cytoplasm was either degraded by cytosolic enzymes or was released out of the cells. Also for (sC18)2, whereof almost nothing was left after 6 h, it can be assumed, that the peptide left the endosomes and got degraded within the cytoplasm or was also released in the medium. These results confirm the idea that the internalization in MCF‐7 cells happens mainly via direct penetration.
Figure 3.
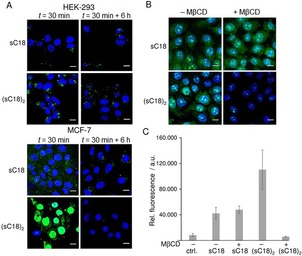
A) CLSM images of MCF‐7 and HEK‐293 cells after 30 min with 10 μm CF‐sC18 or CF‐(sC18)2, and after 6 h in peptide‐free medium. Blue: Hoechst nuclear stain, green: CF‐labeled peptides. Scale bar: 10 μm. B) CLSM images of MCF‐7 incubated with 5 μm CF‐sC18 or 1 μm CF‐(sC18)2 for 30 min after cholesterol depletion using 10 mm methyl‐β‐cyclodextrin (MβCD) for 1 h. Blue: Hoechst nuclear stain, green: CF‐labeled peptides. Scale bar: 10 μm. C) Flow cytometric analysis of cellular uptake of CF‐labeled peptides sC18 and (sC18)2 in MCF‐7 cells incubated for 30 min at 37 °C with or without cholesterol depletion. Experiments were performed in triplicate with n=3.
Relevance of membrane composition
Further insight on the interaction with biological membranes were obtained by using artificial membrane‐like systems. Recently, essential roles of guanidinium groups and two cell components, such as fatty acids and the cell membrane pH gradient, were accounted to an efficient peptide–lipid interaction and uptake of cationic CPPs.15 In addition, the interaction with proteoglycans and their role in cationic CPP‐mediated uptake has been discussed, but not yet delineated.16 For α‐helical anticancer peptides the selectivity for cancer cells was shown to depend mainly on the targeting of their anionic membranes, although other factors not yet elucidated might be involved.1 Definitely the interaction with membrane lipids is one crucial step for peptide internalization. Owing to this, we were particularly interested in membrane attack activity of (sC18)2, and if it penetrates better in more anionic cancer cell lines. Thereby, we used large and giant unilamellar vesicles (LUVs and GUVs), respectively, with different lipid compositions. Zwitterionic, non‐charged, phospholipid vesicles (made from DOPC/DOPE, 50:50) resemble the asymmetric distribution within electrostatically neutral mammalian membranes.2, 5 Neoplastic cells often comprise a more negatively charged membrane than normal cells,17 and therefore, vesicles with negatively charged membranes made from DOPC, DOPG, DOPE (40:30:30) were investigated, too. To not only compare the effect of peptide on zwitterionic vs. anionic lipid membranes, also the effect of different membrane fluidities were studied by adding sphingomyelin and cholesterol to the latter mixture. The interaction of both peptides with neutral charged LUVs or GUVs was negligible after an incubation time of ∼20 or 90 min, respectively. No green fluorescent signal was observed inside the vesicles as well as no intravesicular stain disappeared out of the vesicles (Figure 4 A,B). This observation points to only low peptide‐lipid interactions and no lytic activities of both peptide variants using this setup.
Figure 4.
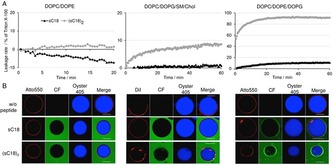
A) Peptide‐induced CF leakage experiments from LUVs composed of DOPC/DOPE (50:50), DOPC/DOPG/SM/Chol (25:5:20:20), DOPC/DOPE/DOPG (40:30:30) as a function of time. The results shown are representative of three independent preparations of LUVs. LUVs were treated with peptides (1 μm) for 60 min. B) CLSM analysis of GUVs composed of DOPC/DOPE (50:50), DOPC/DOPG/SM/Chol (25:5:20:20), DOPC/DOPE/DOPG (40:30:30) treated with sC18 and (sC18)2 (10 μm) for 90 min. Red: membrane stain Atto550 or DiI, green: CF‐labeled peptides, blue: Oyster 405. Scale bar: 30 μm.
Not surprisingly, the experiments with negatively charged vesicles let assume a more tight interaction of (sC18)2 with the negatively charged membranes, presumably leading to pore‐forming events. The results perfectly match with previous reports about the interaction of cationic CPPs with negatively charged membranes.5 Interestingly, by adding cholesterol and sphingomyelin this strong membrane attack can be somehow lowered. On the one hand this may be a consequence of the presence of cholesterol that has profound effects on the membrane fluidity and regulates, among other things, endocytosis. On the other hand sphingomyelin together with cholesterol are reported to be enriched in distinct membrane domains, often referred to as rafts.18 Notably, in contrast to the monomer, the dimeric peptide accumulated especially within the DiI‐labeled disordered phases of the vesicle membranes, which are containing less sphingomyelin and cholesterol (Figure 4 B). Therefore, it is likely that the positively charged CPP interacts with the anionic DOPG but the ordered phase of the lipid rafts in the cell membrane hinders cell membrane disruption or stronger pore‐forming processes. Furthermore, the results showed that the CPPs were not able to overcome neutral charged membranes corroborating the importance of negatively charged constituents at the outer leaflet of the membrane.
For proving this idea, a cholesterol depletion assay was performed using methyl‐β‐cyclodextrin (MβCD), before observation on the microscope or flow cytometer (Figure 3 B,C). It was visible that the internalization and intracellular distribution of the monomeric sC18 was not harmed when cholesterol was sequestered, confirming the observation that was made with the vesicles, namely that the cholesterol in the membrane is not that important for the sC18 membrane interaction. In contrast, the uptake of the dimeric (sC18)2 was extremely decreased when depleting the cholesterol from the membranes of MCF‐7 cells (***p<0.001). Thereby it was visible, that the peptides just accumulate in the nuclei and almost no CPP could be found in the cytoplasm. Notably, for the non‐cancer cell line HEK‐293, a similar result could be obtained (data not shown). Cholesterol depletion has previously shown to inhibit the uptake of other CPPs in two cell lines.19
Hence, our studies suggest that the selectivity of (sC18)2 might depend on the composition of the target membrane and probably also its associated outer layer. Other factors that may be involved in this strong interplay are currently examined by our group.
Uptake and cytotoxicity screening on various cell lines
Subsequently, we were interested in screening the cellular uptake and the influence on cell viability in a set of various cancer cell lines. Next to HEK‐293 and MCF‐7, three other cancer cell lines, namely PC‐3, HeLa and HCT‐15 were used and incubated with the peptides. As shown in Figure 5, both CPPs internalize in all cell lines, but the dimer (sC18)2 still showed higher uptake rates than the monomer. Furthermore, a decreased fluorescent signal was obtained for sC18 when in contact with the cancer cell line HCT‐15. Moreover, (sC18)2 differs strongly in uptake amount and intracellular localization. Notably, all cancer cell lines were characterized by diffuse fluorescent signals next to the punctual distribution in the cytoplasm as well as nuclear accumulation, whereas in the non‐cancer cells just a punctual distribution in the cells but hardly any peptides in the nuclei could be observed. Interestingly, nuclear accumulation was not that strong in PC‐3 cells for (sC18)2.
Figure 5.
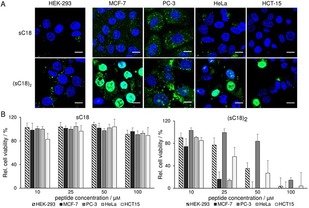
A) CLSM images of HEK‐293, MCF‐7, PC‐3, HeLa and HCT‐15 cells after 30 min with 10 μm CF‐sC18 and CF‐(sC18)2, respectively. Blue: Hoechst nuclear stain, green: CF‐labeled peptides. Scale bar: 10 μm. B) Cell viability assay based on resazurin after 24 h with the CPPs at various concentrations, untreated cells served as negative control, cells treated for 10 min with 70 % ethanol as positive control, experiments were conducted in triplicate with n=3.
Furthermore, the cytotoxic profile of (sC18)2 was evaluated when in contact with these cell lines. The results are in agreement to former published results showing a more distinct action in the cancer versus HEK‐293 cells.10 However, a clear distinction in activity was elucidated, whereas (sC18)2 turned out to be most active against MCF‐7 and HeLa cells (Figure 5 B).
Lower cytotoxic activity was observed when in contact with PC‐3, and HEK‐293 cells. This might point to a mechanism of action that is located or has its initiation in the cell nuclei rather than based only on cell lysis events, what might be the case for (sC18)2 when in contact with HeLa and MCF‐7 cells. Here, only low concentrations of the peptides are responsible for membrane disruption (Supporting Information, Figure S3).
We further evaluated the efficacy of (sC18)2 in a total of 16 cancer cell lines as well as one normal human cell line (fibroblast cells) using slightly different assay conditions (see experimental part). As can be seen from the different IC50 values displayed in Table 2, again, (sC18)2 acted more active against almost all cancer cell lines tested, and the fibroblast cell line was significantly less affected in comparison. Particularly effective was the peptide against the melanoma cell line A2058 with an averaged IC50 value of <4 μm. All in all, the results let assume a high therapeutic potency of (sC18)2 for several cancer models.
Table 2.
IC50 values determined after incubating (sC18)2 with various cell lines.
| Tumor type | Cell line | IC50 [μm][a] |
|---|---|---|
| Melanoma | A2058 | 3.9±0.8 |
| HT168‐M1/9 | 9.2±0.3 | |
| M24 | 16.2±9.8 | |
| Liver | HepG2 | 5.4±0.1 |
| Prostate | PC3 | 8.8±1.4 |
| DU145 | 12.3±1.3 | |
| Pancreas | Panc‐1 | 23.2±5.0 |
| Breast | MDA‐MB‐231 | 30.9±1.2 |
| MCF‐7 | 6.2±1.0 | |
| Lung | A549 | 27.3±2.0 |
| H1975 | 6.5±0.1 | |
| H1650 | 4.5±0.3 | |
| Oral (head and neck) | PE/CA‐PJ15 | 39.6±4.5 |
| PE/CA‐PJ41 | 36.2±1.1 | |
| Fibrosarcoma | HT1080 | 7.4±0.6 |
| Colon | HT29 | 9.5±0.3 |
| Normal fibroblasts | MRC‐5 | >50 |
[a] Values are the mean±SD of n=2 experiments conducted in triplicate.
Cargo delivery and synergistic effects
Driven by these promising results, our aim was to further study how (sC18)2 would contribute to or trigger the uptake of additionally applied drugs. Our previous study already approved the drug transport ability for (sC18)2 for covalently attached cargos.10 Although very effective, this strategy is of course highly laborious and time‐consuming. Because lytic effects play a role in the activity of (sC18)2, we hypothesized that the co‐incubation of anticancer drugs might lead to a higher cytotoxicity. In a preliminary experiment, we used actinomycin D, an antitumor antibiotic that we incubated with the peptides for 24 h on MCF‐7 cells. Clearly, the monomeric sC18 does not efficiently transport the actinomycin D inside the cell nuclei, because no significant decrease of cell viability could be detected (Figure 6). However, incubation with (sC18)2 indicates a high decrease in viable cells up to 70 % exhibiting a successful transport of the cargo over the cell membrane, or owing to pore‐forming events, facilitating the entry of the drug (Figure 6). In any case, when inside the cells actinomycin D targets to the DNA, where it can intercalate and inhibit the RNA synthesis.20
Figure 6.
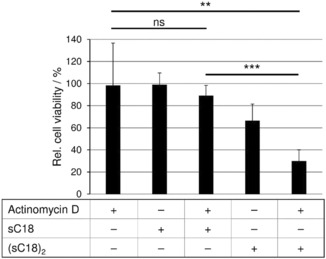
Cells were incubated for 24 h with actinomycin D (0.1 μm), sC18 or (sC18)2 (10 μm) co‐incubated with actinomycin D (0.1 μm). Untreated cells served as negative control, cells treated for 10 min with 70 % ethanol as positive control. Experiments were conducted in triplicate with n=2.
Conclusions
In conclusion, we have presented functional and activity studies that demonstrate the effectiveness of the CPP (sC18)2 as a lytic anticancer peptide. Evidence suggests that (sC18)2 first accumulates preferentially on anionic membranes, where it might induce pores for a direct entry, but endocytotic events may also play a role. However, it still remains to be investigated if other factors are involved in this interaction. Furthermore, it is not yet elucidated if (sC18)2 may possibly have any pro‐apoptotic effects.
Experimental Section
Peptide synthesis: In general, the peptides used were synthesized on Rink amide resin by automated solid‐phase peptide synthesis (SPPS) on a multiple Syro II peptide synthesizer (MultiSynTech, Witten, Germany). Fmoc/tBu‐strategy using a double‐coupling procedure and in situ activation with Oxyma/DIC was followed. Synthesis of the branched versions of (sC18)2 was performed as described previously.10 N‐terminal coupling of 5,6‐carboxyfluorescein (CF) or the chelator NODA‐Ga(tBu)3 (4‐(4,7‐bis(2‐tert‐butoxy‐2‐oxoethyl)‐1,4,7‐triazonan‐1‐yl)‐5‐tert‐butoxy‐5‐oxopentaoic acid) was carried out with 3 equiv of the substance to be coupled and activating with 3 equiv HATU/DIPEA in DMF under vigorous shaking for 2 h. For the successful coupling of NODA‐Ga(tBu)3 this step was sufficient, for the CF‐ and amino acid coupling, another step using 3 equiv of the substance and Oxyma/DIC under shaking overnight was performed, respectively. CF‐polymers were cleaved by treatment with 20 % piperidine for 45 min. Peptides were analyzed by LC–MS using an Agilent instrument with parallel detection at 220 nm UV‐absorption and electrospray‐ionization mass spectrometry (ESI‐MS). Reversed‐phase (RP) analytical HPLC was performed at 1.2 mL min−1 flow rate on a 4.6×100 mm Kinetex 2.6u C18 100A column (Phenomenex, Aschaffenburg, Germany). Gradient of 10–60 % acetonitrile in water over 15 min (with constant 0.1 % formic acid) was used. Purification was achieved by preparative RP‐HPLC using a Hitachi Elite LaChrom instrument (VWR, Darmstadt, Germany) at 6 mL min−1 flow rate and 220 nm detection. Chromatography was performed on a 15×250 mm Jupiter 4u Proteo 90A column (Phenomenex, Aschaffenburg, Germany) and acetonitrile/water with 0.1 % TFA gradients as needed. The collected fractions were evaporated, analyzed with LC–MS and lyophilized to obtain the purified peptides (purity >95 %).
Cell and culture conditions: HEK‐293 (human embryonic kidney), MCF‐7 (human breast adenocarcinoma), PC‐3 (human prostate cancer), HeLa (human cervix carcinoma) and HCT‐15 (human colorectal adenocarcinoma) were cultured in sterile 10 cm petri dishes at 37 °C and 5 % CO2 in a humidified atmosphere. For HEK‐293 cells, complete MEM supplemented with 15 % FBS and 4 mm l‐glutamine was used. HeLa, HCT‐15 and MCF‐7 cells were grown in complete RPMI 1640 supplemented with 10 % FBS and 4 mm glutamine. PC‐3 cells DMEM F‐12 Ham supplemented with 4 mm l‐glutamine and 5 % FBS was used. Cells for the MTT assay were cultured as follows: A2058, M24, HT168‐M1/9 (melanoma), A549, H1975, H1650 (lung), MCF‐7, MDA‐MB‐231 (breast), PC‐3, DU145 (prostate), HT‐29 (colon), HepG2 (liver), PANC‐1 (pancreas), HT1080 (fibrosarcoma), PE/CA‐PJ15 and PE/CA‐PJ41 (head and neck squamous cell carcinoma) cells were hold in RPMI‐1640 medium containing 10 % FBS, 4 mm l‐glutamine and 1 % Pen‐Strep. PE/CA cells were cultured in Iscove's Modified Dulbecco's Medium supplemented with 10 % FBS, 2 mm l‐glutamine and 1 % Pen‐Strep. MRC‐5 (normal fibroblast) cell were cultured in Dulbecco's Modified Eagle's Medium containing 4500 mg l −1 glucose.
Resazurin‐based cell viability assay: Cells were seeded in a 96‐well plate (HEK‐293 70 000, MCF‐7 45 000, HeLa, PC‐3 and HCT‐15 40 000 cells per well), grown to 70–80 % confluency and incubated with various concentrations of peptides in appropriate serum‐free medium for 24 h under standard growth conditions. For the positive control, cells were treated with 70 % ethanol for 10 min. After washing with phosphate buffered saline, resazurin stock solution was diluted with appropriate serum‐free medium (1:10, v/v) and 100 μL of this solution was incubated with the cells for 1 h. Subsequently, the cell viability was determined relative to untreated cells by measurement of the resorufin product at 595 nm (λ ex=550 nm) on a Tecan infinite M200 plate reader. The experiments were done in triplicate.
LDH release assay: For the membrane leakage assay provided by Promega (CytoTox‐ONETM Homogenous Membrane Integrity Assay), cells were seeded in a 96‐well plate (HEK‐293 70 000, MCF‐7 45 000, HeLa, HCT‐15 40 000 cells per well), grown to 70–80 % confluency and incubated with various concentrations of peptides in appropriate medium for 1 h in the absence of serum under standard growth conditions. Afterward, the assay was conducted according to the manufacturer's protocol including the provided cell lysis positive control. Subsequently, the cell lysis was determined relative to untreated cells by measurement at 590 nm (λ ex=560 nm) on a Tecan infinite M200 plate reader. The experiments were done in triplicate.
MTT cytotoxicity assay: For the screening on cell viability of different human tumor cell lines (A2058, M24, HT168‐M1/9 (melanoma), A549, H1975, H1650 (lung), MCF‐7, MDA‐MB‐231 (breast), PC‐3, DU145 (prostate), HT‐29 (colon), HepG2 (liver), PANC‐1 (pancreas), HT1080 (fibrosarcoma), PE/CA‐PJ15, PE/CA‐PJ41 (head and neck squamous cell carcinoma) an MTT assay (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl‐tetrazolium bromide) obtained from Sigma Aldrich Ltd. St. Louis, MO, USA was carried out. For control, MRC‐5 cells were used. Cells were seeded in a 96‐well plate (4000 cells per well), grown to 40–60 % confluency and incubated with various concentrations of (sC18)2 in appropriate serum‐free medium for 48 h under standard growth conditions. Afterward, the MTT assay was performed by adding 20 μL of MTT solution (5 mg mL−1 in serum‐free RPMI‐1640 medium) to each well and after 4 h of incubation at 37 °C, the supernatant was removed. The formazan crystals were dissolved in 100 μL of a 1:1 solution of DMSO (Sigma Aldrich) and EtOH (Molar Chemicals Kft. Hungary) and the absorbance was determined after 15 min at λ=570 nm by using a microplate reader (BIO‐RAD, model 550). Background value (absorbance of DMSO‐EtOH only) was subtracted from measured values and the percentage decrease in cell viability was determined relative to untreated cells.
Quantification of cellular uptake: For peptide‐uptake studies by flow cytometry, cells were seeded in a 24‐well plate (HEK‐293 500 000, MCF‐7 200 000 cells per well) and grown to 70–80 % confluency. After incubation at either 4 °C or 37 °C for 30 min with CF‐labeled peptides in serum‐free medium, the cells were washed twice with PBS, detached with indicator‐free trypsin and resuspended in appropriate, indicator‐free medium. Until measurement the samples were stored on ice. Analyses were performed on a BD Accuri C6 flow cytometer whereas 20 000 viable cells were counted. Cellular autofluorescence was subtracted. The experiments were performed in triplicate. For cholesterol depletion, 400 μL of 10 mm fresh prepared methyl‐β‐cyclodextrin solution dissolved in appropriate serum‐free medium was added to the cells and incubated for 1 h at 37 °C. Afterward, the cells were washed once with PBS and then treated as described above.
Microscopy studies of peptide uptake: For confocal microscopic uptake studies, cells were seeded in a μ‐slide eight‐well (Ibidi) plate (HEK‐293 120 000, MCF‐7, Hela and HCT‐15 50 000, PC‐3 80 000 cells per well), and grown to 70–80 % confluency. The cells were then incubated with CF‐labeled peptides in serum‐free medium for the requested time at either 4 °C or 37 °C. The nuclei were stained for 10 min with Hoechst33342 nuclear dye prior to the end of peptide incubation. Finally, the solution was removed and the cells were treated with 200 μL trypan blue solution (150 μm in 0.1 m acetate buffer, pH 4.15) for 30 s. After washing twice and adding fresh, appropriate medium, images were taken by using a Nikon Eclipse Ti confocal laser scanning microscope (pinhole S, Offset green: 100, blue: 38) equipped with a 60× oil‐immersion objective. Images were recorded with Nikon EZ‐C1 3.91 software and adjusted equally with ImageJ 1.43 m software. For cholesterol depletion, 300 μL of 10 mm fresh prepared methyl‐β‐cyclodextrin solution dissolved in appropriate serum free medium was added to the cells and incubated for 1 h at 37 °C. Afterward, the cells were washed once with PBS and then treated as described above.
Preparation and examination of giant unilamellar vesicles (GUVs): 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC), 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine (DOPE) and 1,2‐dioleoyl‐sn‐glycero‐3‐[phospho‐rac‐(1‐glycerol)] (DOPG) were purchased from Avanti Polar Lipids (Alabaster, USA) and Atto550‐labeled DOPE was from Atto‐Tec (Siegen, Germany). The cholesterol was purchased from Sigma Aldrich. GUVs were prepared as described previously.9, 21 Briefly, super low melting agarose (1 %, w/v) was coated on a clean glass slide and dried on a hot plate (∼50 °C) for 30 min. Afterward, two droplets of the respective lipid solutions (10 μL each) were spread on the agarose film and dried in vacuo for at least 1 h to remove residual chloroform. To visualize the membranes, the lipid solution was prior doped with 0.2 mol % Atto550‐DOPE. Then, a seal ring was placed onto the lipid coated areas on the slide to obtain two sealed chambers. For the preparation of GUVs encapsulating Oyster 405 (Luminaris GmbH, Münster, Germany), a buffer containing 10 mm HEPES; pH 7.4, 50 mm KCl, 50 mm NaCl, 1 mg mL−1 dextran (from Leuconostoc spp., 6 kDa) and 5 μm Oyster 405 (300 μL each) was added to the hybrid film. The glass slide was left in the dark for 2 h to allow hydration and swelling of the lipids. To harvest the GUV suspension, the glass slide was gently tilted in all directions to detach the liposomes from the surface. The giant liposomes were then stored in LoBind tubes (1.5 mL, Eppendorf, Hamburg, Germany) at RT and used within three days. Microscopic studies with GUVs were performed as recently described by us.9 Briefly, to remove untrapped Oyster 405, liposomes were centrifuged two times at 14 000 g for 10 min at RT. A 40 μL aliquot of the GUV solution was diluted in 50 μL of the respective buffer without Oyster 405 and was then transferred into a tissue culture vessel (FlexiPERM slide, eight wells, Sarstedt, Germany). CF‐labeled peptides diluted in buffer containing 10 mm HEPES; pH 7.4, 50 mm KCl, 50 mm NaCl, 1 mg mL−1 dextran (from Leuconostoc spp., 6 kDa) were added to the outer solution of GUVs at a final concentration of 20 μm. The GUV‐peptide interaction was analyzed using a confocal laser scanning system (Nikon d‐Eclipse C1) consisting of an inverted microscope (Nikon Eclipse Ti) equipped with a 20× objective (NA 0.45, Plan Fluor; Nikon). Microscope pictures were recorded in 16‐bit grayscale, pseudocolored in red (channel 1), green (channel 2), and blue (channel 3) followed by processing with ImageJ.
Peptide‐induced CF‐leakage: CF‐containing large unilamellar vesicles (LUVs) were prepared by hydrating a dried lipid film of desired compositions with a buffer containing 100 mm CF. The fluorescence intensity in the presence of 100 mm CF is low due to self‐quenching but increases upon dilution. Free CF outside the LUVs was separated by size exclusion chromatography using a PD10 column (GE Healthcare). Then, peptides were added to LUVs and the release of CF from vesicles was monitored by an increase in the fluorescence intensity using a fluorescence Tecan infinite M200 plate reader (λ ex=485 nm, λ em=538 nm). At the end of each experiment, Triton X‐100 (0.4 % (w/v) final concentration) was applied to measure the maximum of dequenching that will be used to normalize data. The percentage of CF release was determined by [% CF=F (t)−F 0/F f−F 0×100] where F (t) is the fluorescence intensity at time t, F 0 is the fluorescence intensity before peptide addition, and F f is the fluorescence intensity after the final addition of Triton X‐100. Each experiment was carried out with n=3 in duplicate.
Radiolabeling of the NODAGA‐coupled peptides and uptake experiments: The radioisotope [68Ga]Ga3+ was eluted in a 0.05 m ultrapure HCl from a 68Ge/68Ga generator (Isotope Technologies Garching GmbH). For the labeling of [68Ga]Ga‐NODAGA‐sC18, a stock solution was prepared with 1 mg of peptide dissolved in 1235 μL of ultrapure water; 30 μL of a stock solution (10 nmol) was mixed with 15 μL of 2 m sodium acetate buffer; 200 μL of the eluted 68Ga (50–70 MBq) was added to the reaction vial resulting in a solution at pH 4.5. For the labeling of [68Ga]Ga‐NODAGA‐(sC18)2, a stock solution was prepared with 2 mg of peptide dissolved in 1339 μL of ultrapure water; 30 μL of a stock solution (10 nmol) was mixed with 15 μL of 2 m sodium acetate buffer; 200 μL of the eluted [68Ga]Ga3+ (50–70 MBq) was added to the reaction vial resulting in a solution at pH 4.5. The labeling mixture was incubated for 30 min at room temperature, respectively. The radiochemical yield was determined by HPLC, which was performed with Azura ASM 2.1L equipped with two pumps P4.1S with pressure transducer and 10 mL titanium pump head, a degasser DG 2.1S 2‐channels, a Smartmix 350 mixer and a manual 6‐port/3‐channel injection valve with a sample loop of 20 μL. The compounds were determined by a Nucleodur C18 Gravity, 250 mm×4 mm, 5 μm column and a linear A–B gradient (80 % A to 70 % A in 15 min) at a flow rate of 1 mL min−1. Solvent A consisted of water+0.1 % TFA, and solvent B was acetonitrile+0.1 % TFA. The compounds were analyzed by a UV detector UVD2.1L (λ=254 nm) and the radioactive ones were determined by a radioactivity counter STEFFI Raytest. The peak analyses were done by OpenLAB CDS EZChrom edition version A.04.05. For the uptake studies, HEK‐293 and MCF‐7 cells were seeded in 24‐well plates in appropriate serum containing medium. The next day, when they were grown up to ∼80 % confluency, the peptides were added to the cells in serum‐free medium and incubated for 30 min. Afterward, the cells were washed twice with PBS and detached with trypsin. The cells were centrifuged and the radioactivity in the cell pellet was determined in a gamma spectrometer (LB2045 Berthold technologies).
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
A.G. acknowledges financial support by the Jürgen‐Manchot Stiftung (Germany). Financing by the European Union within the MSCA‐ITN‐2014‐ETN MAGICBULLET (grant agreement number 642004) is kindly acknowledged by I.N., I.R., and J.T. This work was supported by the Hungarian National Science Fund (OTKA K116295, J.T.).
A. Gronewold, M. Horn, I. Ranđelović, J. Tóvári, S. Muñoz Vázquez, K. Schomäcker, I. Neundorf, ChemMedChem 2017, 12, 42.
References
- 1. Dennison S. R., Whittaker M., Harris F., Phoenix D. A., Curr. Protein Pept. Sci. 2006, 7, 487–499. [DOI] [PubMed] [Google Scholar]
- 2. Zachowski A., Biochem. J. 1993, 294, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Ran S., Downes A., Thorpe P. E., Cancer Res. 2002, 62, 6132–6140; [PubMed] [Google Scholar]
- 3b. Utsugi T., Schroit A. J., Connor J., Bucana C. D., Fidler I. J., Cancer Res. 1991, 51, 3062–3066. [PubMed] [Google Scholar]
- 4. Reinhardt A., Neundorf I., Int. J. Mol. Sci. 2016, 17, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Jobin M. L., Alves I. D., Biochimie 2014, 107, 154–159; [DOI] [PubMed] [Google Scholar]
- 5b. Raucher D., Ryu J. S., Trends Mol. Med. 2015, 21, 560–570; [DOI] [PubMed] [Google Scholar]
- 5c. Zahid M., Robbins P. D., Molecules 2015, 20, 13055–13070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neundorf I., Rennert R., Hoyer J., Schramm F., Löbner K., Kitanovic I., Wölfl S., Pharmaceuticals 2009, 2, 49–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Geldmacher Y., Splith K., Kitanovic I., Alborzinia H., Can S., Rubbiani R., Nazif M. A., Wefelmeier P., Prokop A., Ott I., Wolfl S., Neundorf I., Sheldrick W. S., J. Biol. Inorg. Chem. 2012, 17, 631–646; [DOI] [PubMed] [Google Scholar]
- 7b. Hu W., Splith K., Neundorf I., Merz K., Schatzschneider U., J. Biol. Inorg. Chem. 2012, 17, 175–185. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Richter S., Bouvet V., Wuest M., Bergmann R., Steinbach J., Pietzsch J., Neundorf I., Wuest F., Nucl. Med. Biol. 2012, 39, 1202–1212; [DOI] [PubMed] [Google Scholar]
- 8b. Splith K., Bergmann R., Pietzsch J., Neundorf I., ChemMedChem 2012, 7, 57–61. [DOI] [PubMed] [Google Scholar]
- 9. Reinhardt A., Horn M., Schmauck J. P., Brohl A., Giernoth R., Oelkrug C., Schubert A., Neundorf I., Bioconjugate Chem. 2014, 25, 2166–2174. [DOI] [PubMed] [Google Scholar]
- 10. Hoyer J., Schatzschneider U., Schulz-Siegmund M., Neundorf I., Beilstein J. Org. Chem. 2012, 8, 1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rennert R., Neundorf I., Jahnke H. G., Suchowerskyj P., Dournaud P., Robitzki A., Beck-Sickinger A. G., ChemMedChem 2008, 3, 241–253. [DOI] [PubMed] [Google Scholar]
- 12. Szczepanski C., Tenstad O., Baumann A., Martinez A., Myklebust R., Bjerkvig R., Prestegarden L., Genes Cancer 2014, 5, 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Guterstam P., Madani F., Hirose H., Takeuchi T., Futaki S., El Andaloussi S., Graslund A., Langel U., Biochim. Biophys. Acta Biomembr. 2009, 1788, 2509–2517; [DOI] [PubMed] [Google Scholar]
- 13b. Madani F., Lindberg S., Langel U., Futaki S., Graslund A., J. Biophys. 2011, 2011, 414729; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Verdurmen W. P., Bovee-Geurts P. H., Wadhwani P., Ulrich A. S., Hallbrink M., van Kuppevelt T. H., Brock R., Chem. Biol. 2011, 18, 1000–1010. [DOI] [PubMed] [Google Scholar]
- 14. Huotari J., Helenius A., EMBO J. 2011, 30, 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herce H. D., Garcia A. E., Cardoso M. C., J. Am. Chem. Soc. 2014, 136, 17459–17467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lönn P., Dowdy S. F., Expert Opin. Drug Delivery 2015, 12, 1627–1636. [DOI] [PubMed] [Google Scholar]
- 17. Dobrzyńska I., Szachowicz-Petelska B., Sulkowski S., Figaszewski Z., Mol. Cell. Biochem. 2005, 276, 113–119. [DOI] [PubMed] [Google Scholar]
- 18. Brown D. A., London E., J. Biol. Chem. 2000, 275, 17221–17224. [DOI] [PubMed] [Google Scholar]
- 19. Wadia J. S., Stan R. V., Dowdy S. F., Nat. Med. 2004, 10, 310–315. [DOI] [PubMed] [Google Scholar]
- 20. Perry R. P., Kelley D. E., J. Cell. Physiol. 1970, 76, 127–129. [DOI] [PubMed] [Google Scholar]
- 21. Horger K. S., Estes D. J., Capone R., Mayer M., J. Am. Chem. Soc. 2009, 131, 1810–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary