

Abstract
Herein, a systematic study of a series of molecular iron model complexes has been carried out using Fe L2,3-edge X-ray absorption (XAS) and X-ray magnetic circular dichroism (XMCD) spectroscopies. This series spans iron complexes of increasing complexity, starting from ferric and ferrous tetrachlorides ([FeCl4]−/2–), to ferric and ferrous tetrathiolates ([Fe(SR)4]−/2–), to diferric and mixed-valent iron–sulfur complexes [Fe2S2R4]2–/3–. This test set of compounds is used to evaluate the sensitivity of both Fe L2,3-edge XAS and XMCD spectroscopy to oxidation state and ligation changes. It is demonstrated that the energy shift and intensity of the L2,3-edge XAS spectra depends on both the oxidation state and covalency of the system; however, the quantitative information that can be extracted from these data is limited. On the other hand, analysis of the Fe XMCD shows distinct changes in the intensity at both L3 and L2 edges, depending on the oxidation state of the system. It is also demonstrated that the XMCD intensity is modulated by the covalency of the system. For mononuclear systems, the experimental data are correlated with atomic multiplet calculations in order to provide insights into the experimental observations. Finally, XMCD is applied to the tetranuclear heterometal–iron–sulfur clusters [MFe3S4]3+/2+ (M = Mo, V), which serve as structural analogues of the FeMoco and FeVco active sites of nitrogenase. It is demonstrated that the XMCD data can be utilized to obtain information on the oxidation state distribution in complex clusters that is not readily accessible for the Fe L2,3-edge XAS data alone. The advantages of XMCD relative to standard K-edge and L2,3-edge XAS are highlighted. This study provides an important foundation for future XMCD studies on complex (bio)inorganic systems.
Short abstract
A systematic Fe L2,3-edge X-ray absorption (XAS) and X-ray magnetic circular dichroism (XMCD) study of iron tetrachlorides ([FeCl4]−/2−), iron tetrathiolates ([Fe(SR)4]−/2−), diferric and mixed-valent iron−sulfur dimers [Fe2S2R4]2−/3− and heterometal−iron−sulfur tetramers [MFe3S4]3+/2+ (M = Mo, V) is reported. The changes in XAS and XMCD energies and intensities across this set of complexes are presented together with atomic multiplet calculations. The advantages of XMCD as an electronic structure probe of complex clusters are highlighted.
1. Introduction
Nature utilizes a wide variety of metalloprotein active sites to perform an array of biological processes, ranging from electron transfer to catalysis. Perhaps the most complex biological cofactors are those found in the nitrogenase family of enzymes. Mo-, V-, and Fe-dependent nitrogenase enzymes are known, all of which contain complex iron−sulfur active sites for the reduction of N2 to ammonia.1−3 The best studied are the MoFe7S9C (FeMoco) and VFe7S9C (FeVco) cofactors of these enzymes, which both contain an unusual central carbon atom4,5 and possess an S = 3/2 ground state in their resting forms.6,7 However, despite countless studies to detail the atomic composition and active site structure of these enzymes, the exact electronic structure and in particular the details of the magnetic coupling remain open questions.4,5,8−18 With the aim to obtain a more detailed understanding of the complex electronic structure of these enigmatic cofactors, our research group has been motivated to develop and apply novel spectroscopic approaches to this enzyme family. This has included both valence to core X-ray emission spectroscopy (VtC XES) and high energy resolution fluorescence detected X-ray absorption spectroscopy (HERFD XAS). VtC XES and HERFD XAS have provided increased ligand selectivity and greater metal oxidation state sensitivity, respectively.4,5,9,13,19 However, these methods do not provide detailed information on the magnetic coupling within these clusters. It is here that X-ray magnetic circular dichroism (XMCD) may provide an attractive experimental means to obtain more detailed electronic structural insights into complex metal clusters.
XMCD measurements for first row transition metals are typically performed at the metal L-edge, which corresponds to a dipole-allowed 2p → 3d transition. The L-edge is split into the L3 and L2 edges, as a result of spin–orbit coupling due to the 2p core hole. Due to the longer core hole lifetimes at these energies, the intrinsic spectral resolution is higher than that observed at the corresponding metal K-edge XAS. Additionally in XMCD measurements, polarized X-rays are utilized. The right (CR) and left circularly (CL) polarized photons transfer the opposite angular momenta to the excited photoelectron. Since the L3 (L + S) and L2 (L – S) edges have opposite spin–orbit coupling, the spin polarization and the absorption properties for CR and CL are also opposite. This results in preferential absorption of spin-down and spin-up electrons at the L3 and L2 edges, respectively. The resultant XMCD spectrum is a subtraction of the L-edge data at two different polarizations and hence affords information about the majority/minority of the spins based inter alia on the sign of the final spectrum.20,21
Although XMCD has been widely applied in solid state physics to numerous magnetic systems22−30 or multimetallic thin layers,31−37 there are relatively limited applications of this technique to molecular systems reported in the literature.38−56 In the 1990s Cramer and co-workers pioneered the application of XMCD to both molecular model complexes and protein active sites (including FeMoco). Later van Elp et al. built upon the work of Cramer and co-workers by incorporating zero-field splitting parameters in the XMCD analysis.57,58 However, despite the progress made in these early studies of molecular and bioinorganic systems there are, to our knowledge, no reported applications of XMCD to bioinorganic systems since 2007. In our view, this may be attributed to an incomplete understanding of the information content of these spectra as it relates to the electronic structure of molecular systems.
To this end, and with the broader goal of applying XMCD to iron–sulfur proteins (including nitrogenase), we have undertaken a systematic study of 10 small molecular iron complexes of increasing complexity. Fe L2,3-edge XAS and XMCD data have been obtained for ferrous and ferric tetrachlorides ([FeCl4]−/2–), ferrous and ferric tetrathiolates ([Fe(SR)4]−/2–), diferric and mixed-valent iron–sulfur dimers [Fe2S2R4]2–/3–, and heterometal–iron–sulfur tetramers [MFe3S4]3+/2+ (where M = Mo, V), as summarized in Table 1. The heterometal–iron–sulfur cubanes have been shown previously to be good electronic structural analogues for half of the FeMoco and FeVco active sites of nitrogenase.8,9,59−63 Across this series, the changes in L2,3-edge energy and intensity, as well as the XMCD energies and intensities, are assessed as a function of metal oxidation state, metal–ligand covalency, and spin state. Through correlation of the experimental data to atomic multiplet calculations further insight into the observed experimental trends are made. The advantages of XMCD relative to standard XAS measurements are highlighted. The results of this study form the foundation for the application of XMCD to more complex systems such as the FeMo and FeV cofactors of nitrogenase.
Table 1. Schematic View and Spin Ground State Information for the Iron Complexes Described in This Work.
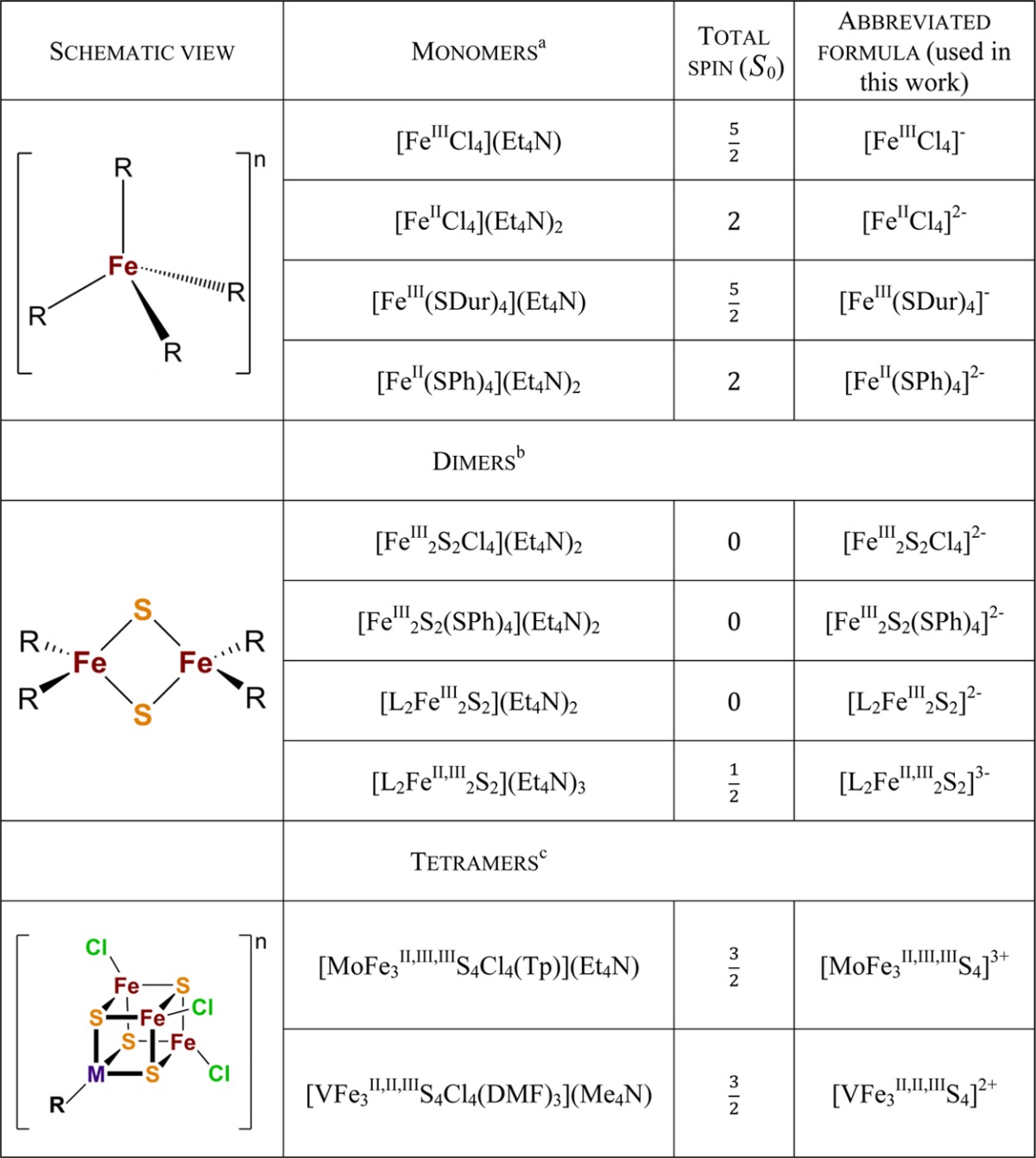
Abbreviations: Et4N, tetraethylammonium; SDur (durenethiolate), 2,3,5,6-tetramethylbenzenethiolate; SPh, benzenethiolate.
Abbreviation: L , bis(benzimidazolato).
Abbreviations: Tp, tris(pyrazolyl)borate; DMF, N,N-dimethylformamide; Me4N, tetramethylammonium.
2. Materials and Methods
2.1. Sample Preparation
All samples (Table 1) were synthesized according to published procedures59,63−71 and were utilized as powders, which were finely ground using a mortar and pestle. [FeIIICl4](Et4N) was purchased from Sigma-Aldrich and used without further purification (www.sigmaaldrich.com). [FeII(SPh)4](Et4N)2 was prepared by salt metathesis using (Et4N)2[FeCl4] and NaSPh via a procedure analogous to that for the synthesis of (Et4N)2[Fe(SEt)4] as reported in ref (70). The light green crystalline product was isolated directly from the filtered reaction solution by volume reduction, recrystallized by MeCN/Et2O diffusion, and characterized by comparison with reported spectroscopic properties (see ref (66)). All sample preparation was done in an inert N2 atmosphere glovebox attached directly to the beamline ultra-high vacuum (UHV) chamber system. The ground samples were spread on carbon tape, attached to copper sample holders, and introduced directly from the glovebox into the system of beamline vacuum chambers.
2.2. Data Collection and Processing
Iron L2,3-edge X-ray absorption spectroscopy (XAS) and X-ray magnetic circular dichroism (XMCD) measurements were carried out at the DEIMOS beamline at the SOLEIL synchrotron facility (France). SOLEIL is a 2.75 GeV ring operating in a top-up mode with a 450 mA electron current. The DEIMOS beamline uses an APPLE II undulator as a polarized light source and a plane grating monochromator (PGM) equipped with a variable groove depth grating (VGD) optimized for 350–2500 eV energy range. The end station consists of a superconducting magnet equipped with a variable-temperature insert (VTI), allowing high magnetic field and low temperature measurements.72,73
All measurements were performed at a temperature of 4 K within an ultrahigh vacuum environment (∼10–10 mbar). The L2,3-edge data for the XAS spectra were obtained at 0 T magnetic field, while the L2,3-edge data used to obtain the XMCD spectra were obtained at ±6 T, to maximize the XMCD intensity. The required magnetic field was estimated on the basis of obtained magnetic hysteresis loops. For most of the compounds full magnetization was achieved at ∼4.5 T. The applied magnetic field and temperature parameters are consistent with those previously used to study XMCD on paramagnetic samples.47,50,52,74 The signal from the sample was detected in total electron yield (TEY) mode using a drain current detector, while the incoming X-rays were monitored by a photocurrent of a gold grid with 25% transmission reference monitor. A beam spot size of 800 × 800 μm2 on the sample was used. The spectra were monitored for any changes due to radiation-induced damages during the measurements. For all of the compounds no changes in the signal due to radiation damage were observed for at least seven spectra obtained at the same sample spot. This allowed us to measure sets with three polarization changes (3 × CR/CL) at every fresh spot of the sample. This beamline was previously shown to be well optimized for radiation-sensitive samples.75
The Fe L2,3-edge XAS spectra were obtained by averaging the spectra obtained with circularly polarized right (CR) and circularly polarized left (CL) light at 0 T magnetic field. In the case of the monomers and dimers, 12 scans were averaged (6CR and 6CL). However, for [L2Fe2S2]n (n = 2–, 3–; see Table 1) only 6 scans (3CR and 3CL) were averaged for each compound. For the tetranuclear systems, 18 scans (9CR and 9CL) were averaged for each heterometal–iron cubane. The background subtraction and normalization of the spectra were performed using BlueprintXAS ver. 2.7.7 software.76 Shallow second-order polynomials were subtracted in the 690–702 eV energy range before the L3-edge region and in the 725–740 eV energy range in the post L2-edge region. The spectra were normalized according to an edge-jump procedure, setting the L3 pre-edge region to 0 and the postedge L2 region to 1, as shown in Figure S7 in the Supporting Information. The reliability of this procedure was verified for all the data by processing all single spectra and averaged spectra obtained for a particular compound. The energy reproducibility is within 0.02 eV, and the errors in intensity are estimated at less than 1%. This is also illustrated in Figures S8 and S9 in the Supporting Information. The energy of the incoming X-ray beam was calibrated on the basis of the spectrum of K3[Fe(CN)6], setting the energies of the first sharp feature of the L3 edge to 705.8 eV and the first feature of the L2 edge to 722.5 eV.77 The energy positions of the features in the XAS spectra were determined on the basis of the second-derivative spectra.
XMCD spectra were obtained as a difference between the spectra obtained with right circularly polarized X-rays (CR) and left circularly polarized X-rays (CL), both with nearly 100% polarization rate, at ±6 T. The signal was additionally corrected by subtracting the 0 T magnetic field spectrum (obtained by subtracting circular right and circular left L-edge data obtained with no applied magnetic field). Overall the process for extracting the XMCD spectra is described by eq 1.
| 1 |
The circularly left-polarized photons carry angular momentum, which excites a primary fraction of spin-down electrons, while the circularly right-polarized photons carry the opposite momentum, exciting a greater fraction of spin-up electrons. The energy positions for all XMCD features are based on the peak maxima, and a complete list of all peak positions is provided in Table S1 in the Supporting Information. The presented L2,3-edge XAS spectra used to obtain XMCD spectra were detected by changing the polarization of photons for both ±6 T magnetic fields. This approach gave 6CR and 6CL spectra for each ±6 T magnetic field for the mononuclear and dinuclear systems; however, only 3CR and 3CL spectra were obtained for each magnetic field for the [L2Fe2S2]n (n = 2–, 3−) complexes and 12CR and 12CL spectra for each magnetic field in the case of the tetranuclear clusters.
2.3. XAS and XMCD Multiplet Calculations
Crystal field multiplet calculations were carried out using the theoretical developments of Thole, Cowan, and Butler,78−83 as implemented within the CTM4XAS code (version 5.5) developed by de Groot and co-workers.84 Fe L2,3-edge XAS and XMCD spectra were calculated in Td symmetry with the exchange field B (corresponding to physical application of the magnetic field) set to 100 meV. In order to reproduce the full shape of the experimental spectra, a Lorentzian broadening of 0.2 eV and a Gaussian broadening of 0.3 eV was used. An energy shift of 2.39 eV was applied in order to align the calculated spectra to experiment. More details about the parameters of the simulations are provided in sections 3.2 and 3.4.
3. Results
3.1. Experimental L2,3-Edge XAS and XMCD for the Tetrachloride Compounds
Figure 1A shows the normalized L2,3-edge XAS spectra for the ferrous and ferric tetrachloride complexes. The maximum of the L3-edge is 1.2 eV higher in energy for the more oxidized complex ([FeIIICl4]−), which is consistent with a 1 unit change in oxidation state.85 In addition, the intensity of the L3-edge decreases for the [FeIIICl4]− complex relative to the [FeIICl4]2– complex. In a simple picture, the L-edge intensity should increase as the number of d holes increases, implying that a ferric complex should have greater L-edge intensity than an analogous ferrous complex. However, the L-edge intensity is also modulated by increasing metal–ligand covalency, which delocalizes Fe 3d character onto the ligands and decreases the intensity of the 2p to 3d transition. The present observation thus appears to reflect that the increase in covalency upon oxidation dominates the observed spectral intensity. We note that the L2,3-edge XAS spectra of both [FeIIICl4]− and [FeIICl4]2– were previously reported,86 indicating an ∼2 eV energy difference between the maximum of the L3-edge spectral features for these complexes, which seems to be inconsistent with our results. However, our data agree with the published Fe K-edge XAS spectroscopic data by the group of Solomon, showing a similar energy shift for these complexes on the basis of the changes in Fe 3d manifold.87
Figure 1.
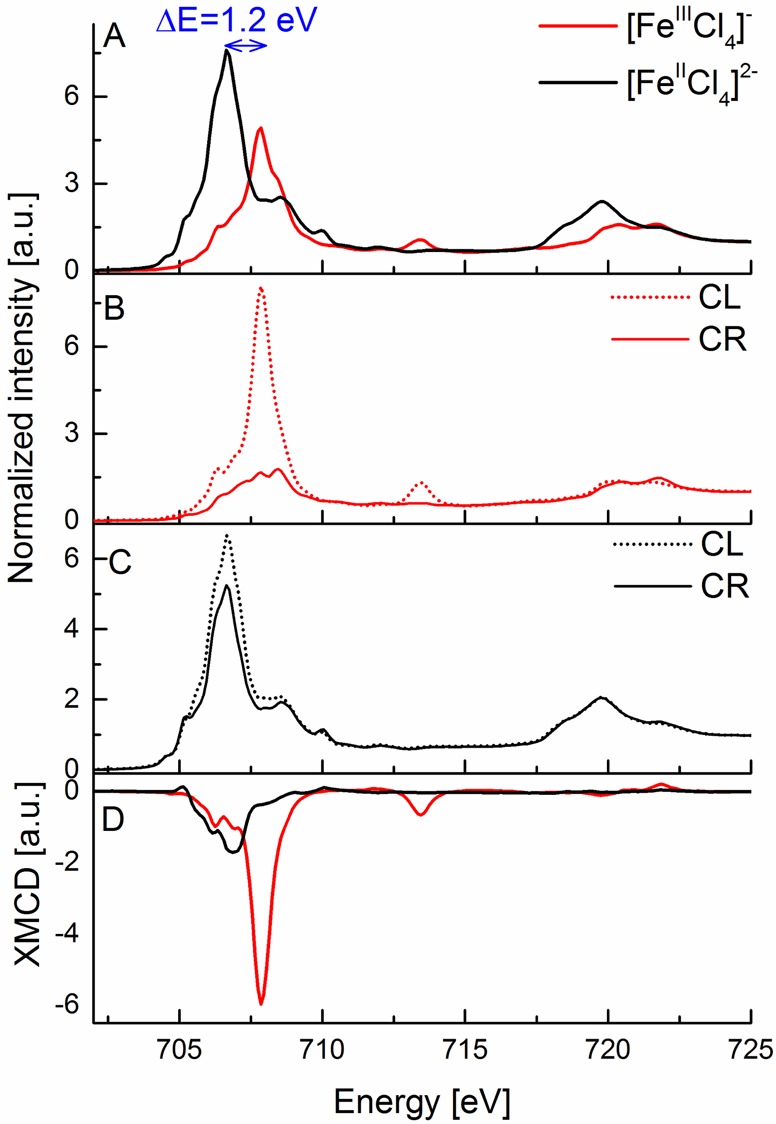
(A) Experimental Fe L2,3-edge X-ray absorption spectra of [FeIIICl4]− (red) and [FeIICl4]2– (black) complexes. (B, C) Experimental Fe L2,3-edge X-ray absorption spectra at 6 T magnetic field with circularly right (solid line) and circularly left (dashed line) polarized light for [FeIIICl4]− (red, B) and [FeIICl4]2– (black, C) complexes. (D) Experimental Fe XMCD spectra of [FeIIICl4]− (red) and [FeIICl4]2– (black) complexes. The blue arrow in (A) indicates the shift between the maxima of the L3-edge peaks for these complexes.
Figure 1B,C shows the normalized L2,3-edge XAS spectra for both X-ray polarizations for the tetrachloride complexes. Here it is of interest to note that for the [FeIIICl4]− spectra the dominant L3-edge intensity is in the CL polarization, consistent with the fact that for a high-spin d5 ground state only β transitions are allowed, if one assumes conservation of spin. In contrast, for the CR polarization, in which α excitations are preferred with a greater probability, the allowed L3-edge intensity decreases significantly. Moreover, the fact that the L3-edge maximum in the CR spectrum is at higher energy than the L3 maximum in the CL spectrum is consistent with spin-flip transitions requiring more energy. At the L2-edge where sextet and quartet contributions to the final state are likely to be more equal, the differences between the two polarizations are diminished.88 The differences in the CR and CL absorption result in an intense negative L3-edge XMCD signal (Figure 2D) for the high-spin ferric compound and a weak positive L2-edge XMCD signal, reflecting the opposite sign of the spin–orbit coupling parameter at the two edges. Further, we note that the ∼714 eV feature in [FeIIICl4]− which is typically associated with a ligand to metal charge transfer86 event occurs only in the CL absorption spectrum, suggesting that no spin-flip is involved in the charge transfer process (i.e., a ligand β electron is transferred to a β hole on the metal).
Figure 2.
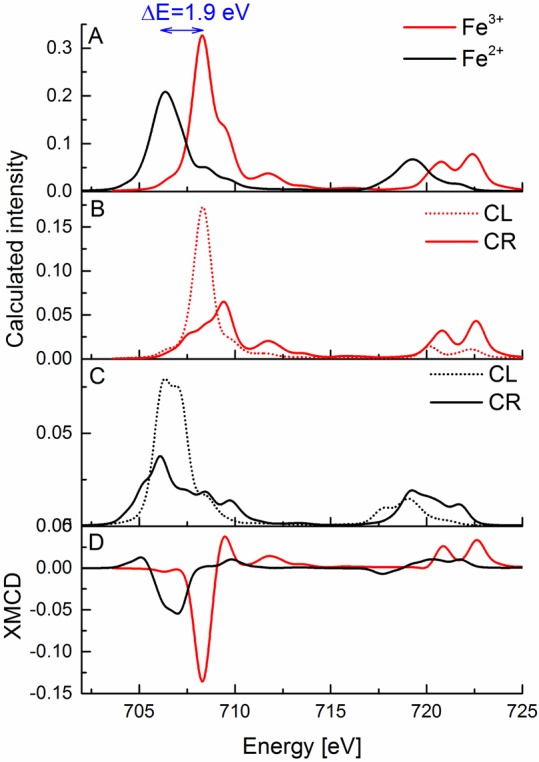
(A) Calculated Fe L2,3-edge X-ray absorption spectra of Fe3+ (red) and Fe2+ (black) systems. (B, C) Calculated Fe L2,3-edge X-ray absorption spectra with circularly right (solid line) and circularly left (dashed line) polarized light for Fe3+ (red, B) and Fe2+ (black, C) systems. (D) Calculated Fe XMCD spectra of Fe3+ (red) and Fe2+ (black) complex systems. The blue arrow in (A) indicates the shift between the maxima of the L3-edge peaks for the calculated spectra.
At this point, it is perhaps instructive to note that the XMCD spectra of low-spin ferric complexes are notably different than the high-spin species presented above. As an example the XMCD of S = 1/2 ferricyanide is provided in Figure S1 in the Supporting Information. Here one observes that the CL and CR spectra are very similar to each other. This is due to the fact that both α and β transitions into the eg set of orbitals are equally allowed. This results in a decrease in the XMCD intensity, reflecting an S = 1/2 ground state. In our view, such correlations are helpful in establishing a more intuitive picture of these complex spectra.
The differences discussed above also allow us to establish some intuition for how the spectra of S0 = 2 [FeIICl4]2– should change relative to the ferric counterpart discussed above. Namely, in a simple picture, we expect that the reduced total spin should be manifested in reduced XMCD intensity. As shown in Figure 1D, this is clearly what is observed. The differences between CR and CL polarizations are greatly reduced (Figure 1C), resulting in an overall decrease in the XMCD intensity, consistent with the reduction in spin ground state from S0 = 5/2 to S0 = 2. Further, we note that in the ferrous case there is effectively no XMCD signal observable at the L2-edge, suggesting roughly equal contribution of spin-allowed and spin-flip contributions and hence no net preference on either the CR or CL absorption channel. In general, we note that the weaker and broader L2-edge XMCD signals observed for both the ferric and ferrous species may be further attributed to the additional Coster–Kronig/Auger decay processes that are possible at the L2-edge. In the high-spin ferrous case, both the reduced XMCD intensity at the L3-edge and the very weak L2-edge XMCD feature seem to be general features which are apparent in all ferrous models in the present study, as well as in previously published data.46,53 We note that in the case of a low-spin S = 0 ferrous complex, there is no mechanism for XMCD intensity, as the α and β excitation channels will fully cancel each other.
3.2. Multiplet Simulations of Fe L2,3-Edge XAS and XMCD for the Monomeric Ferrous and Ferric Tetrachloride
In order to obtain a deeper understanding of the observed differences in the XMCD spectra of ferrous and ferric tetrachlorides, it is useful to simulate the experimental spectra using multiplet calculations. For simplicity, crystal field parameters were chosen on the basis of optical spectroscopy. In the first set of calculations we included only the ligand field splitting of the d-orbitals (10Dq) without any additional parameters.67,86,89
In the next step metal–ligand covalency was introduced through the systematic inclusion of charge transfer parameters. The explored parameters included Δ, which refers to the energy separation between the ground state electron configuration and the ligand to metal charge transfer (LMCT) excited state before configuration interaction, and the interaction matrix element T (hopping integral) for both the t2 and e set of d orbitals T(e) and T(t2). We note that changing the parameter Δ does not change either the L-edge XAS or the XMCD simulated data in a significant way (Figure S2 in the Supporting Information). The calculated spectra shown here correspond to a Δ = 2 eV, which is the same value previously utilized to simulate the Fe L2,3-edge XAS spectra of iron tetrachloride complexes.86 Modifying the hopping parameters T yielded larger changes in the calculated spectra. As shown in Figure S3 in the Supporting Information, when the hopping parameters are set to 0, both the Fe L-edge and XMCD intensities are much greater. By allowing for reasonable interaction matrix element values, however, the intensities of the L-edge and XMCD signals decrease by a factor of ∼5. However, interestingly, smaller systematic variations in the T values result in only minor modulations in spectral intensities. Further, we note that excluding the charge transfer parameters shifts the energy of the features in the spectra by about 0.5 eV toward lower energy. This clearly shows that the presence of charge transfer can dramatically modulate the spectra. However, the small observed differences with different T and Δ values suggest that quantitative differences due to changes in metal–ligand covalency may be more difficult to capture. This point is explored in greater detail in section 3.4.
Figure 2 shows the calculated spectra using an optimized parameter set (Table 2) which includes charge transfer. As shown in Figure 2A, the increase in the L3-edge energy upon oxidation is well modeled within a simple multiplet approach. However, the intensity of the L-edge XAS spectra is reversed relative to experiment. As discussed above, competing contributions due to changes in oxidation state and covalency complicate a simple evaluation of the intensities. Better agreement with experiment could be obtained by further scaling of the Slater–Condon–Shortley integrals in the ferric case (see Figure S4 in the Supporting Information). However, for simplicity, we have chosen to simply present the calculated spectra with the same Slater–Condon–Shortly integrals, as the XMCD spectra are generally well reproduced regardless. The exception is that in the ferric case, where the calculation predicts a weak positive feature at ∼710 eV, which is absent in experiment. However, by uniformly decreasing the Slater–Condon–Shortley integrals, as shown in Figure S4, the positive feature vanishes. Unfortunately, this occurs only when the Slater–Condon–Shortley integrals are reduced to an unphysically small value of 0.1.
Table 2. Crystal Field Parameters Used To Simulate the L-Edge XAS and XMCD Spectra of Fe2+ and Fe3+ Systems Presented in Figure 2.
| parameter | Fe2+ system | Fe3+ system |
|---|---|---|
| 10Dq (eV) | –0.5a | –0.4 |
| Δ (eV) | 2 | 2 |
| hopping integral (eV) | ||
| T(e) set | 2.1 | 2.1 |
| T(t2) set | 2.5 | 2.5 |
| Slater–Condon–Shortley reduction parameter (%) (Fdd = Fpd = Gpd) | 1b | 1b |
As reported previously.86
A value of 1 corresponds to 80% reduction of the electron–electron repulsion of the Hartree–Fock calculated value of a free ion.
Figure 2B–D shows the CR/CL L2,3-edge XAS and the corresponding XMCD spectra calculated within a multiplet-based approach. We note that overall, the XMCD spectra are well simulated with the CL channel dominating the L3 intensity, as also observed experimentally (Figure 1). In addition, the XMCD spectra (Figure 2D) clearly show the reduction in L3-edge intensity upon reduction to Fe(II), as well as a diminished (albeit not fully absent) L2-edge XMCD signal. The generally good agreement with experiment thus allows us to use the multiplet approach in subsequent sections (vide infra) to understand the general trends in the XMCD of the tetrathiolate systems.
3.3. Experimental L2,3-Edge XAS and XMCD for the Tetrathiolate Compounds
With an understanding of the changes that occur upon going from ferric to ferrous tetrachlorides, we now extend our test set to ferric and ferrous tetrathiolates, allowing for the contributions of metal–ligand covalency to the Fe L2,3-edge and XMCD spectra to be evaluated. Figure 3 shows the experimental Fe L2,3-edge XAS and XMCD spectra of the tetrathiolate complexes. Here, the L3-edge energy increases by ∼0.7 eV upon oxidation, which while consistent with an increase in effective charge, is ∼0.5 eV smaller than the shift observed for the tetrachlorides. This likely reflects a diminished change in Zeff at Fe upon oxidation due to the strong metal–ligand covalency in the thiolate-ligated complexes. In addition, as also observed for the tetrachlorides, the L3-edge intensity decreases upon one electron oxidation of the iron atom. Again the changes are less pronounced than what was observed for the tetrachlorides and likely reflects the substantial delocalization of iron 3d character due to the covalent thiolate ligands in both the ferrous and ferric case. The contributions of covalency also appear to be manifested in the XMCD, where the ferric tetrathiolate has XMCD intensity much lower than that observed for the ferric tetrachloride (Figure 4B). Nonetheless, a similar trend is observed upon reduction to the ferrous complex with the L3-edge XMCD intensity decreasing (albeit modestly) and the L2-edge XMCD intensity being absent in the ferrous tetrathiolate.
Figure 3.
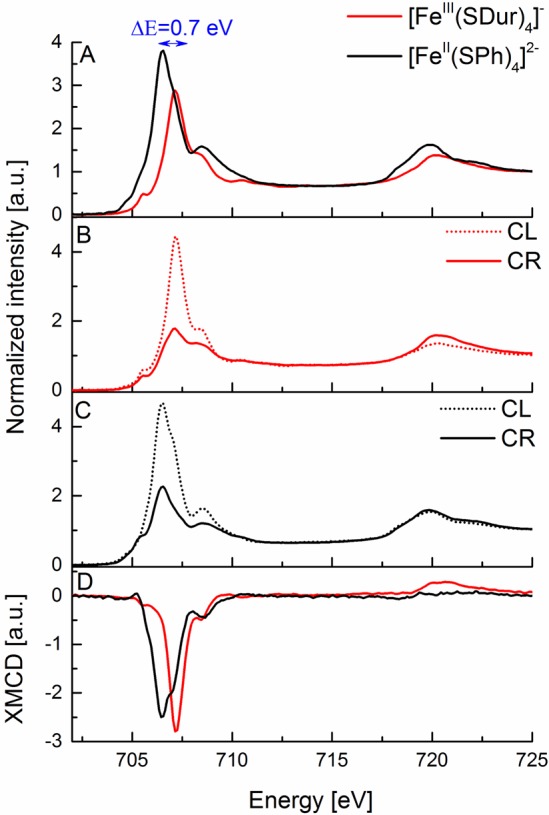
(A) Experimental Fe L2,3-edge X-ray absorption spectra of [FeIII(SDur)4]− (red) and [FeII(SPh)4]2– (black) complexes. (B, C) Experimental Fe L2,3-edge X-ray absorption spectra at 6 T magnetic field with circularly right (solid line) and circularly left (dashed line) polarized light for [FeIII(SDur)4]− (red, B) and [FeII(SPh)4]2– (black, C) complexes. (D) Experimental Fe XMCD spectra of [FeIII(SDur)4]− (red) and [FeII(SPh)4]2– (black) complexes. The blue arrow in (A) indicates the shift between the maxima of the L3-edge peaks for these complexes.
Figure 4.
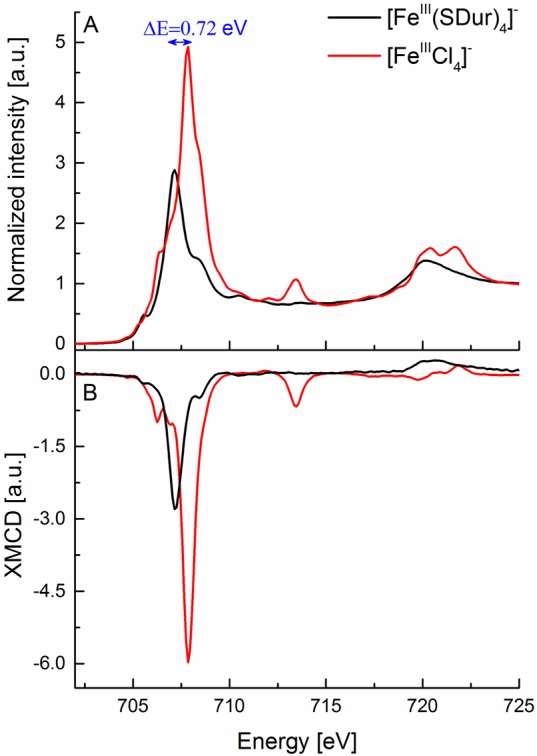
Experimental L2,3-edge XAS (A) and XMCD (B) spectra of [FeIIICl4]− (red) and [FeIII(SDur)4]− (black) complexes.
Further, it is also of interest to note that the ferric tetrachloride has an L3-edge energy that is 0.72 eV higher than that of the ferric tetrathiolate (Figure 4A), despite both complexes being Fe(III). This is consistent with previous optical and MCD spectroscopic data, which have shown that the average energy of the d–d transitions decreases by ∼0.8 eV for ferric tetrathiolate relative to ferric tetrachloride.67 This implies that due to covalency the Racah B value is reduced, and hence the average energy position of the multiplets decreases. This is an important caveat to be aware of when utilizing the Fe L-edge energy as a fingerprint for oxidation state, as covalent contributions can further modify the L-edge energy to an extent that approximates a change in redox state.
3.4. Multiplet Simulations of Fe L2,3-Edge XAS and XMCD for the Thiolate Compounds
Previously (section 3.2) we have shown that a simple multiplet-based approach could capture the general spectroscopic trends for both ferric and ferrous tetrachlorides. In order to more systematically understand the possible origins of the reduced XMCD intensity in the ferric tetrathiolate, we examined the effects of (1) uniformly reducing all Slater–Condon–Shortley integrals, (2) reducing only the d–d repulsion parameters (F2(dd) and F4(dd)), and (3) modulating the charge transfer parameters. As presented in Figure S4 in the Supporting Information, uniform scaling of the Slater–Condon–Shortley integrals actually increases the L-edge intensity and only modestly decreases the XMCD intensity. Similarly, one can scale only the Fdd integrals (Figure S5 in the Supporting Information) in an attempt to model the covalent dilution of d character onto the ligand. Here, the desired decrease in the intensity of the L-edge and XMCD spectra is observed, but only for integral values below 20% (<16% reduction of the Hartree–Fock calculated values for the free ion), which is an unphysical value for the d–d repulsion parameter. We note, however, that similar observations were made by Solomon and co-workers in their analysis of the optical data for ferric tetrathiolate. Their ligand field analysis gave a B value of only 22 cm–1, which corresponds to an unreasonably large nephelauxetic reduction.67 Nevertheless, the multiplet-based approach does allow us to understand the general trends. As demonstrated in section 3.3, the presence or absence of CT results in a 5-fold change in intensity of both the Fe L-edge and XMCD intensity. While in principle one expects both the tetrachloride and tetrathiolate to have CT contributions to the spectra, more quantitative insights are at this point unfortunately not possible.
3.5. Experimental Fe L2,3-Edge XAS and XMCD of the Dinuclear Systems
In the preceding sections, we have evaluated the contributions of oxidation state and covalency to Fe L2,3-edge XAS and XMCD spectra of mononuclear iron complexes. These data form the basis for an extension of these approaches to dinuclear (and subsequently cubane) clusters. Here, we note that the crystal field multiplet approach used to evaluate the mononuclear complexes is no longer applicable to the dinuclear clusters, as this approach does not allow one to model the exchange interaction between the two iron sites. Hence, in the subsequent sections, we focus only on the experimental data and the empirical observations that can be made, building on the lessons learned from the mononuclear studies.
Figure 5 depicts the Fe L2,3-edge XAS spectra of a series of sulfide-bridged iron dimers which include [FeIII2S2(SPh)4]2–, [FeIII2S2Cl4]2–, [L2FeIII2S2]2–, and [L2FeII,III2S2]3– (as detailed in Table 1). The first three complexes are all antiferromagentically coupled diferric S0 = 0 complexes, while the last is a mixed-valent partially delocalized FeIIFeIII complex with an S0 = 1/2 ground state. It is of interest to note that already for the diferric complexes there are subtle shifts in the L3-edge energies, with the [FeIII2S2(SPh)4]2– L3-edge maxima appearing 0.3 eV lower in energy than that of the [FeIII2S2Cl4]2– complex (Figure 5A). This observation is consistent with the Fe L2,3-edge data for the mononuclear species, which showed that [FeIIICl4]− had an L3-edge to higher energy (by ∼0.7 eV) than the [FeIII(SDur)4]−. In the present case, only half of the ligation sphere has been modulated, and hence the effect on the L-edge energy is reduced by a similar amount. This again shows that covalency can modify the Fe L-edge energy in a manner that is similar to a redox event. The present data also demonstrate the latter effect, as the one electron reduction of [L2FeIII2S2]2– to [L2FeII,III2S2]3– yields an ∼0.2 eV shift of the L3-edge to lower energy (Figure 5B, inset). We note that this shift is smaller than what one might expect from typical fingerprinting estimates, where a shift of ∼1 eV per unit change in average iron oxidation state is predicted.85 Hence in the present case, one would predict a shift of ∼0.5 eV on going from [L2FeIII2S2]2– to [L2FeII,III2S2]3–, since the average oxidation state changes from +3 to +2.5. The fact that only an ∼0.2 eV shift is observed provides further evidence that the presence of covalent ligands diminishes the change in Zeff at Fe.90 This is also consistent with previous observations utilizing Fe 1s core electron spectroscopy.90 The present data unfortunately indicate that Fe L2,3-edge XAS become a less sensitive probe of metal oxidation state as the covalency of a complex increases. It is thus of interest to further investigate the added information content of XMCD spectra for the quantitative evaluation of electronic structure changes in highly covalent complexes.
Figure 5.
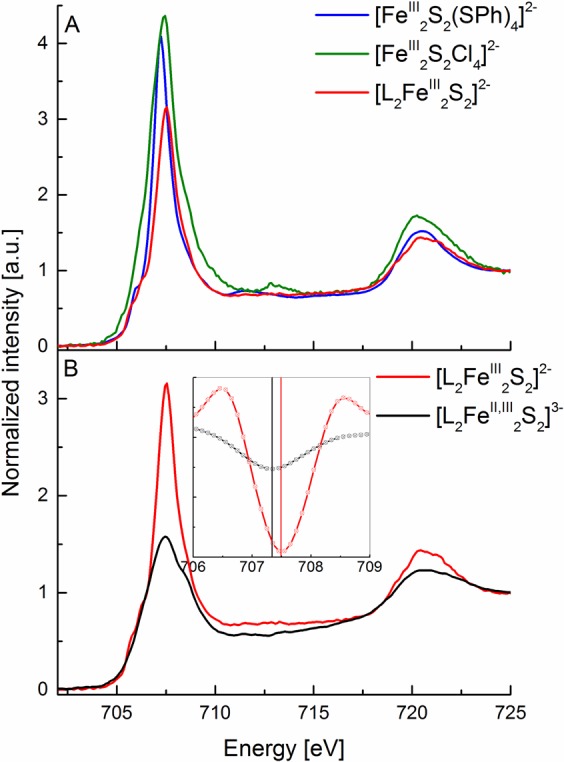
(A) Experimental Fe L2,3-edge X-ray absorption spectra of [FeIII2S2(SPh)4]2− (blue), [FeIII2S2Cl4]2− (green), [L2FeIII2S2]2− (red). (B) Experimental Fe L2,3-edge X-ray absorption spectra of [L2FeIII2S2]2– (red) and [L2FeII,III2S2]3– (black) complexes (B). The inset plot shows the ∼0.2 eV energy difference in the second-derivative spectra (smoothed using the second-order polynomial Savitzky–Golay algorithm in order to obtain better peak definition) in the L3-edge maximum between the [L2FeIII2S2]2– (red) and [L2FeII,III2S2]3– (black) complexes.
However, in the case of the antiferromagnetically coupled dinuclear ferric complexes the S0 = 0 ground state yields no net XMCD intensity. Measurement for [L2FeIII2S2]2– (Figure 6) clearly demonstrates that this is the case. Upon reduction to [L2FeII,III2S2]3–, however, a clear negative L3-edge XMCD feature is observed with minima at ∼707.5 and ∼708.3 eV. While naively, one may wish to think of this spectrum as a simple sum of the monomeric ferrous (S0 = 2) and ferric (S0 = 5/2) sites (as shown in Figure S6 in the Supporting Information), such a simple model does not properly capture the coupling between the two irons, as also previously noted by Cramer et al.91 Further, it is important to note that the overall XMCD intensity is reduced by almost 1 order of magnitude relative to the mononuclear thiolates, reflecting the lower total spin of the dinuclear clusters (S0 = 1/2).
Figure 6.
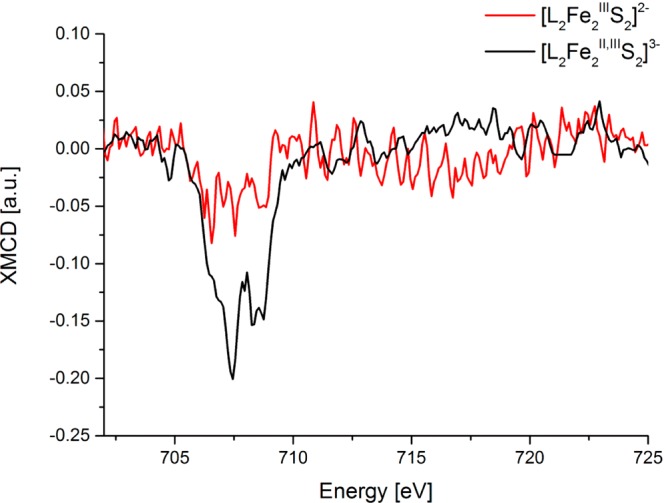
Experimental Fe XMCD spectra of [L2Fe2S2]3− (black) and [L2Fe2S2]2− (red) complexes.
3.6. Experimental Fe L2,3-Edge XAS, XMCD, and Fe Kα Pre-Edge XAS of the Heterometal–Iron–Sulfur Systems
As stated in section 1, the goal of the present systematic series of measurements was to assess the information content of Fe L2,3-edge XAS and XMCD spectra for evaluation of the electronic structure of nitrogenase. As such, we have chosen two cubane model complexes, [MoFe3II,III,IIIS4]3+ and [VFe3II,II,IIIS4]2+, which previous spectroscopic studies have shown to be good electronic structural analogues for FeMoco and FeVco, respectively.8,9,59−63 As indicated in Table 1, both complexes are S0 = 3/2 total spin systems, indicating that there should be observable XMCD intensity. On the basis of previous magnetic Mössbauer studies the oxidation states are also known, with the [VFe3II,II,IIIS4]2+ cubane possessing the greater complement of reduced iron (see Table 1).62 As shown in Figure 7A, one immediately sees that the changes in the Fe L2,3-edge XAS spectra are very subtle. This emphasizes the point also made above that for highly covalent complexes the changes in the Fe L-edge spectral energies become very small and the method is hence intrinsically less sensitive to changes in redox state. It is thus of great interest here to note that the XMCD (Figure 7B) shows much clearer changes between the two cubane model complexes. Using the metrics established for the mononuclear complexes, one notes that both the decrease in the L3-edge intensity and the very weak L2-edge intensity in the [VFe3II,II,IIIS4]2+ cubane is consistent with a greater complement of reduced iron. It is important to note that the electronic structural changes in the [MoFe3II,III,IIIS4]3+ and [VFe3II,II,IIIS4]2+ cubanes could not be assessed on the basis of the Fe L2,3-edge XAS spectra alone, and thus these data clearly highlight the added information content of XMCD.
Figure 7.
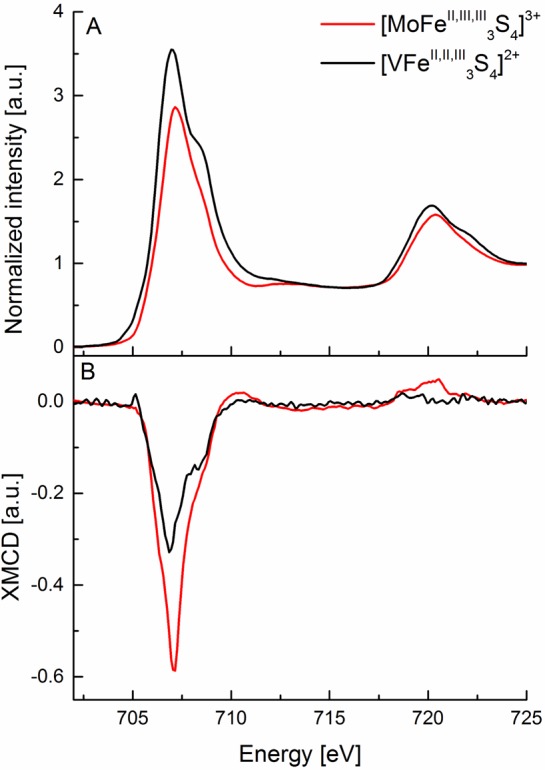
(A) Experimental Fe L2,3-edge X-ray absorption spectra. (B) Fe XMCD spectra of [VFe3II,II,IIIS4]2+ (black) and [MoFe3II,III,IIIS4]3+ complexes.
In this present context, it is also of interest to compare the Fe L2,3-edge XAS data to previously published Fe Kα-detected HERFD XAS data in the pre-edge region.19 Here one notes that the 1s to 3d pre-edge energies for the [MoFe3II,III,IIIS4]3+ and [VFe3II,II,IIIS4]2+ cubanes are at almost identical energies (Figure 8, inset). This further emphasizes the need for a magnetic measurement such as XMCD to distinguish the subtle changes that occur in the electronic structures of these highly covalent heterometal cubane clusters. Interestingly, however, the Fe Kα-detected HERFD XAS spectra do show changes to higher energies. This includes the observation of a metal to metal charge transfer (MMCT, Figure 8 inset) feature at ∼7115 eV in the [MoFe3II,III,IIIS4]3+ cubane, which is not clearly resolved in the vanadium analogue. No similar MMCT feature can be clearly assigned at the Fe L2,3-edge XAS spectra. This is likely attributed to the more complex multiplet structure engendered by the 2p core hole, which makes the assignment of discrete “3d” and “MMCT” envelopes prohibitive. While naively one may generally think that Fe L2,3-edge XAS spectra should provide higher resolution spectra due to the longer 2p core hole lifetime, the added complexity due to 2p spin–orbit coupling may in fact limit the information that can be extracted. Further, we note that while neither the Fe L-edge 2p to 3d or Fe Kα-detected HERFD XAS 1s to 3d transition energies shift between the two cubane complexes, the Fe Kα-detected HERFD XAS spectra do show a shift in the onset of the rising edge and in the intensity of the white line at ∼7125 eV (as marked in Figure 8). This shows that, while transitions to bound states may not effectively probe the changes in Zeff at Fe, the rising edge energies can still provide information as to the change in the 1s core ionization energies. Hence, the present data argue that in some cases Fe K-edge HERFD XAS data are as informative (or perhaps even more) as the Fe L2,3-edge XAS data. In closing, however, we emphasize that the largest spectral differences are observed in the XMCD data, emphasizing the potential this method has for providing more detailed electronic structural insights in complex metal clusters.
Figure 8.
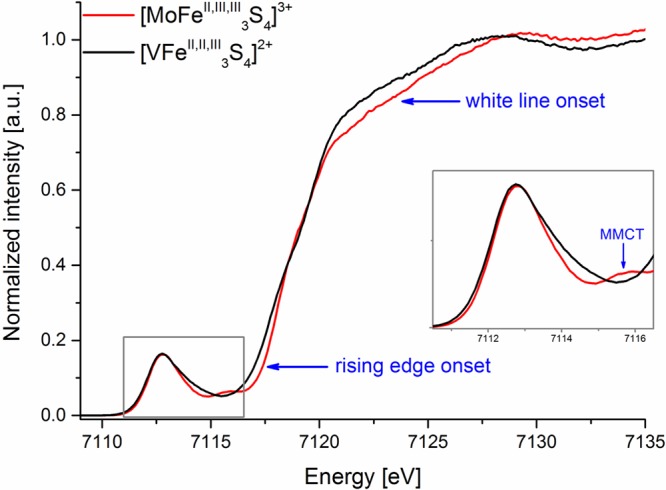
Experimental Fe Kα-detected high energy resolution fluorescence detected X-ray absorption spectra of [VFe3II,II,IIIS4]2+ (black) and [MoFe3II,III,IIIS4]3+ (red) complexes. The inset shows an enlargement at the pre-edge region marked with a gray rectangle. The blue arrows point toward the rising edge and white line onsets as well as indicate the MMCT feature. Adapted from ref (19), published by The Royal Society of Chemistry. See open access article: http://pubs.rsc.org/en/content/articlehtml/2017/dt/c7dt00128b.
4. Summary
Herein a systematic Fe L2,3-edge XAS and XMCD study has been presented on molecular iron model complexes, spanning from simple mononuclear to dinuclear clusters and finally to heterometal-incorporated cubane clusters, which serve as synthetic analogues for the FeMoco and FeVco active sites of molybdenum and vanadium nitrogenases. On the basis of our studies of monomeric model complexes, the primary XMCD spectral signatures, which result upon reduction of a high-spin ferric site to a high-spin ferrous site, have been established. Namely, the L3-edge XMCD intensity is reduced and the L2-edge XMCD intensity is effectively absent. In highly covalent complexes, the redox-dependent changes become much smaller; however, it is still possible to obtain insight into electronic structural changes by comparing complexes with similar ligation environments. The XMCD spectra for mononuclear tetrachlorides are readily modeled within a multiplet-based approach. In the case of the tetrathiolates, however, an unreasonable reduction in the d–d repulsion parameters is required to model the experimental spectra. This parallels a previous ligand field analysis of the optical data in which unreasonably small B values were needed to model the data.67 The present study thus motivates the need for further theoretical developments to more quantitatively understand XMCD spectra. We note that while restricted open shell configuration interaction with singles (ROCIS) approaches have shown great progress in the calculation of transition metal L2,3-edge XAS spectra, the extension of these approaches to XMCD has not yet been possible.88,92
Building on lessons learned from the mononuclear systems, we extended the XMCD studies to both dinuclear and heterometal-incorporated cubane clusters. Importantly, on the basis of the XMCD spectra, we were able to show evidence for an electronic structural change between [MoFe3II,III,IIIS4]3+ and [VFe3II,II,IIIS4]2+, which could not be determined on the basis of the Fe L2,3-edge XAS data alone. The present data form an important foundation for future XMCD studies of molecular complexes, particularly for the study of complex cofactors such as the FeMoco and FeVco active sites of nitrogenase. As nitrogenase contains both the FeMoco (or FeVco) cluster and an additional 8 Fe P-cluster, the study of the cofactors is often limited by the abundance of iron. An advantage of XMCD in going forward with such studies is that the all-ferrous P-cluster has an S0 = 0 ground state, which thus will not contribute to the XMCD spectral intensity. Hence, this should further enhance the electronic structural information that can be obtained from XMCD spectroscopy. Perhaps the greatest challenge in performing such experiments is the introduction of an enzyme sample into ultrahigh vacuum. Previous studies have utilized thin films57,85,93 or lyophilized proteins;94,95 however, recent developments with in-vacuum liquid jet systems96−99 should enable a greater range of applications.
These studies are ongoing in our laboratories and show promise for furthering the quantitative electronic insight that XMCD can bring to complex bioinorganic active sites.
Acknowledgments
J.K.K. and S.D. acknowledge the Max Planck Society for funding. S.D. acknowledges funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013) ERC Grant Agreement No. 615414. S.D. and F.M. are grateful to the DFG for financial support (projects DE 1877/1-1 and ME 1313/13-1, respectively, within the SPP 1927 “Iron–Sulfur for Life”). J.A.R. acknowledges a graduate study scholarship from the German Academic Exchange Service (DAAD). B.N. and S.C.L. acknowledge support from the NSERC. J.A.K. acknowledges funding from the NIH (R01-GM45881). Aleksandr Forov is thanked for assistance during the beam time. Mario Ulises Delgado-Jaime is acknowledged for help with data analysis and fruitful discussions. We acknowledge SOLEIL for provision of synchrotron radiation facilities, and we thank Dr. Kai Chen and the DEIMOS staff for assistance in using beamline DEIMOS. The research leading to these results has partially received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement n.°312284.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.7b00852.
Figures and a table as described in the text (PDF)
Author Present Address
∇ Chemical Sciences Division, Lawrence Berkeley National Laboratory, One Cyclotron Road, MS 70A-1150, Berkeley, CA 94720, USA.
The authors declare no competing financial interest.
Supplementary Material
References
- Hu Y. L.; Ribbe M. W. Decoding the Nitrogenase Mechanism: The Homologue Approach. Acc. Chem. Res. 2010, 43, 475–484. 10.1021/ar900254x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eady R. R. Structure-function relationships of alternative nitrogenases. Chem. Rev. 1996, 96, 3013–3030. 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]
- Hu Y. L.; Ribbe M. W. Nitrogenase and homologs. JBIC, J. Biol. Inorg. Chem. 2015, 20, 435–445. 10.1007/s00775-014-1225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster K. M.; Roemelt M.; Ettenhuber P.; Hu Y. L.; Ribbe M. W.; Neese F.; Bergmann U.; DeBeer S. X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334, 974–977. 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees J. A.; Bjornsson R.; Schlesier J.; Sippel D.; Einsle O.; DeBeer S. The Fe-V Cofactor of Vanadium Nitrogenase Contains an Interstitial Carbon Atom. Angew. Chem., Int. Ed. 2015, 54, 13249–13252. 10.1002/anie.201505930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. E.; Eady R. R.; Lowe D. J.; Gormal C. The Vanadium Iron Protein of Vanadium Nitrogenase from Azotobacter-Chroococcum Contains an Iron Vanadium Cofactor. Biochem. J. 1988, 250, 299–302. 10.1042/bj2500299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess B. K. The Iron Molybdenum Cofactor of Nitrogenase. Chem. Rev. 1990, 90, 1377–1406. 10.1021/cr00106a002. [DOI] [Google Scholar]
- Bjornsson R.; Delgado-Jaime M. U.; Lima F. A.; Sippel D.; Schlesier J.; Weyhermuller T.; Einsle O.; Neese F.; DeBeer S. Molybdenum L-Edge XAS Spectra of MoFe Nitrogenase. Z. Anorg. Allg. Chem. 2015, 641, 65–71. 10.1002/zaac.201400446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson R.; Lima F. A.; Spatzal T.; Weyhermuller T.; Glatzel P.; Bill E.; Einsle O.; Neese F.; DeBeer S. Identification of a spin-coupled Mo(III) in the nitrogenase iron-molybdenum cofactor. Chem. Sci. 2014, 5, 3096–3103. 10.1039/C4SC00337C. [DOI] [Google Scholar]
- Corbett M. C.; Hu Y. L.; Fay A. W.; Ribbe M. W.; Hedman B.; Hodgson K. O. Structural insights into a protein-bound iron-molybdenum cofactor precursor. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1238–1243. 10.1073/pnas.0507853103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay A. W.; Blank M. A.; Lee C. C.; Hu Y. L.; Hodgson K. O.; Hedman B.; Ribbe M. W. Characterization of Isolated Nitrogenase FeVco. J. Am. Chem. Soc. 2010, 132, 12612–12618. 10.1021/ja1019657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay A. W.; Blank M. A.; Lee C. C.; Hu Y. L.; Hodgson K. O.; Hedman B.; Ribbe M. W. Spectroscopic Characterization of the Isolated Iron-Molybdenum Cofactor (FeMoco) Precursor from the Protein NifEN. Angew. Chem., Int. Ed. 2011, 50, 7787–7790. 10.1002/anie.201102724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska J.; DeBeer S. The role of X-ray spectroscopy in understanding the geometric and electronic structure of nitrogenase. Biochim. Biophys. Acta, Mol. Cell Res. 2015, 1853, 1406–1415. 10.1016/j.bbamcr.2014.11.027. [DOI] [PubMed] [Google Scholar]
- Ribbe M. W.; Hu Y. L.; Guo M. L.; Schmid B.; Burgess B. K. The FeMoco-deficient MoFe protein produced by a nifH deletion strain of Azotobacter vinelandii shows unusual P-cluster features. J. Biol. Chem. 2002, 277, 23469–23476. 10.1074/jbc.M202061200. [DOI] [PubMed] [Google Scholar]
- Spatzal T.; Schlesier J.; Burger E. M.; Sippel D.; Zhang L. M.; Andrade S. L. A.; Rees D. C.; Einsle O. Nitrogenase FeMoco investigated by spatially resolved anomalous dispersion refinement. Nat. Commun. 2016, 7, 10902. 10.1038/ncomms10902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M.; Lukoyanov D.; Dean D. R.; Seefeldt L. C. Nitrogenase: A Draft Mechanism. Acc. Chem. Res. 2013, 46, 587–595. 10.1021/ar300267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Aksoyoglu M.; Zhang L. M.; Andrade S. L. A.; Schleicher E.; Weber S.; Rees D. C.; Einsle O. Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940–940. 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson R.; Neese F.; DeBeer S. Revisiting the Mössbauer Isomer Shifts of the FeMoco Cluster of Nitrogenase and the Cofactor Charge. Inorg. Chem. 2017, 56, 1470–1477. 10.1021/acs.inorgchem.6b02540. [DOI] [PubMed] [Google Scholar]
- Rees J. A.; Bjornsson R.; Kowalska J. K.; Lima F. A.; Schlesier J.; Sippel D.; Weyhermüller T.; Einsle O.; Kovacs J. A.; DeBeer S. Comparative electronic structure of nitrogenase FeMoco and FeVco. Dalton Trans. 2017, 46, 2445–2455. 10.1039/C7DT00128B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer S. P. In Paramagnetic Resonance of Metallobiomolecules; Telser J., Ed.; American Chemical Society: Washington, DC, 2003; Chapter 20, pp 358–402. [Google Scholar]
- de Groot F.; Kotani A.. Core Level Spectroscopy of Solids Introduction; CRC Press: Boca Raton, FL, 2008. [Google Scholar]
- Tietze T.; Gacic M.; Schutz G.; Jakob G.; Bruck S.; Goering E. XMCD studies on Co and Li doped ZnO magnetic semiconductors. New J. Phys. 2008, 10, 055009. 10.1088/1367-2630/10/5/055009. [DOI] [Google Scholar]
- Baltic R.; Pivetta M.; Donati F.; Wackerlin C.; Singha A.; Dreiser J.; Rusponi S.; Brune H. Superlattice of Single Atom Magnets on Graphene. Nano Lett. 2016, 16, 7610–7615. 10.1021/acs.nanolett.6b03543. [DOI] [PubMed] [Google Scholar]
- Martins M.; Wurth W. Magnetic properties of supported metal atoms and clusters. J. Phys.: Condens. Matter 2016, 28, 503002. 10.1088/0953-8984/28/50/503002. [DOI] [PubMed] [Google Scholar]
- Dupuis V.; Khadra G.; Linas S.; Hillion A.; Gragnaniello L.; Tamion A.; Tuaillon-Combes J.; Bardotti L.; Tournus F.; Otero E.; Ohresser P.; Rogalev A.; Wilhelm F. Magnetic moments in chemically ordered mass-selected CoPt and FePt clusters. J. Magn. Magn. Mater. 2015, 383, 73–77. 10.1016/j.jmmm.2014.12.061. [DOI] [Google Scholar]
- Guillou F.; Ollefs K.; Wilhelm F.; Rogalev A.; Yaresko A. N.; Yibole H.; van Dijk N. H.; Bruck E. Electronic and magnetic properties of phosphorus across the first-order ferromagnetic transition of (Mn,Fe)2(P,Si,B) giant magnetocaloric materials. Phys. Rev. B: Condens. Matter Mater. Phys. 2015, 92, 224427. 10.1103/PhysRevB.92.224427. [DOI] [Google Scholar]
- Brice-Profeta S.; Arrio M. A.; Tronc E.; Menguy N.; Letard I.; Moulin C. C. D.; Nogues M.; Chaneac C.; Jolivet J. P.; Sainctavit P. Magnetic order in gamma-Fe2O3 nanoparticles: a XMCD study. J. Magn. Magn. Mater. 2005, 288, 354–365. 10.1016/j.jmmm.2004.09.120. [DOI] [Google Scholar]
- Ohldag H.; Regan T. J.; Stohr J.; Scholl A.; Nolting F.; Luning J.; Stamm C.; Anders S.; White R. L. Spectroscopic identification and direct imaging of interfacial magnetic spins. Phys. Rev. Lett. 2001, 87, 247201. 10.1103/PhysRevLett.87.247201. [DOI] [PubMed] [Google Scholar]
- Gambardella P.; Rusponi S.; Veronese M.; Dhesi S. S.; Grazioli C.; Dallmeyer A.; I C.; Zeller R.; Dederichs P. H.; Kern K.; Carbone C.; Brune H. Giant magnetic anisotropy of single cobalt atoms and nanoparticles. Science 2003, 300, 1130–1133. 10.1126/science.1082857. [DOI] [PubMed] [Google Scholar]
- Vogel J.; Fontaine A.; Cros V.; Petroff F.; Kappler J. P.; Krill G.; Rogalev A.; Goulon J. Structure and magnetism of Pd in Pd/Fe multilayers studied by x-ray magnetic circular dichroism at the Pd L2,3 edges. Phys. Rev. B: Condens. Matter Mater. Phys. 1997, 55, 3663–3669. 10.1103/PhysRevB.55.3663. [DOI] [Google Scholar]
- Vivas L. G.; Rubin J.; Figueroa A. I.; Bartolome F.; Garcia L. M.; Deranlot C.; Petroff F.; Ruiz L.; Gonzalez-Calbet J. M.; Pascarelli S.; Brookes N. B.; Wilhelm F.; Chorro M.; Rogalev A.; Bartolome J. Perpendicular magnetic anisotropy in granular multilayers of CoPd alloyed nanoparticles. Phys. Rev. B: Condens. Matter Mater. Phys. 2016, 93, 174410. 10.1103/PhysRevB.93.174410. [DOI] [Google Scholar]
- Caminale M.; Ghosh A.; Auffret S.; Ebels U.; Ollefs K.; Wilhelm F.; Rogalev A.; Bailey W. E. Spin pumping damping and magnetic proximity effect in Pd and Pt spin-sink layers. Phys. Rev. B: Condens. Matter Mater. Phys. 2016, 94, 014414. 10.1103/PhysRevB.94.014414. [DOI] [Google Scholar]
- Stohr J. X-ray magnetic circular dichroism spectroscopy of transition metal thin films. J. Electron Spectrosc. Relat. Phenom. 1995, 75, 253–272. 10.1016/0368-2048(95)02537-5. [DOI] [Google Scholar]
- Vogel J.; Fontaine A.; Cros V.; Petroff F.; Kappler J. P.; Krill G.; Rogalev A.; Goulon J. Palladium magnetism in Pd/Fe multilayers studied by XMCD at the Pd L2,3 edges. J. Magn. Magn. Mater. 1997, 165, 96–99. 10.1016/S0304-8853(96)00480-5. [DOI] [Google Scholar]
- Wende H. Recent advances in x-ray absorption spectroscopy. Rep. Prog. Phys. 2004, 67, 2105–2181. 10.1088/0034-4885/67/12/R01. [DOI] [Google Scholar]
- Poulopoulos P. X-ray magnetic circular dichroism on Pt L-edges in Co-based materials. Int. J. Mod. Phys. B 2005, 19, 4517–4523. 10.1142/S0217979205032905. [DOI] [Google Scholar]
- Stohr J.; Nakajima R. Magnetic properties of transition metal multilayers studied with X-ray magnetic circular dichroism spectroscopy. IBM J. Res. Dev. 1998, 42, 73–88. 10.1147/rd.421.0073. [DOI] [Google Scholar]
- Coker V. S.; Pearce C. I.; Lang C.; van der Laan G.; Pattrick R. A. D.; Telling N. D.; Schuler D.; Arenholz E.; Lloyd J. R. Cation site occupancy of biogenic magnetite compared to polygenic ferrite spinels determined by X-ray magnetic circular dichroism. Eur. J. Mineral. 2007, 19, 707–716. 10.1127/0935-1221/2007/0019-1758. [DOI] [Google Scholar]
- Kalirai S. S.; Lam K. P.; Bazylinski D. A.; Lins U.; Hitchcock A. P. Examining the chemistry and magnetism of magnetotactic bacterium Candidatus Magnetovibrio blakemorei strain MV-1 using scanning transmission X-ray microscopy. Chem. Geol. 2012, 300-301, 14–23. 10.1016/j.chemgeo.2012.01.005. [DOI] [Google Scholar]
- Staniland S.; Williams W.; Telling N.; van der Laan G.; Harrison A.; Ward B. Controlled cobalt doping of magnetosomes in vivo. Nat. Nanotechnol. 2008, 3, 158–162. 10.1038/nnano.2008.35. [DOI] [PubMed] [Google Scholar]
- Lam K. P.; Hitchcock A. P.; Obst M.; Lawrence J. R.; Swerhone G. D. W.; Leppard G. G.; Tyliszczak T.; Karunakaran C.; Wang J.; Kaznatcheev K.; Bazylinski D. A.; Lins U. Characterizing magnetism of individual magnetosomes by X-ray magnetic circular dichroism in a scanning transmission X-ray microscope. Chem. Geol. 2010, 270, 110–116. 10.1016/j.chemgeo.2009.11.009. [DOI] [Google Scholar]
- Zhu X. H.; Hitchcock A. P.; Bazylinski D. A.; Denes P.; Joseph J.; Lins U.; Marchesini S.; Shiu H. W.; Tyliszczak T.; Shapiro D. A. Measuring spectroscopy and magnetism of extracted and intracellular magnetosomes using soft X-ray ptychography. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E8219–E8227. 10.1073/pnas.1610260114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalirai S. S.; Bazylinski D. A.; Hitchcock A. P. Anomalous Magnetic Orientations of Magnetosome Chains in a Magnetotactic Bacterium: Magnetovibrio blakemorei Strain MV-1. PLoS One 2013, 8, e53368. 10.1371/journal.pone.0053368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staniland S.; Ward B.; Harrison A.; van der Laan G.; Telling N. Rapid magnetosome formation shown by real-time x-ray magnetic circular dichroism. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 19524–19528. 10.1073/pnas.0704879104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannini M.; Pineider F.; Sainctavit P.; Danieli C.; Otero E.; Sciancalepore C.; Talarico A. M.; Arrio M. A.; Cornia A.; Gatteschi D.; Sessoli R. Magnetic memory of a single-molecule quantum magnet wired to a gold surface. Nat. Mater. 2009, 8, 194–197. 10.1038/nmat2374. [DOI] [PubMed] [Google Scholar]
- Peng G.; van Elp J.; Jang H.; Que L.; Armstrong W. H.; Cramer S. P. L-Edge X-Ray-Absorption and X-Ray Magnetic Circular-Dichroism of Oxygen-Bridged Dinuclear Iron Complexes. J. Am. Chem. Soc. 1995, 117, 2515–2519. 10.1021/ja00114a014. [DOI] [Google Scholar]
- Funk T.; Friedrich S.; Young A.; Arenholz E.; Cramer S. P. Requirements for x-ray magnetic circular dichroism on paramagnetic biological systems and model compounds. Rev. Sci. Instrum. 2002, 73, 1649–1651. 10.1063/1.1445829. [DOI] [Google Scholar]
- Jafri S. F.; Koumousi E. S.; Sainctavit P.; Juhin A.; Schuler V.; Bunau O.; Mitcov D.; Dechambenoit P.; Mathoniere C.; Clerac R.; Otero E.; Ohresser P.; Moulin C. C. D.; Arrio M. A. Large Orbital Magnetic Moment Measured in the [TpFeIII(CN)3]− Precursor of Photomagnetic Molecular Prussian Blue Analogues. Inorg. Chem. 2016, 55, 6980–6987. 10.1021/acs.inorgchem.6b00664. [DOI] [PubMed] [Google Scholar]
- van Elp J.; Peng G.; Zhou Z. H.; Adams M. W. W.; Baidya N.; Mascharak P. K.; Cramer S. P. Nickel L-Edge X-Ray-Absorption Spectroscopy of Pyrococcus-Furiosus Hydrogenase. Inorg. Chem. 1995, 34, 2501–2504. 10.1021/ic00114a006. [DOI] [Google Scholar]
- Cramer S. P.; Peng G.; Christiansen J.; Chen J.; van Elp J.; George S. J.; Young A. T. Fluorescence detected soft XAS and MCD - Applications to bioinorganic systems. J. Electron Spectrosc. Relat. Phenom. 1996, 78, 225–229. 10.1016/S0368-2048(96)80067-3. [DOI] [Google Scholar]
- Wang H. X.; Patil D. S.; Gu W. W.; Jacquamet L.; Friedrich S.; Funk T.; Cramer S. P. L-edge X-ray absorption spectroscopy of some Ni enzymes: probe of Ni electronic structure. J. Electron Spectrosc. Relat. Phenom. 2001, 114-116, 855–863. 10.1016/S0368-2048(00)00370-4. [DOI] [Google Scholar]
- Funk T.; Deb A.; George S. J.; Wang H. X.; Cramer S. P. X-ray magnetic circular dichroism - a high energy probe of magnetic properties Coordin. Coord. Chem. Rev. 2005, 249, 3–30. 10.1016/j.ccr.2004.05.017. [DOI] [Google Scholar]
- van Elp J.; Peng G.; Zhou Z. H.; Mukund S.; Adams M. W. W. Soft-x-ray magnetic circular dichroism on a paramagnetic two-iron system. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 53, 2523–2527. 10.1103/PhysRevB.53.2523. [DOI] [PubMed] [Google Scholar]
- Wende H.; Bernien M.; Luo J.; Sorg C.; Ponpandian N.; Kurde J.; Miguel J.; Piantek M.; Xu X.; Eckhold P.; Kuch W.; Baberschke K.; Panchmatia P. M.; Sanyal B.; Oppeneer P. M.; Eriksson O. Substrate-induced magnetic ordering and switching of iron porphyrin molecules. Nat. Mater. 2007, 6, 516–520. 10.1038/nmat1932. [DOI] [PubMed] [Google Scholar]
- Dreiser J.; Pedersen K. S.; Birk T.; Schau-Magnussen M.; Piamonteze C.; Rusponi S.; Weyhermuller T.; Brune H.; Nolting F.; Bendix J. X-ray Magnetic Circular Dichroism (XMCD) Study of a Methoxide-Bridged DyIIICrIII Cluster Obtained by Fluoride Abstraction from cis-[(CrF2)-FIII(phen)2]+. J. Phys. Chem. A 2012, 116, 7842–7847. 10.1021/jp303512a. [DOI] [PubMed] [Google Scholar]
- Dreiser J.; Piamonteze C.; Nolting F.; Pedersen K. S.; Bendix J.; Rusponi S.; Brune H. XMCD study of the magnetic exchange coupling in a fluoride-bridged Dy-Cr molecular cluster. J. Korean Phys. Soc. 2013, 62, 1368–1371. 10.3938/jkps.62.1368. [DOI] [Google Scholar]
- van Elp J.; George S. J.; Chen J.; Peng G.; Chen C. T.; Tjeng L. H.; Meigs G.; Lin H. J.; Zhou Z. H.; Adams M. W. W.; Searle B. G.; Cramer S. P. Soft-X-Ray Magnetic Circular-Dichroism - a Probe for Studying Paramagnetic Bioinorganic Systems. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 9664–9667. 10.1073/pnas.90.20.9664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Elp J.; George S. J.; Peng G.; Searle B. G.; Zhou Z. H.; Adams M. W. W.; Chen C. T.; Cramer S. P. Soft-X-Ray Magnetic Circular-Dichroism on a Paramagnetic Bioinorganic System. Proc. SPIE 1993, 2010, 181–189. 10.1117/12.168579. [DOI] [Google Scholar]
- Kovacs J. A.; Holm R. H. Assembly of Vanadium Iron Sulfur Cubane Clusters from Mononuclear and Linear Trinuclear Reactants. J. Am. Chem. Soc. 1986, 108, 340–341. 10.1021/ja00262a050. [DOI] [Google Scholar]
- George G. N.; Coyle C. L.; Hales B. J.; Cramer S. P. X-Ray Absorption of Azotobacter-Vinelandii Vanadium Nitrogenase. J. Am. Chem. Soc. 1988, 110, 4057–4059. 10.1021/ja00220a066. [DOI] [Google Scholar]
- Arber J. M.; Dobson B. R.; Eady R. R.; Hasnain S. S.; Garner C. D.; Matsushita T.; Nomura M.; Smith B. E. Vanadium K-Edge X-Ray-Absorption Spectroscopy of the Functioning and Thionine-Oxidized Forms of the Vfe-Protein of the Vanadium Nitrogenase from Azotobacter-Chroococcum. Biochem. J. 1989, 258, 733–737. 10.1042/bj2580733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney M. J.; Kovacs J. A.; Zhang Y. P.; Papaefthymiou G. C.; Spartalian K.; Frankel R. B.; Holm R. H. Comparative Electronic-Properties of Vanadium-Iron-Sulfur and Molybdenum-Iron-Sulfur Clusters Containing Isoelectronic Cubane-Type [VFe3S4]2+ and [MoFe3S4]3+ Cores. Inorg. Chem. 1987, 26, 719–724. 10.1021/ic00252a016. [DOI] [Google Scholar]
- Fomitchev D. V.; McLauchlan C. C.; Holm R. H. Heterometal cubane-type MFe3S4 clusters (M = Mo, V) trigonally symmetrized with hydrotris(pyrazolyl)borate(1-) and tris(pyrazolyl)methanesulfonate(1-) capping ligands. Inorg. Chem. 2002, 41, 958–966. 10.1021/ic011106d. [DOI] [PubMed] [Google Scholar]
- Hagen K. S.; Reynolds J. G.; Holm R. H. Definition of Reaction Sequences Resulting in Self-Assembly of [Fe4S4(SR)4]2- Clusters from Simple Reactants. J. Am. Chem. Soc. 1981, 103, 4054–4063. 10.1021/ja00404a013. [DOI] [Google Scholar]
- Do Y.; Simhon E. D.; Holm R. H. Improved Syntheses of [Fe2S2Cl4]2- and [Fe2OCl6]2- and Oxo Sulfido Ligand Substitution by Use of Silylsulfide Reagents. Inorg. Chem. 1983, 22, 3809–3812. 10.1021/ic00167a027. [DOI] [Google Scholar]
- Millar M.; Lee J. F.; Koch S. A.; Fikar R. Synthetic Models for the Iron Sulfur Protein Rubredoxin - Synthesis, Structure, and Properties of a Highly Symmetric Iron(Iii) Tetrathiolate Anion. Inorg. Chem. 1982, 21, 4105–4106. 10.1021/ic00141a048. [DOI] [Google Scholar]
- Gebhard M. S.; Deaton J. C.; Koch S. A.; Millar M.; Solomon E. I. Single-Crystal Spectral Studies of Fe(SR)4– [R = 2,3,5,6-(Me)4C6H]: the Electronic-Structure of the Ferric Tetrathiolate Active-Site. J. Am. Chem. Soc. 1990, 112, 2217–2231. 10.1021/ja00162a023. [DOI] [Google Scholar]
- Hagen K. S.; Watson A. D.; Holm R. H. Synthetic Routes to Fe2s2, Fe3S4, Fe4S4, and Fe6S9 Clusters from the Common Precursor [Fe(SC2H5)4]2- - Structures and Properties of [Fe3S4(SR)4]3- and [Fe6S9(SC2H5)2]4-, Examples of the Newest Types of Fe-S-Sr Clusters. J. Am. Chem. Soc. 1983, 105, 3905–3913. 10.1021/ja00350a028. [DOI] [Google Scholar]
- Albers A.; Demeshko S.; Dechert S.; Bill E.; Bothe E.; Meyer F. The Complete Characterization of a Reduced Biomimetic [2Fe-2S] Cluster. Angew. Chem., Int. Ed. 2011, 50, 9191–9194. 10.1002/anie.201100727. [DOI] [PubMed] [Google Scholar]
- Styczen E.; Pattek-Janczyk A.; Gazda M.; Jozwiak W. K.; Wyrzykowski D.; Warnke Z. Thermal analysis of bis(tetraethylammonium) tetrachloroferrate(II). Thermochim. Acta 2008, 480, 30–34. 10.1016/j.tca.2008.09.022. [DOI] [Google Scholar]
- Albers A.; Demeshko S.; Propper K.; Dechert S.; Bill E.; Meyer F. A Super-Reduced Diferrous [2Fe-2S] Cluster. J. Am. Chem. Soc. 2013, 135, 1704–1707. 10.1021/ja311563y. [DOI] [PubMed] [Google Scholar]
- Ohresser P.; Otero E.; Choueikani F.; Chen K.; Stanescu S.; Deschamps F.; Moreno T.; Polack F.; Lagarde B.; Daguerre J. P.; Marteau F.; Scheurer F.; Joly L.; Kappler J. P.; Muller B.; Bunau O.; Sainctavit P. DEIMOS: A beamline dedicated to dichroism measurements in the 350–2500 eV energy range. Rev. Sci. Instrum. 2014, 85, 013106. 10.1063/1.4861191. [DOI] [PubMed] [Google Scholar]
- Ohresser P.; Otero E.; Choueikani F.; Stanescu S.; Deschamps F.; Ibis L.; Moreno T.; Polack F.; Lagarde B.; Marteau F.; Scheurer F.; Joly L.; Kappler J. P.; Muller B.; Sainctavit P. Polarization characterization on the DEIMOS beamline using dichroism measurements. J. Phys.: Conf. Ser. 2013, 425, 212007. 10.1088/1742-6596/425/21/212007. [DOI] [Google Scholar]
- Funk T.; Friedrich S.; Young A. T.; Arenholz E.; Delano R.; Cramer S. P. Soft x-ray magnetic circular dichroism at 2 K: A tool in biological inorganic chemistry. Rev. Sci. Instrum. 2004, 75, 756–759. 10.1063/1.1645635. [DOI] [Google Scholar]
- van Schooneveld M. M.; DeBeer S. A close look at dose: Toward L-edge XAS spectral uniformity, dose quantification and prediction of metal ion photoreduction. J. Electron Spectrosc. Relat. Phenom. 2015, 198, 31–56. 10.1016/j.elspec.2014.12.001. [DOI] [Google Scholar]
- Delgado-Jaime M. U.; Mewis C. P.; Kennepohl P. Blueprint XAS: a Matlab-based toolbox for the fitting and analysis of XAS spectra. J. Synchrotron Radiat. 2010, 17, 132–137. 10.1107/S0909049509046561. [DOI] [PubMed] [Google Scholar]
- Hocking R. K.; Wasinger E. C.; de Groot F. M. F.; Hodgson K. O.; Hedman B.; Solomon E. I. Fe L-edge XAS studies of K4[Fe(CN)6] and K3[Fe(CN)6]: A direct probe of back-bonding. J. Am. Chem. Soc. 2006, 128, 10442–10451. 10.1021/ja061802i. [DOI] [PubMed] [Google Scholar]
- van der Laan G. In Hitchhiker’s Guide to Multiplet Calculations, Beaurepaire E., Bulou H., Scheurer F., Kappler J.-P., Eds.; Springer: Berlin, 2006. [Google Scholar]
- Butler P. H.Point Group Symmetry: Applications, Methods and Tables; Plenum: New York, 1981. [Google Scholar]
- Cowan R. T.The Theory of Atomic Structure and Spectra; University of California Press: Berkeley, CA, 1981. [Google Scholar]
- de Groot F.; Kotani A. Core Level Spectroscopy of Solids Introduction. Adv. Condens Mat Sci. 2008, 6, 512. 10.1201/9781420008425. [DOI] [Google Scholar]
- de Groot F. M. F.; Fuggle J. C.; Thole B. T.; Sawatzky G. A. 2p X-Ray Absorption of 3d Transition-Metal Compounds - an Atomic Multiplet Description Including the Crystal-Field. Phys. Rev. B: Condens. Matter Mater. Phys. 1990, 42, 5459–5468. 10.1103/PhysRevB.42.5459. [DOI] [PubMed] [Google Scholar]
- Thole B. T.; van der Laan G.; Butler P. H. Spin-Mixed Ground-State of Fe Phthalocyanine and the Temperature-Dependent Branching Ratio in X-Ray Absorption-Spectroscopy. Chem. Phys. Lett. 1988, 149, 295–299. 10.1016/0009-2614(88)85029-2. [DOI] [Google Scholar]
- Stavitski E.; de Groot F. M. F. The CTM4XAS program for EELS and XAS spectral shape analysis of transition metal L edges. Micron 2010, 41, 687–694. 10.1016/j.micron.2010.06.005. [DOI] [PubMed] [Google Scholar]
- DeBeer George S.; Metz M.; Szilagyi R. K.; Wang H. X.; Cramer S. P.; Lu Y.; Tolman W. B.; Hedman B.; Hodgson K. O.; Solomon E. I. A quantitative description of the ground-state wave function of Cu-A by X-ray absorption spectroscopy: Comparison to plastocyanin and relevance to electron transfer. J. Am. Chem. Soc. 2001, 123, 5757–5767. 10.1021/ja004109i. [DOI] [PubMed] [Google Scholar]
- Wasinger E. C.; de Groot F. M. F.; Hedman B.; Hodgson K. O.; Solomon E. I. L-edge X-ray absorption spectroscopy of non-heme iron sites: Experimental determination of differential orbital covalency. J. Am. Chem. Soc. 2003, 125, 12894–12906. 10.1021/ja034634s. [DOI] [PubMed] [Google Scholar]
- Westre T. E.; Kennepohl P.; DeWitt J. G.; Hedman B.; Hodgson K. O.; Solomon E. I. A multiplet analysis of Fe K-edge 1s->3d pre-edge features of iron complexes. J. Am. Chem. Soc. 1997, 119, 6297–6314. 10.1021/ja964352a. [DOI] [Google Scholar]
- Maganas D.; Roemelt M.; Weyhermuller T.; Blume R.; Havecker M.; Knop-Gericke A.; DeBeer S.; Schlogl R.; Neese F. L-edge X-ray absorption study of mononuclear vanadium complexes and spectral predictions using a restricted open shell configuration interaction ansatz. Phys. Chem. Chem. Phys. 2014, 16, 264–276. 10.1039/C3CP52711E. [DOI] [PubMed] [Google Scholar]
- Gebhard M. S.; Koch S. A.; Millar M.; Devlin F. J.; Stephens P. J.; Solomon E. I. Single-Crystal Spectroscopic Studies of Fe(SR)4– (R = 2-(Ph)C6H4): Electronic-Structure of the Ferrous Site in Rubredoxin. J. Am. Chem. Soc. 1991, 113, 1640–1649. 10.1021/ja00005a030. [DOI] [Google Scholar]
- Kowalska J. K.; Hahn A. W.; Albers A.; Schiewer C. E.; Bjornsson R.; Lima F. A.; Meyer F.; DeBeer S. X-ray Absorption and Emission Spectroscopic Studies of [L2Fe2S2]n Model Complexes: Implications for the Experimental Evaluation of Redox States in Iron-Sulfur Clusters. Inorg. Chem. 2016, 55, 4485–4497. 10.1021/acs.inorgchem.6b00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer S. P.; Ralston C. Y.; Wang H. X.; Bryant C. Bioinorganic applications of X-ray multiplets - The impact of Theo Thole’s work. J. Electron Spectrosc. Relat. Phenom. 1997, 86, 175–183. 10.1016/S0368-2048(97)00064-9. [DOI] [Google Scholar]
- Roemelt M.; Maganas D.; DeBeer S.; Neese F. A combined DFT and restricted open-shell configuration interaction method including spin-orbit coupling: Application to transition metal L-edge X-ray absorption spectroscopy. J. Chem. Phys. 2013, 138, 204101. 10.1063/1.4804607. [DOI] [PubMed] [Google Scholar]
- George S. J.; van Elp J.; Chen J.; Ma Y.; Chen C. T.; Park J. B.; Adams M. W. W.; Searle B. G.; Degroot F. M. F.; Fuggle J. C.; Cramer S. P. L-Edge X-Ray Absorption-Spectroscopy of Pyrococcus-Furiosus Rubredoxin. J. Am. Chem. Soc. 1992, 114, 4426–4427. 10.1021/ja00037a073. [DOI] [Google Scholar]
- Cao L.; Jiang Z. X.; Du Y. H.; Yin X. M.; Xi S. B.; Wen W.; Roberts A. P.; Wee A. T. S.; Xiong Y. M.; Liu Q. S.; Gao X. Y. Origin of Magnetism in Hydrothermally Aged 2-Line Ferrihydrite Suspensions. Environ. Sci. Technol. 2017, 51, 2643–2651. 10.1021/acs.est.6b04716. [DOI] [PubMed] [Google Scholar]
- George G. N.; Cleland W. E.; Enemark J. H.; Smith B. E.; Kipke C. A.; Roberts S. A.; Cramer S. P. L-Edge Spectroscopy of Molybdenum Compounds and Enzymes. J. Am. Chem. Soc. 1990, 112, 2541–2548. 10.1021/ja00163a010. [DOI] [Google Scholar]
- Mitzner R.; Rehanek J.; Kern J.; Gul S.; Hattne J.; Taguchi T.; Alonso-Mori R.; Tran R.; Weniger C.; Schroder H.; Quevedo W.; Laksmono H.; Sierra R. G.; Han G. Y.; Lassalle-Kaiser B.; Koroidov S.; Kubicek K.; Schreck S.; Kunnus K.; Brzhezinskaya M.; Firsov A.; Minitti M. P.; Turner J. J.; Moeller S.; Sauter N. K.; Bogan M. J.; Nordlund D.; Schlotter W. F.; Messinger J.; Borovik A.; Techert S.; de Groot F. M. F.; Fohlisch A.; Erko A.; Bergmann U.; Yachandra V. K.; Wernet P.; Yano J. L-Edge X-ray Absorption Spectroscopy of Dilute Systems Relevant to Metalloproteins Using an X-ray Free-Electron Laser. J. Phys. Chem. Lett. 2013, 4, 3641–3647. 10.1021/jz401837f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saes M.; Bressler C.; Abela R.; Grolimund D.; Johnson S. L.; Heimann P. A.; Chergui M. Observing photochemical transients by ultrafast x-ray absorption spectroscopy. Phys. Rev. Lett. 2003, 90, 047403. 10.1103/PhysRevLett.90.047403. [DOI] [PubMed] [Google Scholar]
- Sellberg J. A.; McQueen T. A.; Laksmono H.; Schreck S.; Beye M.; DePonte D. P.; Kennedy B.; Nordlund D.; Sierra R. G.; Schlesinger D.; Tokushima T.; Zhovtobriukh I.; Eckert S.; Segtnan V. H.; Ogasawara H.; Kubicek K.; Techert S.; Bergmann U.; Dakovski G. L.; Schlotter W. F.; Harada Y.; Bogan M. J.; Wernet P.; Fohlisch A.; Pettersson L. G. M.; Nilsson A. X-ray emission spectroscopy of bulk liquid water in ″no-man’s land. J. Chem. Phys. 2015, 142, 044505. 10.1063/1.4905603. [DOI] [PubMed] [Google Scholar]
- Soldatov M. A.; Lange K. M.; Gotz M. D.; Engel N.; Golnak R.; Kothe A.; Aziz E. F. On the origin of dips in total fluorescence yield X-ray absorption spectra: Partial and inverse partial fluorescence yield at the L-edge of cobalt aqueous solution. Chem. Phys. Lett. 2012, 546, 164–167. 10.1016/j.cplett.2012.07.067. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.