

Abstract
While significant advances have been made in the diagnosis and treatment of prostate cancer, each year tens of thousands of men still die from prostate cancer in the United States. Thus greater understanding of cellular pathways and molecular basis of prostate cancer progression in the development of androgen resistance is needed to treat these lethal phenotypes. To dissect the mechanism of androgen resistance, we utilize a proteomics approach to study the development of androgen resistance in LNCaP prostate cancer cells. Our results showed the predominant involvement of metabolic pathways that were elevated in androgen resistance phenotype. We further found the amplification of PI3K/AKT pathway and the overexpression of proteasome proteins while the mitochondrial oxidation phosphorylation was severely hampered in castration-resistant LNCaP-95 cells compared to LNCaP cells. Interestingly, we also found the induction of Dicer, a cytoplasmic endoribonuclease microRNA regulator in the androgen ablated LNCaP-95 prostate cancer cells. We verified some of these data by orthogonal methods including western blot analysis and in castrated animal xenograft studies. To our knowledge, this is the first report showing induced expression of proteasome proteins in androgen ablation prostate cancer cells. If validated in clinical studies, the findings will have significant implications in understanding the complexity of biochemical recurrence in prostate cancer.
Keywords: Androgen resistance, PI3K/AKT, LC-MS/MS, Oxidative Phosphorylation, Proteasome
Introduction
Despite intense research, prostate cancer still remains the most common cancer in American-men [1]. Clinically, organ confined prostate cancer is managed through surgery or localized radiation therapy; however, for some patients whose disease recur systemically following treatment or in advance high risk prostate cancer patients (Gleason 7 or higher, T2 or higher) or metastatic disease, the mainstay of treatment is androgen-deprivation therapy (ADT) [2]. Initially (18–24 month), androgen ablation does seem to work by suppressing the PSA levels and tumor size; however, this effect is temporary and inevitably the disease progress to castration resistant phenotype which is fatal [3]. This transformation from a clinically localized hormone-dependent state to the androgen resistance phenotype may involves several signaling pathways that include Androgen receptor (AR) amplification [4], AR mutation with gain of function, overexpression of the AR cofactors, and Ligand independent AR activation by growth factors [5] which is crucial for androgen resistance. Recently, it was believed that the AR signaling might not contribute in the biology of castration resistant prostate cancer. However, current advances in technology has led to our understanding that residual androgens remain after castration and AR itself continue to be crucial both for the progression to castration resistant prostate cancer (CRPC) and for its continued growth. An early indication that AR might be contributing in the development and progression of CRPC came from observations showing that around 30% of patients with CRPC harbored genomic amplification of the AR locus in late tumor stage [6]. Similarly, in the preclinical prostate cancer cell lines, it was further confirmed that overexpression of the androgen receptor (AR) was enough to drive the progression of prostate cancer to the CRPC that acquired resistance to both ADT and bicalutamide [7]. These studies provided a rationale of targeting AR for drug discovery that could inhibit AR signaling [8].
The canonical pathway for AR activation solely depends upon the testosterone, the main male sex hormone circulating within the blood, which is converted to a more potent dihydrotestosteron (DHT) by the action of 5 α-reductases inside the prostatic stromal and basal cells. Based on its higher binding affinity to the AR, the DHT bound AR translocate into the nucleus where it binds to the androgen responsive elements (ARE) inside the DNA sequences inducing the transcriptional activities of genes such as prostate-specific antigen (PSA). In this process, an interaction between the N and C terminals of the Androgen receptor has been shown to play a role in stabilizing the ligand bond which is facilitated by several AR coactivators, including CBP, pCAF, SRC-1, and ARA70 [9] [10]. While androgens remained the main cause of prostate cancer progression, the presence of mutations in the AR receptor has been linked to the AR function and activation by non-steroidal anti-androgens [3]. The current findings about the splicing variants of the AR which can translocate to the nucleus in ligand independent manner has been shown to be responsible for the resistance mechanism to novel hormonal therapies (abiraterone and enzalutamide) in prostate cancer patient [11] [12]. Beside androgens cell signaling receptors such as insulin growth factors [13], tyrosine kinase receptor and epidermal growth factor receptor [14] have been shown to enhance the activity of the AR and its co-activators even in the presence of very low levels or even absence of androgens leading to castration resistance. Similarly, the bypass pathways which include the upregulation of the anti-apoptotic bcl2 family of proteins can overcome cell death after androgen depravation therapies leading to resistance development [15].
Advances in the field of proteomics has enabled us to use state of the art technologies to studies mechanisms and the pathways associated with the phenotypes of prostate cancer acquired androgen resistance. By using a global proteomics approach to study the hormonal resistance in LNCaP cells, we were able to identify protein changes in androgen-independent cells that were not previously reported. In addition to several metabolic proteins that were highly deranged, we found that the amplification of PI3K/AKT pathway and overexpression of the proteasome proteins while also observing a significant reduction of the mitochondrial oxidation phosphorylation was severely hampered in castration-resistant LNCaP-95 cells.
Materials and Methods
Protein and peptide extraction from cells for proteomic analysis
The cell pellet for LNCaP wild type (WT) or LNCaP-95 from three T-75 cm flasks was first denatured in 1 mL of 8M urea and 0.4 M NH4HCO3 and sonicated thoroughly. The protein concentration was measured using a BCA protein assay kit (Thermo Fisher Scientific Inc). The proteins were then reduced by incubating in 120 mM Tris (2-carboxyethyl) phosphine for 30 min and alkylated by addition of 160 mM iodoacetamide at room temperature for 30 min in the dark. Sample was diluted with buffer (100 mM Tris-HCl, pH 7.5) containing 0.5ug/ul trypsin and incubated at 37°C overnight. The digested proteins were checked for completion of trypsin digestion using SDS-PAGE and silver staining. Peptides were purified with C18 desalting columns and dried using SpeedVac.
iTRAQ labeling of global tryptic peptides from cell lines
Each iTRAQ (isobaric tags for relative and absolute quantitation) 8-plex reagent was dissolved in 70 μl of methanol. 1 mg of each tryptic peptide sample was added into 250 μl of iTRAQ dissolution buffer, then mixed with iTRAQ 6 of the 8-plex labeling tags and incubated for one hour at room temperature. iTRAQ channel 113, 115 and 117 were used to label LNCaP WT from triplicate samples, iTRAQ 114, 116 and 118 were used for labelling the LNCaP-95 cells in triplicate in order to determine the analytical reproducibility. The 6 sets of tagged peptides were combined and purified by SCX column. Then, 10% of the labeled peptides were dried and re-suspended into 0.4% acetic acid solution prior to fractionation for mass spectrometry analysis.
LC-MS/MS analysis
The peptide samples were separated utilizing a Dionex Ultimate 3000 RSL C nano system (Thermo Scientific) with a 75 μm × 50 cm C18 PepMap RSLC column (Thermo Scientific) protected by a 5 mm guard column (Thermo Scientific). The flow rate was 0.250 μl/min with 0.1% formic acid and 2% acetonitrile in water (A) and 0.1% formic acid, 90% acetonitrile (B). The peptides were separated with a 7–40% B gradient in 81mins. MS analysis was performed using a Thermo Q Exactive mass spectrometer (Thermo Scientific). The AGC target for MS1 was set for 1 × 106 for MS1 in 60ms maximum injection time (IT). The AGC target for MS/MS was 1 × 105 (resolution of 17,500, High-energy collisional dissociation (HCD) of 33 and maximum IT of 50ms) of the 20 most abundant ions. Charge state screening was enabled to reject ions with z = 1, ≥8 and unassigned protonated ions. A dynamic exclusion time of 15 s was used to discriminate against previously selected ions.
Data analysis
The raw data derived from the tandem mass spectrometry were searched against the RefSeq protein database, downloaded on August 13, 2014 that comprise around 55,926 proteins using the SEQUEST(R) search engine in Proteome Discoverer v1.4. We specified oxidation of methionine, carbarmidomethylation of cysteine, N-terminal iTRAQ modification as fixed residue modifications. We specified lysine (K) and tyrosine (Y) iTRAQ modifications as well as carbamidomethylation of cysteine and oxidation at methionine as dynamic modifications. Peptides were searched with two tryptic termini allowing one missed cleavage.
The search parameters used were 10ppm precursor tolerance and 0.8Da fragment ion tolerance, fragmentation using HCD. Filters used for global data analysis included peptide rank 1, two peptides per protein, and 1% FDR threshold. Peptide spectrum match (PSM) and protein quantifications were based on the ratios of iTRAQ reporter ions.
Statistical and pathway analyses
Our specified reporter ion quantification ratios for replicate one was 114/113 (LNCaP-95/LNCaP WT), replicate two was 116/115 (LNCaP-95/LNCaP WT) and for replicate three was 118/117 LNCaP-95/LNCaP WT). Proteome Discoverer (PD) reports coefficient of variations (CVs) for unique peptides spectrum match reporter ion ratios. As a second level quality control measure, we filtered-out PSM and associated protein with reporter ion ratio CVs greater than 30%. All experiments data were done in biological triplicate. All statistical analyses were performed using Microsoft Excel running on an IBM-PC compatible computer on the Windows 7 operating system. Statistical significance was defined as a P-value < 0.05. The differentially expressed proteins with at least two fold changes were chosen for bioinformatics analysis using Kyoto Encyclopedia of Gene and Genome (KEGG) enrichment analysis.
Results
Evaluation of cell morphological differences of the LNCaP and LNCaP-95 cells
Androgen signaling plays a vital role in the proliferation of prostate cells. In order to evaluate the androgen-independent cell line, LNCaP-95, which was grown and maintained in the charcoal strip fetal bovine serum (cFBS) containing media, we performed the immunofluorescence microscopy. As a control, we included LNCaP WT and LNCaP-95 cells in normal FBS containing growth medium. Cells grown on the glass coverslips were subjected to immunofluorescence assay for the androgen receptor. As shown in the Figure 1A the androgen receptor (AR) in the LNCaP WT cells has a complete nuclear localization when grown in complete media, while AR in in the LNCaP WT cells grown in the cFBS containing media with androgen removed predominantly displayed cytoplasmic staining (Figure 1A). In contrast, AR localization in the LNCaP-95 cells grown in the cFBS containing media predominantly remained nuclear with partial cytoplasmic staining (Figure 1A) with a distinct smaller cellular morphology compared to the wildtype LNCaP cells (Figure 1C). We also observed the presence of AR in the nucleus of LNCaP-95 cells by nuclear and cytoplasmic fractionation in cFBS containing medium (Supplemental Figure 1A). These observations prompted us to evaluate the signaling pathways in LNCaP95 cells. Using western blot analysis, we showed that PSA expression was decreased to undetectable level in the LNCaP-95 cells cultured in the charcoal stripped medium while PSA expression was detectable in wildtype LNCaP cells. These results were consistent with results observed by the AR reporter firefly luciferase assay, which showed reduced AR activity in the LNCaP-95 cells grown in androgen-depleted media (Supplemental Figure 1B & C). Cell cycle arrest at the G0/G1 phase has been previously shown to significantly alter and compromise both the intracellular and the secreted forms of PSA expression in prostate cancer cells [16] [17]. In order to investigate that depleted PSA was not because of the proliferation status, we evaluated the PCNA expression in these cell which was upregulated in the LNCaP-95 cells (Figure 1A). We further evaluated the proliferation status of the LNCaP-95 cells by MTS and compared it to the wild type LNCaP cells in charcoal stripped media where we found around 40% increase in the number of LNCaP-95 cells by 48Hrs (Figure 1E), together these data suggest that the depleted expression of PSA in LNCaP-95 cells was not likely due to the lower proliferation status of the LNCaP-95 cells. Interestingly, we also observed lower levels of PSMA and higher level of total cellular AR protein in LNCaP-95 cells compared to the wild type LNCaP cells (Figure 1B). The higher expression of the AR might be because of the induced expression of PI3K/AKT pathway which has been shown to inhibit the AR transcriptional activity [18]. Further studies are needed to confirm and validate the PI3K/AKT and AR nexus in androgen ablated conditions in prostate cancer cell lines.
Figure 1.
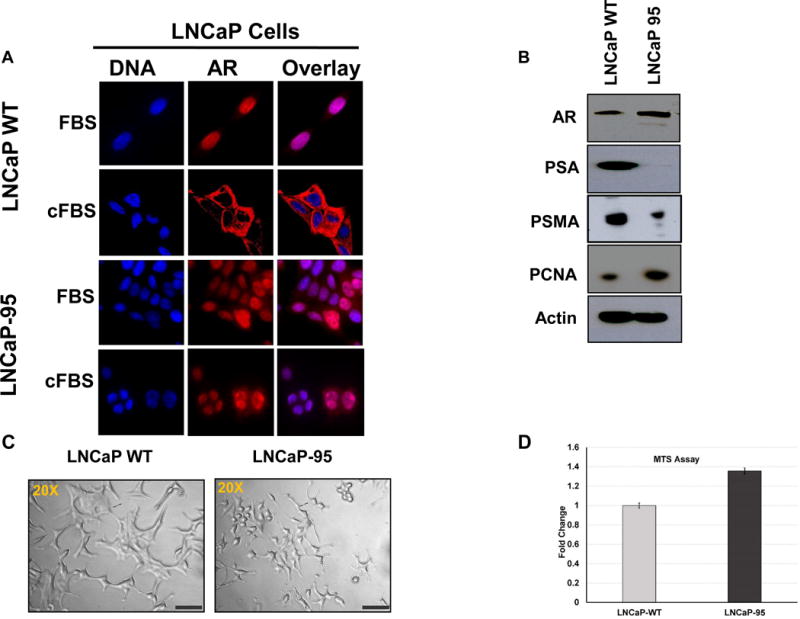
Differential subcellular localization of the AR in prostate cancer LNCaP cells and the androgen resistant LNCaP-95 cells in normal or charcoal stripped containing media (A). Western blot showing expression of the AR and upregulation of the PCNA proteins in the LNCaP-95 cells and down regulation of PSA and PSMA compared to the wildtype LNCaP cells (B). A bright field (20×) microscopy image of the two cell lines showing distinct changes in the size and morphology of the androgen resistant LNCaP-95 cells (C). MTS cell proliferation assay in charcoal stripped FBS containing media showing a statistical difference between the wild type and LNCaP-95 cells at 48h (D).
Global proteomics approach to identify alterations in pathways leading to androgen resistant model
To better understand key pathways that might be playing role in the development of hormone resistance in LNCaP-95 cells, we selected a global proteomic analysis to compare protein expression between wild type and LNCaP-95 cells. LNCaP wildtype and the LNCaP-95 cells were cultured in T75 flask (n=3) with three biological replicates. Cells from each flask were harvested individually at confluency of 80 to 85% and were subjected to iTRAQ labelling Figure 2, as described in the material and methods section. After fractionation by bRPLC each fraction was analyzed by LC-MS/MS to obtained quantitative data. At a 1% FDR, we were able to identify 2,888 protein groups with a minimum of two peptides per protein. (Supplemental Table S1 and Table S2). Based on the ratios for iTRAQ labelled proteins that were used to quantify protein expression changes between the two cell lines, we determined the significance by the student t-test analysis. There were a total of 1883 proteins that displayed significantly altered expression between the two cell types (p <0.05).
Figure 2.
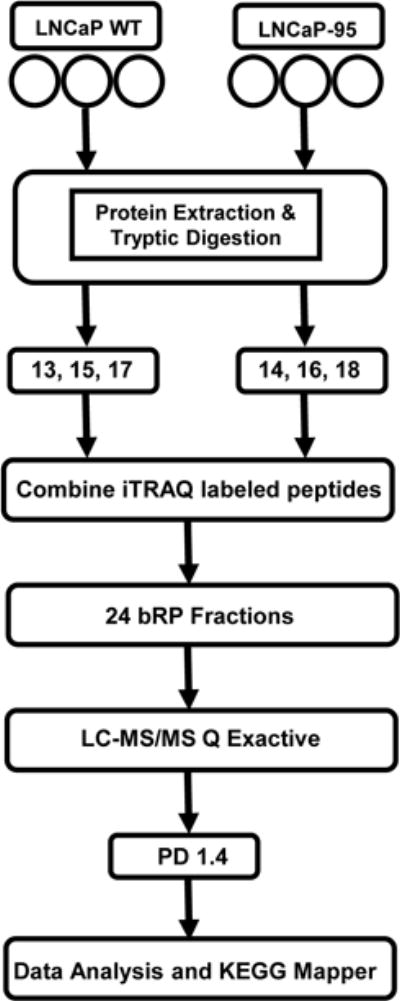
A schematic representation of the workflow using LC-MS/MS for the global proteomic analyses of the LNCaP-WT and androgen resistant LNCaP-95 prostate cancer cell lines.
To further stratify the dysregulated proteins identified in the LNCaP-95 cells into pathways that might be playing role in the development of the castration resistance, we analyze these proteins with the help of KEGG Mapper pathway analysis. Beside the predominant involvement of known metabolic pathways [19], we identified several novel pathways that might be playing role in the development of castration resistance, some of these pathway are described below.
The PI3K-AKT pathway
The PI3K/AKT pathway plays a fundamental role in the regulation of cell growth, division, survival and tumorigenesis [20] [21]. Cancer cells utilize this pathway to adapt to cellular stress. Signaling through this pathway activates the Ser/Thr protein kinase AKT through phosphorylation which in turn phosphorylates and regulates downstream effector proteins and promotes cell proliferation and survival. The tumor suppressor PTEN protein (Phosphatase and tensin homolog deleted from chromosome 10) negatively regulates this pathway; however in LNCaP cells the PTEN gene has been shown to harbor mutation in one allele and a deletion of other allele [22] [23]. In the absence of PTEN the basal level of AKT remained high in wild type LNCaP cells [24], but the PI3K/AKT pathway was induced in LNCaP-95 cells which might compensate for hormonal withdrawal through the upregulating of signal transduction pathways. Based on the global proteomics data obtained by comparing the wildtype LNCaP and LNCaP-95, we identified a total of 15 proteins that were upregulated in this PI3K-AKT signaling pathway (Figure 3A and Supplemental Table 3). Overexpression of the heat shock protein 90 and the mTORC2 in LNCaP-95 cells which directly target the AKT might be playing a role in the phosphorylation of AKT. To further confirm that phosphorylation of the AKT, is involved in the development of castration resistance, we evaluate the LNCaP and LAPC4 tumor xenografts in animals that were either castrated or normal for the pAKT expression using immunohistochemistry. As shown in the Figure 3B, C we found a significantly higher expression of phosphorylated AKT in LNCaP and in the LAPC4 xenografts from the castrated animals. These data independently confirm the activation of pAKT in castration resistance.
Figure 3.
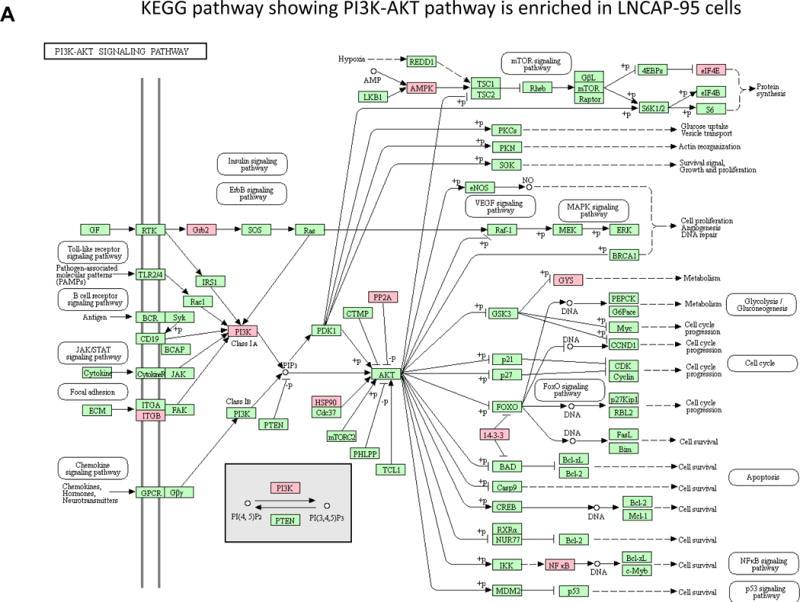
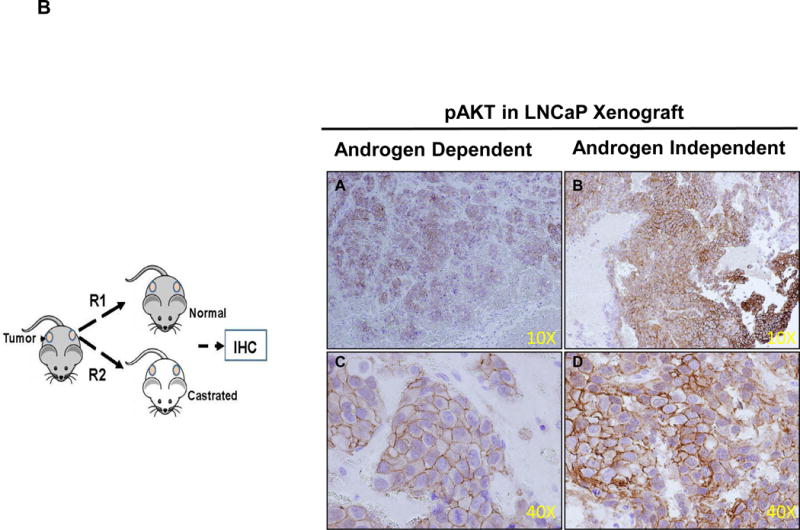
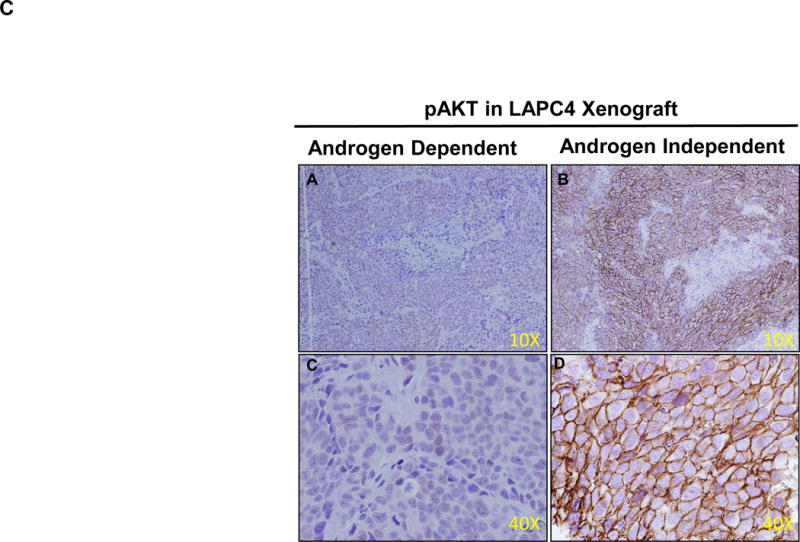
The PI3K-AKT pathway from the KEGG mapper showing several upregulated proteins indicated in the pink based on the two fold change in the protein expression between LNCaP-95 androgen resistant and wildtype LNCaP prostate cancer cells (A). Immunohistochemistry showing overexpression of pAKT in castration resistant LNCaP xenograft tumors when compared to the LNCaP hormonal dependent tumors from the normal animal (B) and in LAPC4 xenograft and castration resistant LAPC4 xenograft model (C).
Proteasome and its subset proteins
The large multi-catalytic 26S Proteasome is the most common proteasomes that recognizes and digests the misfolded proteins that are marked for degradation by an ubiquitin moiety [25]. This enzyme complex comprise of the 20S proteolytic core particle (CP) which is bound by a cap on one or either ends with a 19S regulatory particle (RP) [26]. The 20S proteasome core particle is the focal proteolytic structure consisting of two pairs of rings each containing seven subunits. Three of these subunits are crucial for proteolytic activity, the β1 subunit with “caspase-like” activity, the β2 subunit with trypsin-like activity, and the β5 subunit with chymotrypsin-like activity [27]. Because protein turnover is very critical for cell survival and program cell death, an intact ubiquitin-proteasome system is important for cellular homeostasis. Based on our quantitative iTRAQ data we identified several subunits of the proteasome that were enriched in the androgen ablated LNCaP-95 cells. From the 19S regulatory cap region, we identify seven of the ten particles Rpn3, Rpn5, Rpn6, Rpn8, Rpn10, Rpn11, Rpn12 and seven out of nine from the 19S base regulatory particles that were overexpressed in LNCaP-95 cells (Figure 4, Supplemental Table 4, Supplemental Figure 2A). Similarly, eleven (α1, α3, α4, α5, α6, α7, β2, β3, β4, β6, β7) out of fourteen 20S proteasome core particles were identified by the LC-MS/MS to be upregulated in the androgen ablated model. These data clearly demonstrate the involvement of the UPS pathway in androgen ablated prostate cancer model. These findings of overexpression of proteasome in androgen ablated LNCAP-95 cells have significant clinical implication in terms of biomarker discovery for castration resistance and as a therapeutic target in combination with anti-androgens (bicalutamide and enzalutamide) for additive or synergist effects.
Figure 4.
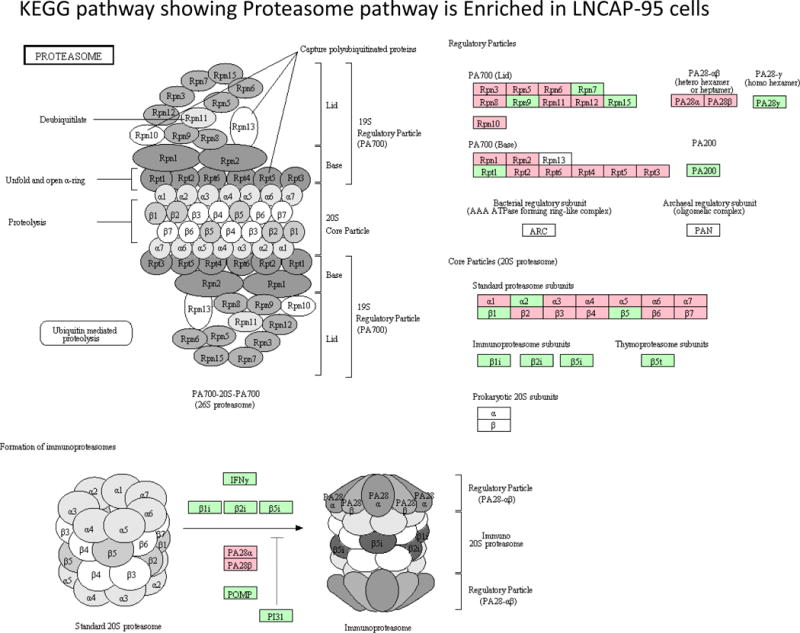
Androgen ablation in LNCaP-95 cells induced the proteasome expression. Several subunits of the 26S proteasome identified by the LC-MS/MS are highlight in pink.
Dysregulated Expression of Oxidative phosphorylated complexes
Defects in the oxidative phosphorylation were first reported by Warburg when he postulated that unlike normal cells that primarily rely on oxidative phosphorylation to generate ATP for energy, cancer cells have impaired respiratory chain functions which are very glycolytic [28]. While the list of mutations in mtDNA affecting OXPHOS in cancer cells is steadily increasing, there are only few reports that studied the function of OXPHOS in androgen ablated prostate cancer cells [29] [30]. In order to evaluate the effect of castration on the multimeric enzyme complexes of OXPHOS in LNCaP-95 cell line model, we compared the wild type and androgen ablated LNCaP-95 cells by LC-MS/MS mass spectrometry using iTRAQ labelling. As shown in the Figure. 5, a large number of proteins in the OXPHOS complexes were downregulated in the LNCaP-95 castrated resistant cells. Complex I, which is the major entry point for electrons to the respiratory chain and is also the rate-limiting step in overall respiration was found to have many downregulated protein subunits suggesting the dysfunction of NADH dehydrogenase (Figure 4, Supplemental Table 5,). A similar lower expression of cytochrome c oxidase, cytochrome c reductase in complex IV (Figure 4, Supplemental Figure 2C) and the ATPase in complex V were also identified in the LNCaP-95 androgen resistant prostate cancer cells. Taken together these observation bring a culmination, that androgen starvation in prostate cancer cells may cause the dysfunctioning of the mitochondrial dependent pathways, which in turn might provide cancer cells with a growth advantage by overproduction of ROS to cause more DNA damage and to accommodate several mutations in the course of high grade aggressive prostate disease.
Figure 5.
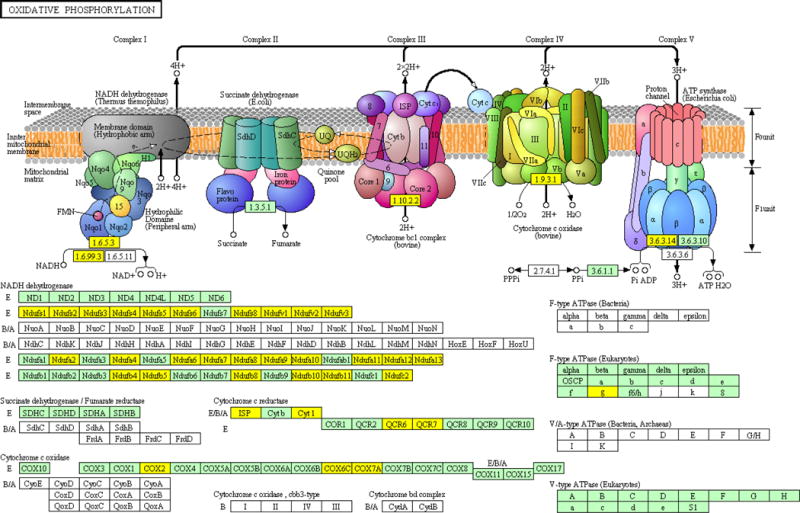
Downregulation of several proteins in the oxidative phosphorylation pathway identified in the global proteomic data for LNCaP-95 androgen resistant prostate cancer cells. Proteins subunits in yellow represent a twofold or more change in the LNCaP-95 compared to the wildtype LNCaP cells.
Discussion
Our data suggest that androgen resistance is a multiple pathway dependent disease which is therefore difficult to avoid in advance prostate cancer patients that are treated via monotherapies. The data provide insights into the components of signal transduction in castration resistance model that beside the overexpression of the PI3K-AKT pathway that involves activation of the NF-κB signaling to modulate cell proliferation and hormone resistance, the mitochondrial dysfunctioning of oxidation phosphorylation and the hyper-expression of the UPS pathway goes hand in hand in making prostate cancer cells androgen independent. The androgen ablated LNCaP-95 cell line model has been previously reported and was specifically used in exploring the AR splicing variants in castration resistant prostate cancer [31] [32]. Studying the AR expression and its transcriptional regulation of the PSA in LNCaP-95 cells, we found that in the androgen ablated condition the AR localization is predominately nuclear with some exceptions where the AR was found inside the cytoplasm (Figure 1). Interestingly, the AR levels in the androgen ablated LNCaP-95 cells were amplified when compared to the wild type LNCaP cells however, the PSA expression which is a direct downstream transcriptional target of AR was significantly suppressed suggesting that the AR in these cells was not functional. This increase in the AR expression might be due to other pathways, including the overexpression of the NF-κB which was amplified in the PI3K pathway in LNCaP-95 cells (Figure 3, Supplemental Table 3). Both mTOR and the PI3K/AKT are important in tumorigenesis by promoting cell growth and suppression of cell death [33] [34]. In a recent phase 2 clinical study for castration resistant prostate cancer, to assess the efficacy and tolerability of mTOR inhibitor (everolimus) in combination with the bicalutamide to evaluate the simultaneous blockade of mTOR and AR pathway showed very encouraging clinical activity in 75% of the patients [35]. Our data support a similar notion for castration resistant prostate cancer that involves multiple signaling pathways, a combinatory therapeutic approach to achieve maximum synergy. We have also identified the proteasome 26S protein to be significantly over-expressed in androgen ablated LNCaP-95 cells compared to the wildtype cells. The 26S proteasome regulates many cellular functions, the most prominent of which includes the cell cycle advancement through mitosis, growth, antigen presentation, and apoptosis. Several proteasome inhibitors have been shown to induce apoptosis and overcome drug resistance in many different type of cancer cells. A pre-clinical screen of 60 cancer cell lines by the national cancer institute with a proteasome inhibitor (PS-341) have demonstrate its anti-proliferative role in majority of these cell lines [36] emphasizing the potential of this pathway for novel anticancer agents.
Oxidative phosphorylation (OXPHOS) is an efficient form of mitochondrial respiration by which cells meet their energy demands, however, beside OXPHOS, the mitochondria is known to regulate multiple cellular signaling pathways including cell death and metabolism. The cellular mitochondria dock includes several pro-apoptotic proteins, such as cytochrome c and Smac/DIBALO which are released from mitochondria in response to anticancer agents. These mitochondrial proteins have been known to act and activate the caspases cascades thus promoting programmed cell death. Many of the tumor cells possess defects in mitochondrial DNA which may provide them with opportunity to overcome stress dependent apoptotic cascade. While exploring the castration resistant pathways, we found a significant down regulated expression of the OXPHOS proteins in LNCaP-95 cells when compared to the wildtype LNCaP cells (Figure 5). Interestingly, many of the identified subunits that were down regulated in LNCaP-95 castration resistant cells were from the NADH dehydrogenase belonging to the complex 1, which is the major entry point for electrons to the respiratory chain and is also the rate-limiting step in overall respiration. We also identified downregulation of several subunits of the cytochrome c oxidase and reductase proteins confirming the dysregulation of this pathway in castration resistant prostate cancer cells. Further studies will be needed to confirm these findings in prostate cancer derived patient specimens.
Based on our model, we have identify and defined several possible pathways that are engaged in driving the castration resistant phenotype in LNCaP-95 cell line model. These pathways have the potential to be exploited for future anticancer strategies or in development of more efficacious anticancer agents.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Health, National Cancer Institute, the Early Detection Research Network (EDRN, U01CA152813), the Clinical Proteomics Tumor Analysis Consortium (CPTAC, U24CA160036), National Heart Lung and Blood Institute, Program of Excellence in Glycosciences (PEG, P01HL107153). We are very thankful to Dr. Michael Haffner (Dept. of Pathology, Johns Hopkins University) for providing the tumor xenografts and Ms. Alvina Pan and Dr. David Clark for editing and proof reading of the manuscript.
Abbreviations
- AR
Androgen Receptor
- PSA
Prostate Specific antigen
- PSMA
Prostate specific membrane antigen
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.American Society of Clinical, O. The State of Cancer Care in America, 2016: A Report by the American Society of Clinical Oncology. J Oncol Pract. 2016 doi: 10.1200/JOP.2015.010462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schroder F, et al. Androgen deprivation therapy: past, present and future. BJU Int. 2012;109(Suppl 6):1–12. doi: 10.1111/j.1464-410X.2012.11215.x. [DOI] [PubMed] [Google Scholar]
- 3.Attard G, et al. Improving the outcome of patients with castration-resistant prostate cancer through rational drug development. Br J Cancer. 2006;95(7):767–74. doi: 10.1038/sj.bjc.6603223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chmelar R, et al. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int J Cancer. 2007;120(4):719–33. doi: 10.1002/ijc.22365. [DOI] [PubMed] [Google Scholar]
- 5.Waltering KK, Urbanucci A, Visakorpi T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol Cell Endocrinol. 2012;360(1–2):38–43. doi: 10.1016/j.mce.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 6.Visakorpi T, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9(4):401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 7.Chen CD, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 8.Tran C, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agoulnik IU, Weigel NL. Androgen receptor coactivators and prostate cancer. Adv Exp Med Biol. 2008;617:245–55. doi: 10.1007/978-0-387-69080-3_23. [DOI] [PubMed] [Google Scholar]
- 10.Daniels G, et al. Androgen receptor coactivators that inhibit prostate cancer growth. Am J Clin Exp Urol. 2014;2(1):62–70. [PMC free article] [PubMed] [Google Scholar]
- 11.Maughan BL, Antonarakis ES. Clinical Relevance of Androgen Receptor Splice Variants in Castration-Resistant Prostate Cancer. Curr Treat Options Oncol. 2015;16(12):57. doi: 10.1007/s11864-015-0375-z. [DOI] [PubMed] [Google Scholar]
- 12.Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011;18(5):R183–96. doi: 10.1530/ERC-11-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sayeed A, et al. Insulin-like growth factor 1 stimulation of androgen receptor activity requires beta(1A) integrins. J Cell Physiol. 2012;227(2):751–8. doi: 10.1002/jcp.22784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherwood ER, et al. Epidermal growth factor receptor activation in androgen-independent but not androgen-stimulated growth of human prostatic carcinoma cells. Br J Cancer. 1998;77(6):855–61. doi: 10.1038/bjc.1998.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dutt SS, Gao AC. Molecular mechanisms of castration-resistant prostate cancer progression. Future Oncol. 2009;5(9):1403–13. doi: 10.2217/fon.09.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zi X, Agarwal R. Silibinin decreases prostate-specific antigen with cell growth inhibition via G1 arrest, leading to differentiation of prostate carcinoma cells: implications for prostate cancer intervention. Proc Natl Acad Sci U S A. 1999;96(13):7490–5. doi: 10.1073/pnas.96.13.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, et al. Indole-3-carbinol induces a G1 cell cycle arrest and inhibits prostate-specific antigen production in human LNCaP prostate carcinoma cells. Cancer. 2003;98(11):2511–20. doi: 10.1002/cncr.11844. [DOI] [PubMed] [Google Scholar]
- 18.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32(49):5501–11. doi: 10.1038/onc.2013.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006;9(3):230–4. doi: 10.1038/sj.pcan.4500879. [DOI] [PubMed] [Google Scholar]
- 20.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 21.Robbins HL, Hague A. The PI3K/Akt Pathway in Tumors of Endocrine Tissues. Front Endocrinol (Lausanne) 2015;6:188. doi: 10.3389/fendo.2015.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 23.van Duijn PW, et al. PTEN-mediated G1 cell-cycle arrest in LNCaP prostate cancer cells is associated with altered expression of cell-cycle regulators. Prostate. 2010;70(2):135–46. doi: 10.1002/pros.21045. [DOI] [PubMed] [Google Scholar]
- 24.Nesterov A, et al. Elevated AKT activity protects the prostate cancer cell line LNCaP from TRAIL-induced apoptosis. J Biol Chem. 2001;276(14):10767–74. doi: 10.1074/jbc.M005196200. [DOI] [PubMed] [Google Scholar]
- 25.Baumeister W, et al. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92(3):367–80. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85(1):12–36. doi: 10.2183/pjab.85.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–68. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 28.Warburg O. Origin of cancer cells. Oncologia. 1956;9(2):75–83. [PubMed] [Google Scholar]
- 29.Sharifi N, et al. Effects of manganese superoxide dismutase silencing on androgen receptor function and gene regulation: implications for castration-resistant prostate cancer. Clin Cancer Res. 2008;14(19):6073–80. doi: 10.1158/1078-0432.CCR-08-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shiota M, Yokomizo A, Naito S. Oxidative stress and androgen receptor signaling in the development and progression of castration-resistant prostate cancer. Free Radic Biol Med. 2011;51(7):1320–8. doi: 10.1016/j.freeradbiomed.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 31.Hu R, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72(14):3457–62. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu C, Luo J. Decoding the androgen receptor splice variants. Transl Androl Urol. 2013;2(3):178–186. doi: 10.3978/j.issn.2223-4683.2013.09.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cargnello M, Tcherkezian J, Roux PP. The expanding role of mTOR in cancer cell growth and proliferation. Mutagenesis. 2015;30(2):169–76. doi: 10.1093/mutage/geu045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. 2005;11(8):353–61. doi: 10.1016/j.molmed.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 35.Chow H, et al. A phase 2 clinical trial of everolimus plus bicalutamide for castration-resistant prostate cancer. Cancer. 2016 doi: 10.1002/cncr.29927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6(10):813–23. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.