

Calcium control of exocytosis has been reconstituted in a hybrid system with purified DCVs and supported target membranes.
Abstract
Regulated exocytosis is a process by which neurotransmitters, hormones, and secretory proteins are released from the cell in response to elevated levels of calcium. In cells, secretory vesicles are targeted to the plasma membrane, where they dock, undergo priming, and then fuse with the plasma membrane in response to calcium. The specific roles of essential proteins and how calcium regulates progression through these sequential steps are currently incompletely resolved. We have used purified neuroendocrine dense-core vesicles and artificial membranes to reconstruct in vitro the serial events that mimic SNARE (soluble N-ethylmaleimide–sensitive factor attachment protein receptor)–dependent membrane docking and fusion during exocytosis. Calcium recruits these vesicles to the target membrane aided by the protein CAPS (calcium-dependent activator protein for secretion), whereas synaptotagmin catalyzes calcium-dependent fusion; both processes are dependent on phosphatidylinositol 4,5-bisphosphate. The soluble proteins Munc18 and complexin-1 are necessary to arrest vesicles in a docked state in the absence of calcium, whereas CAPS and/or Munc13 are involved in priming the system for an efficient fusion reaction.
INTRODUCTION
Regulated exocytosis is used by cells to export stored secretory products—proteins, neurotransmitters, hormones, and small molecules—in response to calcium-mediated signaling (1). In neurons and neuroendocrine cells, the final step of exocytosis is membrane fusion, which is catalyzed by the SNARE [soluble N-ethylmaleimide–sensitive factor attachment protein (SNAP) receptor] proteins syntaxin-1 and SNAP-25 in the plasma membrane and synaptobrevin-2/VAMP-2 (vesicle-associated membrane protein 2) in the secretory vesicle membrane (1–3). Whereas SNARE proteins are sufficient to catalyze membrane fusion (4), several other proteins are involved in regulating the assembly of SNARE complexes and in coupling the calcium signal to the fusion process (2, 3). These include synaptotagmin (syt), complexin, Munc18, Munc13, and CAPS (calcium-dependent activator protein for secretion) (2, 3, 5). Numerous studies have identified prospective roles for these proteins in the vesicle docking, priming, and triggering steps that contribute to exocytosis in situ (6–11), whereas others have analyzed how their interactions affect reconstituted SNARE-mediated fusion in vitro (12–16). However, significant gaps remain in a precise molecular understanding of how and in what order the specific interactions among these proteins relate to the steps of the exocytotic process and interface with calcium’s actions in triggering release. Although several in vitro studies have reported an enhancement of fusion by calcium, complexin, and syt under specialized conditions (12, 17), it has not been possible, so far, to reconstitute a fusion-arrested docked state in vitro in which a robust fusion response is triggered by calcium under conditions containing all known in situ requirements for exocytosis.
RESULTS
To build on current knowledge concerning how the complex molecular machinery of exocytosis might function in vivo, we have sought to use a reconstitution strategy in which individual steps in the process can be resolved and analyzed and the full range of candidate molecular machinery can be evaluated through addition/subtraction approaches. Therefore, we have devised a hybrid system of reconstituted exocytosis in which native neuroendocrine dense-core vesicles (DCVs) and either liposomes or supported planar bilayers containing plasma membrane SNARE proteins serve as fusion partners. DCVs were purified from rat pheochromocytoma (PC12) cells under iso-osmotic conditions using velocity sedimentation and subsequent fractionation on a discontinuous iodixanol gradient (Fig. 1A; see Materials and Methods). The DCVs are functionally active in an ensemble fusion assay in which lipid mixing between DCVs and liposomes was monitored by fluorescence dequenching of NBD-DOPE [1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl)] (Fig. 1B). Moreover, DCVs specifically labeled with a fluorescent secretory protein, neuropeptide Y (NPY) fused to Ruby (expressed in the PC12 cells before fractionation), enable observation of both docking and fusion events with reconstituted supported planar bilayers containing the plasma membrane SNARE proteins (Fig. 1C and fig. S1). DCV fusion was SNARE-dependent in all conditions tested (Fig. 1D and table S1). Lipid mixing in the ensemble fusion assay was accelerated by calcium (fig. S2), consistent with the results for other purified secretory vesicle preparations (18, 19). The amount of docking and the rate and probability of fusion of DCVs to supported bilayers were enhanced by calcium (Fig. 1, E to G, and fig. S3). Calcium-dependent docking did not require SNARE proteins (Fig. 1D), whereas both docking and fusion in the presence of calcium were enhanced by phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] and reduced in the absence of negatively charged lipids (Fig. 1H).
Fig. 1. Reconstitution of DCV membrane fusion.
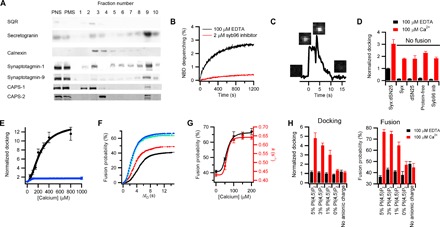
(A) Western blots of the postnuclear supernatant (PNS), postmitochondrial supernatant (PMS), and individual fractions of the iodixanol gradient. Fraction 9 containing the DCV content marker secretogranin was collected as DCVs. DCVs are also enriched in the calcium-interacting proteins syt-1, syt-9, CAPS-1, and CAPS-2. Contamination by mitochondria [succinate-ubiquinone oxidoreductase (SQR)] and endoplasmic reticulum (calnexin) is minimally detected in the DCV fraction. (B) Fusion of purified DCVs with liposomes containing syntaxin-1a (residues 183 to 288):SNAP-25 at a lipid/protein ratio of 500 (black) and a lipid composition of porcine brain PC (bPC):porcine brain PE (bPE):porcine brain PS (bPS):cholesterol (Chol):PI:PI(4,5)P2:Rh-DOPE:NBD-DOPE (23.5:23.5:15:30:4:1:1.5:1.5). Lipid mixing (NBD dequenching) traces shown are averages of four repeated experiments. The presence of synaptobrevin-2 (residues 1 to 96) (syb96) inhibitor peptide (2 mM) abolishes lipid mixing (red). See Materials and Methods for expanded definition of the abbreviated lipid names used here. (C) Single fusion event of NPY-Ruby–labeled DCV with a planar supported bilayer containing syntaxin-1a (syx) (residues 183 to 288):SNAP-25 (lipid/protein = 3000). The black line is an intensity trace of NPY-Ruby fluorescence. The initial increase is caused by binding of the DCV to the planar bilayer within the total internal reflection fluorescence (TIRF) field of the microscope. This intensity is constant during docking; fusion begins with an abrupt decrease, sharp increase, and then following decay in fluorescence to baseline. As described in detail in fig. S1 and the two-step fusion/diffusion model in the Supplementary Materials, this profile reflects collapse of the DCV into the planar bilayer, where NPY-Ruby initially moves forward in the TIRF field and then diffuses away beneath the bilayer. (D) Docking of DCVs to planar supported bilayers that differ in target SNARE (t-SNARE) content in the absence (black) and presence (red) of Ca2+. Syb96 inb refers to the presence of the inhibitor peptide. The total number of events in each case is shown in table S1. dSN25 refers to dodecylated SNAP-25. (E) Docking as a function of [Ca2+] (black) or [Mg2+] (blue) in the single-vesicle planar supported bilayer assay with a K1/2 [Ca2+] of 236 ± 46 μM. Table S2 contains a summary of the total number of docking and fusion events. (F) Delay time between docking and fusion (ΔtD) at different [Ca2+] (black, 100 μM EDTA; red, 50 μM Ca2+; green, 100 μM Ca2+; cyan, 150 μM Ca2+; blue, 200 μM Ca2+) shown as cumulative distribution functions of single DCV fusion events normalized to the fusion probability. The kinetics were fit with a parallel reaction model N(t) = N(1 − e−kt)m, where N is the fusion probability, k is the rate constant, and m is the number of parallel reactions occurring (40). Summary of fit values is shown in table S3. Cumulative distribution functions for additional (higher) [Ca2+] and [Mg2+] are shown in fig. S3. (G) The fusion probabilities (black) and rate constants (red) for parallel reactions are shown as functions of [Ca2+] with K1/2 [Ca2+] of 60 ± 8 μM and 61 ± 8 μM, respectively. For membranes containing syntaxin-1a (residues 183 to 288):SNAP-25 (C to G), the lipid composition was bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:4:1) for all experiments. (H) The effect on docking and fusion of DCVs when PI(4,5)P2 is increased or decreased in the planar bilayers containing bPC:bPE:bPS:Chol:[PI + PI(4,5)P2] (25:25:15:30:5) or in the absence of charged lipids [bPC:bPE:Chol (35:35:30)]. Summaries of docking and fusion events and fitting results of fusion kinetics are shown in tables S5 and S6, respectively.
Both syt (a resident integral protein of synaptic vesicles and DCVs) and CAPS [a soluble but also vesicle-associated protein (Fig. 1A)] have been implicated in the regulation of exocytosis by calcium (5). Numerous studies have identified syt as a calcium sensor that triggers fusion (3, 20). Notably, syt has been shown to accelerate fusion by interacting with PI(4,5)P2 (18, 19, 21). Some of these effects could be due to increased cross-linking of vesicles by syt, although single-particle fusion assays have indicated that syt may have little effect on docking in the presence of SNARE proteins (14). CAPS, which was originally identified as a fusion trigger (22), includes a PI(4,5)P2-binding pleckstrin homology (PH) domain, a C2 domain, and a syntaxin-1a–interacting MUN domain (5). Although it has received less attention than syt and has been questioned previously as a prospective fusion trigger (23), recent studies have shown convincing roles of CAPS in the more proximal steps of docking and priming of DCVs for fusion (5, 8, 11). The abilities to examine both docking and fusion of DCVs to supported bilayers and to manipulate the levels and functions of DCV-associated syt and CAPS have provided us a unique opportunity to examine the roles of these proteins comparatively. We have used two approaches toward this goal: (i) adding function-blocking antibodies to the in vitro assay and (ii) using short hairpin RNA (shRNA)–mediated depletion before purification of DCVs. In the case of syt, the anti-syt antibodies, which bind to the C2 domains, had no effect on the extent of calcium-enhanced docking (Fig. 2A). In addition, shRNA-mediated knockdown of syt-1 and syt-9 [the DCV-associated syts that control exocytosis (24)] to >85% for both isoforms (fig. S4) did not affect the extent of docking (Fig. 2A). However, both treatments profoundly inhibited calcium-accelerated fusion (Fig. 2B and fig. S5A). Notably, the effect on fusion was rescued by expressing RNA interference (RNAi)–resistant syt-1 during knockdown (Fig. 2, A and B).
Fig. 2. Molecular origin of DCV calcium response.
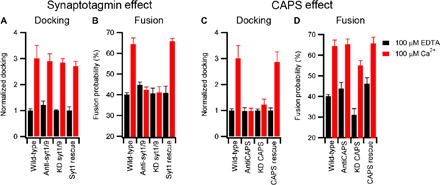
The role of synaptotagmin (A and B) and CAPS (C and D) on DCV docking and fusion in the presence of 100 μM EDTA (black) or 100 μM Ca2+ (red). Wild-type preparations of DCVs were compared either to those that were treated with function-blocking antibodies or to those that were purified from cells subjected to shRNA-mediated knockdown (KD). In the latter case, expression of RNAi-resistant syt1 or CAPS-1 was used as a control. Black and red bar data were obtained in the absence and presence of Ca2+, respectively. Table S7 contains a summary of events, and table S8 contains fitting results. As indicated in Materials and Methods, docking values for preparations of DCVs from wild-type, knockdown, and RNAi rescue cell lines were individually normalized to the value obtained in the presence of 100 μM EDTA, enabling comparison of the relative effects elicited by calcium among the different preparations. This strategy does not enable us to rule out the possible effects of syt or CAPS knockdown on docking in the absence of calcium. For antibody-treated samples, we observed no significant effect on docking in the absence of calcium.
Strikingly, blocking CAPS function using antibodies that bind near the PH and C2 domains (of both isoforms) or shRNA-mediated CAPS depletion (>90% for both isoforms; fig. S5) gave results that significantly contrasted with those for syt. Here, the calcium-dependent increase of DCV docking was inhibited (Fig. 2C and table S5). However, for fusion, the CAPS antibodies had no effect, whereas knockdown decreased the amount of fusion in the absence and presence of calcium as compared to CAPS-containing DCVs (Fig. 2D and table S5). CAPS deficiency also substantially increased the delay time between docking and fusion (fig. S5), likely indicating a role for CAPS in SNARE assembly. Coexpression of RNAi-resistant CAPS-1 during knockdown rescued the effects on both docking and fusion (Fig. 2, C and D, and table S5). As shown by the cumulative distribution for elapsed time between docking and fusion, syt knockdown had no other kinetic effect, although it blocked calcium-accelerated fusion (fig. S5).
The protein machinery necessary for fusion to respond to calcium appears to be present on the secretory vesicle (14, 18, 25, 26), but a large amount of SNARE-dependent fusion occurs in the absence of calcium (~40%; Fig. 1F), which is consistent with the SNAREs representing the minimal fusion machinery (4). Other proteins are well known to regulate the interaction between fusion partners in situ and affect SNARE complex assembly. Thus, we sought to modify the initial hybrid system by replacing the truncated version of syntaxin (residues 183 to 288, containing the transmembrane and SNARE domains) with the full-length protein containing, in addition, the N-terminal and Habc domains that are thought to regulate SNARE complex function. Further, we included the SNARE regulatory proteins Munc18 and complexin-1 to test the effects on docking and fusion in the absence and presence of calcium. The overall goals of this strategy were to suppress calcium-independent fusion and distinguish separate states of vesicle docking and calcium-triggered fusion analogous to those that occur in situ. The soluble proteins Munc18 and complexin-1 are known to be essential in vivo, but it is debated how they positively or negatively regulate fusion (2, 3). Munc18 interacts with the regulatory Habc domain and N-peptide of syntaxin-1a (27–29), and its deletion completely blocks exocytosis (30). Recently, it was proposed that a Munc18 homolog serves as a template for SNARE zippering (31). Complexin-1 has been reported to block spontaneous synaptic vesicle exocytosis in the absence of calcium while increasing synchronized release (32, 33), and earlier in vitro studies claimed that it binds the fully assembled synaptic SNARE complex and serves as a clamp to prevent fusion in advance of calcium signaling (34, 35). However, very recent observations show that complexin-1 may act more proximally by binding a binary complex of syntaxin-1a and SNAP-25, thereby reducing the assembly of synaptobrevin-2 into the fully zippered SNARE complex (36). As a consequence of these observations, Munc18 and complexin-1 were our first priorities for addition during reconstitution.
In our hybrid assay, docking and fusion differed depending on whether the supported bilayers were reconstituted with a truncated syntaxin-1a (residues 183 to 288) or full-length syntaxin-1a (residues 1 to 288). As compared to truncated syntaxin-1a, full-length syntaxin-1a reduced docking in the absence of calcium and reduced fusion in the absence and presence of calcium (Fig. 3, A to D, No Additions columns). When Munc18 was added to the assay, it had little effect on docking or fusion to bilayers containing truncated syntaxin-1a (Fig. 3, A and B). However, it increased docking in the absence of calcium and increased fusion in the absence and presence of calcium when the bilayers contained full-length syntaxin-1a (Fig. 3, C and D), consistent with its ability to bind the full-length protein. When complexin-1 was added to our assay (without Munc18), docking was abolished in the absence of calcium regardless of which syntaxin-1a construct was present (Fig. 3, A and C). Inclusion of calcium with complexin-1 enabled docking and fusion for both truncated and full-length syntaxin-1a, although fusion was decreased when the full-length syntaxin-1a was used (Fig. 3, B and D). When Munc18 and complexin-1 were added together in assays containing truncated syntaxin-1a, docking was blocked in the absence of calcium, but both docking and fusion occurred in the presence of calcium (Fig. 3, A and B). Notably, when full-length syntaxin-1a was used, addition of Munc18 and complexin-1 allowed DCV docking but no fusion in the absence of calcium, whereas the inclusion of calcium allowed fusion to occur readily (Fig. 3, C and D). Using these conditions, which closely approximate those that occur physiologically, this last set of experiments achieved, for the first time, the reconstitution of the calcium dependence of docking and fusion as distinct observable events under full control of this key set of regulatory proteins.
Fig. 3. Effects of Munc18, complexin, and the regulatory Habc domain of syntaxin-1a on docking and fusion.
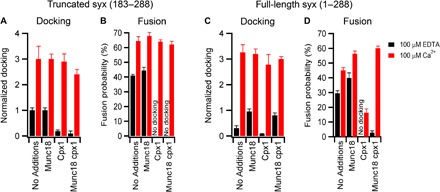
Docking and fusion probability for DCVs to planar supported bilayers containing truncated syntaxin-1a (residues 183 to 288):SNAP-25 (A and B) or full-length syntaxin-1a (1 to 288):SNAP-25 (C and D) in the presence of 0.5 μM Munc18, 2 μM complexin-1 (cpx1), or both. Black and red bar data were obtained in the absence and presence of Ca2+, respectively. Summary of events and fitting results are presented in tables S9 and S10. Western blots show that Munc18 and complexin-1 were not detectable in DCV-enriched fractions prepared by centrifugation (fig. S6).
To complement this demonstration, we used various combinations of syntaxin-1a, SNAP-25, Munc18, and complexin-1 to reveal that syntaxin-1a and Munc18 were sufficient to dock DCVs in the absence of SNAP-25, albeit at levels that were decreased compared to when plasma membrane SNAREs were present (fig. S7). Further, we showed that the soluble synaptobrevin-2 (residues 1 to 96) inhibitor peptide blocked this docking as well as that occurring with both Munc18 and complexin-1 present, thereby indicating that reconstituted docking is synaptobrevin-2–dependent (fig. S7). This outcome supports the prospective role of Munc18 in dual binding to syntaxin-1a and synaptobrevin-2, which may be analogous to the roles of Munc18’s yeast homolog Vps33 (31).
Moreover, addition of complexin-1 in the absence of Munc18 and calcium inhibits docking under conditions with the truncated syntaxin-1a (residues 183 to 288):SNAP-25 or the full-length syntaxin-1a (residues 1 to 288):SNAP-25 complexes (Fig. 3), which agrees with recent results showing that complexin binds quite strongly to the plasma membrane SNAREs that are assembled in a strict 1:1 stoichiometry and thus blocks the interaction with synaptobrevin-2 (36). In the presence of Munc18, there is no effect of complexin-1 on docking to binary syntaxin-1a (residues 1 to 288):SNAP-25 complexes, whereas fusion in the absence of calcium is markedly inhibited by complexin-1, and this response depends strongly on increasing concentrations of complexin (Fig. 3, C and D, and fig. S8). Therefore, Munc18 likely facilitates SNARE assembly, allowing DCVs to be docked, whereas complexin-1 acts within this complex to prevent fusion in the absence of calcium. The data in Fig. 3D also provide a good molecular explanation of the mysterious dual function of complexin, which physiologically inhibits fusion at basal calcium levels but enhances fusion when stimulated by calcium (37). In the absence of calcium, complexin (with Munc18) reduces fusion about 10-fold, and in the presence of calcium, complexin and Munc18 increase fusion by 35%, compared to fusion levels without these proteins, in our system.
Within cells, a subset of secretory vesicles exists in a readily releasable pool, where vesicles are docked at the plasma membrane and primed to undergo rapid fusion in response to elevated levels of calcium (38, 39). The condition where DCVs are docked on planar supported bilayers containing syntaxin-1a (residues 1 to 288) and lipid-anchored SNAP-25 in the presence of Munc18 and complexin-1 but in the absence of calcium likely mimics this primed releasable state. To confirm that these DCVs are fusion-competent, we first allowed them to dock under these conditions for ~30 min and then washed them extensively with a buffer containing the same amounts of Munc18 and complexin-1 to remove undocked DCVs. Then, we added a buffer containing calcium (concentrations are indicated in the figures) and a fluorescent dye (Cy5) to track calcium-containing buffer arrival, which allowed the ensuing calcium-dependent fusion kinetics to be monitored (Fig. 4A). Calcium increased the rate and probability of docked DCV fusion, thus serving as an authentic trigger (Fig. 4B and fig. S9). The observed fusion kinetics from the arrested state (Fig. 4B) was notably different from that observed between docking and fusion in the unarrested pathway (fit of data in Fig. 3D shown in table S10). The kinetics of proceeding from docking to fusion showed a delay that is characteristic of the presence of intermediate(s) (40), whereas the calcium-triggered fusion followed first-order kinetics, indicating that the final intermediate had already been populated. At 1 mole percent (mol %) PI(4,5)P2, 80 to 100 μM calcium was sufficient to fully trigger a response from a ~3% spontaneous constitutive fusion to 60 to 70% fusion probability (fig. S9).
Fig. 4. Calcium-triggered DCV fusion.
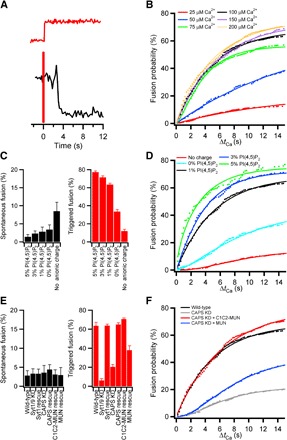
(A) Intensity trace of a single DCV calcium-triggered fusion event (black line) for a DCV docked to a planar supported bilayer in the presence of 0.5 μM Munc18 and 2 μM complexin-1. Fusion was triggered with a buffer [120 mM potassium glutamate, 20 mM potassium acetate, and 20 mM Hepes (pH 7.4)] containing Ca2+. A soluble fluorescent dye (Cy5) was added to the buffer as an indicator for Ca2+ arrival (red line). (B) Cumulative distribution functions for the time delay of fusion following the arrival of calcium (ΔtCa) at different [Ca2+]. Summary of the data is shown in table S11. The probability of triggering DCV fusion as a function of [Ca2+] with K1/2 [Ca2+] of 40 μM is shown in fig. S9. (C) Effect of PI(4,5)P2 and the absence of membrane anionic charge on spontaneous fusion of DCVs in the absence of Ca2+ (black bars) or upon triggering of fusion with 100 μM Ca2+ (red bars). (D) The cumulative distribution function for calcium-triggered fusion as a function of PI(4,5)P2 is shown with the summary of data for spontaneous and triggered fusion in tables S12 and S13, respectively. (E) Effects on spontaneous fusion (black bars) or fusion triggered by 100 μM Ca2+ (red bars) of RNAi-mediated knockdowns of syt and CAPS, of corresponding knockdown/rescue using RNAi-resistant constructs, and of addition of recombinant Munc13-derived C1C2-MUN or the MUN domain alone. Summary of data for spontaneous and triggered fusion of knockdowns is shown in tables S14 and S15, respectively. (F) Cumulative distribution functions of fusion probability beginning at the time of Ca2+ arrival for DCVs from wild-type (wt), CAPS knockdown, and CAPS knockdown rescued by the two Munc13 constructs.
Because PI(4,5)P2 is a key constituent in priming of exocytosis (41–43), we investigated its effect on our reconstituted docking and triggered fusion as well as its influence on the function of the PI(4,5)P2-binding proteins syt and CAPS. The concentration of PI(4,5)P2 included in the supported bilayer had no effect on spontaneous fusion in the absence of calcium but greatly enhanced triggered release (Fig. 4, C and D). The effect of PI(4,5)P2 appeared near maximal from 1 to 5 mol % of total lipid in the planar bilayer, which approximates the estimated plasmalemmal levels supporting DCV exocytosis in PC12 cells (44). The absence of anionic charge [both PI(4,5)P2 and phosphatidylserine] in the target membrane increased spontaneous fusion in the absence of calcium and abolished calcium-triggered fusion (Fig. 4C).
As a final issue in evaluating reconstituted two-step docking and fusion in the presence of regulatory proteins, we examined the effects of depleting syt or CAPS from the DCVs. Loss of neither syt nor CAPS appeared to affect spontaneous fusion (Fig. 4E). However, without syt, calcium-triggered fusion was completely inhibited, whereas without CAPS, triggered fusion was greatly reduced (Fig. 4, E and F). In the latter case, reduced fusion appears to be caused by a kinetic delay (evident in the cumulative distribution curve in Fig. 4F), likely indicating a role of CAPS in priming of the fusion reaction. In situ, the function of CAPS may be closely coupled to or even partially overlap with that of Munc13 (11), a protein that is essential for priming of synaptic fusion (45). Thus, we were interested whether the reduced fusion incurred upon depletion of CAPS might be rescued by adding to our reconstitution a polypeptide containing the C2 and MUN domains that are common to CAPS and Munc13. For this purpose, we used purified recombinant polypeptides containing MUN domain alone or the MUN domain preceded by the C1 and C2 domains (C1C2-MUN) of Munc13. As shown in Fig. 4 (E and F), inclusion of MUN partially rescues, whereas C1C2-MUN fully rescues, the kinetic delay in fusion caused by the CAPS knockdown. This result indicates that we have apparently repopulated the final (primed) step leading to fusion. While under these precise conditions the Munc13 C1C2-MUN domain seems to be interchangeable with CAPS in the kinetic priming of the docked DCV state, these two proteins are not redundant players in vivo and more detailed studies of the respective interactions and effects on priming will need to be addressed in the future.
DISCUSSION
Our hybrid system using neuroendocrine DCVs and planar supported bilayers has enabled us to reconstruct in vitro a stepwise docking, priming, and fusion process that bears a remarkable resemblance to exocytosis in situ and to clarify the roles of key supporting proteins. Using this approach, we have made the following new findings (see also Table 1 for more details): (i) Munc18 and complexin-1 in the presence of syntaxin-1a (residues 1 to 288) and SNAP-25 are able to arrest DCVs in a docked state in the absence of calcium; (ii) at 1 mol % PI(4,5)P2, the fusion probability is triggered by 80 to 100 μM calcium from a ~3% spontaneous constitutive fusion probability to 60 to 70% fusion probability, whereas at 5 mol % PI(4,5)P2, this dynamic range is expanded from 2% spontaneous to 80% triggered fusion; this calcium trigger for fusion is strictly dependent on synaptotagmin; (iii) CAPS appears to kinetically increase the rate of the calcium response from the Munc18/complexin trans-SNARE complex docked state while also being able to dock DCVs to PI(4,5)P2-containing membranes in response to calcium; (iv) the C1C2-MUN domain of Munc13 can replace CAPS priming under the conditions investigated here; and (v) the depletion of PI(4,5)P2 kinetically reduces the rate of calcium triggering from the docked state in a manner that closely resembles the effects of CAPS knockdown.
Table 1. Summary of results on the role of soluble and DCV-associated proteins and regulatory lipids obtained by the hybrid reconstitution approach of docking and fusion of single DCVs on planar supported bilayers with reconstituted SNAREs under different target membrane, vesicle membrane, and added protein conditions. 0, no effect; +, positive effect; +++, strong positive effect; −−−, strong negative effect; N/E, not examined.
| Starting membrane condition | Vesicle condition/ additions |
Docking probability |
Fusion probability | kfusion | |||
| EDTA | Ca2+ | EDTA | Ca2+ | EDTA | Ca2+ | ||
| Syntaxin-1a (183–288):SNAP-25 | Syt | 0 | 0 | 0 | +++ | 0 | +++ |
| CAPS | 0 | +++ | + | + | + | + | |
| Complexin-1 | −−− | 0 | No docking | 0 | No docking | 0 | |
| Munc18 | 0 | 0 | 0 | 0 | + | + | |
| PI(4,5)P2 | 0 | +++ | 0 | +++ | 0 | +++ | |
| Syntaxin-1a (1–288):SNAP-25 | Complexin-1 | −−− | 0 | No docking | 0 | No docking | 0 |
| Munc18 | + | + | + | + | + | + | |
| Syntaxin-1a (183–288):SNAP-25, Munc18 | Complexin-1 | 0 | 0 | −−− | 0 | −−− | 0 |
| Syntaxin-1a (183–288):SNAP-25, Munc18, complexin-1 | Syt | 0 | N/E | 0 | +++ | 0 | +++ |
| CAPS | 0 | N/E | 0 | + | 0 | +++ | |
| PI(4,5)P2 | 0 | N/E | 0 | + | 0 | +++ | |
These findings have led us to propose a model (Fig. 5) where DCVs can dock to membranes in a stable nonfusing state when syntaxin-1a (residues 1 to 288), SNAP-25, Munc18, and complexin-1 are present in a process that is dependent on synaptobrevin-2. CAPS can prime these docked DCVs to fuse efficiently when triggered with calcium. The actual fusion triggering is caused by synaptotagmin-1/9 in a process that requires an anionic lipid in the target membranes with a strong preference for PI(4,5)P2. The calcium-dependent docking of DCVs facilitated by DCV-associated CAPS (11) could then function to recruit more DCVs to PI(4,5)P2-containing membranes, allowing more fusion events to occur (Fig. 5). This calcium-dependent recruitment of DCVs to the target membrane agrees with the electrophysiological results showing that CAPS is necessary for sustained secretion in chromaffin cells in the presence of elevated calcium (46, 47). With respect to this last point, we are encouraged by earlier cellular studies showing calcium-dependent recruitment of synaptic vesicles after depletion of docked vesicles (48) and similar behavior noted for DCVs (49) that may depend at least in part on DCV-associated CAPS (11). The direct binding of calcium by CAPS [dissociation constant (Kd) ~270 μM] (6) was similar to what was observed for calcium’s Kd for docking in our reconstituted system [~240 μM in the presence of 1% PI(4,5)P2]. These calcium concentrations are well above the physiological level of intracellular calcium present under stimulatory conditions, but the docking had a large dynamic range with increases in DCV docking to planar supported bilayers observed under 100 μM calcium. Previously, PI(4,5)P2 was shown to be enriched around syntaxin-1a (50) and act as a recruitment site for secretory vesicle docking (51). Our results indicate that calcium-dependent DCV docking increased in the presence of higher concentrations of PI(4,5)P2, suggesting that local enrichments of PI(4,5)P2 around syntaxin-1a could drive calcium-dependent CAPS-mediated docking to physiologically relevant calcium concentrations. Whether this is a meaningful correlation or the relatively high Kd reflects a current limitation of our in vitro system will require further study. In the future, it will be important to determine whether additional proteins that have been implicated in docking [for example, granuphilin (52), rabphilin-3a, and Munc13-4 (53, 54) as well as lipid microdomains] will, when included in our analyses, reduce the calcium Kd for vesicle docking to micromolar or even submicromolar levels.
Fig. 5. Model of DCV docking and fusion during exocytosis.
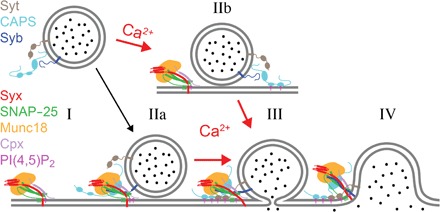
Calcium-dependent release of DCV content requires an acceptor complex consisting of syntaxin-1a, SNAP-25, Munc18, and complexin as well as PI(4,5)P2 in the plasma membrane. The complex interacts with synaptobrevin-2 (syb) and synaptotagmin-1/9 (syt) in the vesicle membrane as well as CAPS that may or may not be vesicle-associated (I). DCVs are able to dock to the complex in the absence of calcium in a SNARE-dependent fashion (IIa). The presence of complexin “clamps” the resulting pre-fusion (trans-SNARE) complex, preventing progression to an open fusion pore, and allows priming of the fusion machinery. Priming depends on CAPS [and/or Munc13 (not shown)] and PI(4,5)P2 and might involve a spatial organization of multiple copies of trans-SNARE complexes and accessory proteins as well as the organization of a specific local nanoscale lipid environment. The primed intermediate state resembles granules in the readily releasable pool in situ. Calcium influx triggers fusion pore opening catalyzed by syts (III) and eventually the collapse of the vesicle membrane into the plasma membrane (IV). Calcium also facilitates CAPS-dependent docking of DCVs to the plasma membrane (IIb). These granules proceed through some intermediates that might resemble the priming steps in the absence of calcium to a fusion pore (III) and eventually to the complete merger of the two membranes (IV).
The hybrid approach combining biological DCVs and reconstituted target membranes in a robust and efficient single-vesicle fusion assay enabled us to dissect and assign precise mechanistic roles to a large number of proteins (and some of their subdomains) that regulate calcium-triggered exocytosis, which has previously eluded experimental confirmation. Our results have demonstrated that complexin, Munc18, and the regulatory Habc domain of syntaxin are all necessary and sufficient to create a fusion-arrested acceptor SNARE complex capable of specifically docking secretory vesicles in the absence of calcium. CAPS or Munc13 coordinates the system to be primed for subsequent efficient calcium-triggered syt-dependent fusion. This work also demonstrated that PI(4,5)P2 in the target membrane plays critical roles in both enhancing CAPS/Munc13–dependent priming and syt-dependent fusion and that the lipid context is of critical importance in defining the protein and lipid interactions that are physiologically relevant in exocytosis. Future work using this powerful system will certainly further refine the proposed model and permit further dissection of the specific interactions and interplays between the different protein modules and lipids that lead to calcium-triggered exocytosis.
MATERIALS AND METHODS
Materials
The following materials were purchased and used without further purification: porcine brain L-α-phosphatidylcholine (bPC), porcine brain L-α-phosphatidylethanolamine (bPE), porcine brain L-α-phosphatidylserine (bPS), L-α-phosphatidylinositol (liver, bovine) (PI), phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2], 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rh-DOPE), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD-DOPE) were from Avanti Polar Lipids; cholesterol, sodium cholate, EDTA, calcium (Ca2+), OptiPrep Density Gradient Medium, sucrose, Mops, L-glutamic acid potassium salt monohydrate, potassium acetate, and glycerol were from Sigma; CHAPS were from Anatrace; Hepes was from Research Products International; chloroform, ethanol, Contrad detergent, all inorganic acids and bases, and hydrogen peroxide were from Fisher Scientific. Water was purified first with deionizing and organic-free 3 filters (Virginia Water Systems) and then with a NANOpure system from Barnstead to achieve a resistivity of 18.2 megohms/cm.
Antibodies for syt-1 (mouse monoclonal), syt-5/9 (rabbit polyclonal), CAPS-1 (rabbit polyclonal), CAPS-2 (rabbit polyclonal), complexin-1/2 (mouse monoclonal), Munc18 (mouse monoclonal), and Munc13 (mouse monoclonal) were all purchased from Synaptic Systems. The calnexin antibody (rabbit polyclonal) was from Enzo Life Sciences, the secretogranin II antibody (mouse monoclonal) was from Biodesign International, and the succinate ubiquinone antibody (mouse monoclonal) was from Molecular Probes.
Protein purification
Syntaxin-1a (both constructs of residues 183 to 288 and 1 to 288), wild-type SNAP-25, soluble synaptobrevin-2 (residues 1 to 96), Munc18, and complexin-1 from Rattus norvegicus were expressed in Escherichia coli strain BL21(DE3) cells under the control of the T7 promoter in the pET28a expression vector and purified as described previously (55–57). Briefly, all proteins were purified using the nickel–nitrilotriacetic acid (Ni-NTA) affinity chromatography. After the removal of N-terminal His-tags by thrombin cleavage, proteins were further purified by subsequent ion-exchange or size-exclusion chromatography, when necessary. Wild-type SNAP-25 was quadruply dodecylated through disulfide bonding of dodecyl methanethiosulfonate (Toronto Research Company) to its four native cysteines (58). All SNAP-25 used in this work refers to this lipid-anchored form of SNAP-25. To express Munc13 in E. coli, DNA of Munc13 (C1C2BMUN, residues 529 to 1407; EF, residues 1453 to 1531) from R. norvegicus was cloned into pET28a vector through the use of restriction enzyme sites Nde I and Hind III. Munc13 expression was carried out in BL21(DE3) cells by IPTG (isopropyl-β-d-thiogalactopyranoside) induction. Harvested cell pellets were resuspended and then lysed in the extraction buffer [20 mM Hepes (pH 7.4), 500 mM NaCl, and 8 mM imidazole]. The supernatant of lysed cells was collected and subjected to binding of Ni-NTA beads. After extensive washing of protein-bound Ni-NTA beads with the wash buffer [20 mM Hepes (pH 7.4), 500 mM NaCl, 20 mM imidazole, and 10% glycerol], Munc13 was eluted from Ni-NTA with the elution buffer [20 mM Hepes (pH 7.4) and 400 mM imidazole]. After the His-tag cleavage by bovine thrombin, Munc13 was further purified by subsequent Mono Q ion-exchange chromatography. Purities of all proteins were verified by SDS–polyacrylamide gel electrophoresis (PAGE).
Reconstitution of SNAREs into proteoliposomes
Proteoliposomes with a lipid composition of bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:4:1) were prepared. All t-SNARE proteins were reconstituted using sodium cholate, as previously described (40, 59). The desired lipids were mixed, and organic solvents were evaporated under a stream of N2 gas followed by vacuum desiccation for at least 1 hour. The dried lipid films were dissolved in 25 mM sodium cholate in a buffer [20 mM Hepes (pH 7.4) and 150 mM KCl] followed by the addition of an appropriate volume of syntaxin-1a and SNAP-25 in their respective detergents to reach a final lipid/protein ratio of 3000 for each protein. After 1 hour of equilibration at room temperature, the mixture was diluted below the critical micellar concentration by adding more buffer to the desired final volume. The sample was then dialyzed overnight against 1 liter of buffer, with one buffer change after ~4 hours.
Preparation of planar supported bilayers containing SNARE acceptor complexes
Planar supported bilayers with reconstituted plasma membrane SNAREs were prepared by the Langmuir-Blodgett/vesicle fusion technique as described in previous studies (60–62). Quartz slides were cleaned by dipping in sulfuric acid/hydrogen peroxide (3:1) for 15 min using a Teflon holder. Slides were then rinsed thoroughly in water. The first leaflet of the bilayer was prepared by Langmuir-Blodgett transfer directly onto the quartz slide using a Nima 611 Langmuir-Blodgett trough by applying the lipid mixture of bPC:Chol:DPS (1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-PEG3400-triethoxysilane) (70:30:3) from a chloroform solution. After allowing the solvent to evaporate for 10 min, the monolayer was compressed at a rate of 10 cm2/min to reach a surface pressure of 31 mN/m. After equilibration for 5 to 10 min, a clean quartz slide was rapidly (200 mm/min) dipped into the trough and slowly (5 mm/min) withdrawn while a computer maintained a constant surface pressure and monitored the transfer of lipids with head groups down onto the hydrophilic substrate. Proteoliposomes reconstituted with 1:1 syntaxin-1a (residues 183 to 288 or 1 to 288):SNAP-25 at a lipid/protein ratio of 3000 were incubated with the Langmuir-Blodgett monolayer to form the outer leaflet of the planar supported bilayer. A concentration of 77 mM total lipid in a total volume of 1.3 ml was used. The lipid composition of the outer leaflet was bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:4:1), unless otherwise noted in the text. After incubation of the proteoliposomes for 2 hours, the excess proteoliposomes were removed by perfusion with 10 ml of buffer [120 mM potassium glutamate, 20 mM potassium acetate, and 20 mM Hepes (pH 7.4)], with the calcium concentration indicated for each experiment or with the presence of 100 μM EDTA when calcium was absent.
Plasmids and shRNA constructs
pEGFP-N1–NPY (a gift from W. Almers) was used to subclone NPY into a pmCherry-N1 and a pmRuby vector. Results from NPY-mRuby were slightly brighter in our experimental setup and used in all subsequent experiments, but initial experiments revealed no differences in results obtained with NPY-mCherry or NPY-mRuby.
For simultaneous shRNA knockdown of multiple syt isoforms, we created a modular vector platform similar to the work by Xu et al. (63) based on pLKO.5. pLKO.5 (Sigma-Aldrich) was digested with Eco RI and Ppu MI, blunted with T4 DNA polymerase, and recircularized. To obtain the empty vector plasmid pLKO.P-emtpy, Eco RI and Bst Z17I sites were introduced by site-directed mutagenesis (64) using primer pairs 1 and 2 (table S16). shRNA expression cassettes targeting syt1 (TRCN0000093258) and syt9 (TRCN0000379591) were amplified by polymerase chain reaction (PCR) from MISSION shRNA plasmids (Sigma-Aldrich) using primer pairs 3 and 5 or 4 and 5, respectively, and subsequently digested with Mfe I and Bst Z17I. These digested fragments were sequentially ligated into pLKO.P-empty digested with Eco RI and Bst Z17I, eventually yielding the double-knockdown plasmid pLKO.P-syt9-syt1.
For the CAPS1 knockdown, oligonucleotide 6 (11) was assembled into pLKO.5 digested with Kpn I and Eco RI using the NEBuilder HiFi DNA Assembly Reaction (New England Biolabs).
To express shRNA-resistant syt1, pHluorin was first removed from pCI-pHluorin-syt1 [a gift from A. Anantharam described by Rao et al. (65)] by overlap extension PCR cloning (66) using primer pairs 7 and 8. Subsequently, five silent mutations were introduced into the shRNA target region of syt1 by site-directed mutagenesis using primer pairs 9 and 10.
For the CAPS1 rescue, pmKate2-resCAPS1 containing eight silent mutations in the shRNA target site [a gift from T. Martin described by Kabachinski et al. (11)] was digested with Bam HI and Not I, blunted with T4 DNA polymerase, and recircularized to remove mKate2. All plasmid sequences were verified by Sanger DNA sequencing (GENEWIZ).
Cell culture
Pheochromocytoma cells (PC12) were cultured on 10-cm plastic cell culture plates at 37°C in 10% CO2 in 1× Dulbecco’s modified Eagle’s medium, high glucose (Gibco) supplemented with 10% horse serum (CellGro), 10% calf serum (Fe+) (HyClone), and 1% penicillin/streptomycin mix. Medium was changed every 2 to 3 days, and cells were passed after reaching 90% confluency by incubating 5 min in Hank’s balanced salt solution and replating in fresh medium. Cells were transfected by electroporation using ECM 830 Electro Square Porator (BTX). After harvesting and sedimentation, cells were suspended in a small volume of sterile cytomix electroporation buffer [120 mM KCl, 10 mM KH2PO4, 0.15 mM CaCl2, 2 mM EGTA, 25 mM Hepes-KOH, 5 mM MgCl2, 2 mM adenosine triphosphate, and 5 mM glutathione (pH 7.6)] (67) and then counted and diluted to ~14 × 106 cells/ml. Cell suspension (~10 × 106 cells) (700 μl) and 30 μg of DNA were placed in an electroporator cuvette with a 4-mm gap, and two 255-V, 8-ms electroporation pulses were applied. Cells were then transferred to a 10-cm cell culture dish with 10 ml of normal growth medium. NPY-Ruby–transfected cells were cultured under normal conditions for 3 days after transfection and then used for fractionation. Transient knockdown cell lines were generated by transfecting shRNA as described above and maintaining cells under puromycin (2 μg/ml) selection beginning 24 hours after transfection. Cells were later transfected with NPY-Ruby and, when relevant, shRNA-resistant plasmids 3 days before fractionation.
DCV purification
DCVs were purified using an iso-osmotic medium as follows. PC12 cells (15 to 30 10-cm plates depending on experiments) were scraped into phosphate-buffered saline (PBS), pelleted by centrifugation, resuspended, and washed once in the homogenization medium (0.26 M sucrose, 5 mM Mops, and 0.2 mM EDTA). Following resuspension in 3 ml of homogenization medium containing protease inhibitor (Roche Diagnostics), the cells were cracked open using a ball bearing homogenizer with a 0.6368 cm bore and 0.6340 cm diameter ball. The homogenate was then spun at 4000 rpm (1000g) for 10 min at 4°C in fixed-angle microcentrifuge to pellet nuclei and larger debris. The postnuclear supernatant was collected and spun at 11,000 rpm (8000g) for 15 min at 4°C to pellet mitochondria. The PMS was then collected, adjusted to 5 mM EDTA, and incubated for 10 min on ice. A working solution of 50% OptiPrep (iodixanol) [five volumes of 60% OptiPrep (one volume of 0.26 M sucrose, 30 mM Mops, and 1 mM EDTA)] and homogenization medium were mixed to prepare solutions for discontinuous gradients in Beckman SW 55 tubes: 0.5 ml of 30% iodixanol on the bottom and 3.8 ml of 14.5% iodixanol, above which 1.2 ml of EDTA-adjusted PMS was layered. Samples were spun at 45,000 rpm (190,000gav) for 5 hours. A clear white band at the interface between the 30% iodixanol and the 14.5% iodixanol was collected as the DCV sample (fraction 9 of Western blot shown in Fig. 1A). The DCV sample was then extensively dialyzed in a cassette with 10,000-kDa molecular weight cutoff (24 to 48 hours, 3 × 5 liters) into the fusion assay buffer [120 mM potassium glutamate, 20 mM potassium acetate, and 20 mM Hepes (pH 7.4)].
TIRF microscopy
Experiments examining single-vesicle docking and fusion events were performed on a Zeiss Axiovert 35 fluorescence microscope (Carl Zeiss), equipped with a 63× water immersion objective (Zeiss; numerical aperture, 0.95) and a prism-based TIRF illumination. The light source was an OBIS 532 LS laser from Coherent Inc. Fluorescence was observed through a 610-nm band pass filter (D610/60, Chroma) by an electron-multiplying charge-coupled device (CCD) (DU-860E, Andor Technology). The electron-multiplying CCD (EMCCD) was cooled to −70°C, and the gain was set between 200 and 220. The prism-quartz interface was lubricated with glycerol to allow easy translocation of the sample cell on the microscope stage. The beam was totally internally reflected at an angle of 72° from the surface normal, resulting in an evanescent wave that decays exponentially with a characteristic penetration depth of ~100 nm. An elliptical area of 250 μm × 65 μm was illuminated. The laser intensity, shutter, and camera were controlled by a homemade program written in LabVIEW (National Instruments).
Experiments triggering DCV fusion with calcium were done on a Zeiss Axiovert 200 fluorescence microscope (Carl Zeiss), with objective and TIRF setups as described above. The light source was a 514-nm beamline of an argon ion laser (Innova 90C, Coherent), controlled through an acousto-optic modulator (Isomet), and a diode laser (Cube 640, Coherent) emitting light at 640 nm. The characteristic penetration depth was ~102 and ~130 nm for the 514- and 640-nm lasers, respectively. An OptoSplit (Andor Technology) was used to separate the fluorescence from the NPY-Ruby and Cy5 fluorescence. Fluorescence signals were recorded by an EMCCD (iXon DV887ESC-BV, Andor Technology). The EMCCD camera was cooled to −70°C, and the electron gain factor was set between 200 and 240.
Single DCV docking and fusion assay
Acceptor t-SNARE proteins containing planar supported bilayers were washed with fusion buffer containing EDTA or divalent metal (Ca2+ or Mg2+), as indicated in the text. They were then perfused with DCV (50 to 100 μl depending on preparation) diluted into 2 ml of fusion buffer [120 mM potassium glutamate, 20 mM potassium acetate, and 20 mM Hepes (pH 7.4)] with additions to buffer as indicated in the text. The fluorescence from DCVs was recorded with a 532-nm laser using an EMCCD camera gain of 200 to 220. After injection of the DCV sample, the microscope was focused within no more than 30 s and then 5000 images were taken with 200-ms exposure times and spooled directly to the hard drive. One spooling set was taken for each bilayer.
Single-vesicle fusion data were analyzed using a homemade program written in LabVIEW (National Instruments). Stacks of images were filtered by a moving average filter. The maximum intensity for each pixel over the whole stack was projected on a single image. Vesicles were located in this image by a single-particle detection algorithm described in by Kiessling et al. (68). The peak (central pixel) and mean fluorescence intensities of a 5-pixel × 5-pixel area around each identified center of mass were plotted as a function of time for all particles in the image series. The exact time points of docking and fusion were determined from the central pixel similar to previous work (40). Cumulative distributions were determined from the time of docking to the time of fusion for individual fusion events, and the fusion efficiency was determined from the number of vesicles that underwent fusion compared with the total number of vesicles that docked within 15 s of DCV docking.
DCV docking was normalized from each DCV preparation by running two bilayers in 100 μM EDTA with syntaxin-1a (residues 183 to 288):SNAP-25 and using the average number of docked events in these experiments to normalize all other data collected for that preparation. Docking values from preparations of DCVs from wild-type, knockdown, and RNAi rescue cell lines cannot be directly compared to each other because normalizations were done within each preparation. However, trends of calcium acceleration are comparable. Experiments to inhibit proteins by antibody binding showed no major effect on docking in the absence of calcium, but an effect of syt or CAPS knockdown on docking in the absence of calcium cannot be completely ruled out.
Single DCV Ca2+-triggered assay
Acceptor t-SNAREs containing planar supported bilayers were washed with fusion buffer containing 100 μM EDTA and then incubated for 30 min with 0.5 μM Munc18, 2 μM complexin-1, and 100 μM EDTA. The DCV sample was then perfused in while keeping the concentrations of Munc18 and complexin-1 constant. After 30 min, fusion buffer containing the indicated [Ca2+] and 0.5 μM soluble Cy5 dye was injected, and fusion was monitored. The presence or absence of Munc18 and complexin-1 in the fusion buffer containing Ca2+ had no effect on the triggered fusion results.
Western blotting
Samples were separated by SDS-PAGE and transferred to nitrocellulose filters (Invitrogen); membranes were washed with PBS and blocked with Odyssey Blocking Buffer (PBS). Primary antibodies were incubated in Odyssey Blocking Buffer with 0.1% (v/v) Tween 20. The appropriate secondary antibody coupled with IRDye 800 was incubated in a dilution of 1 to 10,000 with the nitrocellulose filter. After extensive washing with PBS, 0.1% Tween 20 blots were imaged on an Odyssey Imaging System (LI-COR).
Ensemble DCV lipid mixing assay
Proteoliposomes containing t-SNAREs (syntaxin-1 and SNAP-25) with fluorescence resonance energy transfer–paired lipid probes (1.5% each of Rh-DOPE and NBD-DOPE) were incubated at 37°C in fusion buffer with either EDTA or divalent metal (Ca2+ or Mg2+) at an indicated concentration and with any additional protein, as indicated in the text. After 10 min of incubation, fluorimeter recording was started, DCVs (25 to 50 μl) were added, and NBD dequenching was monitored. At the end of each experiment, 0.1% Triton X-100 was added, and the maximum NBD dequenching was recorded and used to normalize each experiment.
Supplementary Material
Acknowledgments
We would like to thank C. Stroupe, D. Cafiso, P. Kasson, and S.-T. Yang for their suggestions and comments; C. Doyle and R. D’Sousa for their assistance with cell culture and creating shRNA-mediated knockdown cell lines; and A. Anantharam, T. Martin, and W. Almers for plasmid gifts that are acknowledged in Materials and Methods. Funding: This work was supported by NIH (grant P01 GM072694) to L.K.T. and R.J. Author contributions: A.J.B.K. designed and performed the research, analyzed the data, and wrote the article. V.K. designed the research, contributed analytical tools, and analyzed the data. B.L. performed the research and analyzed the data. P.S. performed the research and analyzed the data. S.J. developed a protein purification method, performed the research, and analyzed the data. R.J. developed a protein purification method and analyzed the data. J.D.C. designed the research, analyzed the data, and wrote the article. L.K.T. designed the research, analyzed the data, and wrote the article. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1603208/DC1
Supplementary Text
fig. S1. Line shape of intensity traces from single DCV fusion events.
fig. S2. Ensemble lipid mixing of DCVs with reconstituted proteoliposomes containing syntaxin-1a (residues 183 to 288) (lipid/protein = 500) with a lipid composition of bPC:bPE:bPS:Chol:PI:PI(4,5)P2:Rh-DOPE:NBD-DOPE (23.5:23.5:15:30:4:1:1.5:1.5) at increasing [Ca2+].
fig. S3. Single DCV fusion response.
fig. S4. Quantification of shRNA-mediated knockdowns.
fig. S5. DCV fusion kinetics of syt and CAPS knockdowns.
fig. S6. Western blots of complexin-1/2, Munc18, and Munc13 in fractions generated during DCV purification.
fig. S7. Controls to determine the effects of Munc18 and complexin-1 on docking to full-length syntaxin-1a (residues 1 to 288) and SNAP-25 and distinguish which of these conditions are inhibited by the soluble domain of synaptobrevin-2 (residues 1 to 96).
fig. S8. Complexin-1 inhibits fusion to planar supported bilayers containing syntaxin-1a (residues 1 to 288):SNAP-25 in the presence of 0.5 μM Munc18 in the absence of calcium, whereas there is no effect on DCV docking.
fig. S9. Fusion probability as a function of calcium for fusion of DCVs that were docked to planar supported bilayers with syntaxin-1a (residues 1 to 288):SNAP-25 in the presence of Munc18 and complexin-1 upon triggering with calcium.
table S1. Event statistics for DCV docking and fusion to planar supported bilayers with syntaxin-1a (residues 183 to 288):SNAP-25 (lipid/protein = 3000), syntaxin-1a (residues 183 to 288) (lipid/protein = 3000), dodecylated SNAP-25 (lipid/protein = 3000), no protein, or syntaxin-1a (residues 183 to 288):SNAP-25 (lipid/protein = 3000) incubated with 2 μM synaptobevin-2 (residues 1 to 96) inhibitor peptide.
table S2. Event statistics for DCV docking and fusion to planar supported bilayers with syntaxin-1a (residues 183 to 288):SNAP-25 (lipid/protein = 3000) with increasing concentration of divalent metals (Ca2+ and Mg2+).
table S3. Fit values of a parallel reaction model [N(t) = N(1 − e−kt)m, where N is the fusion probability, k is the rate, and m is the number of parallel reactions occurring] for the cumulative distribution function of delay times between docking and fusion for single-particle DCV events with different concentrations of divalent metals.
table S4. Fits of ensemble lipid mixing data shown in fig. S3.
table S5. Event statistics for docking and fusion of DCVs to planar supported bilayers with 5% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI(4,5)P2 (25:25:15:30:5)], 3% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:2:3)], 1% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:4:1)], 0% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI (25:25:15:30:5)], and no charged lipids [bPC:bPE:Chol (35:35:30)] in the presence of 100 μM EDTA or 100 μM Ca2+.
table S6. Event statistics for docking and fusion of DCVs to planar supported bilayers with 5% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI(4,5)P2 (25:25:15:30:5)], 3% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:2:3)], 1% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:4:1)], 0% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI (25:25:15:30:5)], and no charged lipids [bPC:bPE:Chol (35:35:30)] in the presence of 100 μM EDTA or 100 μM Ca2+.
table S7. Event statistics for DCV docking and fusion to planar supported bilayers with syntaxin-1a (residues 183 to 288):SNAP-25 (lipid/protein = 3000) with wild-type DCVs, DCVs inhibited with antibodies for either syt or CAPS, or DCVs that have had syt or CAPS knocked down using shRNA before purification.
table S8. Fit values of a parallel reaction model [N(t) = N(1 − e−kt)m, where N is the fusion probability, k is the rate, and m is the number of parallel reactions occurring] for the cumulative distribution function of delay times between docking and fusion for single-particle DCV events for conditions described in Fig. 2.
table S9. Event statistics for DCV docking and fusion to planar supported bilayers containing either syntaxin-1a (residues 183 to 288) or syntaxin-1a (residues 1 to 288) with SNAP-25 (lipid/protein = 3000), with the addition of complexin-1 and/or Munc18 in the presence of 100 μM EDTA or 100 μM Ca2+.
table S10. Fit values of a parallel reaction model [N(t) = N(1 − e−kt)m, where N is the fusion probability, k is the rate, and m is the number of parallel reactions occurring] for the cumulative distribution function of delay times between docking and fusion for single-particle DCV events for conditions described in Fig. 3.
table S11. Event statistics of triggered fusion of DCVs at different calcium concentrations from data shown in Fig. 4B.
table S12. Event statistics of spontaneous fusion of DCVs docked in triggering conditions with planar supported bilayers of different lipid compositions.
table S13. Event statistics of triggered fusion of DCVs with planar supported bilayers of different lipid compositions.
table S14. Event statistics of spontaneous fusion of DCVs docked in triggering conditions with knockdowns of syt-1/9 or CAPS with subsequent recoveries.
table S15. Event statistics of triggered fusion of DCVs with knockdowns of syt-1/9 or CAPS and subsequent recoveries.
table S16. Table of primers described in the “Plasmids and shRNA constructs” section of Materials and Methods.
table S17. Event statistics for DCV docking and fusion to planar supported bilayers with the indicated combination of proteins syntaxin-1a (residues 183 to 288), syntaxin-1a (residues 1 to 288), and SNAP-25 (lipid/protein = 3000) incubated, as indicated, with Munc18 (0.5 μM), complexin-1 (2 μM), and synaptobrevin-2 (residues 1 to 96) inhibitor peptide.
table S18. Event statistics for DCV docking and fusion to planar supported bilayers containing syntaxin-1a (residues 1 to 288):SNAP-25, 0.5 μM Munc18, and indicated amounts of complexin-1.
REFERENCES AND NOTES
- 1.Martens S., McMahon H. T., Mechanisms of membrane fusion: Disparate players and common principles. Nat. Rev. Mol. Cell Biol. 9, 543–556 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Jahn R., Fasshauer D., Molecular machines governing exocytosis of synaptic vesicles. Nature 490, 201–207 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Südhof T. C., Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber T., Zemelman B. V., McNew J. A., Westermann B., Gmachl M., Parlati F., Söllner T. H., Rothman J. E., SNAREpins: Minimal machinery for membrane fusion. Cell 92, 759–772 (1998). [DOI] [PubMed] [Google Scholar]
- 5.James D. J., Martin T. F. J., CAPS and Munc13: CATCHRs that SNARE vesicles. Front. Endocrinol. 4, 187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ann K., Kowalchyk J. A., Loyet K. M., Martin T. F. J., Novel Ca2+-binding protein (CAPS) related to UNC-31 required for Ca2+-activated exocytosis. J. Biol. Chem. 272, 19637–19640 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Gerber S. H., Rah J.-C., Min S.-W., Liu X., de Wit H., Dulubova I., Meyer A. C., Rizo J., Arancillo M., Hammer R. E., Verhage M., Rosenmund C., Südhof T. C., Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science 321, 1507–1510 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Truong C. Q. N., Nestvogel D., Ratai O., Schirra C., Stevens D. R., Brose N., Rhee J., Rettig J., Secretory vesicle priming by CAPS is independent of its SNARE-binding MUN domain. Cell Rep. 9, 902–909 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Tang J., Maximov A., Shin O.-H., Dai H., Rizo J., Südhof T. C., A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126, 1175–1187 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Borisovska M., Zhao Y., Tsytsyura Y., Glyvuk N., Takamori S., Matti U., Rettig J., Südhof T., Bruns D., V-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J. 24, 2114–2126 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kabachinski G., Kielar-Grevstad D. M., Zhang X., James D. J., Martin T. F. J., Resident CAPS on dense-core vesicles docks and primes vesicles for fusion. Mol. Biol. Cell 27, 654–668 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diao J., Grob P., Cipriano D. J., Kyoung M., Zhang Y., Shah S., Nguyen A., Padolina M., Srivastava A., Vrljic M., Shah A., Nogales E., Chu S., Brunger A. T., Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. eLife 1, e00109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai Y., Diao J., Cipriano D. J., Zhang Y., Pfuetzner R. A., Padolina M. S., Brunger A. T., Complexin inhibits spontaneous release and synchronizes Ca2+-triggered synaptic vesicle fusion by distinct mechanisms. eLife 3, e03756 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiessling V., Ahmed S., Domanska M. K., Holt M. G., Jahn R., Tamm L. K., Rapid fusion of synaptic vesicles with reconstituted target SNARE membranes. Biophys. J. 104, 1950–1958 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoon T.-Y., Lu X., Diao J., Lee S.-M., Ha T., Shin Y.-K., Complexin and Ca2+ stimulate SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 15, 707–713 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma C., Su L., Seven A. B., Xu Y., Rizo J., Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339, 421–425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kyoung M., Srivastava A., Zhang Y., Diao J., Vrljic M., Grob P., Nogales E., Chu S., Brunger A. T., In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc. Natl. Acad. Sci. U.S.A. 108, E304–E313 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park Y., Hernandez J. M., van den Bogaart G., Ahmed S., Holt M., Riedel D., Jahn R., Controlling synaptotagmin activity by electrostatic screening. Nat. Struct. Mol. Biol. 19, 991–997 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park Y., Seo J. B., Fraind A., Pérez-Lara A., Yavuz H., Han K., Jung S.-R., Kattan I., Walla P. J., Choi M., Cafiso D. S., Koh D.-S., Jahn R., Synaptotagmin-1 binds to PIP2-containing membrane but not to SNAREs at physiological ionic strength. Nat. Struct. Mol. Biol. 22, 815–823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brose N., Petrenko A. G., Südhof T. C., Jahn R., Synaptotagmin: A calcium sensor on the synaptic vesicle surface. Science 256, 1021–1025 (1992). [DOI] [PubMed] [Google Scholar]
- 21.Wang Z., Liu H., Gu Y., Chapman E. R., Reconstituted synaptotagmin I mediates vesicle docking, priming, and fusion. J. Cell Biol. 195, 1159–1170 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hay J. C., Martin T. F. J., Resolution of regulated secretion into sequential MgATP-dependent and calcium-dependent stages mediated by distinct cytosolic proteins. J. Cell Biol. 119, 139–151 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grishanin R. N., Kowalchyk J. A., Klenchin V. A., Ann K., Earles C. A., Chapman E. R., Gerona R. R. L., Martin T. F. J., CAPS acts at a prefusion step in dense-core vesicle exocytosis as a PIP2 binding protein. Neuron 43, 551–562 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Lynch K. L., Martin T. F. J., Synaptotagmins I and IX function redundantly in regulated exocytosis but not endocytosis in PC12 cells. J. Cell Sci. 120, 617–627 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Scheuner D., Logsdon C. D., Holz R. W., Bovine chromaffin granule membranes undergo Ca2+-regulated exocytosis in frog oocytes. J. Cell Biol. 116, 359–365 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheuner D., Holz R. W., Evidence that the ability to respond to a calcium stimulus in exocytosis is determined by the secretory granule membrane: Comparison of exocytosis of injected bovine chromaffin granule membranes and endogenous cortical granules in Xenopus laevis oocytes. Cell. Mol. Neurobiol. 14, 245–257 (1994). [DOI] [PubMed] [Google Scholar]
- 27.Dulubova I., Sugita S., Hill S., Hosaka M., Fernandez I., Südhof T. C., Rizo J., A conformational switch in syntaxin during exocytosis: Role of munc18. EMBO J. 18, 4372–4382 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Misura K. M. S., Scheller R. H., Weis W. I., Three-dimensional structure of the neuronal-Sec1–syntaxin 1a complex. Nature 404, 355–362 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Toonen R. F. G., Verhage M., Munc18-1 in secretion: Lonely Munc joins SNARE team and takes control. Trends Neurosci. 30, 564–572 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Verhage M., Maia A. S., Plomp J. J., Brussaard A. B., Heeroma J. H., Vermeer H., Toonen R. F., Hammer R. E., van den Berg T. K., Missler M., Geuze H. J., Südhof T. C., Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287, 864–869 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Baker R. W., Jeffrey P. D., Zick M., Phillips B. P., Wickner W. T., Hughson F. M., A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science 349, 1111–1114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X., Kaeser-Woo Y. J., Pang Z. P., Xu W., Südhof T. C., Complexin clamps asynchronous release by blocking a secondary Ca2+ sensor via its accessory α helix. Neuron 68, 907–920 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X., Cao P., Südhof T. C., Deconstructing complexin function in activating and clamping Ca2+-triggered exocytosis by comparing knockout and knockdown phenotypes. Proc. Natl. Acad. Sci. U.S.A. 110, 20777–20782 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giraudo G. C., Eng W. S., Melia T. J., Rothman J. E., A clamping mechanism involved in SNARE-dependent exocytosis. Science 313, 676–680 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Giraudo C. G., Garcia-Diaz A., Eng W. S., Yamamoto A., Melia T. J., Rothman J. E., Distinct domains of complexins bind SNARE complexes and clamp fusion in vitro. J. Biol. Chem. 283, 21211–21219 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zdanowicz R., Kreutzberger A. J. B., Liang B., Kiessling V., Tamm L. K., Cafiso D. S., Complexin binding to membranes and acceptor t-SNAREs explains its clamping effect on fusion. Biophys. J. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Südhof T. C., Rothman J. E., Membrane fusion: Grappling with SNARE and SM proteins. Science 323, 474–477 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin T. F. J., Tuning exocytosis for speed: Fast and slow modes. Biochim. Biophys. Acta 1641, 157–165 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Sørensen J. B., Formation, stabilisation and fusion of the readily releasable pool of secretory vesicles. Pflugers Arch. 448, 347–362 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Domanska M. K., Kiessling V., Stein A., Fasshauer D., Tamm L. K., Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J. Biol. Chem. 284, 32158–32166 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holz R. W., Axelrod D., Localization of phosphatidylinositol 4,5-P2 important in exocytosis and a quantitative analysis of chromaffin granule motion adjacent to the plasma membrane. Ann. N. Y. Acad. Sci. 971, 232–243 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Martin T. F. J., Role of PI(4,5)P2 in vesicle exocytosis and membrane fusion. Subcell. Biochem. 59, 111–130 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milosevic I., Sørensen J. B., Lang T., Krauss M., Nagy G., Haucke V., Jahn R., Neher E., Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J. Neurosci. 25, 2557–2565 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.James D. J., Khodthong C., Kowalchyk J. A., Martin T. F. J., Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion. J. Cell Biol. 182, 355–366 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Varoqueaux F., Sigler A., Rhee J.-S., Brose N., Enk C., Reim K., Rosenmund C., Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc. Natl. Acad. Sci. U.S.A. 99, 9037–9042 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y., Schirra C., Stevens D. R., Matti U., Speidel D., Hof D., Bruns D., Brose N., Rettig J., CAPS facilitates filling of the rapidly releasable pool of large dense-core vesicles. J. Neurosci. 28, 5594–5601 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y., Schirra C., Edelmann L., Matti U., Rhee J., Hof D., Bruns D., Brose N., Rieger H., Stevens D. R., Rettig J., Two distinct secretory vesicle–priming steps in adrenal chromaffin cells. J. Cell Biol. 190, 1067–1077 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zenisek D., Steyer J. A., Almers W., Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature 406, 849–854 (2000). [DOI] [PubMed] [Google Scholar]
- 49.Karatekin E., Tran V. S., Huet S., Fanget I., Cribier S., Henry J.-P., A 20-nm step toward the cell membrane preceding exocytosis may correspond to docking of tethered granules. Biophys. J. 94, 2891–2905 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van den Bogaart G., Meyenberg K., Risselada H. J., Amin H., Willig K. I., Hubrich B. E., Dier M., Hell S. W., Grubmuller H., Diederichsen U., Jahn R., Membrane protein sequestering by ionic protein–lipid interactions. Nature 479, 552–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Honigmann A., van den Bogaart G., Iraheta E., Risselada H. J., Milovanovic D., Mueller V., Müllar S., Diederichsen U., Fasshauer D., Grubmüller H., Hell S. W., Eggeling C., Kühnel K., Jahn R., Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat. Struct. Mol. Biol. 20, 679–686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gomi H., Mizutani S., Kasai K., Itohara S., Izumi T., Granuphilin molecularly docks insulin granules to the fusion machinery. J. Cell Biol. 171, 99–109 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chung S.-H., Song W.-J., Kim K., Bednarski J. J., Chen J., Prestwich G. D., Holz R. W., The C2 domains of Rabphilin3A specifically bind phosphatidylinositol 4,5-bisphosphate containing vesicles in a Ca2+-dependent manner. In vitro characteristics and possible significance. J. Biol. Chem. 273, 10240–10248 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Boswell K. L., James D. J., Esquibel J. M., Bruinsma S., Shirakawa R., Horiuchi H., Martin T. F. J., Munc13-4 reconstitutes calcium-dependent SNARE-mediated membrane fusion. J. Cell Biol. 197, 301–312 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pabst S., Hazzard J. W., Antonin W., Südhof T. C., Jahn R., Rizo J., Fasshauer D., Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J. Biol. Chem. 275, 19808–19818 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Liang B., Kiessling V., Tamm L. K., Prefusion structure of syntaxin-1A suggests pathway for folding into neuronal trans-SNARE complex fusion intermediate. Proc. Natl. Acad. Sci. U.S.A. 110, 19384–19389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dawidowski D., Cafiso D. S., Allosteric control of syntaxin 1a by Munc18-1: Characterization of the open and closed conformations of syntaxin. Biophys. J. 104, 1585–1594 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kreutzberger A. J. B., Liang B., Kiessling V., Tamm L. K., Assembly and comparison of plasma membrane SNARE acceptor complexes. Biophys. J. 110, 2147–2150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wagner M. L., Tamm L. K., Reconstituted syntaxin1a/SNAP25 interacts with negatively charged lipids as measured by lateral diffusion in planar supported bilayers. Biophys. J. 81, 266–275 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalb E., Frey S., Tamm L. K., Formation of supported planar bilayers by fusion of vesicles to supported phospholipid monolayers. Biochim. Biophys. Acta 1103, 307–316 (1992). [DOI] [PubMed] [Google Scholar]
- 61.Wagner M. L., Tamm L. K., Tethered polymer-supported planar lipid bilayers for reconstitution of integral membrane proteins: Silane-polyethyleneglycol-lipid as a cushion and covalent linker. Biophys. J. 79, 1400–1414 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kiessling V., Liang B., Tamm L. K., Reconstituting SNARE-mediated membrane fusion at the single liposome level. Methods Cell Biol. 128, 339–363 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu X.-M., Yoo M.-H., Carlson B. A., Gladyshev V. N., Hatfield D. L., Simultaneous knockdown of the expression of two genes using multiple shRNAs and subsequent knock-in of their expression. Nat. Protoc. 4, 1338–1348 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zheng L., Baumann U., Reymond J.-L., An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rao T. C., Passmore D. R., Peleman A. R., Das M., Chapman E. R., Anantharam A., Distinct fusion properties of synaptotagmin-1 and synaptotagmin-7 bearing dense core granules. Mol. Biol. Cell 25, 2416–2427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van den Ent F., Löwe J., RF cloning: A restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 67, 67–74 (2006). [DOI] [PubMed] [Google Scholar]
- 67.van den Hoff M. J., Moorman A. F., Lamers W. H., Electroporation in “intracellular” buffer increases cell survival. Nucleic Acids Res. 20, 2902 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kiessling V., Crane J. M., Tamm L. K., Transbilayer effects of raft-like lipid domains in asymmetric planar bilayers measured by single molecule tracking. Biophys. J. 91, 3313–3326 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Z., Wu Y., Wang Z., Dunning F. M., Rehfuss J., Ramanan D., Chapman E. R., Jackson M. B., Release mode of large and small dense-core vesicles specified by different synaptotagmin isoforms in PC12 cells. Mol. Biol. Cell 22, 2324–2336 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kiessling V., Tamm L. K., Measuring distances in supported bilayers by fluorescence interference-contrast microscopy: Polymer supports and SNARE proteins. Biophys. J. 84, 408–418 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zareh S. K., DeSantis M. C., Kessler J. M., Li J.-L., Wang Y. M., Single-image diffusion coefficient measurements of proteins in free solution. Biophys. J. 102, 1685–1691 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fromherz P., Kiessling P., Kottig K., Zeck G., Membrane transistor with giant lipid vesicle touching a silicon chip. Appl. Phys. A 69, 571–576 (1999). [Google Scholar]
- 73.Kiessling V., Bernt M., Fromherz M., Extracellular resistance in cell adhesion measured with a transistor probe. Langmuir 16, 3517–3521 (2000). [Google Scholar]
- 74.Albillos A., Dernick G., Horstmann H., Almers W., Alvarez de Toledo G., Lindau M., The exocytotic event in chromaffin cells revealed by patch amperometry. Nature 389, 509–512 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1603208/DC1
Supplementary Text
fig. S1. Line shape of intensity traces from single DCV fusion events.
fig. S2. Ensemble lipid mixing of DCVs with reconstituted proteoliposomes containing syntaxin-1a (residues 183 to 288) (lipid/protein = 500) with a lipid composition of bPC:bPE:bPS:Chol:PI:PI(4,5)P2:Rh-DOPE:NBD-DOPE (23.5:23.5:15:30:4:1:1.5:1.5) at increasing [Ca2+].
fig. S3. Single DCV fusion response.
fig. S4. Quantification of shRNA-mediated knockdowns.
fig. S5. DCV fusion kinetics of syt and CAPS knockdowns.
fig. S6. Western blots of complexin-1/2, Munc18, and Munc13 in fractions generated during DCV purification.
fig. S7. Controls to determine the effects of Munc18 and complexin-1 on docking to full-length syntaxin-1a (residues 1 to 288) and SNAP-25 and distinguish which of these conditions are inhibited by the soluble domain of synaptobrevin-2 (residues 1 to 96).
fig. S8. Complexin-1 inhibits fusion to planar supported bilayers containing syntaxin-1a (residues 1 to 288):SNAP-25 in the presence of 0.5 μM Munc18 in the absence of calcium, whereas there is no effect on DCV docking.
fig. S9. Fusion probability as a function of calcium for fusion of DCVs that were docked to planar supported bilayers with syntaxin-1a (residues 1 to 288):SNAP-25 in the presence of Munc18 and complexin-1 upon triggering with calcium.
table S1. Event statistics for DCV docking and fusion to planar supported bilayers with syntaxin-1a (residues 183 to 288):SNAP-25 (lipid/protein = 3000), syntaxin-1a (residues 183 to 288) (lipid/protein = 3000), dodecylated SNAP-25 (lipid/protein = 3000), no protein, or syntaxin-1a (residues 183 to 288):SNAP-25 (lipid/protein = 3000) incubated with 2 μM synaptobevin-2 (residues 1 to 96) inhibitor peptide.
table S2. Event statistics for DCV docking and fusion to planar supported bilayers with syntaxin-1a (residues 183 to 288):SNAP-25 (lipid/protein = 3000) with increasing concentration of divalent metals (Ca2+ and Mg2+).
table S3. Fit values of a parallel reaction model [N(t) = N(1 − e−kt)m, where N is the fusion probability, k is the rate, and m is the number of parallel reactions occurring] for the cumulative distribution function of delay times between docking and fusion for single-particle DCV events with different concentrations of divalent metals.
table S4. Fits of ensemble lipid mixing data shown in fig. S3.
table S5. Event statistics for docking and fusion of DCVs to planar supported bilayers with 5% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI(4,5)P2 (25:25:15:30:5)], 3% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:2:3)], 1% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:4:1)], 0% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI (25:25:15:30:5)], and no charged lipids [bPC:bPE:Chol (35:35:30)] in the presence of 100 μM EDTA or 100 μM Ca2+.
table S6. Event statistics for docking and fusion of DCVs to planar supported bilayers with 5% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI(4,5)P2 (25:25:15:30:5)], 3% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:2:3)], 1% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:4:1)], 0% PI(4,5)P2 [bPC:bPE:bPS:Chol:PI (25:25:15:30:5)], and no charged lipids [bPC:bPE:Chol (35:35:30)] in the presence of 100 μM EDTA or 100 μM Ca2+.
table S7. Event statistics for DCV docking and fusion to planar supported bilayers with syntaxin-1a (residues 183 to 288):SNAP-25 (lipid/protein = 3000) with wild-type DCVs, DCVs inhibited with antibodies for either syt or CAPS, or DCVs that have had syt or CAPS knocked down using shRNA before purification.
table S8. Fit values of a parallel reaction model [N(t) = N(1 − e−kt)m, where N is the fusion probability, k is the rate, and m is the number of parallel reactions occurring] for the cumulative distribution function of delay times between docking and fusion for single-particle DCV events for conditions described in Fig. 2.
table S9. Event statistics for DCV docking and fusion to planar supported bilayers containing either syntaxin-1a (residues 183 to 288) or syntaxin-1a (residues 1 to 288) with SNAP-25 (lipid/protein = 3000), with the addition of complexin-1 and/or Munc18 in the presence of 100 μM EDTA or 100 μM Ca2+.
table S10. Fit values of a parallel reaction model [N(t) = N(1 − e−kt)m, where N is the fusion probability, k is the rate, and m is the number of parallel reactions occurring] for the cumulative distribution function of delay times between docking and fusion for single-particle DCV events for conditions described in Fig. 3.
table S11. Event statistics of triggered fusion of DCVs at different calcium concentrations from data shown in Fig. 4B.
table S12. Event statistics of spontaneous fusion of DCVs docked in triggering conditions with planar supported bilayers of different lipid compositions.
table S13. Event statistics of triggered fusion of DCVs with planar supported bilayers of different lipid compositions.
table S14. Event statistics of spontaneous fusion of DCVs docked in triggering conditions with knockdowns of syt-1/9 or CAPS with subsequent recoveries.
table S15. Event statistics of triggered fusion of DCVs with knockdowns of syt-1/9 or CAPS and subsequent recoveries.
table S16. Table of primers described in the “Plasmids and shRNA constructs” section of Materials and Methods.
table S17. Event statistics for DCV docking and fusion to planar supported bilayers with the indicated combination of proteins syntaxin-1a (residues 183 to 288), syntaxin-1a (residues 1 to 288), and SNAP-25 (lipid/protein = 3000) incubated, as indicated, with Munc18 (0.5 μM), complexin-1 (2 μM), and synaptobrevin-2 (residues 1 to 96) inhibitor peptide.
table S18. Event statistics for DCV docking and fusion to planar supported bilayers containing syntaxin-1a (residues 1 to 288):SNAP-25, 0.5 μM Munc18, and indicated amounts of complexin-1.