

Abstract
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is a persistent environmental contaminant which elicits hepatotoxicity through activation of the aryl hydrocarbon receptor (AhR). Male C57BL/6 mice orally gavaged with TCDD (0.01–30 µg/kg) every 4 days for 28 days exhibited bile duct proliferation and pericholangitis. Mass spectrometry analysis detected a 4.6-fold increase in total hepatic bile acid levels, despite the coordinated repression of genes involved in cholesterol and primary bile acid biosynthesis including Cyp7a1. Specifically, TCDD elicited a >200-fold increase in taurolithocholic acid (TLCA), a potent G protein-coupled bile acid receptor 1 (GPBAR1) agonist associated with bile duct proliferation. Increased levels of microbial bile acid metabolism loci (bsh, baiCD) are consistent with accumulation of TLCA and other secondary bile acids. Fecal bile acids decreased 2.8-fold, suggesting enhanced intestinal reabsorption due to induction of ileal transporters (Slc10a2, Slc51a) and increases in whole gut transit time and intestinal permeability. Moreover, serum bile acids were increased 45.4-fold, consistent with blood-to-hepatocyte transporter repression (Slco1a1, Slc10a1, Slco2b1, Slco1b2, Slco1a4) and hepatocyte-to-blood transporter induction (Abcc4, Abcc3). These results suggest that systemic alterations in enterohepatic circulation, as well as host and microbiota bile acid metabolism, favor bile acid accumulation that contributes to AhR-mediated hepatotoxicity.
Introduction
Metabolic syndrome (MetS) is defined as a collection of cardiometabolic factors including obesity, dyslipidemia, hypertension, and hyperglycemia which increase the risk of developing cardiovascular disease, type II diabetes, and hepatocellular carcinoma1. The prevalence of MetS among adults in the United States is ~30%, representing an emerging epidemic of concern as the population ages2. In the liver, MetS can manifest as non-alcoholic fatty liver disease (NAFLD) where hepatic steatosis (lipid accumulation) progresses to steatohepatitis (steatosis with inflammation) with fibrosis (collagen deposition)3. High fat diet, sedentary behavior, and various genetic loci are key risk factors for MetS and NAFLD development. However, accumulating evidence suggests that exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and other environmental contaminants play an underappreciated role4–6.
TCDD is the prototypical ligand for a structurally diverse group of synthetic chemicals, natural products, and endogenous metabolites that activate the aryl hydrocarbon receptor (AhR)7. Ligand binding initiates the dissociation of chaperone proteins, triggering translocation of the cytoplasmic AhR to the nucleus and heterodimerization with the aryl hydrocarbon receptor nuclear translocator (ARNT). In the canonical pathway, the liganded AhR-ARNT complex binds to dioxin response elements (DREs) within the promoter region of target genes, leading to recruitment of transcriptional co-regulators and altered gene expression8. However, an increasing number of studies report DRE-independent mechanisms of differential gene expression9, 10. AhR-mediated gene expression changes elicit a broad spectrum of toxic responses including wasting, tumor promotion, immunosuppression, dermal lesions, teratogenicity, and hepatotoxicity in a species-, sex-, age-, tissue-, and cell-specific manner11, 12. In mice, TCDD-elicits AhR-dependent lipid accumulation, immune cell infiltration, and bile duct proliferation13–15, which progresses to steatohepatitis with fibrosis following repeated treatment16, 17. Beyond AhR activation, the specific target genes and altered biological processes that contribute to TCDD-elicited hepatotoxicity remain poorly understood.
Bile acids, the predominant organic solutes in bile, are synthesized from cholesterol in the liver, stored in the gallbladder, and secreted into the duodenal lumen following consumption of a meal. These amphipathic molecules promote the solubilization and absorption of dietary lipids and lipid-soluble vitamins, and provide an excretion mechanism for excess hepatic cholesterol. Additionally, specific bile acid species are endogenous ligands for a number of receptors including farnesoid X receptor (FXR), G protein-coupled bile acid receptor 1 (GPBAR1), pregnane X receptor (PXR), vitamin D receptor (VDR), liver X receptor α (LXRα), and sphingosine-1-phosphate receptor 2 (S1P2)18, 19. Consequently, they serve as important signaling molecules that regulate not only bile and cholesterol homeostasis, but also glucose and energy metabolism. Moreover, bile acids can elicit hepatocyte and non-parenchymal cell injury by disrupting cell membranes through their detergent action and by contributing to oxidative stress through reactive oxygen species (ROS) generation20. Furthermore, they promote inflammation by inducing pro-inflammatory mediators such as cytokines (IL-1β, CSF1), chemokines (CXCL1, CXCL2, CCL2), and adhesion molecules (VCAM-1)21. The dysregulation of mitochondrial function, depletion of ATP, and activation of signaling cascades can trigger bile acid-induced necrosis20. Therefore, bile acid accumulation and/or disrupted biliary flow (cholestasis) can contribute to hepatotoxicity and impaired liver function, while promoting the development of fibrosis.
Previous studies have reported that TCDD and related AhR ligands increase bile acid levels in serum22, 23, decrease biliary flow24, and induce bile duct epithelial cell proliferation25. TCDD also alters hepatic expression of genes associated with cholesterol metabolism and bile acid biosynthesis26, and exacerbates cholestatic liver disease induced by bile duct ligation (BDL)27. To further investigate AhR-mediated qualitative and quantitative changes in bile acid profiles, liquid chromatography mass spectrometry (LC-MS) was integrated with hepatic and ileal RNA-Seq analyses, along with complementary histopathological assessments. The effects of TCDD on intestinal permeability and motility, and gut microbiota bile acid metabolism were also examined. Despite the coordinated repression of genes involved in cholesterol and primary bile acid biosynthesis, total bile acid levels increased in the liver and serum, while fecal levels decreased. In particular, TCDD increased levels of hepatic taurolithocholic acid (TLCA), a potent GPBAR1 agonist associated with bile duct proliferation and cholestasis28, 29. Overall, TCDD-elicited differential expression of bile acid transporters, increased intestinal permeability, decreased gut motility, and alterations in the gut microbiome suggest disruption of enterohepatic circulation and bile acid metabolism. Collectively, the qualitative and quantitative changes in bile acid homeostasis are consistent with alterations in host and intestinal microbiota metabolism, yielding hepatotoxic species that contribute to the development of steatohepatitis with fibrosis.
Results
Histopathology
Previous studies report hepatic steatosis, immune cell infiltration, fibrosis, and bile duct proliferation in male mice following treatment with TCDD and related AhR agonists14, 17, 25, 30, 31. Comparable effects are reported in female C57BL/6 mice orally gavaged with TCDD every 4 days (d) for either 28 or 92d16, 32. Here, the histopathological features associated with TCDD-elicited hepatotoxicity in both female and male C57BL/6 mice orally gavaged with TCDD every 4d for 28d were compared. Histopathological scoring suggests the incidence and severity of vacuolization, inflammatory cell infiltration, bile duct proliferation, and periportal fibrosis were consistently greater in male mice compared to females (Table 1; Fig. 1). More specifically, hepatocyte vacuolization (fatty change) was first observed in males at 0.3 µg/kg and in females at 10 µg/kg TCDD. Fat accumulation was more severe in males compared to females at ≥3 µg/kg TCDD (Table 1). QuHAnT analysis of ORO-stained sections indicated that hepatic fat accumulation at 30 µg/kg TCDD was 6.2-fold greater in males compared to females (Fig. 1; Supplementary Fig. S1), consistent with the pathologist’s assessment. Minimal to slight hepatocyte necrosis and inflammatory cell infiltration (e.g. neutrophils and lymphocytes) were present in males at ≥3 µg/kg and in females at 30 µg/kg. Both were more severe in males at 30 µg/kg compared to the females (Table 1). F4/80 labeling revealed that lymphocyte foci consisted primarily of macrophages, suggesting infiltration (Fig. 1). Bile duct proliferation was observed in both sexes at 30 µg/kg, with a higher incidence in males, while inflammation surrounding the bile ducts (pericholangitis) was exclusive to males (Table 1; Fig. 2A,B). Similarly, PSR-staining revealed that the incidence of periportal fibrosis at 30 µg/kg was higher in males compared to females (Table 1; Fig. 1). In summary, male mice exhibited greater sensitivity to TCDD-elicited hepatotoxicity compared to females, and exhibited more severe histopathology at lower doses. Subsequent studies therefore focused on responses in male mice.
Table 1.
Comparison of TCDD-elicited histopathological changes between male and female C57BL/6 mice.
| TCDD (µg/kg) | 0.3 | 1 | 3 | 10 | 30 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Male | Female | Male | Female | Male | Female | Male | Female | ||||
| Number of animals | 8 | 8 | 8 | 6 | 8 | 6 | 8 | 4 | 8 | 4 | ||||
| Vacuolation (centrilobular) | Mean Grade | 0.1 ± 0.4 | — | 0.9 ± 0.8 | — | 1.0 ± 0.8 | — | * | 3.0 ± 0.5 | 0.5 ± 0.3 | * | 4.0 ± 0.0 | 2.0 ± 1.2 | * |
| Incidence | 1/8 | 0/8 | 5/8 | 0/6 | 6/8 | 0/6 | 8/8 | 1/4 | 8/8 | 4/4 | ||||
| Necrosis; Inflammatory cell foci | Mean Grade | — | — | — | — | 0.3 ± 0.5 | — | 0.8 ± 0.5 | — | 1.3 ± 0.5 | 0.3 ± 0.5 | * | ||
| Incidence | 0/8 | 0/8 | 0/8 | 0/6 | 2/8 | 0/6 | 6/8 | 0/4 | 8/8 | 1/4 | ||||
| Bile duct proliferation | Mean Grade | — | — | — | — | — | — | — | — | 0.9 ± 0.8 | 0.3 ± 0.5 | |||
| Incidence | 0/8 | 0/8 | 0/8 | 0/6 | 0/8 | 0/6 | 0/8 | 0/4 | 5/8 | 1/4 | ||||
| Fibrosis (periportal) | Mean Grade | — | — | — | — | — | — | — | — | 0.6 ± 0.5 | 0.3 ± 0.5 | |||
| Incidence | 0/8 | 0/8 | 0/8 | 0/6 | 0/8 | 0/6 | 0/8 | 0/4 | 5/8 | 1/4 | ||||
Histopathological grade: 0 = Within normal limits, 1 = Minimal, 2 = Slight, 3 = Moderate, 4 = Marked. No noteworthy hepatic changes were observed in mice treated with sesame oil vehicle or ≤0.1 µg/kg TCDD. Mean Grade = Mean of histopathological grades ±SD. *p ≤ 0.05 (compared between males and females at the same dose, Mann—Whitney rank sum test). Incidence = Number of mice in which the histopathological feature was present.
Figure 1.
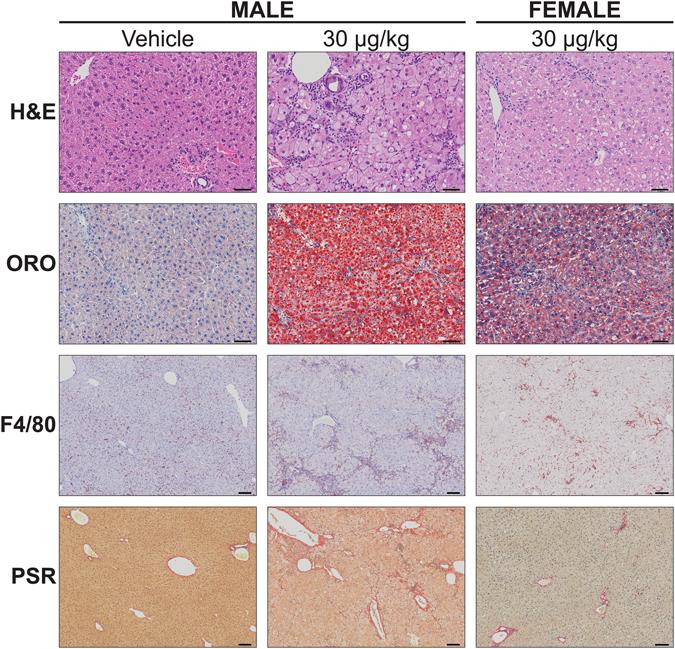
Histological evaluation of livers from male and female C57BL/6 mice orally gavaged with sesame oil vehicle or 30 µg/kg TCDD every 4 days for 28 days. Livers were stained with hematoxylin and eosin (H&E) for general assessment, Oil Red O (ORO) for neutral lipids, F4/80 for macrophages, and Picrosirius Red (PSR) for collagen deposition. Scale bars represent 50 µm for H&E and ORO, and 100 µm for F4/80 and PSR.
Figure 2.
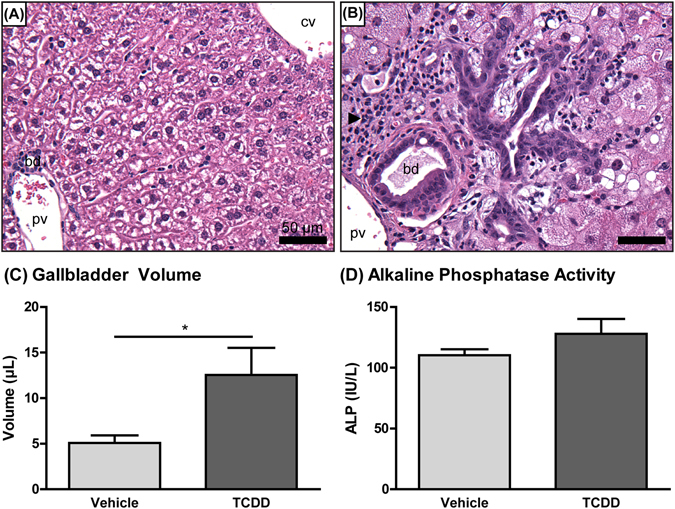
Representative photomicrographs of hematoxylin and eosin-stained livers from male C57BL/6 mice orally gavaged with (A) sesame oil vehicle or (B) 30 µg/kg TCDD every 4 days for 28 days. Bile duct proliferation and pericholangitis (arrow) were identified at 30 µg/kg TCDD. cv = central vein; pv = portal vein; bd = bile duct; bar = 50 µm. (C) Gallbladder volume and (D) serum alkaline phosphatase (ALP) activity of male C57BL/6 mice orally gavaged with sesame oil vehicle or 30 µg/kg TCDD every 4 days for 28 days. Bars represent the average ± standard error of the mean for at least 5 biological replicates. Statistical significance (*p ≤ 0.05) was determined using a Student’s t-test performed in SAS 9.3.
Gross Morphology and Clinical Chemistry
Terminal body weights and tissue weights for the mice used in this study were previously reported33. Gallbladder volume increased 2.5-fold in mice treated with 30 µg/kg TCDD compared to controls (Fig. 2C). The gallbladder distention is consistent with previous studies31. Serum alkaline phosphatase (ALP) activity was unchanged at 30 µg/kg, suggesting TCDD did not damage bile duct epithelial cells (cholangiocytes) (Fig. 2D).
Cholesterol and Primary Bile Acid Biosynthesis
Cholesterol is not only essential for maintaining cell membrane fluidity, but it also serves as the precursor for steroid hormone and bile acid biosynthesis. In mammals, the liver is the primary site of acetyl-CoA conversion to cholesterol, while the intestine also exhibits de novo synthesis capabilities. Hepatic cholesterol (not confirmed by high resolution MS/MS) and cholesterol ester levels increased 9.0- and 11.3-fold, respectively, in male C57BL/6 mice orally gavaged with 30 µg/kg TCDD every 4d for 28d34. The committed step of cholesterol biosynthesis involves the conversion of 3-hydroxy-3-methylglutaryl-CoA to mevalonate catalyzed by the rate-limiting enzyme 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). Hepatic expression of Hmgcr was dose-dependently induced at 0.3–10 µg/kg TCDD (max 3.4-fold), with no significant induction at 30 µg/kg (Fig. 3). Other enzymes involved in cholesterol biosynthesis including mevalonate kinase (Mvk), mevalonate diphosphate decarboxylase (Mvd), squalene epoxidase (Sqle), lanosterol synthase (Lss), methylsterol monoxygenase 1 (Msmo1), and 24-dehydrocholesterol reductase (Dhcr24) exhibited a comparable dose-dependent expression pattern (Fig. 3). This suggests that at low doses, AhR activation induces cholesterol biosynthesis, consistent with a 10.3-fold increase in AhR genomic binding at the Hmgcr loci. However, negative feedback at 30 µg/kg TCDD may limit de novo biosynthesis.
Figure 3.
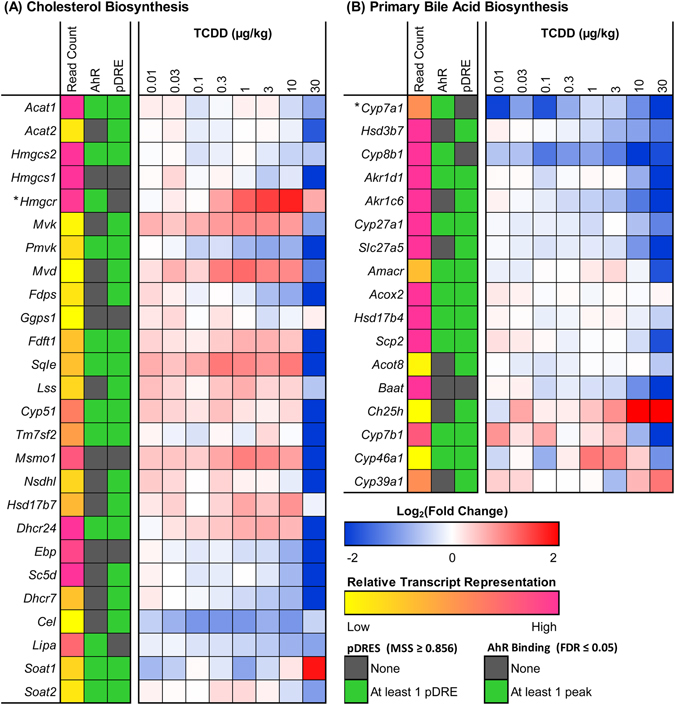
TCDD-elicited hepatic differential expression of genes involved in (A) cholesterol and (B) primary bile acid biosynthesis in male C57BL/6 mice orally gavaged with sesame oil vehicle or TCDD (0.01–30 µg/kg TCDD) every 4 days for 28 days. Color scale represents the log2(fold change) for differential gene expression relative to vehicle controls, as determined through RNA-Seq analysis (n = 3). The presence of pDREs (MSS ≥ 0.856) and AhR enrichment peaks (FDR ≤ 0.05) at 2 h are shown as green boxes. Read count represents the raw number of aligned reads to each transcript indicating potential level of expression, where yellow represents a lower level of expression (≤500 reads) and pink represents a higher level of expression (≥10,000). The rate-limiting enzyme of each pathway is marked with an asterisk (*).
Several genes involved in cholesterol biosynthesis, including 3-hydroxy-3-methylglutaryl-CoA synthase 1 (Hmgcs1), Hmgcr, farnesyl diphosphate synthetase (Fdps), and farnesyl diphosphate farnesyl transferase 1 (Fdft1), are transcriptionally regulated by sterol regulatory element-binding protein 2 (SREBP2) binding to sterol response elements (SREs). Elevated intracellular cholesterol levels prevent intramembrane SREBP2 proteolysis and subsequent nuclear translocation, thereby reducing SRE-regulated gene transcription. Consequently, higher hepatic cholesterol levels may repress SREBP2-mediated transcription. Furthermore, Srebf2 (encodes SREBP2) was repressed 2.7-fold at 30 µg/kg TCDD (Supplementary Table S1), which may further compromise expression of cholesterol biosynthetic genes.
Approximately 90% of excess cholesterol is excreted from the body by hepatic conversion to bile acids. In humans, the two primary bile acids synthesized directly by the liver are cholic acid (CA) and chenodeoxycholic acid (CDCA). Rodent livers hydroxylate CDCA at the 6β-positon yielding α-muricholic acid (α-MCA), which can epimerize at the 7-OH position to form β-muricholic acid (β-MCA), and therefore α/β-MCA are also primary bile acids in mice35. In the classical bile acid biosynthesis pathway, cholesterol is directly 7α-hydroxylated by CYP7A1, representing the rate-limiting step. Alternative pathways involve initial hydroxylation at C24, C25, or C27 catalyzed by CYP46A1, cholesterol 25-hydroxylase (CH25H), and CYP27A1, respectively, followed by 7α-hydroxylation. TCDD repressed Cyp7a1 39.7-fold with a 2.3-fold enrichment in AhR genomic binding (Fig. 3). Furthermore, 9 of the 12 downstream enzymes involved in the classical pathway were also repressed, including hydroxy-delta-5-steroid dehydrogenase 3β7 (Hsd3b7; 2.5-fold), Cyp8b1 (4.4-fold), aldo-keto reductase 1D1 (Akr1d1; 16.3-fold), aldo-keto reductase 1C6 (Akr1c6; 22.1-fold), Cyp27a1 (2.4-fold), Slc27a5 (16.0-fold), α-methylacyl-CoA racemase (Amacr; 3.3-fold), sterol carrier protein 2 (Scp2; 3.4-fold), and bile acid-Coenzyme A: amino acid N-acyltransferase (Baat; 12.0-fold) (Fig. 3). With the exception of CYP8B1, which is unique to CA synthesis, these enzymes are required for synthesis of all murine primary bile acids. Interestingly, AhR binding was enriched 2.2-fold within Cyp8b1, suggesting AhR-mediated regulation of the CDCA to CA ratio. There are four genes unique to the alternative pathway of bile acid synthesis: Cyp46a1 and Cyp7b1, which were repressed 2.1- and 7.6-fold, respectively, as well as Cyp39a1 and Ch25h, which were induced 2.1- and 6.1-fold, respectively, by TCDD (Fig. 3).
Bile acid biosynthesis is primarily regulated by two negative feedback loops that repress Cyp7a1, both involving bile acid-mediated activation of FXR. Hepatic FXR induces short heterodimer partner (Shp, aka Nr0b2), which then antagonizes liver receptor homolog 1 (LRH-1)- and hepatocyte nuclear factor 4α (HNF4α)-mediated Cyp7a1 expression. However, TCDD repressed hepatic Nr0b2 22.2-fold (Supplementary Table S1). Alternatively, activation of intestinal FXR induces ileal fibroblast growth factor 15 (Fgf15) expression. FGF15 binds its hepatic heterodimer receptor consisting of fibroblast growth factor receptor 4 (FGFR4) and klotho β (KLB), which causes Cyp7a1 repression and regulates bile flow. Yet, hepatic Fgfr4 and Klb were repressed 4.7- and 8.1-fold respectively, while ileal Fgf15 was repressed 9.7-fold at 30 µg/kg TCDD (Supplementary Table S1). Therefore, it is unlikely that either feedback loop is responsible for Cyp7a1 repression.
Hepatic, Serum, and Fecal Bile Acid Profiles
HRAM-LC-MS analysis detected a dose-dependent increase in total hepatic bile acids, with 2.1- and 4.6-fold increases at 10 and 30 µg/kg TCDD, respectively (Fig. 4). CA, the predominant primary hepatic bile acid in control samples, increased 4.7-fold following treatment with 30 µg/kg TCDD. α/β-MCA was the second most abundant primary bile acid in controls, while hepatic CDCA levels were negligible. TCDD had no effect on hepatic α/β-MCA or CDCA, but dose-dependently increased total hepatic conjugated primary bile acid levels (max 4.7-fold), including glycocholic acid (GCA; 3.9-fold), taurocholic acid (TCA; 9.7-fold), and tauro-α-muricholic acid (T-α-MCA; 3.7-fold) (Fig. 4). Paradoxically, hepatic expression of Baat, which is responsible for hepatic taurine- and glycine-conjugation of bile acids, was repressed 12.0-fold (Fig. 3). In female mice, TCDD increased hepatic taurine (max 1.4-fold; p = 0.05) and glycine (max 20.3-fold; p = 0.10) levels36. TCDD repressed hepatic expression of taurine biosynthesis genes including cysteine dioxygenase 1 (Cdo1; 2.7-fold) and cysteine sulfinic acid decarboxylase (Csad; 4.5-fold), while the taurine transporter Slc6a6 (encoding TAUT) was induced 1.8-fold (Supplementary Table S1), suggesting hepatic taurine uptake rather than de novo synthesis. Furthermore, TCDD elicited a dose-dependent increase in total hepatic levels of conjugated secondary bile acids (max 77.7-fold), suggesting increased bacterial metabolism. More specifically, TLCA, the most induced hepatic bile acid, was dose-dependently increased 72.1- and 233.8-fold at 10 and 30 µg/kg TCDD, respectively (Fig. 4).
Figure 4.
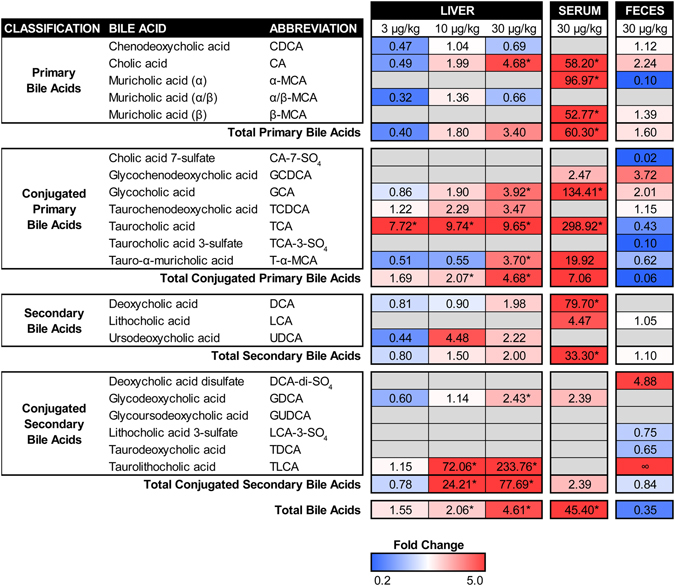
Bile acid profiles for the liver, serum, and feces of male C57BL/6 mice orally gavaged with sesame oil vehicle or TCDD (3–30 µg/kg) every 4 days for 28 days. Individual bile acid species were quantified using high resolution accurate mass liquid chromatography mass spectrometry and expressed as fold changes relative to vehicle controls. Red and blue indicate increased or decreased levels, respectively, while grey indicates ‘not detected’. ‘∞’ indicates an incalculable increase as the bile acid was detected in treated animals but not controls. Statistical significance (*p ≤ 0.05) was determined using one-way ANOVA analysis followed by Dunnett’s post-hoc test (liver) or a Student’s t-test (serum) performed in SAS 9.3. Fecal pellets were collected from two cages of co-housed mice for each treatment group (n = 2) and therefore statistical analysis was not performed.
Individual bile acids differ in their capacity to bind hepatic receptors. TLCA is the most potent endogenous activator of GPBAR1, a metabotropic bile acid receptor located on non-parenchymal cells including Kupffer cells, sinusoidal epithelial cells, and bile duct epithelial cells (cholangiocytes)28. Using qRT-PCR, hepatic Gpbar1 expression was induced 4.3-fold at 30 µg/kg TCDD (Supplementary Fig. S2). Increased hepatic TLCA levels combined with Gpbar1 induction would promote activation of GPBAR1 following TCDD treatment. Likewise, CDCA is the most potent endogenous FXR agonist, while T-α-MCA and T-β-MCA are competitive antagonists37. At low hepatic CDCA levels due to conversion to α-MCA, TCDD-elicited increases in T-α-MCA levels may inhibit FXR activation. Moreover, TCDD repressed hepatic Nr1h4 (encodes FXR) 3.2-fold, further inhibiting the FXR signaling pathway (Supplementary Table S1).
Total bile acids in serum increased 45.4-fold at 30 µg/kg TCDD (Fig. 4). Taurine- and glycine-conjugates of CA exhibited increases of 298.9- and 134.4-fold, respectively. CA, α-MCA, and β-MCA were the most abundant species present in treated serum samples with increases of 58.2-, 97.0-, and 52.8-fold, respectively. Additionally, the secondary bile acid deoxycholic acid (DCA) was increased 79.7-fold.
Total fecal bile acids decreased 2.8-fold at 30 µg/kg TCDD (Fig. 4), suggesting decreased bile secretion from the gallbladder and/or increased intestinal reabsorption. Fecal levels of cholic acid 7-sulfate (CA-7-SO4) and taurocholic acid 3-sulfate (TCA-3-SO4) were decreased 55.2- and 10.3-fold, respectively, while deoxycholic acid disulfate (DCA-di-SO4) levels increased 4.9-fold. The former is consistent with a 267.5-fold repression of Sult2a8, the predominant sulfotransferase in the male liver based on RNA-Seq read counts (Supplementary Table S1). TCDD also decreased α-MCA fecal levels 10.3-fold, while TLCA levels changed from undetectable in controls to detectable in treated mice.
Bile Acid Hydrophobicity
In general, bile acid toxicity increases with hydrophobicity38, 39, which depends on ionization, hydroxylation, and conjugation (taurine, glycine, sulfate, glucoronate)40. The potential toxicity of a bile acid mixture can be estimated by calculating the hydrophilic-hydrophobic balance using the hydrophobicity index (HIx) of each bile acid species in the mixture, as described by Heuman41. The HIx of TCA was arbitrarily set to 0, with increasingly positive and negative values assigned to species exhibiting relatively greater hydrophobicity and hydrophilicity, respectively. TCDD (3–30 µg/kg) shifted the hepatic bile acid mixture HIx toward greater hydrophobicity (Fig. 5), suggesting increased hepatotoxic potential. Each bile acid’s contribution to the overall HIx is dependent on its relative abundance within the mixture, and therefore low abundance species such as TLCA have lower impact. Consequently, the shift towards increased hydrophobicity is primarily driven by a decrease in the relative abundance of T-α-MCA, a highly hydrophilic species. In contrast, the serum bile acid mixture became more hydrophilic, while the fecal mixture exhibited a moderate increase in hydrophobicity (Fig. 5).
Figure 5.
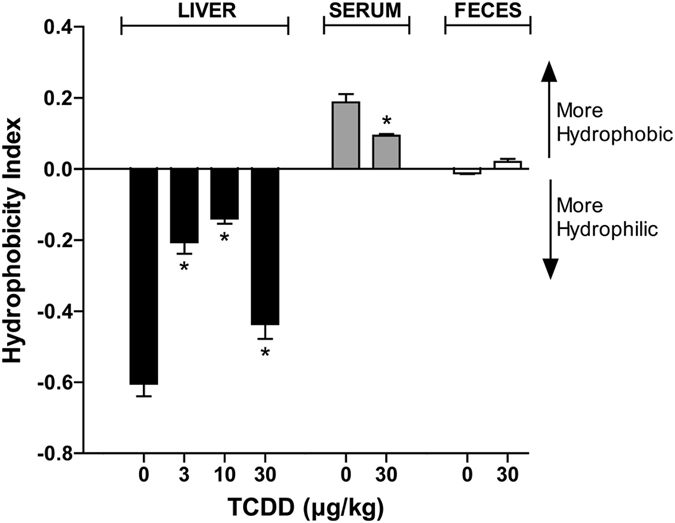
Hydrophobicity indices (HIx) of bile acid mixtures in the liver, serum, and feces of male C57BL/6 mice orally gavaged with sesame oil vehicle or TCDD (3–30 µg/kg TCDD) every 4 days for 28 days. Bile acid toxicity is positively associated with the degree of hydrophobicity, where increasingly positive and negative HIx values indicate greater hydrophobicity and hydrophilicity, respectively. Bars represent the average ± standard error of the mean for at least 4 biological replicates for liver and serum, or 2 pooled samples for feces. Statistical significance (*p ≤ 0.05) was determined using one-way ANOVA analysis followed by Dunnett’s post-hoc test (liver) or a Student’s t-test (serum) performed in SAS 9.3. Fecal pellets were collected from two cages of co-housed mice for each treatment group (n = 2) and therefore statistical analysis was not performed.
CYP3A enzymes play a role in bile acid metabolism by catalyzing 6α-, 7α-, and 6β-hydroxylation42, 43. TCDD repressed all hepatic Cyp3a isoforms including 3a13, 3a25, 3a16, 3a11, 3a59, 3a44, and 3a57 (17.0-, 6.0-, 5.2-, 4.9-, 4.4-, 3.0-, 2.4-fold), as well as ileal Cyp3a44 (2.2-fold) (Supplementary Table S1). Cyp3a repression may decrease bile acid hydroxylation, thereby increasing the hydrophobicity of the bile acid pool.
Enterohepatic Circulation
Following synthesis, conjugated primary bile acids are secreted into the bile canaliculi by canalicular bile salt export pump (BSEP; aka ABCB11) and ABCC2. TCDD repressed hepatic Abcb11 4.7-fold, while Abcc2 was unchanged (Fig. 6). Reduced hepatocyte-to-canaliculi export, combined with increased levels of hepatic conjugated primary bile acids likely caused spill-over into the sinusoidal blood and systemic circulation. Conjugated primary bile acids stored in the gallbladder are released into the duodenum in response to the intestinal hormone cholecystokinin following a meal, and progressively pass through the small intestine. Upon reaching the ileum, they are taken up into enterocytes by apical SLC10A2, and exported to the portal circulation by basal SLC51A and SLC51B. TCDD induced ileal expression of Slc10a2 and Slc51a 1.6- and 1.5-fold, respectively, while ileal Slc51b was unchanged (Fig. 6). Induction of these intestinal transporters likely promotes active reabsorption of bile acids across the ileum. Circulating bile acids are then transported into hepatocytes by Slc10a1, Slco1a1, Slco1a4, Slco1b2, and Slco2b1, which were repressed 17.9-, 1186.3-, 2.5-, 7.9-, and 12.9-fold, respectively, by TCDD (Fig. 6). Specifically, SLC10A1 is primarily responsible for taurine- and glycine-conjugated bile acid uptake, while SLCO1B2 plays a key role in unconjugated bile acid uptake44, 45. Abcc3 and Abcc4, which are responsible for bile acid efflux from hepatocytes into systemic circulation, were induced 1.8- and 58.1-fold at 30 µg/kg TCDD (Fig. 6). Collectively, repression of hepatic importers combined with induction of hepatic exporters likely contributed to the 45.4-fold increase in total serum bile acids.
Figure 6.
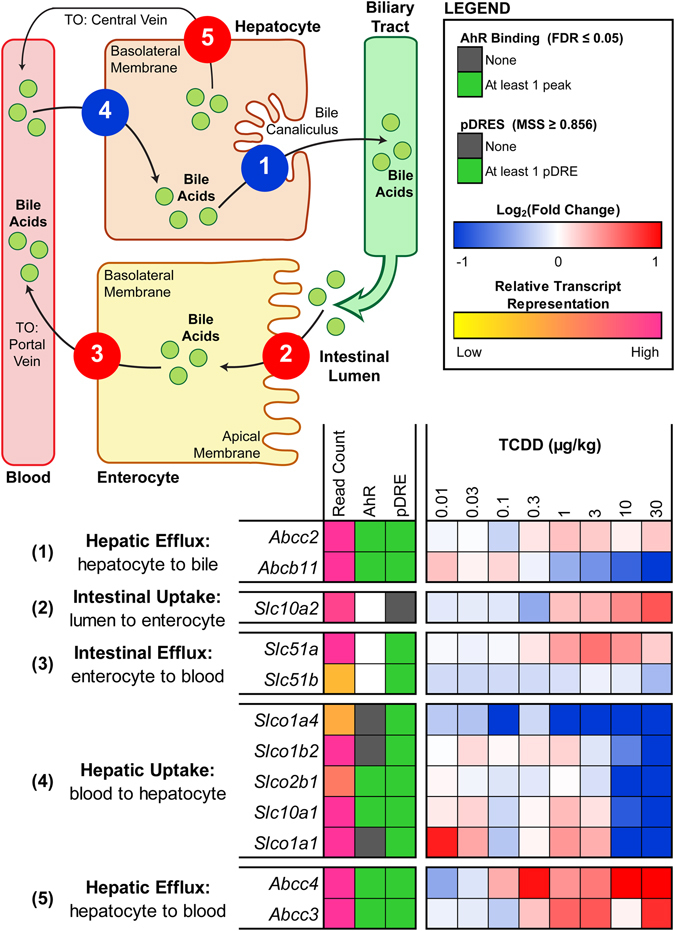
TCDD-elicited differential expression of bile acid transporters in the liver and ileum of male C57BL/6 mice orally gavaged with sesame oil vehicle or 0.01–30 µg/kg TCDD every 4 days for 28 days. Color scale represents the log2(fold change) for differential gene expression relative to vehicle controls, as determined through RNA-Seq analysis (n = 3). The numeric labels to the left of the heat map correspond to the numbered transport functions in the pictorial representation, where the color of each function represents the overall direction of differential expression exhibited by the group of transporters involved (i.e. induced = red vs. repressed = blue). The presence of pDREs (MSS ≥ 0.856) and hepatic AhR enrichment peaks (FDR ≤ 0.05) at 2 h are shown as green boxes. Read count represents the raw number of aligned reads to each transcript indicating potential level of expression, where yellow represents a lower level of expression (≤500 reads) and pink represents a higher level of expression (≥10,000).
Approximately 95% of bile acids secreted into the intestinal lumen are reabsorbed with the remaining 5% lost via fecal excretion18. Highly efficient enterohepatic circulation allows the liver to maintain a low rate of de novo primary bile acid biosynthesis. At 30 µg/kg TCDD, sugar probe analysis revealed a 2.4- and 2.0-fold increase in gastroduodenal and colonic para-cellular (i.e. between enterocytes) permeability, respectively, while the permeability of the small intestine was unaffected (Fig. 7A–C). Furthermore, whole gut transit time increased from 2.5 h in controls to 4.1 h in 30 µg/kg TCDD-treated mice, indicating a 1.6-fold reduction in gut motility (Fig. 7D,E). Induction of active ileal transporters (Slc10a2, Slc51a), increased passive para-cellular duodenal and colonic transport, and decreased gut motility are in accordance with increased intestinal reabsorption and decreased fecal excretion of bile acids. These changes in intestinal function are also consistent with TCDD-elicited increases in total serum bile acids and decreases in total fecal bile acids.
Figure 7.
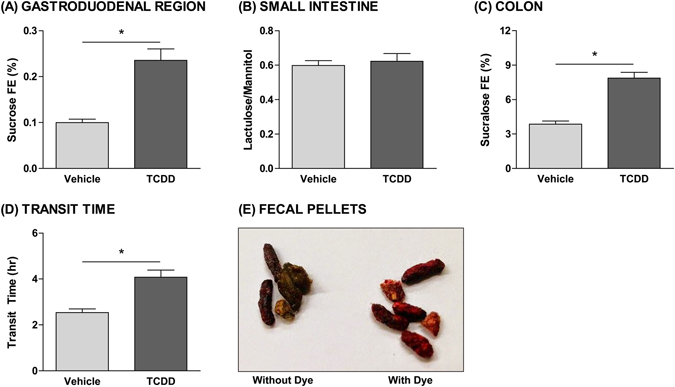
Effects of TCDD on intestinal permeability and motility. Para-cellular permeability of the (A) gastroduodenal region, (B) small intestine, and (C) colon in male C57BL/6 mice orally gavaged with sesame oil vehicle or 30 µg/kg TCDD every 4 days. At 26 days after the initial dose, mice were orally gavaged with sucrose, lactulose, mannitol, and sucralose to assess segment-specific intestinal permeability. Urine was collected over a 5-hour period, and fractional excretion (FE) of the probes was measured using ultra high performance liquid chromatography (UPLC) tandem mass spectrometry (MS/MS). (D) Whole gut transit time of male C57BL/6 mice orally gavaged with sesame oil vehicle or 30 µg/kg TCDD every 4 days. At 27 days after the initial dose, transit time was monitored by orally gavaging mice with Carmine Red and then measuring the interval between the gavage and the first observance of dye in the fecal pellets. (E) Fecal pellets collected from mice following oral gavage with Carmine Red dye (right) compared to pellets lacking the dye (left). Bars represent the average ± standard error of the mean for at least 6 biological replicates. Statistical significance (*p ≤ 0.05) was determined using a Student’s t-test performed in SAS 9.3.
Microbial Bile Acid Metabolism
Bile acids not reabsorbed by ileal enterocytes are metabolized by microbiota in the colon, the intestinal segment with the densest and most diverse microbial population. Various bacterial genera metabolize primary and conjugated primary bile acids to yield “secondary” bile acids. Bacterial bile salt hydrolase (BSH) cleaves the C24 N-acyl amide bond of conjugated bile acids, removing the glycine or taurine moiety. Several gut bacterial genera express bsh including Bacteroides, Bifidobacterium, Clostridium, Lactobacillus, and Listeria 46. The exposed 7-hydroxyl groups of unconjugated CA and CDCA are susceptible to bacterially-catalyzed 7α-dehydroxylation, yielding the secondary bile acids DCA and lithocholic acid (LCA), respectively. Bile acid 7α-dehydroxylation is a multi-step enzymatic process involving a set of genes encoded within the bile acid-inducible (bai) operon including baiCD, the oxidoreductase enzyme which catalyzes 3-dehydro-4-bile acid oxidation47. To date, 7α-dehydroxylation activity has only been identified in the Clostridium and Eubacterium genera46.
As opposed to evaluating changes in the abundance of specific bacterial species associated with bile acid metabolism, we examined changes in the levels of bsh and baiCD, the loci responsible for bile acid deconjugation and dehydroxylation, respectively. Bacterial DNA extracted from fecal pellets was analyzed by qRT-PCR using computationally-designed degenerate primer sets. Overall, bsh gene levels within phylogenetic groups 1, 2, and 3 were increased, while levels within phylogenetic group 4 were unaffected by TCDD (Fig. 8A2). In particular, primer sets 7 and 8 within group 1 increased 21.8- and 7.3-fold, respectively, while primer sets 2 and 11 within group 2 increased 7.6- and 7.5-fold, respectively, and primer set 9 within group 3 increased 4.2-fold at 30 µg/kg TCDD. These primer sets primarily target Lactobacillus (Group1-Primer7; Group2-Primer2; Group2-Primer11), Clostridium (Group1-Primer8), Streptococcus (Group2-Primer11), and Listeria (Group3-Primer9) species, suggesting TCDD increased the relative abundance of several species within these genera. Primers targeting Clostridium baiCD 48, 49 identified a 2.1-fold increase at 30 µg/kg TCDD (Fig. 8B), suggesting increased relative abundance of Clostridium strains known to exhibit 7α-dehydroxylation activity. Similarly, 7 of the 8 degenerate primer sets designed to target baiCD across the gut microbiome exhibited increased levels (1.5- to 2.5-fold). Consistent with these results, TCDD is reported to increase the abundance of several Lactobacillus and Clostridium species in the mouse gut microbiota50. Together, these results suggest that TCDD increased deconjugation and 7α-dehydroxylation activity within the gut microbiota, consistent with increased secondary bile acids such as DCA in the serum and increased conjugated secondary bile acids such as TLCA and GDCA in the liver (Fig. 9).
Figure 8.
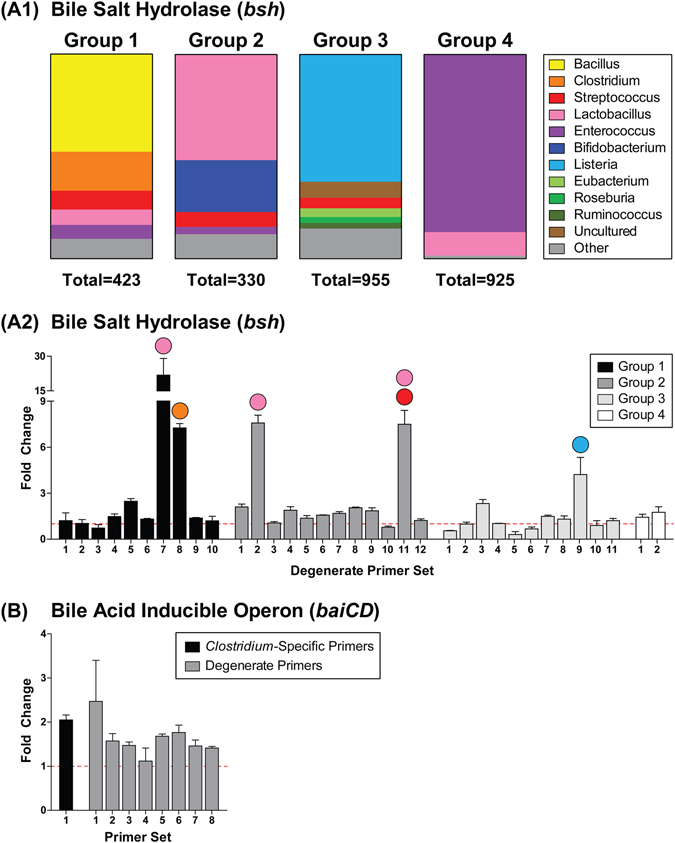
Effects of TCDD on bacterial genes involved in bile acid metabolism. (A1) Bacterial classifications (genus level) of the bile salt hydrolase (bsh) sequences in each phylogenetic group, which were separated based on nucleotide similarity. Degenerate qRT-PCR analysis of (A2) bsh and (B) bile acid-inducible operon (baiCD) gene levels in the fecal pellets of male C57BL/6 mice orally gavaged with sesame oil vehicle or 30 µg/kg TCDD every 4 days for 28 days. Fecal levels of baiCD were also quantified using a primer set previously validated to target several Clostridium species which exhibit 7α-dehydroxylation activity (B: black bar). Colored circles above highly increased bsh primer sets indicate the bacterial genus which is primarily targeted. Values were normalized to levels of the 16S ribosomal RNA gene. Bars represent the average fold change relative to vehicle controls ± standard error of the mean. Fecal pellets were collected from two cages of co-housed mice for each treatment group (n = 2) and therefore statistical analysis was not performed.
Figure 9.
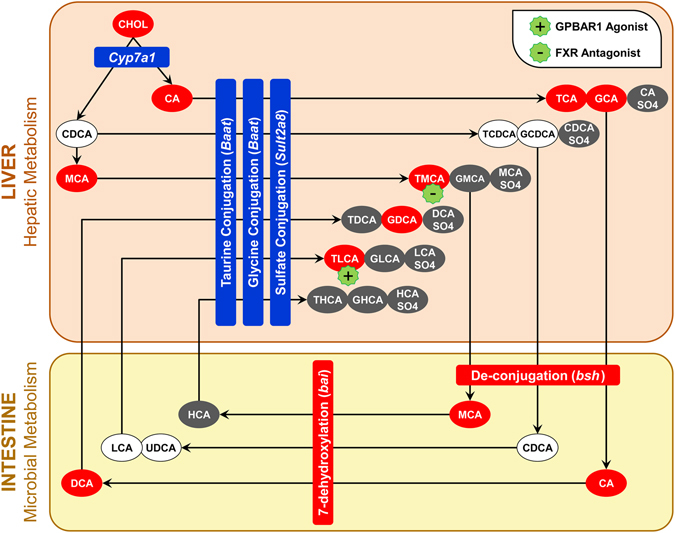
The effect of TCDD on the hepatic and microbial metabolism of individual bile acid species in male C57BL/6 mice orally gavaged with sesame oil vehicle or TCDD every 4 days for 28 days. Ovals represent bile acid species, while rectangles represent genes involved in bile acid metabolism. Placement of genes within the ‘liver’ and ‘intestine’ boxes represents the location of the metabolic reaction and the source of the enzyme involved. Red represents ‘increased levels’, blue represents ‘decreased levels’, white represents ‘no change’, and grey represents ‘not detected’. The color of each bile acid depicts the direction of change within the liver and/or serum relative to vehicle controls.
Discussion
The effects of TCDD and related compounds on bile acid levels have been previously reported in a wide variety of rodent models using various treatment regimens22, 23, 26, 30, 51, 52. This study further elucidates AhR-mediated dysregulation of bile acid homeostasis in C57BL/6 mice by mapping complementary metabolomics, RNA-Seq, histopathology, and phenotypic data to bile acid biosynthesis, transport, and metabolism pathways. We demonstrate that TCDD altered primary and secondary bile acid profiles, consistent with changes in bile acid transport, gut microbiota metabolism, and intestinal permeability and motility.
In this study, TCDD dose-dependently increased total bile acid levels in the liver and serum, as previously reported in rodent studies involving AhR agonists22, 23, 30, 51. This is in accordance with the hepatic accumulation of cholesterol and cholesterol-esters, and may represent an adverse consequence of the liver’s attempt to minimize potential lipotoxicity through the removal of excess cholesterol. Paradoxically, the majority of genes associated with bile acid biosynthesis exhibited dose-dependent repression including the rate-limiting enzyme Cyp7a1, Cyp7b1 of the alternative pathway, and Baat, which catalyzes taurine/glycine-conjugation. AhR-mediated repression of Cyp7a1 has been previously reported13, 26, 27, 52, although TCDF is reported to induce Cyp7a1 mRNA and increase CYP7A1 protein51. Interestingly, AhR binding was enriched upstream of the Cyp7a1 transcription start site in both male and female mice, in the absence of a pDRE.
Bile acid biosynthesis is regulated by negative feedback involving FXR activation. Bile acid-activated FXR induces hepatic Nr0b2 (encodes SHP), which interacts with LRH-1 and inhibits Cyp7a1 expression53. Moreover, in the distal ileum, activated FXR induces Fgf15 which binds to hepatic FGFR4/KLB to inhibit Cyp7a1 expression. However, Nr1h4 (encodes FXR), Nr0b2, Fgfr4, and Klb were all repressed by TCDD, while Fgf15 exhibited no clear expression pattern. Related AhR agonists including TCDF and flutamide also repress Nr1h4 and Nr0b2 51, 54. Furthermore, hepatic CDCA levels, the most potent FXR agonist, were negligible, while TCDD increased hepatic levels of T-α-MCA, a competitive FXR antagonist. Together, these results suggest that TCDD disrupts bile acid-induced feedback inhibition possibly by inhibiting the FXR signaling pathway.
In contrast to biosynthesis, TCDD-elicited differential expression of genes associated with enterohepatic circulation and transport is consistent with serum and hepatic bile acid accumulation. As previously reported27, 55, TCDD repressed hepatic Abcb11 (Bsep), which is responsible for transporting newly synthesized primary bile acids from hepatocytes to bile canaliculi. This would impair bile collection within the gallbladder, causing accumulation within hepatocytes and eventual spill over into sinusoidal blood. Inhibition of FXR signaling may be responsible for repression of Abcb11, a known FXR target gene. TCDD also repressed transporters involved in hepatic uptake from the blood (Slco1a1, Slc10a1, Slco2b1, Slco1b2, and Slco1a4) and induced hepatic efflux transporters (Abcc4, Abcc3). Accordingly, bile acid accumulation was greater in the serum compared to the liver. Serum bile acid accumulation may also be facilitated by enhanced enterocyte uptake (Slc10a2) and efflux (Slc51a), as well as passive reabsorption due to reduced gut motility and increased intestinal para-cellular permeability. As a result, TCDD reduced fecal bile acid levels, although impaired gallbladder emptying may also be a contributing factor. Overall, AhR-mediated differential expression of bile acid transporters is consistent with bile acid accumulation, as well as reduced bile flow and biliary excretion in TCDD-treated rodents24.
Increased secondary bile acid levels provide indirect evidence of microbial bile acid metabolism and changes in the gut microbiota. qRT-PCR analysis of bacterial bile acid metabolism loci revealed that TCDD increased fecal levels of bsh and the bai operon, suggesting increased gut microbiota deconjugation and dehydroxylation activity. Accordingly, serum levels of secondary bile acids such as DCA and hepatic levels of conjugated secondary bile acids including TLCA and GDCA were increased by TCDD. Decreased gut motility may also facilitate bacterial metabolism and the passive reabsorption of hydrophobic secondary bile acids.
TCDD shifted the hepatic HIx towards increased hydrophobicity, which correlates with hepatotoxicity due to membrane disruption, ROS generation, induction of pro-inflammatory mediators, and the activation of necrotic signaling. Specifically, TLCA, a conjugated secondary bile acid species with a hydrophobicity index of 1.00, exhibited the greatest hepatic increase in response to TCDD. TLCA induces cholestasis by impairing hepatobiliary exocytosis, inhibiting the insertion of transport proteins into apical membranes, and disrupting bile flow29, 56, 57. It is also the most potent endogenous agonist of GPBAR1, a metabotropic receptor highly expressed on bile duct cholangiocytes28. TLCA activation of GPBAR1 induces cholangiocyte proliferation through: (i) generation of ROS which activate Rous sarcoma oncogene (cSrc), epidermal growth factor receptor (EGFR), and ERK1/2, and (ii) inhibition of apoptosis through phosphorylation of the CD95 death receptor29. Therefore, increased levels of hepatic TLCA and other hydrophobic bile acids may contribute to TCDD-elicited hepatotoxicity by inducing bile duct proliferation and impairing bile flow.
Collectively, these results suggest that bile acid accumulation, enhanced enterohepatic circulation, and alterations in microbial metabolism contribute to TCDD-elicited hepatotoxicity and the progression of hepatic steatosis to steatohepatitis with fibrosis. Numerous studies involving whole-body AhR knockout mice have demonstrated that AhR activation is required for TCDD-elicited hepatotoxicity including bile duct proliferation15, 58. Other AhR ligands (TCDF, OCDD, PeCDF, and flutamide) also dysregulate bile acid homeostasis22, 23, 51, 54. The presence of DREs, AhR enrichment, and dose-dependent induction/repression of key genes associated with bile acid homeostasis, provide compelling evidence that these effects are AhR mediated. However, hepatocyte-specific conditional AhR knockout studies would be required to unequivocally demonstrate the role of AhR activation in TCDD-elicited dysregulation of bile acid homeostasis.
In addition to effects on bile acid homeostasis, TCDD also disrupts other hepatic processes and metabolic pathways including lipid metabolism13, 59, iron homeostasis and heme metabolism33, extracellular matrix deposition and remodeling16, and antioxidant defenses36. Accumulating evidence suggests AhR-mediated hepatotoxicity is a cumulative response to the overall burden of multiple disrupted metabolic pathways, rather than a single event. Further studies are required to determine whether TCDD and related compounds elicit comparable metabolic effects in human models.
Materials and Methods
Animal Handling and Treatment
Postnatal day 25 (PND25) male C57BL/6 mice weighing within 10% of each other were obtained from Charles River Laboratories (Kingston, NY) and housed in Innovive Innocages (San Diego, CA) containing ALPHA-dri bedding (Shepherd Specialty Papers, Chicago, IL) in a 23 °C environment with 30–40% humidity and a 12-hour light/dark cycle (7am–7 pm). Mice were provided Aquavive water (Innovive) and Harlan Teklad 22/5 Rodent Diet 8940 (Madison, WI) ad libitum, and were acclimated for 4d prior to treatment. Beginning on PND28, animals (n = 8; co-housed 4 per cage) were orally gavaged with sesame oil vehicle (Sigma-Aldrich, St. Louis, MO), 0.01, 0.03, 0.1, 0.3, 1, 3, 10, or 30 μg/kg TCDD (AccuStandard, New Haven, CT) every 4d for a total of 28d (7 exposures; Supplementary Fig. S3). The doses used compensate for the relatively short study duration compared to lifelong cumulative human exposure from diverse AhR ligands, the bioaccumulative nature of halogenated AhR ligands, and differences in TCDD’s metabolism and half-life (humans: 1–11 years60, 61, mice: 8–12d62). TCDD doses between 0.01 and 30 µg/kg result in mouse hepatic tissue levels that span human background serum concentrations reported in the United States, Germany, Spain, and the United Kingdom as well as serum levels in Viktor Yushchenko 4–39 months following intentional poisoning16. For the histopathological comparison, female C57BL/6 mice were orally gavaged with sesame oil vehicle or 0.01–30 µg/kg TCDD every 4d for 28d, as previously described32. All animal handling procedures were performed with the approval of the Michigan State University (MSU) Institutional Animal Care and Use Committee, in accordance with the relevant ethical guidelines and regulations.
Sample Collection
At 25d after the initial dose (PND53), fecal pellets from the previous 24 hours (h) were collected from each cage (4 mice per cage) and stored at −80 °C. At 28d after the initial exposure (PND56), mice (fasted for 6 h) were weighed and blood was collected from the submandibular vein prior to cervical dislocation. The gallbladder length, width, and depth were measured using a micro-caliper. Its volume was calculated assuming an ellipsoid shape: volume (µL) = length (mm) × width (mm) × depth (mm) × π/663, 64. The distal ileum (10 cm section extending from the caecum toward the jejunum) was removed, flushed with Ca2+/Mg2+-free phosphate buffered saline (PBS; Sigma), and opened longitudinally. The epithelial lining was scraped into a vial containing TRIzol (Invitrogen, Carlsbad, CA), frozen in liquid nitrogen, and stored at −80 °C. Liver, gonadal white adipose tissue (gWAT), and brown adipose tissue were removed, weighed, frozen in liquid nitrogen, and stored at −80 °C. Serum alkaline phosphatase (ALP) activity was determined using a commercial kit (Pointe Scientific, Canton, MI).
Histopathology
All processing was performed by the MSU Investigative Histopathology Laboratory (https://humanpathology.natsci.msu.edu/). Formalin-fixed hepatic tissues were vacuum infiltrated with paraffin using a Tissue-Tek VIP 2000 tissue processor (Sakura) and embedded with the Thermo Fisher HistoCentre III Embedding Center (Thermo Fisher, Waltham, Massachusetts). Paraffin blocks were sectioned at 4–5 µm with a Reichert Jung 2030 rotary microtome (Reichert, Depew, New York) and dried for 2–24 h at 56 °C to ensure adherence to the slides. For lipid staining, liver sections frozen in O.C.T. compound were sectioned at 6 µm, fixed in 10% NBF for 5 min, rinsed with water, and immersed in 100% propylene glycol for 5 min. Liver sections were stained with hematoxylin and eosin (H&E) for general histological assessment. Lipids were stained using Oil Red O (ORO) as previously described65. Macrophages were labeled using a monoclonal anti-mouse F4/80 antibody (1:100 dilution; AbD Serotec, Raleigh, NC). Collagen was stained with 0.1% Picro-Sirius Red (PSR) dye, with Weigert’s Hematoxylin nuclear dye. Histological assessment of hepatic tissue from female C57BL/6 mice was performed as previously described16, 32.
Quantitation of ORO staining was performed using the Quantitative Histological Analysis Tool (QuHAnT)66. Briefly, slides were digitized at 20x magnification using an Olympus Virtual Slide System VS110 (Olympus, PA) and images were sampled at 100% coverage using the Visiomorph Microimager (Visiopharm, Denmark). Using ImageJ (http://rsb.info.nih.gov.ij/), optimal hue, saturation, and value (HSV) image segmentation thresholds for feature extraction were determined to be 0 to 50 and 225 to 255 (hue), 125 to 255 (saturation), and 0 to 255 (value), while optimal total tissue feature extraction thresholds were 0 to 255 (hue), 20 to 255 (saturation), and 0 to 255 (value). Volume densities were estimated as the sum of positive hits (Ppositive staining) divided by the total number of tissue hits (Ptissue) for each section: Vv = (Ppositive staining/Ptissue) × 100.
RNA-Seq
Frozen ileal epithelium scrapings and liver samples were homogenized in TRIzol (Ambion, Waltham, MA) using a Mixer Mill 300 tissue homogenizer (Retsch, Germany) and total RNA was isolated as previously described13. RNA was quantified using a Nano-drop spectrophotometer (Thermo Scientific, Wilmington, DE) at 260 nm, and purity was assessed using the A260/A280 ratio and the Caliper LabChip GX (Perkin Elmer, Waltham, MA).
Dose-dependent gene expression was examined using RNA-Seq performed at the MSU Research Technology Support Facility (RTSF) Genomics Core (rtsf.natsci.msu.edu/genomics). Ileal libraries from three independent biological replicates (n = 3) were prepared using the Ovation Mouse RNA-Seq System 1–16 sample preparation kit (NuGen, San Carlos, CA), with an additional DNase step. Libraries were quantified and sequenced as previously described, using a read depth of ~30 M per sample33, 67. Quality was determined using FASTQC v0.11.3 and adaptor sequences were removed using Trimmomatic v0.33. Reads were mapped to the mouse reference genome (GRCm38 release 81) using Bowtie2 v2.2.6 and TopHat2 v2.1.0. Alignments were converted to SAM format using SAMTools v1.2.0. For TCDD-mediated differential gene expression, fold changes were calculated relative to vehicle controls. Hepatic RNA-Seq analysis was previously published33. Genes were considered differentially expressed if |fold change| ≥ 1.5 and statistical P1(t) value ≥ 0.8 at one or more doses. RNA-Seq datasets for the ileal epithelium and liver were deposited in the Gene Expression Omnibus (GEO; accession number GSE89430 and GSE87519, respectively). The RNA-Seq results (fold changes, P1(t) values) for each gene discussed in the manuscript have been compiled in Supplementary Table S1. Dose-response modeling was performed using the ToxResponse Modeler68, and median effective dose (ED50) values for differentially expressed genes exhibiting a sigmoidal response were reported in Supplementary Table S1. Estimation of benchmark dose (BMD) and BMD lower confidence limit (BMDL) was performed using BMDExpress69 as previously described32, and has been included in Supplementary Table S1.
Putative DRE Identification and Hepatic AhR ChIP-Seq
Putative dioxin response elements (pDREs) with matrix similarity scores (MSS) ≥ 0.856 were previously identified36. ChIP-Seq was previously performed on liver samples from male C57BL/6 mice 2 h after a single bolus oral gavage of 30 µg/kg TCDD33, 70. Briefly, cross-linked DNA was immunoprecipitated with either rabbit IgG or anti-AhR (H-211, sc-5579; Santa Cruz, CA). Libraries were prepared using the MicroPlex kit (Diagenode), pooled, and sequenced at a depth of a ~30 M on an Illumina HiSeq 2500 at the MSU RTSF Genomics Core. Reads were mapped to the mouse reference genome (GRCm38 release 81) using Bowtie 2.0.0 and alignments converted to SAM format using SAMTools v0.1.2. Normalization and peak calling was performed using CisGenome71, by comparing IgG control and AhR enriched samples (n = 5) using a bin size (−b) of 25 and boundary refinement resolution (−bw) of 1 with default parameters.
Hepatic Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Given the low (≤10) read counts for Gpbar1 in the RNA-Seq dataset, differential expression was confirmed by qRT-PCR13. Total hepatic RNA was reverse transcribed by SuperScript II (Invitrogen) using oligo dT primer according to the manufacturer’s protocol. PCR amplification was conducted on a Bio-Rad CFX Connect Real-Time PCR Detection System. Gene expression relative to vehicle control was calculated using the 2−∆∆CT method, where each sample was normalized to the geometric mean of 3 housekeeping genes (Actb, Gapdh, and Hprt). Gene expression data are plotted relative to vehicle control. Forward and reverse primer sequences are provided in Supplementary Table S2.
LC-MS Analysis of Bile Acids
Serum (10 µL) was added to 10 µL of a 20 ng/µL stock solution of deuterated (d4) glycochenodeoxycholic acid (GCDCA, from Steraloids, Inc.) in methanol, and extracted with ice-cold methanol as previously described72. Frozen liver samples (50 mg) or dried fecal pellets (50 mg) were combined with 10 µL of 20 ng/µL d4-GCDCA stock solution and 1.0 mL of ice-cold methanol72, and homogenized with 0.5 mm zirconium oxide beads using an air-cooled Bullet Blender (NextAdvance). Extracts were dried under vacuum, reconstituted in 2.0 mL of 50% methanol, filtered through 0.2 µm nylon membranes (Pall Life Sciences), and stored at −80 °C.
Bile acids were analyzed by high resolution/accurate mass (HRAM)-LC-MS using an Agilent 1260 capillary HPLC coupled to a Thermo Scientific LTQ-Orbitrap Velos mass spectrometer. Extracts (2 µL) were injected into water containing 0.1% formic acid, and loaded onto an inline peptide Opti-Trap (Optimize Technologies) for 3.0 min at 5 µL/min for desalting and concentration. Bile acids were then eluted by a 30 min water/acetonitrile gradient from 98% water containing 0.1% formic acid to 95% acetonitrile containing 0.1% formic acid (modified from ref. 73) and separated by a ProntoSil C18AQ 200 µm × 50 mm, 3 u column (nanoLCMS Solutions) at a flow rate of 2 µL/min. The column eluent was introduced to the LTQ-Orbitrap Velos mass spectrometer by an Advance nano-ESI source (Michrom BioResources) at a spray voltage of −1.7 kV. High resolution (R = 100,000 at 400 m/z) negative ion mass spectra were collected over 300–900 m/z. The ion transfer tube of the mass spectrometer was maintained at 250 °C, and the S-lens was set to 50%. Appropriate sample dilution factors were determined empirically to ensure a linear range of detector response74. Chromatographic peak alignment, feature detection, quantitation and bile acid identification were performed using MAVEN75 and SECD-LIMSA76 software. All bile acids were identified by comparison of retention time and accurate mass data against reference standards. Relative quantitation was performed against the deuterated GCDCA internal standard. TCDD-elicited changes in bile acid species are reported as fold changes relative to vehicle controls.
Intestinal Function
At 26d after the initial dose (PND54), mice were fasted for 4 h (without access to food or water) prior to oral gavage with 150 µL of a solution containing 100 mg sucrose, 12 mg lactulose, 8 mg mannitol, and 6 mg sucralose. Urine from each mouse was collected over a 5 h period, flash frozen, and combined prior to analysis. Urine was diluted in 95% acetonitrile/5% water: 100-fold for mannitol detection, 200-fold for sucrose and lactulose detection, and 500-fold for sucralose detection. Sugar probe recovery was measured using ultra high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS), performed at the MSU RTSF Mass Spectrometry and Metabolomics Core (https://rtsf.natsci.msu.edu/mass-spectrometry/). Diluted urine samples were injected onto an Acquity UPLC BEH Amide column (1.7 µm, 2.1 × 100 mm) (Waters, Milford, MA) maintained at 50 °C on a Waters ACQUITY UPLC systems (Waters). Mobile phase A was 50 mM ammonium acetate buffer (pH = 9.6) and mobile phase B was acetonitrile. Chromatographic separation was performed over 8 minutes (min) using a flow rate of 0.2 mL/min in the following gradient: time (t) = 0 min, 95% B; t = 1 min, 95% B; t = 2 min, 70% B; t = 5 min, 30% B; t = 5.01 min, 10% B; t = 6 min, 10% B; t = 6.01 min, 95% B; t = 8 min, 95% B. The autosampler was cooled to 10 °C, with an injection volume of 5 µL. UPLC was coupled with negative-mode electrospray ionization to a Waters Quattro Premier XE Mass Spectrometer (Waters) operating in multiple reaction monitoring (MRM) mode. The capillary voltage was −2.5 kV, while desolvation gas flow rate was 600 L/h. Source temperature and desolvation temperature were 120 °C and 450 °C, respectively. MRM parameters including cone voltage, collision voltage, precursor ion, and product ion were optimized by flow injection of pure standard for each individual compound (Supplementary Table S3).
Data analysis including peak integration was performed using MAVEN75. Waters raw data were converted to mzxml format using msconvert in the ProteoWizard Tools77. Fractional excretion of each probe was calculated using: (probe concentration from MS X total urine volume excreted)/probe input. Fractional sucrose excretion (%) was used to assess gastroduodenal permeability, the lactulose-to-mannitol ratio was used to assess small intestinal permeability, and fractional sucralose excretion (%) was used to assess colonic permeability78, 79.
At 27d after the initial dose (PND55), whole gut transit time (WGTT) was determined using carmine red dye, which cannot be intestinally absorbed. At time 0 (t0), mice were orally gavaged with 150 µL of carmine red solution (6% carmine red dye suspended in 0.5% methylcellulose). Fecal pellets were monitored continuously for the presence of carmine red. WGTT represents the time interval between t0 and the first observance of carmine red dye in the fecal pellets.
Degenerate qRT-PCR of Bacterial Functional Genes
The bsh Hidden Markov Model (HMM) constructed by the Ribosomal Database Project consisted of 12 vetted seed sequences from Lactobacillus, Clostridium, Roseburia, Eubacterium, Butyrivibrio, and Streptococcus species, while the baiCD HMM consisted of 10 vetted seed sequences from Clostridium and Lachnospiraceae species. Using these models, more inclusive sequence sets were created for bsh and baiCD within the ‘FunGene functional gene pipeline & repository’80 by extracting sequences from GenBank. Setting the HMM consensus percent identity cut-off to ≥40%, nucleotide sequences of bsh and baiCD were obtained from 2633 and 129 bacterial organisms, respectively. Given the diversity of bacterial organisms found to express bsh, the 2633 sequences from FunGene were separated into 4 groups based on nucleotide similarity determined through construction of a phylogenetic tree (Group 1: 423, Group 2: 330, Group 3: 955, Group 4: 925). Bacterial classifications (genus level) of the bsh sequences in each phylogenetic group are depicted in Fig. 8A1. In contrast, the 129 baiCD sequences exhibited sufficient similarity and were therefore kept as a single composite group. The Primer Design terminal program developed by the Ribosomal Database Project (https://github.com/rdpstaff/PrimerDesign) was used to design degenerate primer sets which target conserved regions within baiCD and each phylogenetic group of bsh (Supplementary Table S2). To estimate the overall abundance of bile acid metabolism genes within the gut microbiome, primer sets were selected to target ≥75% of the baiCD sequences and the sequences within each bsh group. Fecal levels of baiCD were also quantified using a previously validated primer pair (Supplementary Table S2) which targets a conserved region of the gene within Clostridium scindens VPI 12708 and Clostridium hiranonis TO931, two strains known to exhibit bile acid 7α-dehydroxylation activity48, 49.
Frozen fecal pellets were crushed into a powder and bacterial DNA was isolated using the QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA). To investigate bile acid metabolism loci, qRT-PCR was performed on a Bio-Rad CFX Connect Real-Time PCR Detection System as previously described13. Gene expression relative to vehicle control was calculated using the 2−∆∆CT method, with each fecal sample extract normalized to total bacterial DNA levels using universal primers targeting the 16 S ribosomal RNA gene81. Gene levels are plotted relative to vehicle control.
Data Availability
RNA-Seq datasets for the ileal epithelium and liver are available in the Gene Expression Omnibus (GEO; accession number GSE89430 and GSE87519, respectively). Primer sequences used for qRT-PCR are listed in Supplementary Table S2. MRM parameters used in the sugar probe permeability analysis are provided in Supplementary Table S3.
Electronic supplementary material
Acknowledgements
This work was supported by the National Institute of Environmental Health Sciences Superfund Research Program [NIEHS SBRP P42ES04911] to TRZ. TRZ is partially supported by AgBioResearch at Michigan State University. KAF is supported by the Canadian Institutes of Health Research Doctoral Foreign Study Award [DFS-140386]. RN is supported by the National Institutes of Health Integrative Training in the Pharmacological Sciences Award [5T32GM092715]. The authors would like to thank Dr. James Cole, Dr. Benli Chai, and Leo Tift for their guidance and assistance with the FunGene repository and the Primer Design terminal program. We would also like to thank Dr. Bryan Copple for his critical reading of the manuscript.
Author Contributions
K.A.F., R.N. and T.R.Z. designed the study. K.A.F. and R.N. performed the animal work. K.A.F. performed the experiments and analyzed the data. R.N. performed the RNA-Seq analysis, while C.Z. developed the UPLC-MS/MS method used to quantify the sugar probes in the intestinal permeability assay. K.K. and J.R.H. performed the histopathological assessment and scoring of the male and female liver sections. K.A.F. and T.R.Z. wrote the manuscript. K.A.F. prepared the figures and tables. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05656-8
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Vanni E, et al. From the metabolic syndrome to NAFLD or vice versa? Dig Liver Dis. 2010;42:320–330. doi: 10.1016/j.dld.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 2.Ford ES, Li C, Zhao G. Prevalence and correlates of metabolic syndrome based on a harmonious definition among adults in the US. J Diabetes. 2010;2:180–193. doi: 10.1111/j.1753-0407.2010.00078.x. [DOI] [PubMed] [Google Scholar]
- 3.Trauner M, Arrese M, Wagner M. Fatty liver and lipotoxicity. Biochim Biophys Acta. 2010;1801:299–310. doi: 10.1016/j.bbalip.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Taylor KW, et al. Evaluation of the Association between Persistent Organic Pollutants (POPs) and Diabetes in Epidemiological Studies: A National Toxicology Program Workshop Review. Environ Health Perspect. 2013;121:774–783. doi: 10.1289/ehp.1205502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casals-Casas C, Desvergne B. Endocrine disruptors: from endocrine to metabolic disruption. Annu Rev Physiol. 2011;73:135–162. doi: 10.1146/annurev-physiol-012110-142200. [DOI] [PubMed] [Google Scholar]
- 6.Grun F, Blumberg B. Endocrine disrupters as obesogens. Mol Cell Endocrinol. 2009;304:19–29. doi: 10.1016/j.mce.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 8.Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 9.Huang G, Elferink CJ. A novel nonconsensus xenobiotic response element capable of mediating aryl hydrocarbon receptor-dependent gene expression. Mol Pharmacol. 2012;81:338–347. doi: 10.1124/mol.111.075952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr. 2008;18:207–250. doi: 10.1615/CritRevEukarGeneExpr.v18.i3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Safe S. Polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs), dibenzofurans (PCDFs), and related compounds: environmental and mechanistic considerations which support the development of toxic equivalency factors (TEFs) Crit Rev Toxicol. 1990;21:51–88. doi: 10.3109/10408449009089873. [DOI] [PubMed] [Google Scholar]
- 12.Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol Sci. 2011;124:1–22. doi: 10.1093/toxsci/kfr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boverhof DR, et al. Temporal and dose-dependent hepatic gene expression patterns in mice provide new insights into TCDD-Mediated hepatotoxicity. Toxicol Sci. 2005;85:1048–1063. doi: 10.1093/toxsci/kfi162. [DOI] [PubMed] [Google Scholar]
- 14.Jones G, Greig JB. Pathological-Changes in Liver of Mice Given 2,3,7,8-Tetrachlorodibenzo-P-Dioxin. Experientia. 1975;31:1315–1317. doi: 10.1007/BF01945801. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- 16.Nault R, et al. Dose-Dependent Metabolic Reprogramming and Differential Gene Expression in TCDD-Elicited Hepatic Fibrosis. Toxicol Sci. 2016;154:253–266. doi: 10.1093/toxsci/kfw163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pierre S, et al. Aryl hydrocarbon receptor-dependent induction of liver fibrosis by dioxin. Toxicol Sci. 2014;137:114–124. doi: 10.1093/toxsci/kft236. [DOI] [PubMed] [Google Scholar]
- 18.Chiang JY. Bile acid metabolism and signaling. Compr Physiol. 2013;3:1191–1212. doi: 10.1002/cphy.c120023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Copple BL, Li T. Pharmacology of bile acid receptors: Evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol Res. 2016;104:9–21. doi: 10.1016/j.phrs.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol. 2009;15:1677–1689. doi: 10.3748/wjg.15.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178:175–186. doi: 10.1016/j.ajpath.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brewster DW, Uraih LC, Birnbaum LS. The acute toxicity of 2,3,4,7,8-pentachlorodibenzofuran (4PeCDF) in the male Fischer rat. Fundam Appl Toxicol. 1988;11:236–249. doi: 10.1016/0272-0590(88)90148-0. [DOI] [PubMed] [Google Scholar]
- 23.Couture LA, Elwell MR, Birnbaum LS. Dioxin-like effects observed in male rats following exposure to octachlorodibenzo-p-dioxin (OCDD) during a 13-week study. Toxicol Appl Pharmacol. 1988;93:31–46. doi: 10.1016/0041-008X(88)90023-3. [DOI] [PubMed] [Google Scholar]
- 24.Yang KH, Croft WA, Peterson RE. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on plasma disappearance and biliary excretion of foreign compounds in rats. Toxicol Appl Pharmacol. 1977;40:485–496. doi: 10.1016/0041-008X(77)90075-8. [DOI] [PubMed] [Google Scholar]
- 25.Vos JG, Moore JA, Zinkl JG. Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in C57B1/6 mice. Toxicol Appl Pharmacol. 1974;29:229–241. doi: 10.1016/0041-008X(74)90060-X. [DOI] [PubMed] [Google Scholar]
- 26.Fletcher N, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) alters the mRNA expression of critical genes associated with cholesterol metabolism, bile acid biosynthesis, and bile transport in rat liver: a microarray study. Toxicol Appl Pharmacol. 2005;207:1–24. doi: 10.1016/j.taap.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Ozeki J, et al. Aryl hydrocarbon receptor ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin enhances liver damage in bile duct-ligated mice. Toxicology. 2011;280:10–17. doi: 10.1016/j.tox.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato H, et al. Novel potent and selective bile acid derivatives as TGR5 agonists: biological screening, structure-activity relationships, and molecular modeling studies. J Med Chem. 2008;51:1831–1841. doi: 10.1021/jm7015864. [DOI] [PubMed] [Google Scholar]
- 29.Reich M, et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut. 2016;65:487–501. doi: 10.1136/gutjnl-2015-309458. [DOI] [PubMed] [Google Scholar]
- 30.Birnbaum LS, McDonald MM, Blair PC, Clark AM, Harris MW. Differential toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in C57BL/6J mice congenic at the Ah Locus. Fundam Appl Toxicol. 1990;15:186–200. doi: 10.1016/0272-0590(90)90175-J. [DOI] [PubMed] [Google Scholar]
- 31.McConnell EE, Moore JA, Haseman JK, Harris MW. The comparative toxicity of chlorinated dibenzo-p-dioxins in mice and guinea pigs. Toxicol Appl Pharmacol. 1978;44:335–356. doi: 10.1016/0041-008X(78)90195-3. [DOI] [PubMed] [Google Scholar]
- 32.Fader KA, et al. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Alters Lipid Metabolism and Depletes Immune Cell Populations in the Jejunum of C57BL/6 Mice. Toxicol Sci. 2015;148:567–580. doi: 10.1093/toxsci/kfv206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fader KA, et al. Convergence of hepcidin deficiency, systemic iron overloading, heme accumulation, and REV-ERBalpha/beta activation in aryl hydrocarbon receptor-elicited hepatotoxicity. Toxicol Appl Pharmacol. 2017;321:1–17. doi: 10.1016/j.taap.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nault R, Fader KA, Lydic TA, Zacharewski TR. Lipidomic Evaluation of Aryl Hydrocarbon Receptor-Mediated Hepatic Steatosis in Male and Female Mice Elicited by 2,3,7,8-Tetrachlorodibenzo-p-dioxin. Chem Res Toxicol. 2017;30:1060–1075. doi: 10.1021/acs.chemrestox.6b00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Botham KM, Boyd GS. The metabolism of chenodeoxycholic acid to beta-muricholic acid in rat liver. Eur J Biochem. 1983;134:191–196. doi: 10.1111/j.1432-1033.1983.tb07550.x. [DOI] [PubMed] [Google Scholar]
- 36.Nault R, et al. Pyruvate Kinase Isoform Switching and Hepatic Metabolic Reprogramming by the Environmental Contaminant 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. Toxicol Sci. 2016;149:358–371. doi: 10.1093/toxsci/kfv245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sayin SI, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Billington D, Evans CE, Godfrey PP, Coleman R. Effects of bile salts on the plasma membranes of isolated rat hepatocytes. Biochem J. 1980;188:321–327. doi: 10.1042/bj1880321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmer RH. Bile acids, liver injury, and liver disease. Arch Intern Med. 1972;130:606–617. doi: 10.1001/archinte.1972.03650040130012. [DOI] [PubMed] [Google Scholar]
- 40.Hofmann AF, Roda A. Physicochemical properties of bile acids and their relationship to biological properties: an overview of the problem. J Lipid Res. 1984;25:1477–1489. [PubMed] [Google Scholar]
- 41.Heuman DM. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J Lipid Res. 1989;30:719–730. [PubMed] [Google Scholar]
- 42.Araya Z, Wikvall K. 6alpha-hydroxylation of taurochenodeoxycholic acid and lithocholic acid by CYP3A4 in human liver microsomes. Biochim Biophys Acta. 1999;1438:47–54. doi: 10.1016/S1388-1981(99)00031-1. [DOI] [PubMed] [Google Scholar]
- 43.Xie W, et al. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci USA. 2001;98:3375–3380. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meier PJ, Stieger B. Bile salt transporters. Annu Rev Physiol. 2002;64:635–661. doi: 10.1146/annurev.physiol.64.082201.100300. [DOI] [PubMed] [Google Scholar]
- 45.Csanaky IL, et al. Organic anion-transporting polypeptide 1b2 (Oatp1b2) is important for the hepatic uptake of unconjugated bile acids: Studies in Oatp1b2-null mice. Hepatology. 2011;53:272–281. doi: 10.1002/hep.23984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gérard P. Metabolism of Cholesterol and Bile Acids by the Gut Microbiota. Pathogens. 2014;3:14–24. doi: 10.3390/pathogens3010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ridlon JM, Hylemon PB. Identification and characterization of two bile acid coenzyme A transferases from Clostridium scindens, a bile acid 7alpha-dehydroxylating intestinal bacterium. J Lipid Res. 2012;53:66–76. doi: 10.1194/jlr.M020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wells JE, Williams KB, Whitehead TR, Heuman DM, Hylemon PB. Development and application of a polymerase chain reaction assay for the detection and enumeration of bile acid 7alpha-dehydroxylating bacteria in human feces. Clin Chim Acta. 2003;331:127–134. doi: 10.1016/S0009-8981(03)00115-3. [DOI] [PubMed] [Google Scholar]
- 49.Ou J, et al. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr. 2013;98:111–120. doi: 10.3945/ajcn.112.056689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lefever DE, et al. TCDD modulation of gut microbiome correlated with liver and immune toxicity in streptozotocin (STZ)-induced hyperglycemic mice. Toxicol Appl Pharmacol. 2016;304:48–58. doi: 10.1016/j.taap.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L, et al. Persistent Organic Pollutants Modify Gut Microbiota-Host Metabolic Homeostasis in Mice Through Aryl Hydrocarbon Receptor Activation. Environ Health Perspect. 2015;123:679–688. doi: 10.1289/ehp.1409055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kakizuka S, et al. Dioxin-Produced Alteration in the Profiles of Fecal and Urinary Metabolomes: A Change in Bile Acids and Its Relevance to Toxicity. Biol Pharm Bull. 2015;38:1484–1495. doi: 10.1248/bpb.b15-00235. [DOI] [PubMed] [Google Scholar]
- 53.Wahlstrom A, Sayin SI, Marschall HU, Backhed F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016;24:41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 54.Gao, X. et al. The antiandrogen flutamide is a novel aryl hydrocarbon receptor ligand that disrupts bile acid homeostasis in mice through induction of Abcc4. Biochem Pharmacol, doi:10.1016/j.bcp.2016.08.021 (2016). [DOI] [PMC free article] [PubMed]
- 55.Iwanaga T, Nakakariya M, Yabuuchi H, Maeda T, Tamai I. Involvement of bile salt export pump in flutamide-induced cholestatic hepatitis. Biol Pharm Bull. 2007;30:739–744. doi: 10.1248/bpb.30.739. [DOI] [PubMed] [Google Scholar]
- 56.Javitt NB, Emerman S. Effect of sodium taurolithocholate on bile flow and bile acid exeretion. J Clin Invest. 1968;47:1002–1014. doi: 10.1172/JCI105790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beuers U, et al. Taurolithocholic acid exerts cholestatic effects via phosphatidylinositol 3-kinase-dependent mechanisms in perfused rat livers and rat hepatocyte couplets. J Biol Chem. 2003;278:17810–17818. doi: 10.1074/jbc.M209898200. [DOI] [PubMed] [Google Scholar]
- 58.Gonzalez FJ, Fernandez-Salguero P. The aryl hydrocarbon receptor: studies using the AHR-null mice. Drug Metab Dispos. 1998;26:1194–1198. [PubMed] [Google Scholar]
- 59.Angrish MM, Dominici CY, Zacharewski TR. TCDD-elicited effects on liver, serum, and adipose lipid composition in C57BL/6 mice. Toxicol Sci. 2013;131:108–115. doi: 10.1093/toxsci/kfs277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sorg O, et al. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko: identification and measurement of TCDD metabolites. Lancet. 2009;374:1179–1185. doi: 10.1016/S0140-6736(09)60912-0. [DOI] [PubMed] [Google Scholar]
- 61.Wolfe WH, et al. Determinants of TCDD half-life in veterans of operation ranch hand. J Toxicol Environ Health. 1994;41:481–488. doi: 10.1080/15287399409531858. [DOI] [PubMed] [Google Scholar]
- 62.Gasiewicz TA, Geiger LE, Rucci G, Neal RA. Distribution, excretion, and metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J, DBA/2J, and B6D2F1/J mice. Drug Metab Dispos. 1983;11:397–403. [PubMed] [Google Scholar]
- 63.Everson GT, Braverman DZ, Johnson ML, Kern F., Jr. A critical evaluation of real-time ultrasonography for the study of gallbladder volume and contraction. Gastroenterology. 1980;79:40–46. [PubMed] [Google Scholar]
- 64.Wang HH, et al. Effect of gallbladder hypomotility on cholesterol crystallization and growth in CCK-deficient mice. Biochim Biophys Acta. 2010;1801:138–146. doi: 10.1016/j.bbalip.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kopec AK, et al. PCB153-elicited hepatic responses in the immature, ovariectomized C57BL/6 mice: comparative toxicogenomic effects of dioxin and non-dioxin-like ligands. Toxicol Appl Pharmacol. 2010;243:359–371. doi: 10.1016/j.taap.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nault R, Colbry D, Brandenberger C, Harkema JR, Zacharewski TR. Development of a computational high-throughput tool for the quantitative examination of dose-dependent histological features. Toxicol Pathol. 2015;43:366–375. doi: 10.1177/0192623314544379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nault R, Fader KA, Zacharewski T. RNA-Seq versus oligonucleotide array assessment of dose-dependent TCDD-elicited hepatic gene expression in mice. BMC Genomics. 2015;16:373. doi: 10.1186/s12864-015-1527-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burgoon LD, Zacharewski TR. Automated quantitative dose-response modeling and point of departure determination for large toxicogenomic and high-throughput screening data sets. Toxicol Sci. 2008;104:412–418. doi: 10.1093/toxsci/kfn083. [DOI] [PubMed] [Google Scholar]
- 69.Yang L, Allen BC, Thomas RS. BMDExpress: a software tool for the benchmark dose analyses of genomic data. BMC Genomics. 2007;8:387. doi: 10.1186/1471-2164-8-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lo R, Matthews J. High-resolution genome-wide mapping of AHR and ARNT binding sites by ChIP-Seq. Toxicol Sci. 2012;130:349–361. doi: 10.1093/toxsci/kfs253. [DOI] [PubMed] [Google Scholar]
- 71.Ji H, et al. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26:1293–1300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.John C, et al. A liquid chromatography-tandem mass spectrometry-based method for the simultaneous determination of hydroxy sterols and bile acids. J Chromatogr A. 2014;1371:184–195. doi: 10.1016/j.chroma.2014.10.064. [DOI] [PubMed] [Google Scholar]
- 73.Swann JR, et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4523–4530. doi: 10.1073/pnas.1006734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lydic TA, Busik JV, Reid GE. A monophasic extraction strategy for the simultaneous lipidome analysis of polar and nonpolar retina lipids. J Lipid Res. 2014;55:1797–1809. doi: 10.1194/jlr.D050302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Melamud E, Vastag L, Rabinowitz JD. Metabolomic analysis and visualization engine for LC-MS data. Anal Chem. 2010;82:9818–9826. doi: 10.1021/ac1021166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haimi P, Uphoff A, Hermansson M, Somerharju P. Software tools for analysis of mass spectrometric lipidome data. Anal Chem. 2006;78:8324–8331. doi: 10.1021/ac061390w. [DOI] [PubMed] [Google Scholar]
- 77.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arrieta MC, Bistritz L, Meddings JB. Alterations in intestinal permeability. Gut. 2006;55:1512–1520. doi: 10.1136/gut.2005.085373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Teshima CW, Meddings JB. The measurement and clinical significance of intestinal permeability. Curr Gastroenterol Rep. 2008;10:443–449. doi: 10.1007/s11894-008-0083-y. [DOI] [PubMed] [Google Scholar]
- 80.Fish JA, et al. FunGene: the functional gene pipeline and repository. Front Microbiol. 2013;4:291. doi: 10.3389/fmicb.2013.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Muyzer G, de Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59:695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq datasets for the ileal epithelium and liver are available in the Gene Expression Omnibus (GEO; accession number GSE89430 and GSE87519, respectively). Primer sequences used for qRT-PCR are listed in Supplementary Table S2. MRM parameters used in the sugar probe permeability analysis are provided in Supplementary Table S3.