

Abstract
As cancer has become increasingly more prevalent in our society, cancer prevention research has evolved toward placing a greater emphasis on reducing cancer deaths and minimizing the adverse consequences of having cancer. “Precision cancer prevention” takes into account the collaboration of intrinsic and extrinsic factors for influencing cancer incidence and aggressiveness in the context of the individual, as well as the recognition that such knowledge can improve early detection and more accurate discrimination of cancerous lesions. The premise of this review is that analyses of mouse models can greatly augment precision cancer prevention. However, as of now, mouse models, and particularly genetically-engineered mouse (GEM) models, have yet to be fully integrated into prevention research. Herein we discuss opportunities and challenges for “precision mouse modeling”, including their essential criteria of mouse models for prevention research, representative success stories, and opportunities for the more refined analyses in future studies.
Cancer prevention in the era of precision medicine
Considering the close association of many adult cancers with aging and the continuing rise in the aging population, it seems unlikely that cancer will be completely eradicated anytime in the near future. However, reducing the incidence of cancer deaths and minimizing the adverse life-changing consequences that impact cancer survivors are achievable goals in the near-term. Thus, we can consider cancer prevention research not simply in terms of seeking to prevent the occurrence of cancer, but also seeking to minimize (or prevent) the adverse consequences that result from having cancer. This conceptual framework requires a precise understanding of the genetic, and epigenetic factors that promote cancer, and how these are influenced by environmental factors such as diet, hormones, pharmaceutical agents, or others. Undoubtedly, more cancer deaths could be prevented by early diagnosis and concomitant early intervention. However, early detection often comes at the price of over-diagnosis and overtreatment of non-harmful lesions that are not likely to progress to lethal tumors. This is evident from the rise in breast cancer diagnosis as a consequence of wide-spread mammogram screening, and the analogous increase in prostate cancer diagnosis as a result of screening for prostate specific antigen (PSA)1. Thus, realizing the benefits of early detection will require improved means of distinguishing, at the earliest possible stages, “true” cancers that require intervention from the majority of non-malignant ones. On the other hand, identifying “true” cancers as early as possible would likely allow more effective treatment management and improve cancer outcomes.
In recognition of these challenges, the definition of cancer prevention has been evolving2. According to traditional definitions, Primary Prevention refers to the identification of extrinsic and intrinsic risk factors in healthy individuals with the goal of minimizing their exposure and thereby reducing cancer incidence (Figure 1). Secondary Prevention refers to the identification of pre-neoplastic and/or early stage cancer lesions with the goal of improving their detection and discrimination, and preventing or delaying progression to clinically-relevant disease. Tertiary Prevention refers to controlling the adverse consequences associated with having clinically-relevant cancer, which is not to be confused with cancer treatment; as there are limited opportunities for mouse models to improve on tertiary prevention, this is not a major focus of this review.
Figure 1. New paradigms for cancer prevention using mouse models.
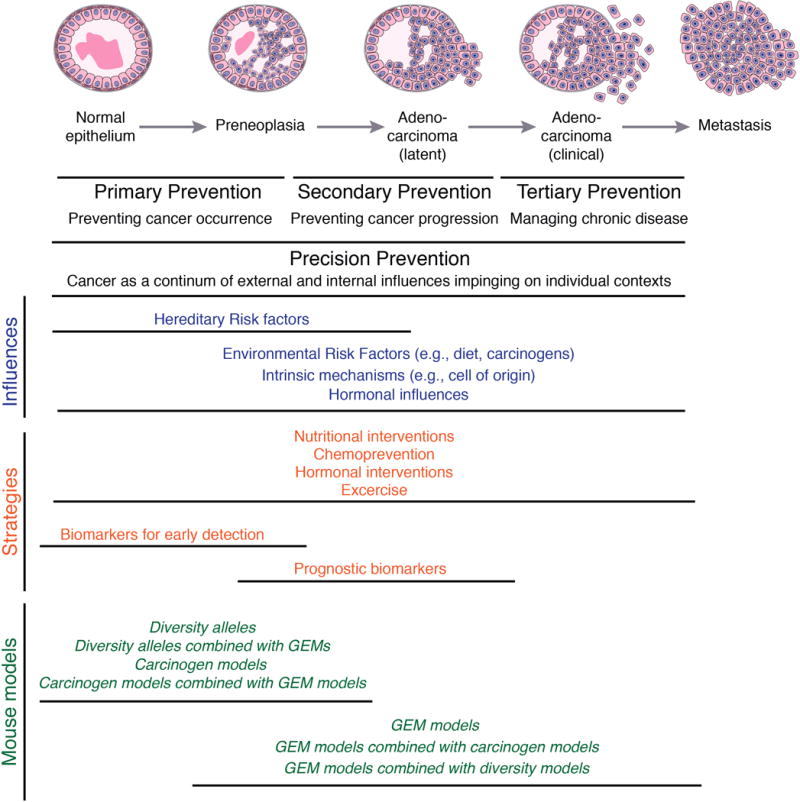
Comparison of traditional cancer prevention and precision prevention paradigms. Shown are representative influences for cancer occurrence or progression, compared with known interventions, and potential applications of mouse models.
While in many respects this traditional framework has proven useful, in fact, such framework may inadvertently detract from the broader goals of minimizing cancer deaths and reducing the adverse consequences of cancer treatment. In reality, primary prevention — which focuses on how cancers arise and how to prevent them from occurring in the first place — may be exceedingly difficult to achieve in the short term. On the other hand, secondary prevention — which focuses on effective early detection and finding ways to discriminate lethal from non-lethal lesions at early stages — while still challenging, may be more feasible in the near-term and, if realized, could have a significant impact on disease outcome, while at the same time providing insights that will ultimate impact primary prevention.
Another missing piece is the context in which cancers arise, meaning that even if it were possible to precisely define the intricacies of the environmental, genetic, and epigenetic factors that give rise to cancer, one would need to consider how these coalesce to influence the emergence of specific cancer phenotypes in any given individual3, as well as in the context of the characteristic “tumor heterogeneity” of human cancers4. Indeed, it is now widely accepted that defining the context of individual patient tumors is likely to have a significant impact on their effective treatment – a concept called “Precision Medicine”5–7. The logical extension of this concept, namely Precision Prevention3, is now being implemented for breast8, 9, esophageal10, and oral cancer11. This framework is intended to convey the importance of considering not only whether various factors (may they be carcinogens or dietary factors or hormones etc.) promote or reduce cancer incidence but also how such factors are interpreted in context of the individual to elicit the wide range of cancer phenotypes observed in the human population (Figure 1).
Modeling cancer prevention in mice in the context of precision prevention
Implicit in this framework is the notion that how factors promote or reduce the incidence or aggressiveness of cancer will be nuanced by individual contexts, which may be difficult to decipher solely by studying cancer in the human population. In this regard, mouse models can provide valuable resources for precision cancer prevention, since they enable both longitudinal and molecular analyses of precancerous and cancerous lesions in defined genetic contexts and in environmentally-controlled conditions, which would be exceedingly difficult to accomplish solely by studying human cancer. However, the relevance of mouse models for cancer prevention has often been called into question, despite the fact that much of our current knowledge about carcinogens as cancer-causing agents has its origins in analysis of mice (see below). Skepticism regarding the value of mouse models is particularly evident for genetically-engineered mice (GEM) (Text box 1)12, 13, which now dominate studies of cancer biology and cancer therapeutics, but are relatively underrepresented in cancer prevention research.
Text box 1: Genetically-engineered mouse (GEM) models of cancer.
Mouse models of human cancer are now dominated by genetically-engineered mice (GEM)12, 13. The earliest GEM models of cancer were the so-called “oncomice”, which refer to transgenic mice expressing oncogenes usually in specific tissue compartments giving rise to tumors in those tissues13. The next-generation of GEM models were based on germline deletion of tumor suppressor genes, so-called “knock-out” mice12, 116, 117, such as p53 and RB mutant mice, which established the potential for mice to develop cancer following loss-of-function (as opposed to gain of function) and provided in vivo evidence for the tumor suppressor functions of these respective genes118–120. Among their limitations, germline mice often do not recapitulate the cancer phenotype expected based on their human counterpart, as exemplified by the absence of retinoblastoma in RB mutant mice118. Additionally, homozygous germline deletion of certain tumor suppressors results in embryonic lethality, as exemplified by germline deletion of the breast cancer susceptibility genes, BRCA1 and BRCA2, which impedes analysis of breast cancer phenotypes in the corresponding mutant mice121.
Nowadays, most GEM models are based on conditional gene targeting wherein genes of interest are inactivated (or activated) in a temporal and tissue-specific manner following expression of Cre recombinase or alternative approaches12. Conditional GEM models can overcome inherent challenges associated with studying cancer phenotypes in germline mutant mice, since they can circumvent embryonic lethality, and enable functional investigations of individual or multiple genes in specific tissue contexts, as exemplified by conditional loss-of-function of Brca1 and Brca2 for breast cancer121, 122. Moreover, the further refinement of GEM models has been augmented by technological advances for gene targeting and/or regulation of gene expression in precise spatial and temporal contexts including: (i) the use of inducible Cre drivers123; (ii) viral delivery of Cre-recombinase into tissues124; (iii) alternative approaches for gene recombination, such as Flp-FRT recombinase125; and (iv) approaches for regulatable gene expression, such as tetracycline-regulated alleles123. Moreover, the generation of sophisticated GEM models has been further advanced by: (i) the use of reporter alleles for in vivo imaging126 and lineage-marking in vivo127; (ii) new approaches for rapid generation of multi-allelic GEM models128, 129; and (iii) new strategies for rapid gene targeting using genome editing130.
So, why have mouse models not been fully integrated in cancer prevention? In our view, this reflects actual disparities in their genetics and physiology relative to humans, as well as perceived misconceptions regarding their suitability for prevention research. In particular, because most mouse models are derived from inbred laboratory strains, they are not well suited to inform on the genetic diversity of the human population (with the notable exception of the diversity strains, discussed below). However, the fact that mice may not effectively model ALL aspects of cancer prevention does not mean that they cannot model ANY aspects of such research. For example, although dissimilarities in the physiology and anatomy of mice relative to humans may hinder effective analyses of the toxicity or pharmacodynamic/pharmacokinetic properties of chemopreventive agents, preclinical studies using mouse models can reveal mechanisms of action of such agents, lead to identification of biomarkers of response, and facilitate evaluation of combinations of agents (see below). Furthermore, analyses of mouse models offer unique opportunities to interrogate aspects of cancer prevention that are difficult to study in humans, such as accessing pre-cancerous and/or early stage cancers with known progressive potential.
Indeed, if we consider the breadth of research encompassed by cancer prevention, it is unlikely that any one model (or type of model) would be suitable to study all aspects of cancer prevention, and it is not surprising that a “one size fits all” approach has not proven to be informative. In fact, the term “mouse models” is often used as a “catch-all” phrase to describe many types of models having a range of suitability for prevention research, the consequence of which has been to undermine the merits of certain types models, while failing to acknowledge the limitations of others. We believe that successful deployment of mouse models will require consideration of the appropriateness of specific model(s) or types of models for specific types of prevention research, and to capitalize on combining modeling approaches for more refined and robust analyses (Figure 1).
Herein we discuss the features of mouse models that would make them suitable (or not) for studying specific aspects of cancer prevention, with a focus on how mouse models can advance precision cancer prevention. We describe what has been learned thus far about cancer prevention by studying mouse models, and consider how we can better capitalize on these resources in the future. Rather than attempting to provide a comprehensive review, we address these broad concepts by highlighting specific examples and refer the reader to previous reviews that have addressed specific topics and/or cancer types in depth (e.g.,14–18).
What constitutes an informative mouse model of cancer prevention?
Although mice rarely develop tumors spontaneously, they can be induced to form tumors following genetic or environmental manipulation, and are now the model of choice for studying cancer in vivo. The two broadest categories are autochthonous and non-autochthonous models (Table 1). Autochthonous models, which are now dominated by genetically-engineered mice (GEM)12, 13, develop tumors de novo in the context of the native microenvironment, thereby enabling analysis of tumorigenesis in the natural milieu (Text box 1). In contrast, the tumors in non-autochthonous models originate from implantation of cells or tissues into heterologous mice. For example, the now-popular patient-derived xenograft (PDX) models are derived from patient tumors that are implanted into heterologous sites of immunodeficient host mice19, 20. Since these features of PDX models are unlikely to capture key events associated with tumor initiation and progression (discussed in21, 22), they may be more appropriate for studying cancer treatment rather than cancer prevention. This exemplifies the important distinction between modeling cancer and modeling cancer prevention, which is a recurring theme of this review.
Table 1.
Mouse models of cancer and applications for precision cancer prevention
| Model types | Description | Applications | Limitations/Challenges |
|---|---|---|---|
| Non-autochthonous models | |||
|
|
|
|
|
|
||
|
|
||
| Autochthonous models | |||
Population diversity strains
|
|
|
|
Carcinogen-induced models
|
|
|
|
GEM models
|
|
|
|
Indeed, notwithstanding their significance for modeling cancer, in order for mouse models to be relevant for cancer prevention, they should have certain “minimal” characteristics that enable them to inform on early events in cancer initiation and progression (Text box 2). First, tumors should arise de novo in the context of the native tumor microenvironment and intact immune system. This includes most (if not all) autochthonous models, as well as some non-germline GEM models23, but excludes most non-autochthonous models. Second, they should model multistage cancer progression from premalignancy to overt tumorigenesis; this common feature of most autochthonous models, including carcinogen and GEM models.
Text box 2: Criteria to assess the suitability of mouse models for cancer prevention.
Essential Criteria
Develop tumors de novo in the context of the native tissue environment and intact immune system
Tumors arise from genetic and/or environmental factors that are relevant for the human cancers they are intended to emulate
Model multistage progression including pre-malignant lesions that have the potential to progress to invasive ones
Model early events in cancer initiation as well as progression to adenocarcinoma
Tumor phenotypes should display histological and biological features in common with their human counterpart
Should have related molecular pathways that are dysregulated and model key molecular events that occur in the human cancers they are intended to emulate
Optimal Criteria
Cancer phenotype should be highly penetrant and arise with reasonable latency
Tumors should originate in appropriate cells of origin
Tumors should display heterogeneity as occurs in human cancer
Mice should model genetic diversity as occurs in humans
Third, cancer phenotypes should arise from genetic and/or environmental factors that have known relevance for the human cancers they are intending to emulate, which are characteristic of GEM and carcinogen models, respectively. For example, many GEM models of prostate cancer are based on loss-of-function of Pten24, which is prevalent in the human disease. Similarly, treatment of mice with tobacco-related carcinogens leads to bladder cancer, reminiscent of human bladder cancer for which a major risk factor is smoking25. Fourth, the histological and molecular properties of mouse tumors should resemble their human counterparts as closely as possible. Notably, while it is often the case that the histological features of mouse tumors may be dissimilar to human tumors, as is the case for prostate cancer26, the molecular pathways that are dysregulated in mouse tumors may be conserved with human cancer (e.g.,27).
Additional criteria that would be highly advantageous include having cancer phenotypes that originate in relevant cell(s) of origin; however, in practice this can be complicated by difficulties in the definitive identification of relevant cell(s) of origin, as well as difficulties in targeting gene recombination specifically in such cell(s)28. Lastly, to truly emulate human cancer, mouse models should capture the genetic diversity that is characteristic of the human population; however, this is by far the most challenging feature of human cancer to model in mice, mainly because it is incompatible with laboratory mice being derived from inbred strains (see below).
While the relative importance of these various criteria may be nuanced by the specific aspect of prevention research being investigated, having well-defined criteria sets a high but achievable bar to gauge the relevance of any given model, to recognize their advantages and limitations, and to discover how limitations can be transformed into opportunities. For example, while the challenges of modeling genetic heterogeneity in mouse models are formidable, the relative homogeneity of cancer in mice relative to humans can provide an opportunity to precisely define the influences of extrinsic factors in well-defined genetic contexts. Moreover, inherent species differences can be a hindrance as well as a benefit. For example, the relatively short life-span of mice compared with humans is problematic given that one of the major risk factors of many human cancers is aging; however, their shorter lifespan makes certain analyses far more feasible to study in mice compared with humans. In other scenarios, species differences can be used creatively to “tease out” specific roles for extrinsic or intrinsic influences for cancer. For example, inherent differences in the immune system in mice represent a key challenge for their effective use29 particularly in light of the importance of the immune system and the potential for immunotherapy for prevention and treatment of human cancer30. However, these species differences may also provide a unique opportunity to systematically evaluate the contribution of specific components of the immune system for tumorigenesis in “humanized” mouse models31, 32. Thus, in our view, optimizing mouse models for precision cancer prevention will require acknowledgment of and adherence to essential criteria as well as an appreciation of inherent species differences that impact the implementation and interpretation of prevention studies; however, such knowledge is also likely to uncover scenarios in which “limitations” or “species differences” can actually provide unique opportunities for investigation.
Modeling population diversity in mice
As discussed above, one of the major criticisms of using mouse models for cancer prevention is that they do not effectively model genetic diversity as is characteristic of human cancer. Indeed, the evolution of the mouse as the model of choice for cancer research has its origins in the pioneering work of Little and others, which led to the generation of “inbred” mouse strains that enabled efficient engineering of their genome (33, and discussed in34). However, the same genetic uniformity that has made mice tractable for genetic manipulation, also contributes to their dissimilarity to humans, which are far from genetically homogeneous.
The intention of modeling genetic diversity has led to the generation of the Collaborative Cross (CC)35–39. This unique resource, which was 10 years in the making, represents a large panel of recombinant inbred mouse lines (~150 lines) that were generated by intercrossing eight common inbred strains to obtain a series of lines that, in aggregate, model a complex outbred population with significant diversity40. These unique features of the CC panel enable analysis of the complex genetic etiology of human disease, as well as interactions between genetic and environmental factors. Notable examples include their use for studying inherited resistance to viral pathogenesis41, and the influence of the immune system for cancer initiation and progression42. Indeed, the CC strains provide a unique opportunity for cancer prevention since they can integrate population genetics with the influence of tumor drivers and/or environmental exposures, as has been demonstrated for melanoma and prostate cancer43, 44. For example, crossing a transgenic mouse model of lethal prostate cancer into the eight founder strains of the CC revealed the modulation of tumor and metastatic phenotypes in the specific strains44; although not directly related to cancer prevention, this example demonstrates the feasibility of modeling population diversity and assessing the consequences of such diversity for tumorigenesis in vivo.
However, the CC is not without its challenges, which include issues related to feasibility (e.g., space, mouse costs, etc.) as well as implementation (e.g., genetic drift and loss of genetic diversity). These challenges have been partially circumvented by the generation of the Diversity Outbred (DO) population, a heterogeneous stock derived from the CC inbred strains that maintains a similar level of diversity as the CC but with fewer challenges for implementation45. Notably, the recent analysis of benzene exposure in the DO strains led the identification of associated genetic factors that influence response to such exposure46.
So, how might these population diversity strains be used to bolster precision cancer prevention? In the simplest scenario, the CC or DO strains can be treated with environmental agents, such as carcinogens, dietary components, or hormones, to directly evaluate the interaction between genetic heterogeneity and such factors for cancer incidence or aggressiveness. Admittedly, the implementation of such studies is likely to be challenging, considering the space and costs associated with housing the large cohorts of mice that would be needed for meaningful analyses, as well as difficulties that are likely to be encountered in identifying presumably subtle and rare phenotypic events. For the latter, improvements in small animal imaging are likely to be beneficial for improving detection of subtle phenotypic alterations47.
Another application draws directly from the example of the prostate cancer model described above44; in particular, the diversity strains can be crossed with GEM models to investigate the influence of genetic context for cancer incidence or aggressiveness. Such analyses are relatively straightforward for dominant-acting alleles, since they entail crossing the diversity strains with a given GEM model and directly analyzing the resulting F1 hybrids. However, such analyses would be more far challenging for GEM models that are not dominantly-acting, in which case it would not be feasible to study the F1 hybrids. We envision that the implementation of CRISPR/Cas9 technology48, 49 will vastly improve the feasibility of using the diversity alleles by making it feasible to introduce genetic alterations into the CC or DO strains and then analyzing the mice directly. Thus, in our view, these diversity strains combined with genetic technologies represent an unparalleled opportunity to evaluate the interaction of genetic and environmental influences for tumorigenesis and thereby vastly expand the repertoire of using mouse models for precision prevention research.
Modeling environmental causes of cancer in mice
Modeling cancer in mice has its origins in carcinogen-induced models in which mice (or other rodents) were treated with suspected agents resulting in tumor phenotypes50. Dating back to the early 1900’s in which aromatic hydrocarbons present in coal tar were shown to produce cancer phenotypes when topically applied to mouse skin51, the general approach of evaluating the consequences of suspected carcinogens has had far-reaching implications for cancer prevention. Analysis of carcinogen-treated mice has provided definitive evidence that suspected agents are indeed cancer causing, which has contributed to recommendations for limiting human exposure to such agents, while simultaneously generating mouse models based on highly relevant environmental perturbations.
Systemic treatment of mice with nitrosamines, aromatic amines, asbestos or other agents has resulted in cancers at many tissue sites, depending on the agent and the mode of delivery, including stomach, kidney, bladder, breast, cervix, colon, skin, and lung17, 50–53. As discussed above, one of the major risk factors for bladder cancer is smoking, and systemic treatment of mice with carcinogens similar to those present in tobacco smoke (e.g., N-butyl-N-4-hydroxybutyl nitrosamine (BBN)) results in mouse bladder tumors that resemble human bladder cancer53. Likewise, local treatment of skin with phorbol esters produces skin cancer in mice51, 54; furthermore, analyses of mice demonstrated the significance of UV light for skin cancer, which ultimately contributed to recommendations for limiting UV exposure55–58.
In a particularly compelling example, studies in mice provided direct evidence of the cancer-causing properties of cigarette smoke and helped to identify the deleterious components in tobacco59–61. Ultimately, these studies contributed to the substantial campaign toward smoking cessation and the reduction of exposure to tobacco smoke, which have had an enormous impact on human health. Furthermore, studies in mice revealed that a major carcinogen in tobacco, namely nicotine-derived nitrosamine ketone (also known as 4-(methylnitro-samino)-1-(3-pyridyl)-1-butanone; NNK), leads to dysregulation of the mTOR/Akt targetable signaling pathway; this pathway is up-regulated by tobacco carcinogens in GEM models as well as in humans that are heavy smokers62–66. Notably, these preclinical and mechanism-based studies have led to clinical trials aimed at evaluating the consequences of inhibiting mTOR/AKT signaling for prevention of lung cancer in high-risk individuals61. This example emphasizes the value multi-disciplinary efforts, which incorporate analyses of carcinogen-based and GEM models with validation to human cancer to elucidate the consequences of suspected carcinogens, to define their underlying cancer-promoting mechanisms, and to ultimately identify interventions for individuals at greatest risk.
This example also highlights the value for precision cancer prevention of combining carcinogen-based and GEM models, which have highly complementary attributes and challenges (Table 1). Since by definition GEM models are derived from genetically-defined alterations and they have relatively homogeneous tumor phenotypes compared with tumors arising in carcinogen-based models. Conversely, carcinogen-based models are more likely to capture the heterogeneity of human cancer and to acquire genomic alterations as is characteristic of human tumors (e.g.,67–69); this is exemplified in a recent study that compared the mutational spectrum of carcinogen-based versus GEM models of lung cancer70. However, although the comparative heterogeneity of carcinogen models relative to GEM models, in terms of incidence, latency, and tumor spectrum may be more reminiscent of human tumors, these features also make it more challenging to utilize carcinogen models as investigatory tools.
Thus, combining carcinogen-based and GEM modeling provides an opportunity to capitalize on the significance of relevant environmental influences yet in the context of genetically-defined tumor phenotypes. Indeed, there are numerous examples in which treatment of p53-mutant mice with cancer-promoting agents has been shown to exacerbate relevant cancer phenotypes in liver (e.g., DMN)71, colon (e.g., AOM)72, and bladder (e.g., BBN)73, 74; similarly, treatment of p27- or p21-mutant mice with relevant agents leads to their enhanced susceptibility to skin cancer75. Notably, although the general concept of combining carcinogen-based and GEM models was introduced several decades ago, this approach has not been widely incorporated in cancer prevention research. We envision that expansion of such studies would have a tremendous impact on precision cancer prevention by facilitating the discovery of how environmental factors promote tumorigenesis in defined genetic contexts.
Modeling chemoprevention in mice
Mouse models have been widely used to evaluate the consequences of a wide variety of synthetic and naturally-occurring chemopreventive agents in many types of cancers (e.g.,14, 16, 18). Nevertheless, considering the expansive body of chemoprevention studies in mice, the corresponding clinical impact for human cancer prevention has been relatively modest. This contrasts with the relatively significant impact that preclinical investigations of therapeutics in mice have had for treatment of human cancer22, 76, 77. In our view, the comparative success of preclinical analyses of therapeutics reflects, at least in part, the use of well-validated mouse models, the implementation of standardized criteria for optimal study design and validation of experimental findings from mouse models to human cancer, and input from teams of investigators with complementary expertise ranging from mouse modelers to clinical investigators76, 78. It would be highly beneficial to standardly incorporate analogous criteria for chemoprevention studies in mouse models.
Indeed, in some cases the use of GEM models for chemoprevention has been opportunistic rather than ideal. For example, one of the earliest and most widely utilized GEM models of prostate cancer is the TRAMP model, which expresses SV40 large T antigen in prostate79. While these mice develop pre-invasive lesions that progress to advanced disease, they primarily model neuroendocrine cancer80, whereas most human prostate tumors are adenocarcinomas. Nonetheless, because of its accessibility and relative simplicity, TRAMP mice have been has been widely used for evaluating chemopreventive agents, such as COX-2 inhibitors (e.g.,81, 82), as well as dietary and nutritional factors, such as lycopene83. However, the limited relevance of their prostate cancer phenotype relative to human prostate cancer raises concerns as to whether chemoprevention studies in TRAMP mice will ultimately be informative for human cancer. Moreover, there are now numerous GEM models of prostate cancer that more closely model the human disease24, including an alternative transgenic model that are similarly accessible and relatively simple as the TRAMP model; for example a transgenic model based on expression of c-Myc in the prostate84, 85 is now being employed for chemoprevention research86.
Nevertheless, the value of pursuing chemoprevention studies in mouse models is highlighted by key “success stories” in which analyses of mice have provided important insights that have impacted clinical practice for human cancer prevention and have provided significant mechanistic insights. For example, mouse models based on loss-of-function of the Adenomatous polyposis coli (Apc) gene, either as a consequence of carcinogen treatment or via gene targeting, have provided valuable models of colon cancer that have enabled many investigations of chemopreventive agents, including COX inhibitors72. Furthermore, the importance of targeting COX2 for cancer prevention was established using a gene targeting approach in which COX2 was deleted in the context of Apc mutant mice, resulting in a reduction in the number of small intestinal polyps87. Accordingly, preclinical chemoprevention studies using a COX2 inhibitor, celecoxib, were shown to protect against intestinal polyps in Apc mutant mice72. Subsequently, celecoxib was found to reduce the number of tumors in patients with familial or sporadic adenomatous polypopsis88, 89, which led to its FDA approval. Although celecoxib was subsequently found to have potentially harmful side effects90, these studies in mouse models provide the foundation for understanding the mechanism of COX2 activity and for the human clinical trials, and also provide the foundation for future studies to evaluate alternative COX2 inhibitors.
Other major “success stories” include the use of mouse models to study the consequences of hormones and hormonal therapy for female and male hormone-driven tumors. In particular, one of the major treatment paradigms for women at high risk for development or progression of breast cancer is treatment with anti-estrogens, and studies in mice played a key role in establishing current clinical practices. Analyses of mice were paramount in demonstrating the consequences of altered estrogen signaling for breast cancer through direct assessment of mice deficient for estrogen receptor, as well as preclinical assessment of the consequences of anti-estrogens for breast cancer91–94. Additionally, studies in mice played an important role in the development of hormone-driven contraceptives as well as establishing clinical guidelines for hormone replacement therapy95, 96. Analogously, studies in mice and other rodents have had a significant impact on our understanding of the consequences of hormone levels and hormone signaling for initiation and progression of prostate cancer97, 98. In summary, these success stories illustrate the value of conducting chemoprevention studies using well-validated mouse models and optimally-designed preclinical studies, and implemented by multidisciplinary investigatory teams.
Using mouse models to improve early detection
A key feature of virtually all cancer types is that detection at early disease stages, when tumors are locally invasive, is associated with better outcomes as compared to diagnosis at more advanced disease stages. Despite a great deal of progress in this area overall, some cancers have eluded early detection and are typically first identified only at advanced stages. This is exemplified for pancreatic cancer, which is rarely detected at early stages, primarily due to the lack of specific symptoms and rapid progression99; indeed, the median overall survival is only six months100. Notably, there are few serum biomarkers for pancreatic cancer currently available, and the primary one, CA19.9, has relatively poor sensitivity and specificity101.
Pancreatic cancer is well represented by informative GEM models102, which have been employed for various aspects of cancer prevention16, including the identification of potential biomarkers for early detection. Notably, a recent study integrated human epidemiological data with experimental studies in mouse models to reveal and mechanistically explain the finding that elevated plasma levels of branched-chain amino acids (BCAAs) are associated with increased risk for pancreatic cancer years prior to disease presentation103. This is a promising application for the use of mouse models in conjunction with human studies to identify new biomarkers for early detection and therefore secondary prevention.
More generally, analysis of GEM models may be informative for identification of biomarkers for early detection because they enable longitudinal sampling of both tumors and body fluids from the earliest stages of cancer initiation throughout the course of cancer progression104, 105. Indeed, analyses of GEM models have led to identification of putative biomarkers for early detection of ovarian106, colon107–109, and lung cancer110, which are at various stages of clinical validation. In particular, the use of mouse models of lung cancer to identify biomarkers for early detection may be particularly advantageous, considering the relatively poor survival of patients with lung cancer and the paucity of existing biomarkers111. Along these lines, profiling tumors and plasma from mouse models of lung cancer has revealed protein signatures that reflect the biology of human lung cancer110, which can be evaluated in proposed clinical trials for early detection of lung cancer. While these studies have promise for using mouse models for the identification of biomarkers and improving the landscape for early cancer detection, it will be imperative for definitive demonstrate of the translation of these findings from mouse models for improving early detection of human cancer.
Using mouse models to discriminate indolent versus aggressive cancers
At the other end of the spectrum are cancers that are readily detected at early stages and in dire need of having better means for determining whether premalignant or early cancerous lesions pose any significant threat. For example, prostate cancer, which is now the most commonly diagnosed cancer in American men, can readily detected at early stages by cancer screening testing for prostate specific antigen (PSA). However, only a minority of such early stage prostate cancers will progress to lethal tumors, and currently there is a lack of accurate means of distinguishing the majority of indolent prostate cancers from the minority of potentially lethal ones1, 112; this has led to a considerable problem of over-diagnosis and consequently to overtreatment. As a result, many men diagnosed with early stage prostate cancer are now opting for “active surveillance”113, which refers to active monitoring for disease progression rather than immediate treatment. While active surveillance is intended to minimize overtreatment, the concern is that avoiding or delaying treatment may mean that the opportunity for early treatment of more aggressive tumors will be missed. Thus, it would be beneficial to identify prognostic markers that distinguish indolent versus aggressive prostate cancer at the earliest possible stages to prevent aggressive disease.
As noted above, prostate cancer is well represented by numerous mouse models that represent the spectrum of disease phenotypes, including the earliest stages of cancer initiation24, which have provided an excellent resource for identification of biomarker panels associated with indolent or lethal prostate cancer27, 114. The identification of robust biomarkers of cancer initiation and progression has been facilitated by systems biology analyses, including cross-species approaches to effectively integrate experimental data from mouse models with human clinical data27, 114. The resulting biomarkers are now being evaluated in clinical studies to assess their prognostic accuracy and specificity for monitoring men on active surveillance. We believe that the general approach of integrate experimental data from mouse models to human cancer using cross-species systems biology analyses will represents an important application of mouse models for precision cancer prevention, and will ultimately improve the identification of biomarker panels for other cancers that, similar to prostate, are in need of effective means of discriminating lethal and non-lethal tumors at the earliest possible stages.
Conclusions and perspectives: Toward “ideal” mouse models for precision cancer prevention
In summary, we foresee a considerable future for investigations of mouse models to advance precision cancer prevention (Figure 2). However, in order for such investigations to have the best chance of having a real impact on human cancer prevention, it will be necessary to establish and adhere to essential criteria for using well-validated models, to have a precise understanding of the advantages and limitations of specific types of models for distinct experimental paradigms, and to optimize study designs that best simulate human clinical studies. In our view, the minimal criteria for mouse models to be relevant for cancer prevention include their ability to capture the de novo evolution of cancer as occurs in its native context, that they progress from premalignancy to overt cancer, and that they capture key biological and molecular features of the human cancers that they are intending to emulate. While we recognize the challenges associated with modeling tumor heterogeneity and genetic diversity in mice, we offer the suggestion that, in fact, the relative “homogeneity” of tumors in mice compared with human tumors can be beneficial in certain contexts, provided that these differences are recognized and accounted for. Furthermore, we envision that combining distinct types of modeling approaches and capitalizing on new technological advances will enable the generation of robust models that can be applied in specific experimental circumstances. In essence, we propose that “precision mouse modeling” will provide the best means of advancing precision cancer prevention.
Figure 2. Applications for mouse models for precision cancer prevention.
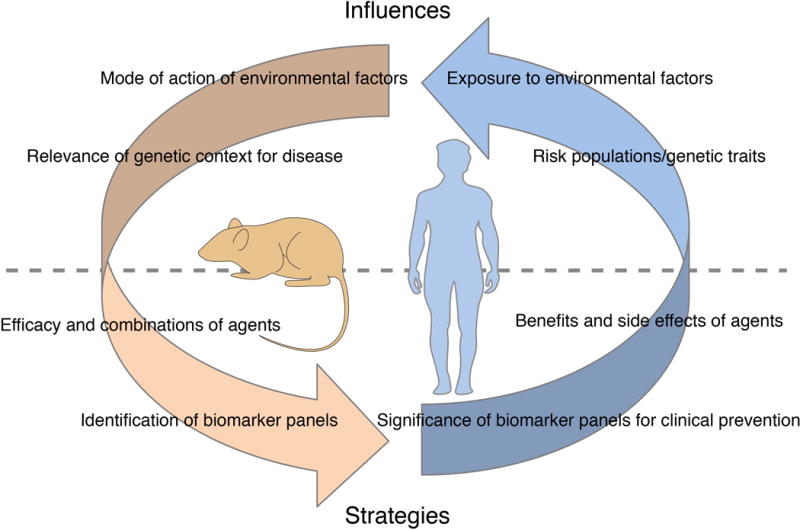
Examples of how studies in mouse models can augment human cancer prevention.
So, how exactly can mouse models help us to advance a precision cancer prevention framework that recognizes the importance of the individual context for how and when cancers will arise, as well as whether they will progress to harmful tumors (Text box 3) ? In our view, the greatest value of mouse models is their ability to provide detailed mechanistic studies and longitudinal analyses in vivo, which would be exceedingly challenging to pursue solely by studying human cancer. Such analyses can empower a breadth of investigations, ranging from assessment of the influence of risk factors to the development of biomarker panels for early detection and prognosis, including investigations of circulating tumor cells and exosomes. These are promising opportunities for using mouse models to inform on human cancer, particularly when combined with systems biology approaches to facilitate cross-species analysis from mouse to man. Indeed, a common theme of most of the success stories discussed in this review is the extrapolation of studies from mouse models to humans, including the recent application of systems biology approaches to integrate experimental data from mice to humans. Notably, as cancer prevention is now approaching the genomic era, genome-wide sequencing efforts focused on early stage cancer are likely to be increasingly important in future studies and complementary analyses of mouse models is likely to play a significant role in their interpretation115.
Text box 3: The path forward for mouse models for precision cancer prevention.
Key question: Mechanistic basis for genetic diversity influencing individualized cancer phenotypes
Approach: Use the diversity mouse strains, potentially in combination with using CRISPR technology to introduce genetic alterations
Challenges: Cost for implementation and challenges associated with analysis of potentially subtle phenotypes
Key Question: Assessment of risk factors in defined genetic contexts
Approach: Use the diversity mouse strains, in combination with GEM models and/or carcinogen-based models
Challenge: Cost for implementation and challenges in analyses
Key Question: Coalescing of environmental and genetic events for cancer initiation and progression
Approach: Combination of GEM models, DO, and carcinogen models
Challenge: Cost for implementation and challenges in analyses
Key Question: Genomic alterations in early stage cancers
Approach: Combination of GEM and carcinogen models
Challenge: GEM models may not capture the genomic changes that occur in human disease
Key Question: Evaluating the interaction of discrete genetic events for tumor evolution and tumor heterogeneity
Approach: Use CRISPR/Cas9 technologies in GEM or other types of models to introduce genetic events
Challenge: Still a relatively novel technology that is in development
Key Question: Elucidating cells of origin for tumor subtypes and relationship to tumor heterogeneity
Approach: GEM models that incorporate lineage tracing
Challenge: Difficult to identify cells of origin and to specifically target them for recombination
Key Question: Advancement of circulating tumor cells (CTCs) and tumor DNA as early prognosis markers
Approach: Longitudinal and molecular analyses of GEM models that incorporate lineage tracing
Challenge: Rare cell populations may be difficult to isolate and study
Key Question: Investigation of exosomes as diagnostic tools
Approach: Longitudinal and molecular analyses of GEM models
Challenge: Exosome levels maybe be low; need to adapt methods for mice
In conclusion, we believe that mouse models have the potential to play a vital role in all aspects of cancer prevention research, and their effective incorporation will undoubtedly be essential in a modern framework that focuses on precision prevention. Thus, in our view, it is time to move beyond wondering whether mouse models can provide insights for cancer prevention, and towards discussions about of mouse models can be used effectively to inform on prevention of human cancer.
Acknowledgments
We are very grateful to our colleagues who provided thoughtful comments on this review including: Nigel Crawford, Andrew Dannenberg, Ronny Drapkin, Edward Gelmann, Jeff Green, Sam Hanash, Kent Hunter, Chris Kemp, Kenneth Olive, Karlyne Reilly, David Threadgill, and Michael Shen.
The research in CAS laboratory is supported in part by funding from the National Cancer Institute (CA154293, CA0141535, and CA084294). CL is supported by the Swiss National Science Foundation (PBBSP3_146959 and P300P3_151158). AD is supported in part by the National Center for Advancing Translational Sciences, National Institutes of Health, Grant Number UL1 TR000040. CAS is an American Cancer Society Research Professor supported in part by a generous gift from the F.M. Kirby Foundation.
Glossary
- Primary prevention
Prevention of cancer occurrence and reducing the risk of cancer in the general population. Includes identification of hazards that promote disease or increase the risk of disease and reducing the exposures to such hazards
- Secondary prevention
Reduction of the impact of cancer after it has occurred and controlling of cancer progression or risk of progression. Includes early detection and early intervention as well as adopting lifestyles to prevent progression or recurrence
- Tertiary prevention
Reduction of the impact of cancer to ensure longer and better quality of life. Includes chronic disease management, but excludes cancer treatment
- Precision Medicine
Customization of health care with medical decisions and practices tailored to the individual patient
- Precision Prevention
Prevention strategies that incorporate precision medicine approaches and consider an individual’s unique risk profile
- Autochthonous mouse model
Models in which tumors arise de novo in the whole organism, as exemplified by GEM models
- Non-autochthonous
Models in which tumors are engrafted into host organisms as exemplified by PDX models
References
- 1.Welch HG, Black WC. Overdiagnosis in cancer. J Natl Cancer Inst. 2010;102:605–13. doi: 10.1093/jnci/djq099. [DOI] [PubMed] [Google Scholar]
- 2.Lippman SM, et al. Cancer prevention and the American Society of Clinical Oncology. J Clin Oncol. 2004;22:3848–51. doi: 10.1200/JCO.2004.07.139. [DOI] [PubMed] [Google Scholar]
- 3.Rebbeck TR. Precision prevention of cancer. Cancer Epidemiol Biomarkers Prev. 2014;23:2713–5. doi: 10.1158/1055-9965.EPI-14-1058. [DOI] [PubMed] [Google Scholar]
- 4.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–37. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–5. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hawgood S, Hook-Barnard IG, O’Brien TC, Yamamoto KR. Precision medicine: Beyond the inflection point. Sci Transl Med. 2015;7:300ps17. doi: 10.1126/scitranslmed.aaa9970. [DOI] [PubMed] [Google Scholar]
- 7.Friedman AA, Letai A, Fisher DE, Flaherty KT. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer. 2015;15:747–56. doi: 10.1038/nrc4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narod SA. Breast cancer prevention in the era of precision medicine. J Natl Cancer Inst. 2015;107 doi: 10.1093/jnci/djv078. [DOI] [PubMed] [Google Scholar]
- 9.Sieh W, Rothstein JH, McGuire V, Whittemore AS. The role of genome sequencing in personalized breast cancer prevention. Cancer Epidemiol Biomarkers Prev. 2014;23:2322–7. doi: 10.1158/1055-9965.EPI-14-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaughan TL, Fitzgerald RC. Precision prevention of oesophageal adenocarcinoma. Nat Rev Gastroenterol Hepatol. 2015;12:243–8. doi: 10.1038/nrgastro.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.William WN, Jr, et al. Erlotinib and the Risk of Oral Cancer: The Erlotinib Prevention of Oral Cancer (EPOC) Randomized Clinical Trial. JAMA Oncol. 2015:1–8. doi: 10.1001/jamaoncol.2015.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Dyke T, Jacks T. Cancer modeling in the modern era: progress and challenges. Cell. 2002;108:135–44. doi: 10.1016/s0092-8674(02)00621-9. [DOI] [PubMed] [Google Scholar]
- 13.Hanahan D, Wagner EF, Palmiter RD. The origins of oncomice: a history of the first transgenic mice genetically engineered to develop cancer. Genes Dev. 2007;21:2258–70. doi: 10.1101/gad.1583307. [DOI] [PubMed] [Google Scholar]
- 14.Green JE, Hudson T. The promise of genetically engineered mice for cancer prevention studies. Nat Rev Cancer. 2005;5:184–98. doi: 10.1038/nrc1565. [DOI] [PubMed] [Google Scholar]
- 15.Abate-Shen C, et al. The untapped potential of genetically engineered mouse models in chemoprevention research: opportunities and challenges. Cancer Prev Res (Phila) 2008;1:161–6. doi: 10.1158/1940-6207.CAPR-08-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grippo PJ, Tuveson DA. Deploying mouse models of pancreatic cancer for chemoprevention studies. Cancer Prev Res (Phila) 2010;3:1382–7. doi: 10.1158/1940-6207.CAPR-10-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenberg DW, Giardina C, Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis. 2009;30:183–96. doi: 10.1093/carcin/bgn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Rouggly L, You M, Lubet R. Animal models of lung cancer characterization and use for chemoprevention research. Prog Mol Biol Transl Sci. 2012;105:211–26. doi: 10.1016/B978-0-12-394596-9.00007-X. [DOI] [PubMed] [Google Scholar]
- 19.Tentler JJ, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–50. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hidalgo M, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. 2007;7:645–58. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- 22.Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. 2006;5:741–54. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- 23.Heyer J, Kwong LN, Lowe SW, Chin L. Non-germline genetically engineered mouse models for translational cancer research. Nat Rev Cancer. 2010;10:470–80. doi: 10.1038/nrc2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irshad S, Abate-Shen C. Modeling prostate cancer in mice: something old, something new, something premalignant, something metastatic. Cancer Metastasis Rev. 2013;32:109–22. doi: 10.1007/s10555-012-9409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi T, Owczarek TB, McKiernan JM, Abate-Shen C. Modelling bladder cancer in mice: opportunities and challenges. Nat Rev Cancer. 2015;15:42–54. doi: 10.1038/nrc3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ittmann M, et al. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013;73:2718–36. doi: 10.1158/0008-5472.CAN-12-4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aytes A, et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell. 2014;25:638–51. doi: 10.1016/j.ccr.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shibata M, Shen MM. The roots of cancer: stem cells and the basis for tumor heterogeneity. Bioessays. 2013;35:253–60. doi: 10.1002/bies.201200101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 30.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. 2012;12:786–98. doi: 10.1038/nri3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zschaler J, Schlorke D, Arnhold J. Differences in innate immune response between man and mouse. Crit Rev Immunol. 2014;34:433–54. [PubMed] [Google Scholar]
- 33.Morse H. Origins of inbred mice. Academic Press; New York: 1978. [Google Scholar]
- 34.Hunter KW. Mouse models of cancer: does the strain matter? Nat Rev Cancer. 2012;12:144–9. doi: 10.1038/nrc3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Churchill GA, et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36:1133–7. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 36.Threadgill DW, Churchill GA. Ten years of the Collaborative Cross. Genetics. 2012;190:291–4. doi: 10.1534/genetics.111.138032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Threadgill DW, Miller DR, Churchill GA, de Villena FP. The collaborative cross: a recombinant inbred mouse population for the systems genetic era. ILAR J. 2011;52:24–31. doi: 10.1093/ilar.52.1.24. [DOI] [PubMed] [Google Scholar]
- 38.Collaborative Cross, C. The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics. 2012;190:389–401. doi: 10.1534/genetics.111.132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Threadgill DW, Hunter KW, Williams RW. Genetic dissection of complex and quantitative traits: from fantasy to reality via a community effort. Mamm Genome. 2002;13:175–8. doi: 10.1007/s00335-001-4001-Y. [DOI] [PubMed] [Google Scholar]
- 40.Roberts A, Pardo-Manuel de Villena F, Wang W, McMillan L, Threadgill DW. The polymorphism architecture of mouse genetic resources elucidated using genome-wide resequencing data: implications for QTL discovery and systems genetics. Mamm Genome. 2007;18:473–81. doi: 10.1007/s00335-007-9045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasmussen AL, et al. Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science. 2014;346:987–91. doi: 10.1126/science.1259595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillippi J, et al. Using the emerging Collaborative Cross to probe the immune system. Genes Immun. 2014;15:38–46. doi: 10.1038/gene.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferguson B, et al. Melanoma susceptibility as a complex trait: genetic variation controls all stages of tumor progression. Oncogene. 2014;0 doi: 10.1038/onc.2014.227. [DOI] [PubMed] [Google Scholar]
- 44.Patel SJ, Molinolo AA, Gutkind S, Crawford NP. Germline genetic variation modulates tumor progression and metastasis in a mouse model of neuroendocrine prostate carcinoma. PLoS One. 2013;8:e61848. doi: 10.1371/journal.pone.0061848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Churchill GA, Gatti DM, Munger SC, Svenson KL. The Diversity Outbred mouse population. Mamm Genome. 2012;23:713–8. doi: 10.1007/s00335-012-9414-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.French JE, et al. Diversity Outbred Mice Identify Population-Based Exposure Thresholds and Genetic Factors that Influence Benzene-Induced Genotoxicity. Environ Health Perspect. 2015;123:237–45. doi: 10.1289/ehp.1408202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Tseng JC, Sun Y, Beck AH, Kung AL. Noninvasive imaging of tumor burden and molecular pathways in mouse models of cancer. Cold Spring Harb Protoc. 2015;2015:135–44. doi: 10.1101/pdb.top069930. [DOI] [PubMed] [Google Scholar]
- 48.Mou H, Kennedy Z, Anderson DG, Yin H, Xue W. Precision cancer mouse models through genome editing with CRISPR-Cas9. Genome Med. 2015;7:53. doi: 10.1186/s13073-015-0178-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer. 2015;15:387–95. doi: 10.1038/nrc3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuspa SH, Poirier MC. Chemical carcinogenesis: from animal models to molecular models in one decade. Adv Cancer Res. 1988;50:25–70. doi: 10.1016/s0065-230x(08)60434-0. [DOI] [PubMed] [Google Scholar]
- 51.Balmain A, Yuspa SH. Milestones in skin carcinogenesis: the biology of multistage carcinogenesis. J Invest Dermatol. 2014;134:E2–7. doi: 10.1038/skinbio.2014.2. [DOI] [PubMed] [Google Scholar]
- 52.Kemp CJ. Animal models of chemical carcinogenesis: driving breakthroughs in cancer research for 100 years. Cold Spring Harb Protoc. 2014:51–60. doi: 10.1101/pdb.top069906. doi:10.1.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen SM. Urinary bladder carcinogenesis. Toxicol Pathol. 1998;26:121–7. doi: 10.1177/019262339802600114. [DOI] [PubMed] [Google Scholar]
- 54.Balmain A, Brown K, Akhurst RJ, Fee FM. Molecular analysis of chemical carcinogenesis in the skin. Br J Cancer Suppl. 1988;9:72–5. [PMC free article] [PubMed] [Google Scholar]
- 55.Bain JA, Rusch HP. Carcinogenesis with Ultraviolet Radiation of Wave Length 2,800– 3,400 A*. Cancer Res. 1943;3:425–430. [Google Scholar]
- 56.Fisher MS, Kripke ML. Suppressor T lymphocytes control the development of primary skin cancers in ultraviolet-irradiated mice. Science. 1982;216:1133–4. doi: 10.1126/science.6210958. [DOI] [PubMed] [Google Scholar]
- 57.Kripke ML. The ABCs of sunscreen protection factors. J Invest Dermatol. 2003;121:VII–VIII. doi: 10.1046/j.1523-1747.2003.12337.x. [DOI] [PubMed] [Google Scholar]
- 58.Walker GJ, Soyer HP, Terzian T, Box NF. Modelling melanoma in mice. Pigment Cell Melanoma Res. 2011;24:1158–76. doi: 10.1111/j.1755-148X.2011.00923.x. [DOI] [PubMed] [Google Scholar]
- 59.Hecht SS, et al. Tobacco-specific nitrosamines: formation from nicotine in vitro and during tobacco curing and carcinogenicity in strain A mice. J Natl Cancer Inst. 1978;60:819–24. doi: 10.1093/jnci/60.4.819. [DOI] [PubMed] [Google Scholar]
- 60.Pfeifer GP, et al. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21:7435–51. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- 61.Memmott RM, Dennis PA. The role of the Akt/mTOR pathway in tobacco carcinogen-induced lung tumorigenesis. Clin Cancer Res. 2010;16:4–10. doi: 10.1158/1078-0432.CCR-09-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.West KA, Linnoila IR, Belinsky SA, Harris CC, Dennis PA. Tobacco carcinogen-induced cellular transformation increases activation of the phosphatidylinositol 3′-kinase/Akt pathway in vitro and in vivo. Cancer Res. 2004;64:446–51. doi: 10.1158/0008-5472.can-03-3241. [DOI] [PubMed] [Google Scholar]
- 63.Wislez M, et al. Inhibition of mammalian target of rapamycin reverses alveolar epithelial neoplasia induced by oncogenic K-ras. Cancer Res. 2005;65:3226–35. doi: 10.1158/0008-5472.CAN-04-4420. [DOI] [PubMed] [Google Scholar]
- 64.Han W, Gills JJ, Memmott RM, Lam S, Dennis PA. The chemopreventive agent myoinositol inhibits Akt and extracellular signal-regulated kinase in bronchial lesions from heavy smokers. Cancer Prev Res (Phila) 2009;2:370–6. doi: 10.1158/1940-6207.CAPR-08-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin RK, et al. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J Clin Invest. 2010;120:521–32. doi: 10.1172/JCI40706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawabata S, et al. Rapamycin prevents the development and progression of mutant epidermal growth factor receptor lung tumors with the acquired resistance mutation T790M. Cell Rep. 2014;7:1824–32. doi: 10.1016/j.celrep.2014.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim SK, et al. Identification of gene expression signature modulated by nicotinamide in a mouse bladder cancer model. PLoS One. 2011;6:e26131. doi: 10.1371/journal.pone.0026131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He Z, Kosinska W, Zhao ZL, Wu XR, Guttenplan JB. Tissue-specific mutagenesis by N-butyl-N-(4-hydroxybutyl)nitrosamine as the basis for urothelial carcinogenesis. Mutat Res. 2012;742:92–5. doi: 10.1016/j.mrgentox.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 69.Nassar D, Latil M, Boeckx B, Lambrechts D, Blanpain C. Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat Med. 2015;21:946–54. doi: 10.1038/nm.3878. [DOI] [PubMed] [Google Scholar]
- 70.Westcott PM, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature. 2015;517:489–92. doi: 10.1038/nature13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harvey M, et al. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat Genet. 1993;5:225–9. doi: 10.1038/ng1193-225. [DOI] [PubMed] [Google Scholar]
- 72.Corpet DE, Pierre F. Point: From animal models to prevention of colon cancer. Systematic review of chemoprevention in min mice and choice of the model system. Cancer Epidemiol Biomarkers Prev. 2003;12:391–400. [PMC free article] [PubMed] [Google Scholar]
- 73.Ozaki K, et al. High susceptibility of p53(+/−) knockout mice in N-butyl-N-(4-hydroxybutyl)nitrosamine urinary bladder carcinogenesis and lack of frequent mutation in residual allele. Cancer Res. 1998;58:3806–11. [PubMed] [Google Scholar]
- 74.Van Batavia J, et al. Bladder cancers arise from distinct urothelial sub-populations. Nat Cell Biol. 2014;16:982–91. doi: 10.1038/ncb3038. [DOI] [PubMed] [Google Scholar]
- 75.Philipp J, Vo K, Gurley KE, Seidel K, Kemp CJ. Tumor suppression by p27Kip1 and p21Cip1 during chemically induced skin carcinogenesis. Oncogene. 1999;18:4689–98. doi: 10.1038/sj.onc.1202840. [DOI] [PubMed] [Google Scholar]
- 76.Abate-Shen C, Pandolfi PP. Effective utilization and appropriate selection of genetically engineered mouse models for translational integration of mouse and human trials. Cold Spring Harb Protoc. 2013;2013 doi: 10.1101/pdb.top078774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Politi K, Pao W. How genetically engineered mouse tumor models provide insights into human cancers. J Clin Oncol. 2011;29:2273–81. doi: 10.1200/JCO.2010.30.8304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Olive KP, Politi K. Translational therapeutics in genetically engineered mouse models of cancer. Cold Spring Harb Protoc. 2014;2014:131–43. doi: 10.1101/pdb.top069997. [DOI] [PubMed] [Google Scholar]
- 79.Greenberg NM, et al. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci U S A. 1995;92:3439–43. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shappell SB, et al. Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64:2270–305. doi: 10.1158/0008-5472.can-03-0946. [DOI] [PubMed] [Google Scholar]
- 81.Gupta S, et al. Suppression of prostate carcinogenesis by dietary supplementation of celecoxib in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2004;64:3334–43. doi: 10.1158/0008-5472.can-03-2422. [DOI] [PubMed] [Google Scholar]
- 82.Narayanan BA, Narayanan NK, Pittman B, Reddy BS. Regression of mouse prostatic intraepithelial neoplasia by nonsteroidal anti-inflammatory drugs in the transgenic adenocarcinoma mouse prostate model. Clin Cancer Res. 2004;10:7727–37. doi: 10.1158/1078-0432.CCR-04-0732. [DOI] [PubMed] [Google Scholar]
- 83.Wan L, et al. Dietary tomato and lycopene impact androgen signaling- and carcinogenesis-related gene expression during early TRAMP prostate carcinogenesis. Cancer Prev Res (Phila) 2014;7:1228–39. doi: 10.1158/1940-6207.CAPR-14-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ellwood-Yen K, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–38. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 85.Iwata T, et al. MYC overexpression induces prostatic intraepithelial neoplasia and loss of Nkx3.1 in mouse luminal epithelial cells. PLoS One. 2010;5:e9427. doi: 10.1371/journal.pone.0009427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blando J, et al. Dietary energy balance modulates prostate cancer progression in Hi-Myc mice. Cancer Prev Res (Phila) 2011;4:2002–14. doi: 10.1158/1940-6207.CAPR-11-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oshima M, et al. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–9. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 88.Steinbach G, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–52. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 89.Arber N, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355:885–95. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 90.Solomon SD, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071–80. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 91.Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2:205–13. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 92.Jordan VC. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr Relat Cancer. 2014;21:R235–46. doi: 10.1530/ERC-14-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jordan VC, Koerner S. Tamoxifen as an anti-tumour agent: role of oestradiol and prolactin. J Endocrinol. 1976;68:305–11. doi: 10.1677/joe.0.0680305. [DOI] [PubMed] [Google Scholar]
- 94.Ali S, Buluwela L, Coombes RC. Antiestrogens and their therapeutic applications in breast cancer and other diseases. Annu Rev Med. 2011;62:217–32. doi: 10.1146/annurev-med-052209-100305. [DOI] [PubMed] [Google Scholar]
- 95.Haslam SZ, Osuch JR, Raafat AM, Hofseth LJ. Postmenopausal hormone replacement therapy: effects on normal mammary gland in humans and in a mouse postmenopausal model. J Mammary Gland Biol Neoplasia. 2002;7:93–105. doi: 10.1023/a:1015726608146. [DOI] [PubMed] [Google Scholar]
- 96.Raafat AM, Hofseth LJ, Li S, Bennett JM, Haslam SZ. A mouse model to study the effects of hormone replacement therapy on normal mammary gland during menopause: enhanced proliferative response to estrogen in late postmenopausal mice. Endocrinology. 1999;140:2570–80. doi: 10.1210/endo.140.6.6634. [DOI] [PubMed] [Google Scholar]
- 97.Bosland MC. Hormonal factors in carcinogenesis of the prostate and testis in humans and in animal models. Prog Clin Biol Res. 1996;394:309–52. [PubMed] [Google Scholar]
- 98.Bosland MC, et al. Multistage prostate carcinogenesis: the role of hormones. Princess Takamatsu Symp. 1991;22:109–23. [PubMed] [Google Scholar]
- 99.Urayama S. Pancreatic cancer early detection: expanding higher-risk group with clinical and metabolomics parameters. World J Gastroenterol. 2015;21:1707–17. doi: 10.3748/wjg.v21.i6.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 101.Poruk KE, et al. The clinical utility of CA 19-9 in pancreatic adenocarcinoma: diagnostic and prognostic updates. Curr Mol Med. 2013;13:340–51. doi: 10.2174/1566524011313030003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Westphalen CB, Olive KP. Genetically engineered mouse models of pancreatic cancer. Cancer J. 2012;18:502–10. doi: 10.1097/PPO.0b013e31827ab4c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mayers JR, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med. 2014;20:1193–8. doi: 10.1038/nm.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hanash SM, Taguchi A. Mouse to human blood-based cancer biomarker discovery strategies. Cold Spring Harb Protoc. 2014;2014:144–9. doi: 10.1101/pdb.top078808. [DOI] [PubMed] [Google Scholar]
- 105.Kucherlapati R. Genetically modified mouse models for biomarker discovery and preclinical drug testing. Clin Cancer Res. 2012;18:625–30. doi: 10.1158/1078-0432.CCR-11-2021. [DOI] [PubMed] [Google Scholar]
- 106.Pitteri SJ, et al. Integrated proteomic analysis of human cancer cells and plasma from tumor bearing mice for ovarian cancer biomarker discovery. PLoS One. 2009;4:e7916. doi: 10.1371/journal.pone.0007916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hung KE, et al. Comprehensive proteome analysis of an Apc mouse model uncovers proteins associated with intestinal tumorigenesis. Cancer Prev Res (Phila) 2009;2:224–33. doi: 10.1158/1940-6207.CAPR-08-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Taguchi A, et al. MAPRE1 as a Plasma Biomarker for Early-Stage Colorectal Cancer and Adenomas. Cancer Prev Res (Phila) 2015;8:1112–9. doi: 10.1158/1940-6207.CAPR-15-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fijneman RJ, et al. Proximal fluid proteome profiling of mouse colon tumors reveals biomarkers for early diagnosis of human colorectal cancer. Clin Cancer Res. 2012;18:2613–24. doi: 10.1158/1078-0432.CCR-11-1937. [DOI] [PubMed] [Google Scholar]
- 110.Taguchi A, et al. Lung cancer signatures in plasma based on proteome profiling of mouse tumor models. Cancer Cell. 2011;20:289–99. doi: 10.1016/j.ccr.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shaw AT, Kirsch DG, Jacks T. Future of early detection of lung cancer: the role of mouse models. Clin Cancer Res. 2005;11:4999s–5003s. doi: 10.1158/1078-0432.CCR-05-9005. [DOI] [PubMed] [Google Scholar]
- 112.Daskivich TJ, et al. Overtreatment of men with low-risk prostate cancer and significant comorbidity. Cancer. 2011;117:2058–66. doi: 10.1002/cncr.25751. [DOI] [PubMed] [Google Scholar]
- 113.Cooperberg MR, Carroll PR, Klotz L. Active surveillance for prostate cancer: progress and promise. J Clin Oncol. 2011;29:3669–76. doi: 10.1200/JCO.2011.34.9738. [DOI] [PubMed] [Google Scholar]
- 114.Irshad S, et al. A molecular signature predictive of indolent prostate cancer. Sci Transl Med. 2013;5:202ra122. doi: 10.1126/scitranslmed.3006408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Robles-Espinoza CD, Adams DJ. Cross-species analysis of mouse and human cancer genomes. Cold Spring Harb Protoc. 2014;2014:350–8. doi: 10.1101/pdb.top078824. [DOI] [PubMed] [Google Scholar]
- 116.Jacks T. Tumor suppressor gene mutations in mice. Annu Rev Genet. 1996;30:603–36. doi: 10.1146/annurev.genet.30.1.603. [DOI] [PubMed] [Google Scholar]
- 117.Wang J, Abate-Shen C. Analyses of tumor-suppressor genes in germline mouse models of cancer. Cold Spring Harb Protoc. 2014;2014:807–12. doi: 10.1101/pdb.top069773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jacks T, et al. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 119.Jacks T, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 120.Donehower LA, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 121.Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 2006;25:5885–97. doi: 10.1038/sj.onc.1209871. [DOI] [PubMed] [Google Scholar]
- 122.Jonkers J, Berns A. Conditional mouse models of sporadic cancer. Nat Rev Cancer. 2002;2:251–65. doi: 10.1038/nrc777. [DOI] [PubMed] [Google Scholar]
- 123.Yeh ES, Vernon-Grey A, Martin H, Chodosh LA. Tetracycline-regulated mouse models of cancer. Cold Spring Harb Protoc. 2014;2014 doi: 10.1101/pdb.top069823. pdb top069823. [DOI] [PubMed] [Google Scholar]
- 124.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4:1064–72. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Branda CS, Dymecki SM. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev Cell. 2004;6:7–28. doi: 10.1016/s1534-5807(03)00399-x. [DOI] [PubMed] [Google Scholar]
- 126.Lyons SK, Patrick PS, Brindle KM. Imaging mouse cancer models in vivo using reporter transgenes. Cold Spring Harb Protoc. 2013;2013:685–99. doi: 10.1101/pdb.top069864. [DOI] [PubMed] [Google Scholar]
- 127.Kretzschmar K, Watt FM. Lineage tracing. Cell. 2012;148:33–45. doi: 10.1016/j.cell.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 128.Premsrirut PK, et al. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell. 2011;145:145–58. doi: 10.1016/j.cell.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Huijbers IJ, Krimpenfort P, Berns A, Jonkers J. Rapid validation of cancer genes in chimeras derived from established genetically engineered mouse models. Bioessays. 2011;33:701–10. doi: 10.1002/bies.201100018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang H, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–8. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]