

Abstract
Hepatocellular carcinoma (HCC) represents the fifth‐most common form of cancer worldwide and carries a high mortality rate attributed to lack of effective treatment. Males are 8 times more likely to develop HCC than females, an effect largely driven by sex hormones, albeit through still poorly understood mechanisms. We previously identified TRIM28 (tripartite protein 28), a scaffold protein capable of recruiting a number of chromatin modifiers, as a crucial mediator of sexual dimorphism in the liver. Trim28hep–/– mice display sex‐specific transcriptional deregulation of a wide range of bile and steroid metabolism genes and development of liver adenomas in males. We now demonstrate that obesity and aging precipitate alterations of TRIM28‐dependent transcriptional dynamics, leading to a metabolic infection state responsible for highly penetrant male‐restricted hepatic carcinogenesis. Molecular analyses implicate aberrant androgen receptor stimulation, biliary acid disturbances, and altered responses to gut microbiota in the pathogenesis of Trim28hep–/–‐associated HCC. Correspondingly, androgen deprivation markedly attenuates the frequency and severity of tumors, and raising animals under axenic conditions completely abrogates their abnormal phenotype, even upon high‐fat diet challenge. Conclusion: This work underpins how discrete polyphenic traits in epigenetically metastable conditions can contribute to a cancer‐prone state and more broadly provides new evidence linking hormonal imbalances, metabolic disturbances, gut microbiota, and cancer. (Hepatology 2017;66:235–251).
Abbreviations
- ADT
androgen deprivation therapy
- ALT
alanine transaminase
- ANOVA
analysis of variance
- AR
androgen receptor
- AREs
AR‐binding elements
- AST
aspartate transaminase
- BA
bile acid
- BMI
body mass index
- Ccnd1
CyclinD1
- CD
chow diet
- ChIP‐seq
chromatin immunoprecipitation sequencing
- Cidec
cell‐death–inducing DFFA‐like effector C
- μ‐CT
microcomputational tomography
- DHT
dihydrotestosterone
- Foxa
forkhead box protein A
- FXR
farnesoid X receptor
- GF
germ free
- GO
Gene Ontology
- gs
glutamine synthase
- GSEA
gene set enrichment analysis
- GSK‐3β
glycogen synthase kinase 3‐beta
- GST
glutathione S‐transferase
- HCA
hepatocellular adenoma
- HCC
hepatocellular carcinoma
- HFD
high‐fat diet
- HS
hepatic steatosis
- IHC
immunohistochemistry
- IL‐6
interleukin‐6
- KZFPs
KRAB‐containing zinc fingers
- LCFAs
low‐chain fatty acids
- LDA
linear discriminant analysis
- LDL
low‐density lipoprotein
- LEfSe
LDA effect size
- LPS
lipopolysaccharide
- MCFAs
medium‐chain fatty acids
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OTUs
operational taxonomic units
- PCa
prostate cancer
- RBCC
RING‐B box‐coiled coil
- SHP
small heterodimer partner
- TAMPs
tumor‐associated molecular patterns
- TCA
taurocholic acid
- T‐αMCA
tauro‐alpha‐muricholic acid
- T‐βMCA
tauro‐beta‐muricholic acid
- Tmprss2
transmembrane protease serine 2
- TRIM28
tripartite motif‐containing 28
- wt
wild type
Adverse dietary habits and aging represent crucial stress‐inducing conditions detrimental to internal homeostasis, ultimately increasing the susceptibility of various tissues to metabolic disorders and fostering oncogenic transformation.1 The burden of malignancies increases progressively in overweight and elderly people, both conditions promoting a cancer‐prone phenotype characterized by systemic inflammation, increased oxidative stress, and altered xenobiotic metabolism.2 For certain organs, including the liver, the overall risk of cancer death is 4.5‐fold higher in men with a body mass index (BMI) exceeding 40 kg/m2 and classified as obese.3 The tumor‐promoting dynamics driven by stress‐inducing factors display a strong sex bias,4 and the resulting imbalance is especially evident for HCC, with a solid male predominance regardless of geographical area and ethnicity.5 Although both the protective role of estrogens and the promoting effect of androgens on HCC development are well documented,6 little is known about the underlying molecular mechanisms, although a recent study implicates forkhead box protein A (Foxa) transcription factors.7
Mammals are endowed with intrinsic homeostatic plasticity, defined as the ability to respond to external stimuli by adjusting their metabolic phenotype to the environment. This leads to adaptive changes aimed at preserving tissue homeostasis through multiple and elaborate organ‐organ interactions. As a downside of this trait, metabolic infection refers to a functional link between the gut and distal organs, such as the liver, whereby unfavorable products of the microbiota and altered xenobiotic and bile acid metabolism become pathogenic forces for the development of nonalcoholic steatohepatitis (NASH) and liver cancer.8
How cells sense and respond to extrinsic cues relies on dynamic transcriptional changes correlating with extensive chromatin remodeling. Correspondingly, the increased susceptibility to stress‐inducing factors and related diseases may arise, at least in part, from epigenetically driven alterations in gene expression. A broad range of chromatin changes can be observed in different hepatocellular carcinoma (HCC) subtypes, and even though they often converge toward the activation of oncogenic pathways such as β‐catenin, the mechanistic connection is largely unexplained.9 Each HCC bears a unique mutational profile, yet up to 50% harbor mutations in some chromatin regulator.10
We previously identified tripartite motif‐containing 28 (TRIM28; also known as transcription intermediary factor 1‐beta, KRAB‐associated protein 1, or KRAB‐A‐interacting protein 1), a scaffold protein capable of recruiting a number of chromatin modifiers, as a crucial mediator of sexual dimorphism in the liver, with hepato‐specific Trim28 deletion (Trim28hep–/–) in the mouse leading to sex‐specific transcriptional deregulation of a wide range of bile and steroid metabolism genes and to male‐restricted development of liver adenomas.11
In the present work, we demonstrate that lack of hepatic Trim28 expression engenders an epigenetically unstable condition that severely compromises stress‐adaptation abilities. Trim28hep–/– mice exhibit alterations of transcriptional dynamics exacerbated by environmental insults, such as obesity and ageing, which lead to altered sex hormone signaling, perturbations of bile acids metabolism, intestinal dysbiosis, and a state of metabolic infection that precipitate the development of HCC.
Materials and Methods
MOUSE AND NUTRITIONAL CHALLENGE
Mice were fed with standard mouse chow (Kliba Nafag 3242 diet, Kilba Nafag, Kaiseraugst, Switzerland, irradiated >25 kGy, 3.85 kcal/g, and 10% kcal from fat) or high‐fat diet (HFD; Open Source Diets, Research Diets, Inc., New Brunswick, NJ, USA, D12331, irradiated twice >25 kGy; 5.56 kcal/g, and 58% kcal from fat) and water provided ad libitum. All animal experiments were approved by the local veterinary office and carried out in accord with the European Community Council Directive (86/609/EEC) for care and use of laboratory animals. Mice were rederived to germ‐free status by two‐cell embryo transfer into pseudopregnant germ‐free recipient females. Germ‐free (GF) mice were bred and maintained in flexible film isolators at the Clean Mouse Facility, University of Bern (Bern, Switzerland). GF mice were routinely monitored by culture‐dependent and culture‐independent methods and were additionally confirmed to be pathogen free.
CHROMATIN IMMUNOPRECIPITATION SEQUENCING ANALYSIS
Trim28 chromatin immunoprecipitation sequencing (ChIP‐seq) was produced in our lab,11 whereas androgen receptor (AR) and Foxa1 ChIP‐seq comes from.7 Reads were mapped to the murine genome assembly (mm9) using Bowtie212 (the exact parameters are: bowtie2 ‐t —sensitive‐local ‐x $index ‐U $reads). The peaks were called using the MACS program.13
RNA SEQUENCING ANALYSIS
Reads were mapped to the murine genome (mm9) using TopHat14 (v2.0.11) in sensitive mode (the exact parameters are: tophat ‐g 1 —no‐novel‐juncs —no‐novel‐indels ‐G $gtf —transcriptome‐index $ transcriptome —b2‐sensitive $index $reads1 $reads2). Gene counts were generated using HTSeq‐count. Normalization for sequencing depth and fold changes (for gene set enrichment analysis [GSEA]) between groups of interest were computed using Voom15 because it has been implemented in the limma package of Bioconductor.
STATISTICAL ANALYSIS
All data are expressed as means with error bars representing the standard error of the means except for the bacterial taxonomic analysis in Figure 7D where the bars represent the 95% coefficient of variation. Outcomes on sufficiently metric scales (mRNA levels, biomarkers levels, and percentage of total input) were assessed with linear models (one‐ or two‐way analysis of variance [ANOVA]), modeling the mean value of each group. Relevant comparisons of mean values between two groups were made with two‐sided pair‐wise t tests; the null hypotheses stated that there was no difference in the means. P values were adjusted for the number of comparisons by using the Bonferroni method. Contingency testing was performed using Fisher's exact test.
Figure 7.
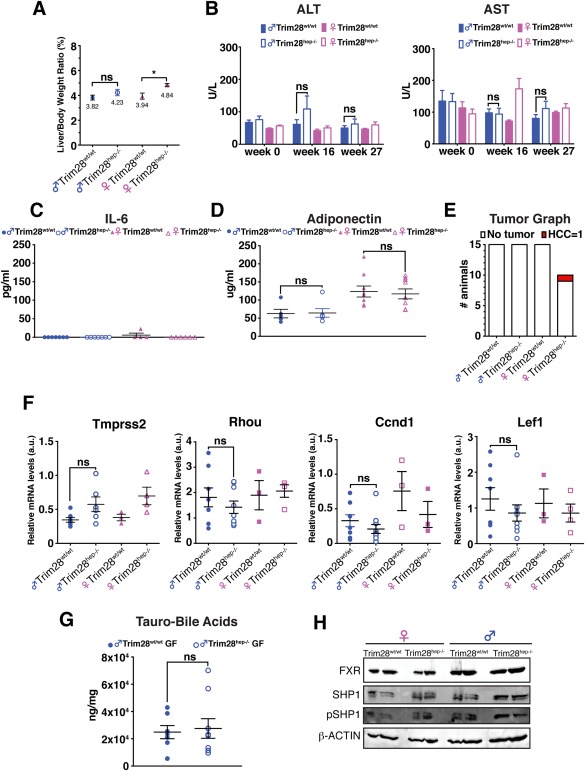
Axenic conditions rescue both the inflammatory and cancer phenotype of Trim28hep–/– animals. (A) Liver‐to‐body weight ratios in germ‐free (GF) HFD‐fed indicated animals (n = 10 for female Trim28hep–/–; n = 15 for all other groups). *P < 0.05; ns (not significant), by one‐way ANOVA followed by Bonferroni's posttest. (B) Serum ALT (left) and AST (right) activity of GF HFD‐fed animals. Ns = not significant, by two‐way ANOVA followed by Bonferroni's posttest. (C,D) Serum IL‐6 (C) and adiponectin (D) levels in GF mice receiving HFD for 27 weeks (n = 7 per group). ns (not significant), by two‐way ANOVA followed by Bonferroni's posttest. (E) Tumor classification graph in livers of GF HFD‐fed animals (n = 10 for female Trim28hep–/–; n = 15 for all other groups). (F) qPCR on liver samples for direct targets of AR (Tmprss2 and Rhou1) and β‐catenin (Cnnd1 and Lef1). Values are calculated as mean relative expression after normalization to three housekeeping genes ± SEM; ns (not significant), by one‐way ANOVA followed by Bonferroni's posttest. (G) Absolute quantification of hepatic BA content in male GF Trim28wt/wt (filled circle) and Trim28hep–/– (open circle) mice after receiving HFD for 27 weeks. Values are normalized per protein content and per mg of tissue and expressed as mean ± SEM; ns (not significant), by two‐way ANOVA followed by Bonferroni's posttest. (H) Western blotting analysis for FXR, phospho‐, and total‐SHP expression in liver samples from GF animals receiving HFD for 27 weeks. β‐actin was used as internal loading control. One experiment of three is shown. Abbreviations: a.u., arbitrary units; pSHP, phosphorylated SHP; Rhou1, Ras homolog family member U1.
Results
AGING AND OBESITY LEAD TO MALE‐RESTRICTED LIVER CANCER IN Trim28hep–/– MICE
Trim28hep–/– animals were generated as reported on a pure C57BL/6 background.11 Deep‐sequencing–based transcriptome profiling of chow diet (CD)‐fed young animals revealed a dysregulation of 862 and 703 genes in male and female mice, respectively (Supporting Fig. S1A), with male‐specific over‐representation of transcripts encoding for proteins involved in xenobiotic, bile acid (BA), and steroid metabolism, as assessed by Gene Ontology (GO) and GSEA (Supporting Fig. S1B,C).
We first assessed the impact of aging on Trim28hep–/– animals by keeping them on a CD for 73 weeks (Fig. 1A). This resulted in deregulation of hepatic enzymes with male‐specific signs of liver dysfunction, such as hepatomegaly and increases in circulating inflammatory cytokines (Supporting Fig. S1D‐G), albeit without frank steatosis (Fig. 1B). Still, 7 of 9 of these mice, all male, bore malignant lesions at the outset of the experiment (Fig. 1C and Supporting Fig. S1H).
Figure 1.
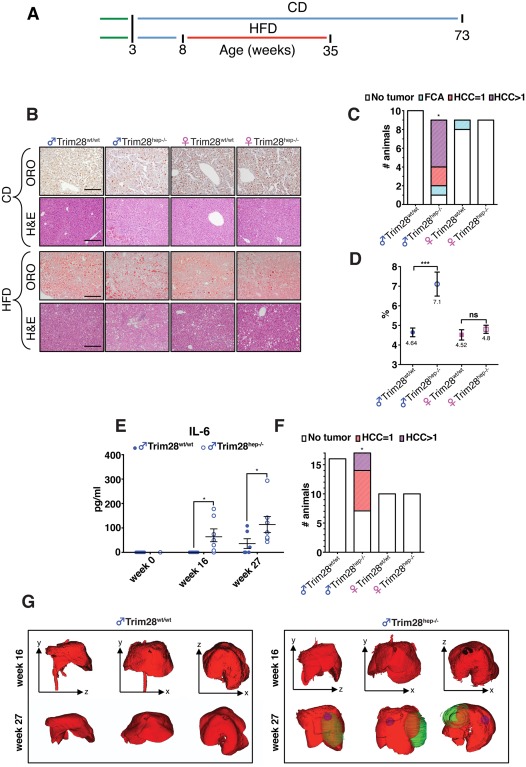
Environmental stress‐inducing factors induce liver damage in Trim28hep–/– mice. (A) Experimental timeline procedure before final analysis. Weaning = green lines; CD = chow diet, in blue lines; HFD = high‐fat diet, in red line (for long‐term CD challenge, n = 10 for male Trim28hep–/– and n = 9 for other groups; for the HFD challenge, n = 16 for male Trim28wt/wt, n = 17 for male Trim28hep‐/–, and n = 10 for female cohorts). (B) Hematoxilin‐eosin (H&E) and Oil Red O (ORO) stainings of liver samples from long‐term CD‐fed (top panels) and HFD‐fed (bottom panels) indicated animals; scale bars, 200 μm. (C) Tumor classification graph in livers of 73 wo CD‐fed animals (FCA [focal cellular adhesion], HCC = 1 single carcinoma, and HCC >1 multiple carcinomas). * P value of male Trim28hep–/– versus Trim28wt/wt comparison = 0.0004114 by Fisher's exact test. (D) Liver‐to‐body weight ratios as measure of hepatomegaly in HFD‐fed animals. ***P < 0.001, by one‐way ANOVA followed by Bonferroni's posttest; ns, not significant. (E) Serum protein levels of proinflammatory cytokine IL‐6 measured at different time points in HFD‐fed male mice (n = 10). *P < 0.01, by two‐way ANOVA followed by Bonferroni's posttest. (F) Tumor classification graph in livers of 35 wo HFD‐fed animals. * P value of male Trim28hep–/– versus Trim28wt/wt comparison = 0.008834 by Fisher's exact test. (G) Representative anatomical plane and three‐dimensional reconstruction from μ‐CT analysis of male livers (red) performed 16 (top) or 27 weeks (bottom) after receiving HFD. Sagittal (y‐x), coronal (y‐x), and transverse (z‐x) planes are represented; tumor masses are indicated in blue and green. Abbreviation: wo, weeks old.
To determine whether the known contribution of obesity to hepatic inflammation may set the stage for HCC development in our model, we subjected mice to a 27‐week‐long metabolic challenge of HFD enriched in medium‐chain fatty acids (MCFAs) derived from hydrogenated coconut oil (58% calories from fat; caloric density, 5.56 Kcal/g), starting at 8 weeks of age (Fig. 1A). Likely attributable to up‐regulation of lipogenic pathways, high doses of dietary MCFAs cause higher degrees of hepatic steatosis (HS) and triglyceride accumulation than lard‐based HFD, which is rather enriched in long‐chain fatty acids (LCFAs). MCFA‐rich HFD triggered weight gain and HS, yet these and other metabolic parameters, such as body fat composition, food intake, and oral glucose tolerance test were comparable between gender‐matched Trim28wt/wt and Trim28hep–/– mice (Fig. 1B and Supporting Fig. S1I‐O). However, absence of TRIM28 led to male‐specific hepatomegaly (Fig. 1D and Supporting Fig. S1P), progressive plasma dyslipidemia, and abnormally elevated levels of hepatic enzymes (Supporting Fig. S1Q). Furthermore, exposure to HFD triggered a low‐grade systemic inflammation state in male Trim28hep–/– mice, characterized by both systemic increase in interleukin‐6 (IL‐6) and decreased levels of the anti‐inflammatory adipokine, adiponectin (Fig. 1E and Supporting Fig. S1R).16, 17 Diet‐induced obesity also drove accelerated oncogenesis and increased tumor burden in male Trim28hep–/– mice, with 10 of 17 of these animals developing HCC whereas all others in the cohort remained tumor free (Fig. 1F). Longitudinal in vivo assessment by microcomputational tomography (μ‐CT) further delineated a temporal window for emergence of macroscopically visible cancer lesions, spanning between the sixteenth week on HFD and the 27‐week endpoint (Fig. 1G).
ABERRANT AR ACTIVATION IN Trim28hep–/– TUMORS
Hepatocarcinogenesis is a complex process caused by the accumulation of genetic and epigenetic changes. In addition to its role during liver development, the Wnt/β‐catenin pathway has been identified as a key player in metabolic zonation, BA homeostasis, and drug detoxification.18 Furthermore, aberrant β‐catenin activation has been detected in HCC from both human and rodents, with a mutation rate ranging between 12% and 34% and anomalous expression detected in up to 90% of neoplastic lesions.9, 10 Correspondingly, immunohistochemistry (IHC)‐based studies revealed the nuclear accumulation of β‐catenin and ectopic expression of its target gene, glutamine synthase (gs),19 in Trim28hep–/– neoplastic lesions (Fig. 2A, top and middle panels), and western blotting analyses confirmed increased levels of stabilized β‐catenin and hyperphosphorylation at serine 9 of its negative regulator, glycogen synthase kinase 3‐beta (GSK‐3β; Fig. 2B). Furthermore, upon confronting our transcriptome results with publicly available data, GSEA analyses positively correlated β‐catenin targets, as documented in liver of Apc hep–/– mice, with the transcript repertoire of male Trim28hep–/– HCC (Fig. 2C), which was also antithetical to β‐catenin repressed transcripts (Supporting Fig. S2A).
Figure 2.
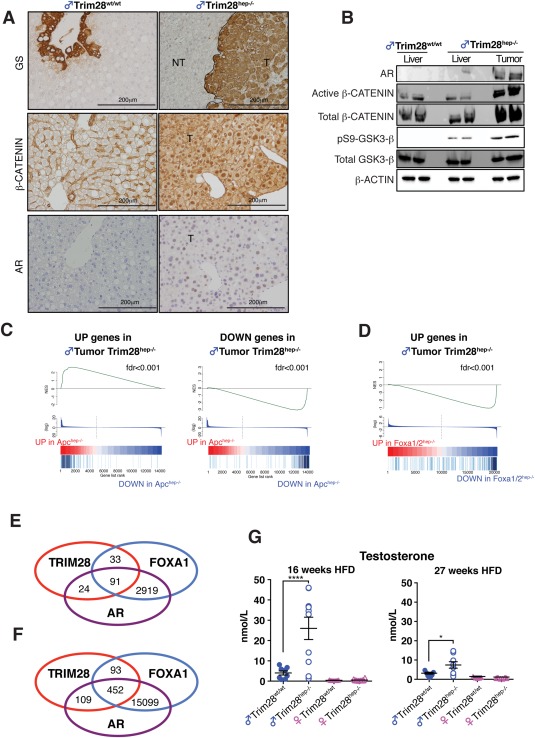
TAMPs characterization of male Trim28hep–/– hepatocarcinogenesis. (A) Diaminobenzidine staining of liver sections for β‐Catenin (top panels), Glutamine Synthase (GS; middle panels), and AR (bottom panels). NT, parenchyma; T, tumor; scale bar, 200 μm. (B) Western blotting analysis of indicated proteins on liver and tumor lysates from HFD‐fed animals. β‐actin was used as internal loading control. One representative experiment of three is represented. (C) Plot of GSEA analysis of up‐regulated (left) and down‐regulated (right) genes in Trim28hep–/– tumors with genes deregulated in Apchep–/– livers (fold change [FC], >5; P < 0.05). NES, normalized enrichment score; fdr, false discovery rate. (D) Plots of GSEA analysis of up‐regulated (left) genes in Trim28hep–/– tumors with genes deregulated in diethylnitrosamine‐treated FoxA1hep–/– livers (FC, >5; P < 0.05), showing the anticorrelation of male Trim28hep–/– transcriptome with a model of AR signaling attenuation. NES, normalized enrichment scores; fdr, false discovery rate. (E,F) Venn diagram illustrating the degree to which TRIM28, FOXA1, and AR peaks are overlapping in liver tissues when limiting the analysis to gene promoter (E; ±5 kbp TSS) or regulatory regions (F; ±50 kbp TSS). (G) Serum testosterone levels of indicated groups measured 16 (left) or 27 weeks (right) after receiving HFD (n = 15 for males and n = 10 for females). *P < 0.05; ***P < 0.001, by one‐way ANOVA followed by Bonferroni's posttest. Values are calculated as mean ± SEM. Abbreviations: kbp, kilobase pairs; TSS, transcription start sites.
Strongly elevated levels of AR were documented by IHC (Fig. 2A, bottom) and western blotting (Fig. 2B) in Trim28hep–/– tumors. This resulted in aberrant activation of AR‐dependent pathways, as revealed by the negative correlation between the transcriptome of these dysplastic lesions and that of mice in which AR signaling is severely compromised by Foxa1/2 knockout (Fig. 2D).7 Accordingly, upon matching our ChIP‐seq results with publicly available data, we found a relevant fraction of overlap between the binding sites of TRIM28, AR, and FOXA1 at both promoters and enhancers (91 and 452 peaks, respectively; Fig. 2E,F). These included the regulatory regions of male‐restricted deregulated genes, such as the β‐catenin target, CyclinD1 (Ccnd1),20 the AR‐direct target transmembrane protease serine 2 (Tmprss2),21 and the prosteatotic factor, cell‐death–inducing DFFA‐like effector C (Cidec; Supporting Fig. S2B).11 We asked whether these deregulations might also partly result from an imbalance of sex hormones, and found that Trim28hep–/– males indeed exhibited elevated plasma testosterone levels after 16 weeks of HFD (Fig. 2G). This is consistent with the observation that biological processes deregulated in male Trim28hep–/– animals include steroid hormone metabolism and genes required for androgen biosynthesis, such as cytochrome P450 family 17 subfamily a member 1, a rate‐limiting enzyme for steroidogenesis and an important target for treatment of prostate cancer (Supporting Fig. S2C).
ANDROGEN DEPRIVATION THERAPY ALLEVIATES TUMOR BURDEN
Abnormal androgen activity underlies many pathological conditions, mainly represented by androgen‐dependent prostate cancer (PCa).22 Androgen deprivation therapy (ADT) is currently the most effective systematic treatment available for PCa patients.23 The increase in plasma testosterone, aberrant AR expression, and activation of AR‐responsive genes in hepatic tumors of Trim28hep–/– animals prompted us to explore the role of this pathway in the oncogenic process. For this, we gelded 4‐week‐old Trim28hep–/– and Trim28wt/wt mice and subjected them to our HFD metabolic challenge (Fig. 3A). Bilateral orchiectomy led to an effective reduction of plasma testosterone levels (Supporting Fig. S3A). Body weight and fat composition increased to the same extent in both arms of the cohort (Supporting Fig. S3B‐D) with comparable levels of daily food consumption (Supporting Fig. S3E), although anorchid animals had a reduced appetite compared to age‐matched controls (see Supporting Fig. S1M), as described.24 Neutered HFD‐fed Trim28hep–/– animals displayed progressive increases in plasma levels of low‐density lipoprotein (LDL)‐cholesterol and markers of hepatic inflammation, compared to wild‐type (wt) Trim28 controls (Fig. 3B,C and Supporting Fig. S3F). Castration impacted on tumor burden with a significant reduction in number and size of the lesions (Fig. 3D,E). Furthermore, histopathological analyses determined that neutered mice only developed hepatocellular adenoma (HCA), contrasting with the high‐frequency HCC observed in gonad‐endowed controls (Supporting Fig. S3G‐L). Nevertheless, despite a marked drop in circulating androgens (Supporting Fig. S3M), anorchid Trim28hep–/– HCA lesions presented the same tumor‐associated molecular patterns (TAMPs) as HCCs found in control animals, with nuclear accumulation of β‐catenin, aberrant AR expression, and up‐regulation of their direct targets, Ccnd1 and Tmprss2, among many (Fig. 3F,G). Thus, Trim28 deletion in the liver appears to induce a cell‐intrinsic defect that plays an important role in the gene deregulations underlying tumorigenesis.
Figure 3.
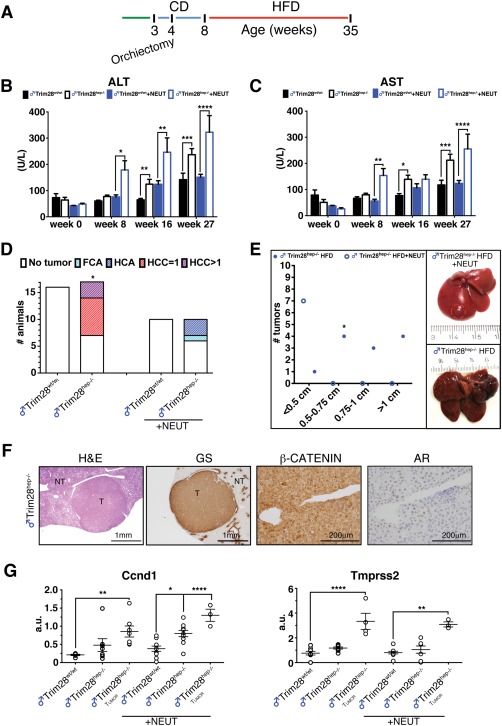
Orchiectomy alleviates tumor burden with persistence of AR signaling. (A) Experimental timeline procedure for the HFD challenge of neutered animals (n = 10). Bilateral orchiectomy was performed at 4 wo and HFD started at 8 wo until final analysis at 35 weeks. (B,C) Serum ALT (B) and AST (C) activity in gonad‐endowed (black‐lined bars) and castrated (+NEUT, blue‐lined bars) animals at different timepoints. *P < 0.05; **P < 0.01; ***P < 0.001; **** P < 0.0001, by two‐way ANOVA followed by Bonferroni's posttest. (D) Tumor classification graph in livers of gonad‐endowed and neutered Trim28hep–/– mice (+NEUT) after HFD challenge. * P value of the Trim28hep–/– versus Trim28hep–/–+NEUT comparison = 0.001611, by Fisher's exact test. (E) Left: tumor size graph in gonad‐endowed (filled circle) and neutered (open circle) Trim28 mutant mice. * P value of the Trim28hep–/– versus Trim28hep–/–+NEUT comparison = 0.0003572, by Fisher's exact test. Right: representative pictures of cancer lesions from gonad‐endowed (bottom) and anorchid (top, adenoma circled in red) mice. (F) H&E and diaminobenzidine stainings for Glutamine Synthase, β‐CATENIN, and AR in HCA of neutered Trim28hep–/– animals. NT, parenchyma; T, tumor. (G) qPCR on liver and tumor biopsies for Ccnd1 and Tmprss2 expression as direct targets of β‐catenin and AR, respectively. Values are calculated as mean relative expression after normalization to three housekeeping genes ± SEM; *P < 0.05; **P < 0.01; ****P < 0.0001, by one‐way ANOVA followed by Bonferroni's posttest. Abbreviations: a.u., arbitrary units; H&E, hematoxylin‐eosin; wo, weeks old.
TRIM28 INTERACTS WITH THE AR/β‐CATENIN AXIS
The persistence of AR signaling in a ligand‐free environment, as observed in HCA lesions of castrated Trim28hep–/– animals, is reminiscent of the fate of ADT‐treated PCa patients, where an initial phase of tumor shrinkage is followed by a hormone‐independent relapse. The molecular mechanisms underlying progression to this castration‐resistant stage are heterogeneous, encompassing altered stability of the AR coregulators with epigenetic alterations exposing AR‐regulated cryptic enhancers.25 Moreover, in cancer‐prone settings, β‐catenin interacts with the AR to establish a dynamic cross‐talk and enhance AR‐transcriptional activity.26
We used the human prostate carcinoma cell line, 22Rv1, to ask whether TRIM28 can serve as a partner for the AR/β‐catenin axis. Upon dihydrotestosterone (DHT) stimulation of these cells, AR translocated to the nucleus where it colocalized with TRIM28 (Supporting Fig. S4A). In vitro glutathione S‐transferase (GST) pull‐down assays using recombinant TRIM28 (Supporting Fig. S4B) revealed a DHT‐dependent association with AR mediated by the RING‐B box‐coiled coil (RBCC) domain (Fig. 4A), but independent from serine residues known to serve as phosphorylation targets (Supporting Fig. S4C). TRIM28 could also be immunoprecipitated in PCa cells with AR, FOXA1, and β‐catenin upon DHT stimulation (Fig. 4B). Furthermore, TRIM28 was recruited in this setting to a subset of AR‐binding elements (AREs) located in genes deregulated in tumors of male Trim28hep–/– animals (Fig. 4C). In contrast, no such TRIM28 recruitment at AREs was detected in the androgen‐independent PC3 cell line (Fig. 4D). Finally, confirming that TRIM28 can interfere with the association of AR/FOXA1 with β‐catenin, the latter interacted with the former two in Trim28hep–/– hepatocellular neoplastic lesions, but not in healthy tissue (Fig. 4E).
Figure 4.
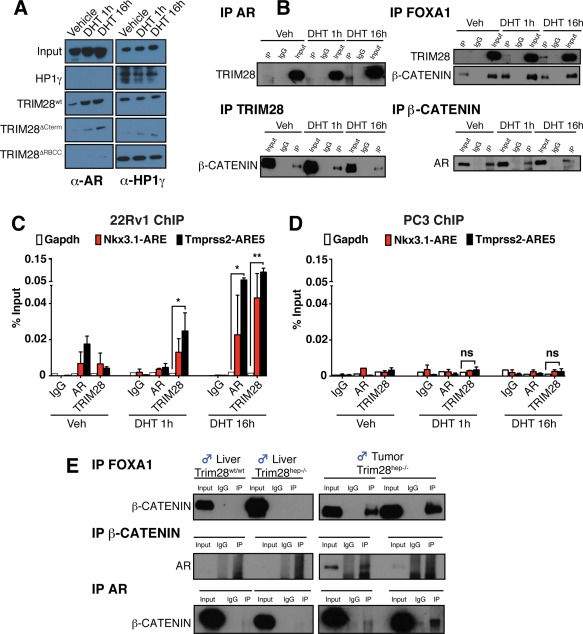
Biochemical characterization of TRIM28‐associating partners. (A) Purified recombinant wt or GST‐TRIM28 deletion mutants were incubated with 22Rv1 cell lysates, and the pull‐down complexes were analyzed by western blotting with the indicated antibodies. As a positive control, pull‐down complexes were incubated with HP1γ antibody. Recombinant GST‐HP1γ was used as negative control for AR interaction. n = 4. (B) IPs were performed using nuclear extracts (NEs) prepared from methanol‐stimulated (Veh) or DHT‐stimulated 22Rv1 cells using antibodies against TRIM28, β‐CATENIN, FOXA1, and AR. IP complexes were analyzed by western blotting using indicated antibodies. (C,D) 22Rv1 (C) or PC3 (D) cells were stimulated for 1 or 16 hours with DHT or vehicle alone (Veh). Chromatin was immunoprecipitated with antibodies recognizing TRIM28, AR, and IgG as a negative control and quantitated by qPCR using primer sets specific for indicated genomic regions. Gapdh promoter region was used as negative control. Values are represented as mean ± SEM as a percentage of the input chromatin; n = 3, *P < 0.05; **P < 0.01; ns (not significant), by two‐way ANOVA followed by Bonferroni's posttest. (E) In vivo IPs were performed on protein extract from livers and tumors of Trim28wt/wt and Trim28hep–/– mice incubated with antibodies against FOXA1 (top panels), β‐CATENIN (middle panels), and AR (bottom panels), and the resulting eluates were analyzed by western blotting with the indicated antibodies. Abbreviations: Gapdh, glyceraldehyde‐3‐phosphate dehydrogenase; HP1γ, heterochromatin protein 1‐gamma; IgG, immunoglobulin G; IPs, immunoprecipitations; Veh, vehicle.
BA DYSREGULATION AND FARNESOID X RECEPTOR ATTENUATION IN Trim28hep–/– MICE
GO analysis of dysregulated transcripts (2‐fold change; P < 0.05) indicated that loss of TRIM28 affected molecular functions and biological processes involved in BA metabolism (see Supporting Fig. S1B,C). BAs are produced and conjugated in hepatocytes, stored in the gallbladder, and, finally, released in the duodenum to facilitate absorption of food and vitamins. They are increasingly being appreciated as complex metabolic integrators and signaling factors, and their aberrant accumulation can promote carcinogenesis.27
Liquid chromatography and mass spectrometry analyses revealed a substantial increase of BAs in the liver of both naïve and anorchid HFD‐fed male Trim28hep–/– mice (Fig. 5A,B). Taken individually, tauro‐alpha‐muricholic acid (T‐αMCA), tauro‐beta‐muricholic acid (T‐βMCA), and taurocholic acid (TCA) were the most abundant species in both HFD‐fed Trim28wt/wt and Trim28hep–/– animals (Fig. 5CD). Consistent with their tumorigenic phenotype, aberrant BA accumulation was also documented in aged Trim28hep–/– animals (Supporting Fig. S5A,B). Livers from male Trim28hep–/– exhibited abnormally elevated levels of T‐βMCA (+51% for HFD‐fed neutered, +102% for HFD‐fed gonad‐endowed, and +314% for aged mice) compared to their wt counterparts. T‐βMCA acts as an antagonist of the nuclear farnesoid X receptor (FXR),28 which has hepatoprotective and tumor‐suppressor effects. Fxr knockout (Fxr–/–) mice present with elevated BA levels, hepatosteatosis, and 100% of late‐onset β‐catenin‐dependent HCC.29 FXR expression was markedly decreased in TRIM28‐less livers and was almost undetectable in cancer tissue lesions (Fig. 5E). This was paralleled by the down‐regulation of its main downstream target, small heterodimer partner (SHP; Fig. 5E). Attributable to their small size, it was impossible to analyze protein expression from Trim28hep–/– HCAs, but we observed a negative correlation with FXR and SHP expression when analyzing the surrounding nontumor tissue (Supporting Fig. S5C). Confirming the phenotypic parallels between liver‐specific Trim28 and Fxr knockouts,30 GSEA analysis revealed a marked correlation between transcripts deregulated in the two settings, whether considering Trim28hep–/– nontumoral liver or HCC lesions (Fig. 5F and Supporting Fig. S5D).
Figure 5.
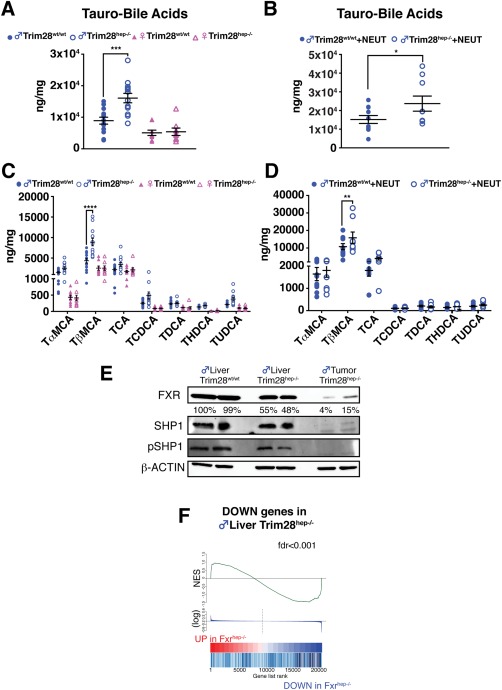
TRIM28 loss causes BA dysregulation and FXR signaling attenuation. (A,B) Absolute quantification of hepatic BA content in HFD‐fed gonad‐endowed (A) and castrated (B) mice (n = 15 for males and n = 10 for other groups). Values are normalized per protein content and per mg of tissue and expressed as mean ± SEM. *P < 0.05; ***P < 0.001, by two‐way ANOVA followed by Bonferroni's posttest. (C,D) Hepatic BA species composition in gonad‐endowed (C) and castrated (D) mice. T– = tauro‐conjugated species; αMCA, alpha‐muricholic acid; βMCA, beta‐muricholic acid; CA, cholic acid; CDCA, chenodeoxycoholic acid; DCA, deoxycholic acid; HDCA, hyodeoxycholic acid; UDCA, ursodeoxycholic acid. Values are normalized per protein content and per mg of tissue and expressed as mean ± SEM. **P<0.01; ****P < 0.0001, by two‐way ANOVA followed by Bonferroni's posttest. (E) Western blotting of liver and tumor lysates of gonad‐endowed HFD‐fed mice incubated with the indicated antibodies. For FXR quantification, band intensities were normalized to β‐actin and calculated as percentage relative to Trim28wt/wt liver sample. (F) Plot of GSEA analysis showing a positive correlation between down‐regulated genes in HFD‐fed Trim28 mutant livers and FXR‐dependent targets (FC, >5; P < 0.05). Abbreviation: FC, fold change; fdr, false discovery rate; NES, normalized enrichment scores; pSHP, phosphorylated SHP.
PHENOTYPE SEVERITY CORRELATES WITH BACTERIAL VARIATIONS AND DYSBIOSIS
The gut microbiota is emerging as a dynamic environmental factor exquisitely sensitive to metabolic perturbations, such as dietary obesity, and detrimental influences through the enterohepatic axis can, in turn, play a role in obesity‐associated cancer.31 We used 16S ribosomal RNA gene amplicon sequencing to characterize the gut microbiota of HFD‐fed wt and Trim28hep–/– mice with or without castration. We found that the obesogenic regimen progressively increased the relative representation of Firmicutes at the expense of the Bacteroidetes as reported,32 without striking differences detected between genotypes (Fig. 6A). However, linear discriminant analysis (LDA) effect size (LEfSe) results revealed different degrees of operational taxonomic unit (OTU) deregulation between HCA‐ and HCC‐bearing Trim28hep–/– mice (Fig. 6BC), with a near 4‐fold increase of OTU variability in cases of more advanced tumors (51 in HCC‐ vs. 14 deregulated OTUs in HCA‐bearing animals). Aging induced levels of microbiota perturbation comparable to obese Trim28hep–/– animals carrying malignant lesions (54 deregulated OTUs; Supporting Fig. S6A,B). When comparing microbial communities of HFD‐fed gonad‐endowed and anorchid mutant mice, intermediate levels of microbial variation (35 deregulated OTUs) were recorded, correlating with the attenuation of the tumorigenic phenotype by castration (Supporting Fig. S6C,D).
Figure 6.
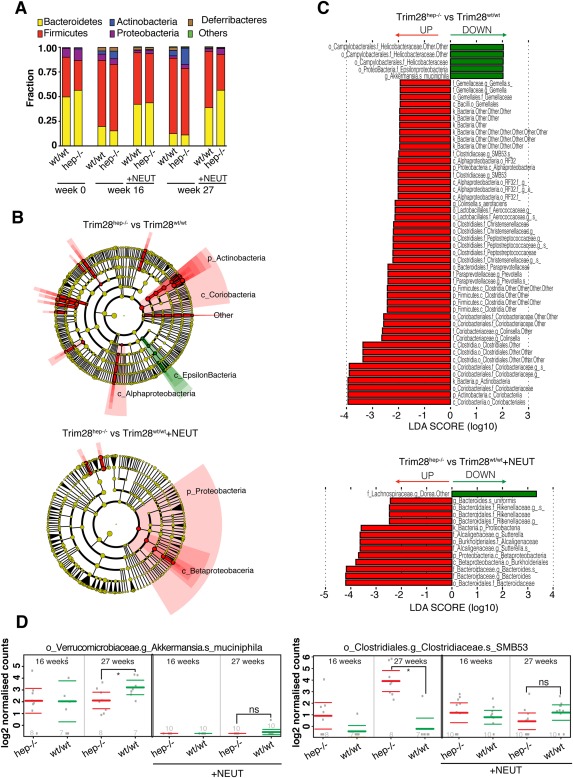
Microbiota dynamics correlates with tmor phenotype. (A) Fecal microbiota composition at the phyla level of Trim28wt/wt and Trim28hep–/– fecal stool at 0, 16, and 27 weeks from HFD diet. +NEUT, anorchid mice. (B) Cladograms generated from LEfSe analysis representing enriched (red) and reduced (green) taxa in fecal microbiota of HCC‐bearing (top) or HCA‐bearing (bottom) Trim28hep–/– mice. The central point represents the root of the tree (bacteria), and each ring represents the next lower taxonomic level (phylum through species). The diameter of each circle represents the relative abundance of the taxon. When full identification was not possible, g_ or s_ alone was used for genus or species, respectively. (C) LDA scores of the differentially abundant taxa shown in cladograms. Taxa enriched or reduced in microbiota from Trim28hep–/– mice are indicated with a positive (red) or negative (green) LDA score, respectively. (D) Relative abundances of Akkermansia muciniphila (left) and Clostridiaceae SMB53 (right) in Trim28wt/wt (green) and Trim28hep–/– (red) mice receiving HFD for 16 and 27 weeks. Values are expressed as mean ± 95% confidence interval. n per group indicated at the bottom of the graph.
Fecal microbial communities from HCC‐bearing Trim28hep–/– mice displayed significant increases in the Prevotellaceae family, which are considered as hallmarks of dysbiosis and frequently altered in patients with cirrhosis. The abundance of Clostridiaceae, Peptostreptococcaceae, and Lactobacillaceae was significantly higher in HCC‐bearing Trim28hep–/– mice, a condition also observed in HFD‐induced intestinal carcinogenesis.33 Tumor severity correlated with the specific down‐regulation of the mucin‐degrading bacteria, Akkermansia muciniphila (Fig. 6D, left), a bacterial strain associated with the improvement of obesity‐related metabolic and immune alterations.34 Conversely, we found that still poorly characterized strains, such as the Clostridiaceae SMB53, were particularly abundant in HCC‐bearing Trim28hep–/– mice (Fig. 6D, right). Once established, overall patterns of alpha diversity across HCA‐ and HCC‐bearing mice were stable and consistent at all the time points analyzed (Supporting Fig. S6E,F). Furthermore, qPCR on genomic liver DNA with universal 16S RNA primer sets indicated no sign of hepatic bacterial invasion (Supporting Fig. S6G). Finally, lipopolysaccharide (LPS) systemic levels measured by enzyme‐linked immunosorbent assay revealed no endotoxemia in obese Trim28hep–/– mice (Supporting Fig. S6H).
HEPATOCARCINOGENESIS IS SUPPRESSED BY AXENIC CONDITIONS
Dysbiosis can contribute to liver disease and compelling evidence links the enteric microbiome with inflammation‐ and obesity‐associated hepatocarcinogenesis.8, 35 Based on this premise, we repeated the HFD challenge in mice raised in GF conditions. No difference in body weight was observed between sex‐matched genotypes (Supporting Fig. S7A,B). In line with previous reports,36 GF animals gained less weight than their conventionally raised counterparts, and in this environment Trim28hep–/– males displayed neither hepatomegaly (Fig. 7A and Supporting Fig. S7C) nor elevation of plasma transaminases, although a significant accumulation of LDL‐cholesterol was noted (Fig. 7B and Supporting Fig. S7D). Furthermore, they presented no increase in serum levels of proinflammatory cytokine IL‐6 and no decrease in anti‐inflammatory adipokine adiponectin (Fig. 7C,D). Finally, 15 of 15 Trim28hep–/– male GF mice remained tumor free for the entire time of the experiment, only 1 case of liver cancer being observed in 1 of 10 Trim28hep–/– females, albeit without the TAMPs recorded in all other cases of HCA or HCC (Fig. 7E and data not shown). Accordingly, qPCR analysis revealed no significant up‐regulation of AR and β‐catenin gene targets (Fig. 7F), no aberrant hepatic BA content (Fig. 7G and Supporting Fig. S7E), and no deregulation of FXR or SHP (Fig. 7H and Supporting Fig. S7F,G).
Discussion
This study demonstrates that hepatic ablation of a master epigenetic regulator leads to the sex‐specific development of a polyphenic trait fostered by age and obesity and characterized by altered hormonal signaling, dysbiosis, and a state of metabolic infection precipitating the occurrence of HCC. Diverging from the simplistic one gene/one phenotype relationship, the hereby described consequences of hepatic TRIM28 ablation do not imply that this represents a common genetic basis of malignancy, but serve as a remarkable model for increased vulnerability to cancer. Our data indeed support a major role for epigenetic regulation in adaptive metabolic plasticity37, 38 and reveal TRIM28 as an important factor in the ability of higher organisms to cope with environmental stress. Our work also indicates causative links between the gut microbiota and liver cancer and provides not only an attractive preclinical model for studying this dreadful disease, but also leads for research toward its prevention and possibly its treatment.
The phenotype displayed by Trim28hep–/– mice presents many similarities with human liver cancer, whether in terms of clinical course or molecular hallmarks. In contrast with the abrupt pathology observed in preclinical models relying on the administration of carcinogenic substances, the liver‐specific deletion of TRIM28 leads to a slowly progressive, multistep disease, with a detectable role for established or suspected contributors of human nonalcoholic fatty liver disease (NAFLD) and hepatocarcinoma, such as obesity, microbiota, and aging, some of which at least are actionable both in this model and in the clinics. As well, the nuclear accumulation of β‐catenin characterizes the vast majority of human HCCs, and AR signaling is commonly activated in human liver tumors, pointing to the relevance of Trim28hep–/– mice for research aimed at discovering and stratifying new therapies for liver cancer.
We had previously observed that hepato‐specific ablation of Trim28 led to sex‐specific transcriptional deregulation of a wide range of xenobiotic and steroid metabolism genes, and to the male‐limited development of liver adenomas, with low baseline penetrance moderately increased on conventional, lard‐rich HFD.11 Encouraged by these findings, we brought several modifications to our experimental protocol. First, we moved from a genetically mixed to a pure C57BL/6 background. Second, we extended the time of the study to assess the impact of aging. Third, we subjected animals to a more challenging diet, enriched in highly prosteatotic MCFAs.
Time alone had a strong influence on the pathologic phenotype of Trim28hep–/– mice, leading to highly penetrant male‐specific carcinogenesis. This was associated with signs of both local and systemic inflammation, with elevated levels of hepatotoxicity markers such alanine aminotransferase (ALT) and aspartate aminotransferase (AST), the release of proinflammatory cytokines, and a drop in their anti‐inflammatory counterparts. Our study endpoint of 73 weeks corresponds to a mid‐elderly stage, considering an average life span of 110 weeks for conventionally raised C57BL/6 mice.39 It likely reflected the cumulative impact of age‐related factors, such as increased DNA‐damage by reactive oxygen species production.40 A high BMI precipitates the aging of liver tissue, qualifying obesity as an accelerator of the hepatic epigenetic clock, as defined according to the DNA methylation status of a few hundred selected CpG dinucleotides.41 Obesity is also an epidemiologically established risk factor for HCC, and is associated with inflammation and NAFLD. Here, it had a dramatic effect, increasing both the rate of appearance and overall frequency of HCC in Trim28hep–/– mice.
TRIM28 is a member of the RBCC protein family that is devoid of DNA‐binding ability but can be tethered to the genome by a variety of partners, amongst which hundreds of KRAB‐containing zinc fingers (KZFPs).42 It generally serves as a scaffold for the assembly of macromolecular complexes containing diverse chromatin modifiers, including histone methyltransferases, histone acetylases and deacetylases, as well as nucleosome remodeling factors. Initially identified as a corepressor,43 notably found responsible for the early embryonic silencing of KZFP‐targeted endogenous endogenous retroelements,44 TRIM28 has since been demonstrated to be capable of acting also as a coactivator45 and has been implicated not only in transcriptional regulation, but also in DNA repair and maintenance of heterochromatin during genome replication.46, 47
Within the inflammatory context triggered by aging or obesity, the alterations of Trim28‐dependent transcriptional dynamics resulted in activating TAMPs, with attenuation of FXR signaling, hyperactivation of β‐catenin, and altered AR signaling. Our analyses suggest that TRIM28 association with β‐catenin and FOXA1/AR normally prevents the illegitimate activation of this TAMP, a finding corroborated by the aberrant activation of AR signaling in Trim28hep–/– mice, and the partially protective effect of androgen deprivation on their tumorigenic phenotype, with few anorchid mutant males developing small benign neoplasms. Whereas this partly explains the male exclusiveness of the pathology triggered by TRIM28 hepatic ablation, persistently altered AR signaling in this setting is consistent with the long‐term limitations of androgen depletion in both HCC and castration‐resistant prostate cancer; yet it suggests that other approaches interfering with AR signaling might succeed in slowing down liver cancer progression.6, 25
Initially only considered as mediators of food and vitamin absorption, BAs are emerging as important signaling factors along the gut‐liver axis, and perturbation of their homeostasis can have major detrimental consequences, including the promotion of gastrointestinal cancer.27, 31 Consistent with the deregulation of genes encoding for factors involved in BA metabolism and secretion, both obese and aged males lacking TRIM28 in the liver displayed an aberrant accumulation of tauro‐conjugated bile acids, particularly the FXR‐antagonist, T‐βMCA. FXR is a recognized hepatic tumor suppressor,29 and Trim28hep–/– and Fxr–/– mice present striking phenotypic similarities, with deletion of the nuclear‐receptor–inducing elevated triglycerides, ALT, AST, and hepatic BA levels, and spontaneous development of β‐Catenin‐dependent HCCs at >1 year of age.29, 48 This strongly suggests that the synergistic influences of elevated BA concentrations and hepatic FXR inactivation expose TRIM28‐less hepatocytes to a microenvironment that fuels inflammation and promotes tumorigenesis.
Mounting evidence links dietary factors with alterations of the gut microbiota, potentially resulting in dysbiosis and increased intestinal permeability.8, 32, 33 Most xenobiotics are metabolized in the liver, and alteration of this process can impede the detoxification of oncogenic metabolites. The intestinal flora can thus impact on hepatic functions by promoting inflammation and tumorigenesis. In turn, by‐products of altered hepatic metabolism can influence the composition of the gut microbiota.8, 35 Here, we observed that HCC‐bearing TRIM28‐deficient animals, being obese or aged, exhibited deregulation of more than 50 bacterial species compared to their wt, tumor‐less counterparts, 4‐fold more than counted in HCA‐bearing anorchid animals. In Trim28hep–/– mice with HCC, taxonomic species previously related to metabolic dysfunction and inflammation, such as Prevotella, Akkermansia muciniphila, and Bacteroides uniformis,34, 35, 49 were prominently deregulated, as were some other species not previously associated with any pathologies, such as Clostridiaceae SMB53.
Demonstrating the critical role of the gut microbiota in the genesis of HCC in our model, the pathological phenotype of Trim28hep–/– mice was completely abrogated when they were raised in axenic conditions. In this environment, in spite of their mutant genotype, these animals displayed no sign of hepatic inflammation, no perturbation of serum IL‐6 and adiponectin levels, no activation of TAMPs nor drop in FXR and its SHP target, and, most important, no tumor development after 27 weeks on strongly prosteatotic HFD, a time at which 60% of their conventionally raised controls exhibited macroscopically visible HCC lesions. Thus, the intestinal flora represents an obligatory factor in the development of liver cancer in our model. Although our data do not formally identify microbial oncogenic drivers nor determine whether the accumulation of BAs modulates the growth of defined bacterial species, the use of gnotobiotic models or BA‐enriched regimens could help address these questions. More generally, the present work warrants efforts aimed at characterizing the gut microbiota of patients presenting with states known to predispose to liver cancer, as well as at exploring the potential of its manipulations for the prevention and treatment of this disease.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29182/suppinfo.
Supporting Information
Acknowledgments
We thank Julien Duc and Alexis Rapin for technical assistance, Anna Groner for the gift of reagents, and the EPFL core facilities for support.
Potential conflict of interest: Nothing to report.
Supported by grants from the Swiss National Science Foundation and the European Research Council (ERC 268721 and ERC 694658).
REFERENCES
- 1. Font‐Burgada J, Sun B, Karin M. Obesity and cancer: the oil that feeds the flame. Cell Metab 2016;23:48‐62. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57‐70. [DOI] [PubMed] [Google Scholar]
- 3. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer 2004;4:579‐591. [DOI] [PubMed] [Google Scholar]
- 4. Dorak MT, Karpuzoglu E. Gender differences in cancer susceptibility: an inadequately addressed issue. Front Genet 2012;3:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wands J. Hepatocellular carcinoma and sex. N Engl J Med 2007;357:1974‐1976. [DOI] [PubMed] [Google Scholar]
- 6. Ma WL, Hsu CL, Wu MH, Wu CT, Wu CC, Lai JJ, et al. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology 2008;135:947‐955, 955.e1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li Z, Tuteja G, Schug J, Kaestner KH. Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer. Cell 2012;148:72‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity‐induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013;499:97‐101. [DOI] [PubMed] [Google Scholar]
- 9. Nault JC, Zucman‐Rossi J. Genetics of hepatocellular carcinoma: the next generation. J Hepatol 2014;60:224‐226. [DOI] [PubMed] [Google Scholar]
- 10. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr. , Kinzler KW. Cancer genome landscapes. Science 2013;339:1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bojkowska K, Aloisio F, Cassano M, Kapopoulou A, Santoni de Sio F, Zangger N, et al. Liver‐specific ablation of Kruppel‐associated box‐associated protein 1 in mice leads to male‐predominant hepatosteatosis and development of liver adenoma. Hepatology 2012;56:1279‐1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Langmead B, Salzberg SL. Fast gapped‐read alignment with Bowtie 2. Nat Methods 2012;9:357‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA‐seq read counts. Genome Biol 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL‐6 and TNF expression. Cell 2010;140:197‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sharma D, Wang J, Fu PP, Sharma S, Nagalingam A, Mells J, et al. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology 2010;52:1713‐1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gougelet A, Torre C, Veber P, Sartor C, Bachelot L, Denechaud PD, et al. T‐cell factor 4 and beta‐catenin chromatin occupancies pattern zonal liver metabolism in mice. Hepatology 2014;59:2344‐2357. [DOI] [PubMed] [Google Scholar]
- 19. Cadoret A, Ovejero C, Terris B, Souil E, Levy L, Lamers WH, et al. New targets of beta‐catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002;21:8293‐8301. [DOI] [PubMed] [Google Scholar]
- 20. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben‐Ze'ev A. The cyclin D1 gene is a target of the beta‐catenin/LEF‐1 pathway. Proc Natl Acad Sci U S A 1999;96:5522‐5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kondo Y, Kanai Y, Sakamoto M, Genda T, Mizokami M, Ueda R, Hirohashi S. Beta‐catenin accumulation and mutation of exon 3 of the beta‐catenin gene in hepatocellular carcinoma. Jpn J Cancer Res 1999;90:1301‐1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCarthy N. Prostate cancer: studying the classics. Nat Rev Cancer 2011;11:386. [DOI] [PubMed] [Google Scholar]
- 23. Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin 1972;22:232‐240. [DOI] [PubMed] [Google Scholar]
- 24. Asarian L, Geary N. Modulation of appetite by gonadal steroid hormones. Philos Trans R Soc Lond B Biol Sci 2006;361:1251‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013;32:5501‐5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang F, Li X, Sharma M, Sasaki CY, Longo DL, Lim B, Sun Z. Linking beta‐catenin to androgen‐signaling pathway. J Biol Chem 2002;277:11336‐11344. [DOI] [PubMed] [Google Scholar]
- 27. Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res 2005;589:47‐65. [DOI] [PubMed] [Google Scholar]
- 28. Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro‐beta‐muricholic acid, a naturally occurring FXR antagonist. Cell Metab 2013;17:225‐235. [DOI] [PubMed] [Google Scholar]
- 29. Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res 2007;67:863‐867. [DOI] [PubMed] [Google Scholar]
- 30. Zhan L, Liu HX, Fang Y, Kong B, He Y, Zhong XB, et al. Genome‐wide binding and transcriptome analysis of human farnesoid X receptor in primary human hepatocytes. PLoS One 2014;9:e105930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chassaing B, Etienne‐Mesmin L, Gewirtz AT. Microbiota‐liver axis in hepatic disease. Hepatology 2014;59:328‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Serino M, Luche E, Gres S, Baylac A, Berge M, Cenac C, et al. Metabolic adaptation to a high‐fat diet is associated with a change in the gut microbiota. Gut 2012;61:543‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schulz MD, Atay C, Heringer J, Romrig FK, Schwitalla S, Aydin B, et al. High‐fat‐diet‐mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 2014;514:508‐512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dao MC, Everard A, Aron‐Wisnewsky J, Sokolovska N, Prifti E, Verger EO, et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 2016;65:426‐436. [DOI] [PubMed] [Google Scholar]
- 35. Henao‐Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome‐mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012;482:179‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet‐induced obesity in germ‐free mice. Proc Natl Acad Sci U S A 2007;104:979‐984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gluckman PD, Hanson MA, Buklijas T, Low FM, Beedle AS. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol 2009;5:401‐408. [DOI] [PubMed] [Google Scholar]
- 38. Dalgaard K, Landgraf K, Heyne S, Lempradl A, Longinotto J, Gossens K, et al. Trim28 haploinsufficiency triggers bi‐stable epigenetic obesity. Cell 2016;164:353‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goodrick CL. Life‐span and the inheritance of longevity of inbred mice. J Gerontol 1975;30:257‐263. [DOI] [PubMed] [Google Scholar]
- 40. Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, et al. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci U S A 2001;98:10469‐10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schonfels W, Ahrens M, et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci U S A 2014;111:15538‐15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ecco G, Cassano M, Kauzlaric A, Duc J, Coluccio A, Offner S, et al. Transposable elements and their KRAB‐ZFP controllers regulate gene expression in adult tissues. Dev Cell 2016;36:611‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ 3rd. KAP‐1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev 1996;10:2067‐2078. [DOI] [PubMed] [Google Scholar]
- 44. Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010;463:237‐240. [DOI] [PubMed] [Google Scholar]
- 45. Singh K, Cassano M, Planet E, Sebastian S, Jang SM, Sohi G, et al. A KAP1 phosphorylation switch controls MyoD function during skeletal muscle differentiation. Genes Dev 2015;29:513‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. White D, Rafalska‐Metcalf IU, Ivanov AV, Corsinotti A, Peng H, Lee SC, et al. The ATM substrate KAP1 controls DNA repair in heterochromatin: regulation by HP1 proteins and serine 473/824 phosphorylation. Mol Cancer Res 2012;10:401‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rauwel B, Jang SM, Cassano M, Kapopoulou A, Barde I, Trono D. Release of human cytomegalovirus from latency by a KAP1/TRIM28 phosphorylation switch. Elife 2015;4. doi: 10.7554/eLife.06068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor‐null mice. Carcinogenesis 2007;28:940‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cano PG, Santacruz A, Trejo FM, Sanz Y. Bifidobacterium CECT 7765 improves metabolic and immunological alterations associated with obesity in high‐fat diet‐fed mice. Obesity (Silver Spring) 2013;21:2310‐2321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29182/suppinfo.
Supporting Information