

Abstract
Schizophrenia is a severe mental illness that affects almost 1% of the population worldwide. Even though the etiology of schizophrenia is uncertain, it is believed to be a neurodevelopmental disorder that results from a combination of environmental insults and genetic vulnerabilities. Over the past 20 years, there has been a confluence of evidence from many research disciplines pointing to alterations in excitatory signaling, particularly involving hypofunction of the N-methyl-D-aspartate receptor (NMDAR), as a key contributor to the schizophrenia disease process. This review describes the structure-function relationship of the NMDAR channel and how the glycine modulatory site (GMS) acts as an important regulator of its activity. In addition, this review highlights the genetic, pharmacologic, and biochemical evidence supporting the hypothesis that NMDAR hypofunction contributes to the pathophysiology of schizophrenia. Finally, this chapter highlights some of the most recent and promising pharmacological strategies that are designed to either, directly or indirectly, augment NMDAR function in an effort to treat the cognitive and negative symptoms of schizophrenia that are not helped by currently available medications.
Keywords: schizophrenia, serine racemase, D-serine, glycine, NMDA receptor, metabotropic glutamate receptor 5, α7 nicotinic acetylcholine receptors, cognition
1. INTRODUCTION
Schizophrenia is a chronic, disabling mental disorder that affects 0.5–1% of the population worldwide (Perala et al., 2007) that commonly presents in late adolescence and early adulthood. It is characterized clinically by positive symptoms (delusions, hallucinations, and thought disorder), negative symptoms (i.e. social withdrawal, poverty of speech, and anhedonia), and cognitive deficits (i.e attention, working memory, and executive function). While the negative and positive symptoms typically present during late adolescence or early adulthood (Lewis & Lieberman, 2000), the cognitive deficits generally appear years before clinical diagnosis (Lesh, Niendam, Minzenberg, & Carter, 2011).
One leading hypothesis is that the pathophysiology of schizophrenia has a neurodevelopmental component (Lewis & Lieberman, 2000; Rapoport, Giedd, & Gogtay, 2012). Brain development during adolescence, the time when many schizophrenia symptoms appear or worsen, is a dynamic period marked by extensive functional and neuroanatomical changes. This period is characterized by: 1) fine-tuning of excitatory, inhibitory, and monoaminergic neurotransmitter systems, 2) stabilization of synapses to increase efficiency of neural function and diminish redundancy, and 3) beginning of integration between late maturing and early maturing brain structures (Keshavan, Giedd, Lau, Lewis, & Paus, 2014). Therefore, genetic predisposition and environmental disturbances that lead to changes or imbalances in the timing of these developmental processes could increase the risk for developing schizophrenia.
Schizophrenia is associated with various neuroanatomical and structural brain abnormalities. Diffusion tensor magnetic resonance imaging studies show that white matter alterations are present from the preclinical to the chronic stages of schizophrenia, involving the long association white matter tracts, corticospinal tracts, interhemispheric connections, cerebello-thalamo-cortical circuit, and limbic system (Canu, Agosta, & Filippi, 2015). A number of longitudinal structural magnetic resonance imaging studies have shown reduced whole brain volume and increased lateral ventricular volume in the early stages of schizophrenia and in chronically ill patients (Fusar-Poli et al., 2013; Kempton, Stahl, Williams, & DeLisi, 2010; Levitt, Bobrow, Lucia, & Srinivasan, 2010), with some abnormalities already present during the prodromal phase (Fusar-Poli et al., 2011). The decreased brain volume in schizophrenia is especially pronounced in cortical areas, as well as in the hippocampus (Levitt et al., 2010; Tamminga, Stan, & Wagner, 2010). The extent of progressive brain tissue decrease in patients (−0.5%/year) has been estimated as twice that of healthy controls (−0.2%/year) (Hulshoff Pol & Kahn, 2008). Some of these neuroanatomical alterations may be correlated with antipsychotic treatment, as the higher the cumulative exposure to antipsychotic treatment, the greater the progressive gray matter volume decreases in patients over time (Fusar-Poli et al., 2013). Studies using human post-mortem brain tissue suggest that decreased neuropil, including reduced dendritic length and spine density on pyramidal neurons, might be the primary contributor to reduced cortical gray matter volumes in schizophrenia (Glausier & Lewis, 2013; Konopaske, Lange, Coyle, & Benes, 2014).
Schizophrenia is a highly heritable disorder, with the relative risk being 6–17x greater among first-degree relatives and 40–50x greater in the monozygotic twin of an affected sibling compared to the general population (Cardno et al., 1999). Thus, there has been a massive effort in the field of psychiatric genetics to identify genes that confer risk to developing schizophrenia. Unfortunately, the genetics are very complex with non-Mendelian inheritance. Prior to genome-wide association studies (GWAS), candidate gene studies were utilized in an attempt to discover genetic risk factors for schizophrenia. Although these studies were able to identify numerous risk genes, there was often a failure to replicate associations across studies. We now know that these early candidate gene association studies for common genetic variation had inadequate statistical power to detect the small differences seen in schizophrenia (Farrell et al., 2015). Although the largest schizophrenia GWAS to date (~37,000 cases and ~113,000 controls; exponentially larger sample sizes than previous studies) identified SNPs in ~600 brain-enriched genes, almost all of the previously identified candidate risk genes were not found to be associated with schizophrenia in this study (Schizophrenia Working Group of the Psychiatric Genomics, 2014). Copy number variants (stretches of DNA that are either deleted or duplicated) and rare exonic variants, can also lead to increased genetic risk for schizophrenia (Fromer et al., 2014; Kirov et al., 2012). As a whole, these recent large-scale studies have identified both common and rare genetic variants in N-methyl-D-aspartate receptor (NMDAR) genes, as well as components of the postsynaptic density, with increased risk for schizophrenia.
In this chapter, we describe the structure and function of the NMDAR channel, with a particular focus on how the glycine modulatory site (GMS) regulates its activity. We will then discuss how it is believed NMDAR hypofunction contributes to the pathophysiology of schizophrenia. Finally, we will highlight some of the latest pharmacological strategies aimed at both, directly and indirectly, augmenting NMDAR function.
2. NMDA RECEPTOR: STRUCTURE AND FUNCTION
In the brain, glutamate activates metabotropic and ionotropic receptors. The latter family of receptors is comprised of 18 gene products that are further divided into three subtypes based on their agonist preference: α-amino-3-hydroxy-5- methylisoxazole-4-propionate (AMPA) receptors, kainate receptors and NMDARs/GluNs. The heterotetrameric NMDAR is widely distributed throughout most of the brain and is a critical postsynaptic mediator of activity-dependent synaptic plasticity. This receptor is composed of two obligatory GluN1 subunits with either two GluN2 subunits or a combination of GluN2 and GluN3 subunits. The GluN1 subunit is encoded by a single gene (GRIN1), which has eight different splice variants. There are four GluN2 subunits (GluN2A-D) and two GluN3 subunits (GluN3A-B) that are encoded by separate genes, GRIN2A-D and GRIN3A-B, respectively (Paoletti, Bellone, & Zhou, 2013).
GluN1 is the obligatory NMDAR subunit that it is expressed throughout the brain and is present during the entire lifespan (Henson et al., 2008). On the other hand, GluN2A-D subunits have a complex spatial and temporal expression pattern. For example, GluN2B is expressed at low levels embryonically and increases in the hippocampus and cortex after birth. GluN2A begins to be expressed after birth and its levels continue to rise during postnatal development throughout the brain. In the adult brain, GluN2D is found only in the diencephalon and mesencephalon, while GluN2C is confined to the olfactory bulb and cerebellum. GluN3A and GluN3B, similar to GluN2 subunits, are expressed in a developmentally and spatially restricted manner. This large diversity in subunit expression and composition leads to NMDARs with distinct pharmacological and biophysical properties, including the formation of nonconventional diheteromeric (GluN1/GluN3) and triheteromeric (GluN1/GluN2/GluN3) NMDARs (Hansen, Ogden, Yuan, & Traynelis, 2014; Kehoe, Bernardinelli, & Muller, 2013).
Each of the GluN subunits consists of four domains: the intracellular C-terminal domain, the transmembrane domain that contains the ion channel, the agonist binding domain (glycine or D-serine bind to GluN1 or GluN3, while glutamate binds to GluN2), and the N-terminal domain. The C-terminal domain, through protein-protein interactions, regulates receptor trafficking and intracellular signaling. However, the C-terminal domain is the only domain that is not affected by allosteric modulators (Paoletti et al., 2013). The three helices and hairpin that comprise the transmembrane domain form the pore of the channel, giving it ion selectivity. It was only recently that the first crystal structure of the intact heterotetrameric GluN1/GluN2B NMDAR ion channel was described (Karakas & Furukawa, 2014).
NMDARs are unique compared to other members of the ionotropic glutamate receptor family. These receptors have much slower deactivation kinetics, are highly permeable to Ca2+, and act as a molecular coincidence detector. In addition to the binding of its agonist glutamate to the GluN2 subunit, activation of the NMDAR requires 1) post-synaptic depolarization, which relieves the Mg2+ blockade of the channel and 2) either glycine or D-serine must be bound at the GMS on the GluN1 subunit (Figure 1) [Insert Figure 1 here]. It should be noted that although the endogenous high potency co-agonists glycine and D-serine are present in the extracellular space (Johnson & Ascher, 1987), the GMS is not saturated in vivo (Bergeron, Meyer, Coyle, & Greene, 1998). Upon NMDAR channel opening, Ca2+ enters the neuron and triggers a cascade of intracellular events that mediate local, acute functional synaptic plasticity and changes in gene expression that influence long-term neural structural plasticity (Greer & Greenberg, 2008).
Figure 1. Structure of the NMDA receptor.
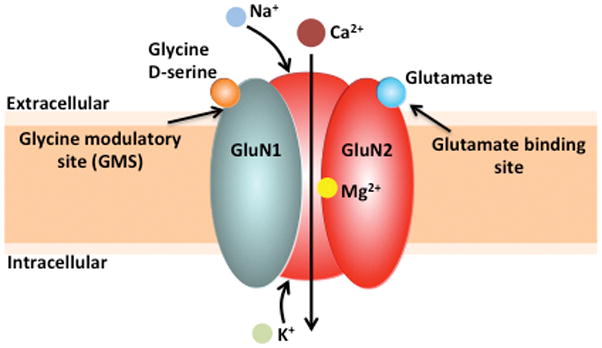
The conventional NMDAR ion channel is heterotetrameric, consisting of two GluN1 and two GluN2 subunits. These receptors act as molecular coincidence detectors, as in addition to the binding of its agonist glutamate to the GluN2 subunit, activation of the NMDAR requires 1) post-synaptic depolarization, which relieves the Mg2+ blockade of the channel and 2) either glycine or D-serine must be bound at the glycine modulatory site (GMS) on the GluN1 subunit. Upon NMDAR channel opening, calcium (Ca2+) and sodium (Na+) enter the neuron, while potassium (K+) exits the neuron.
3. GLYCINE MODULATORY SITE
Work done on both native NMDARs (Johnson & Ascher, 1987) and those expressed by oocytes (Kleckner & Dingledine, 1988) demonstrated the need for concomitant binding of glutamate and glycine for receptor activation. Furthermore, it was shown that D-serine or D-alanine could also act as NMDAR co-agonists at the GMS (Kleckner & Dingledine, 1988). D-amino acids, including D-serine are now well-established modulators of neuronal activity in mammals (Boehning & Snyder, 2003; Wolosker, Dumin, Balan, & Foltyn, 2008).
3.1. Serine racemase and D-serine
Serine racemase (SR) and D-serine were first observed in eukaryotic insects, such as silkworms and earthworms (Corrigan & Srinivasan, 1966). SR is the enzyme responsible for the both the conversion of L-serine to D-serine, as well as the α,β-elimination of water from L- or D-serine to yield pyruvate and ammonia (Figure 2) [Insert Figure 2 here]. SR is classified as a fold II pyridoxal 5′ phosphate (PLP) enzyme (Wolosker, Blackshaw, & Snyder, 1999). However, SR is more structurally similar to bacterial serine/threonine dehydratases, rather than classical amino acid racemases (De Miranda, Santoro, Engelender, & Wolosker, 2000). Even though the PLP attachment sites are conserved and there is structural similarity between SR and type II PLP family members, there are critical differences regarding allosteric regulation and reaction specificity. The crystal structure of SR revealed that the enzyme consists of two identical subunits that function as a dimer. In addition to PLP, SR has binding sites for magnesium (Mg2+) and a Mg2+-ATP complex, both of which lie outside of the catalytic site (Goto et al., 2009). It is believed that ATP is not an energy requirement for enzyme activity because ADP is also equally effective in SR activation (De Miranda, Panizzutti, Foltyn, & Wolosker, 2002).
Figure 2. Enzymatic pathways involved in D-serine synthesis and breakdown.
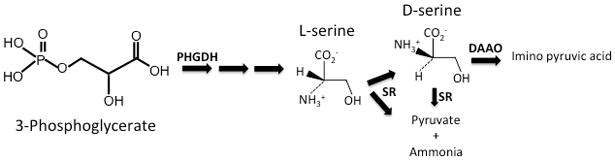
Phosphoglycerate 3-dehydrogenase (PGDH), an astrocyte-enriched enzyme, catalyzes the first step in L-serine biosynthesis. L-serine can then be converted to D-serine by the primarily neuronal enzyme serine racemase (SR). SR can also break down L-serine and D-serine (less efficient; smaller arrow) into pyruvate and ammonia. D-serine can be oxidatively deaminated to imino pyruvic acid by D-amino acid oxidase (DAAO).
In addition to the reversible racemization of L-serine to D-serine, SR also catalyzes the irreversible α,β-elimination of water from both enantiomers to produce ammonia and pyruvate (De Miranda et al., 2002), although the reaction is more efficient with L-serine as the substrate (Foltyn et al., 2005). The exact physiological role SR αβ-elimination activity is unknown. Although pyruvate is an important metabolite, the rate of its formation from other sources, such as glycolysis, are several orders of magnitude faster than the rate of L-serine αβ-elimination, making it unlikely that SR-derived pyruvate plays an important metabolic role (Wolosker & Mori, 2012). The production of pyruvate may be the primary role of SR in the liver, since it lacks NMDARs and where D-serine is believed to not play a functional role (Wolosker et al., 1999). However in the adult forebrain, where D-serine plays an important role in regulating excitatory neurotransmission, the SR αβ-elimination reaction might provide a mechanism by which to control intracellular D-serine levels in certain brain regions (Wolosker, 2011). There is very little D-amino acid oxidase (DAO), the main degradative enzyme for D-serine, in the adult forebrain (Figure 2). Furthermore, forebrain D-serine levels are unaltered in mice that have a catalytically inactive form of DAO, demonstrating the lack of importance for this enzyme in regulating D-serine concentration (Hamase, Konno, Morikawa, & Zaitsu, 2005).
3.1.1 Cellular Localization of Serine Racemase and D-serine
D-Serine is enriched in corticolimbic regions of the brain and is localized to the same areas as NMDARs (Schell, Molliver, & Snyder, 1995). D-serine is believed to be the primary forebrain co-agonist in the forebrain for synaptic, but not extra-synaptic NMDARs (Papouin et al., 2012). Initial in vitro studies suggested that SR was an astrocytic enzyme, and therefore astrocytes were the major source of brain D-serine (Mothet et al., 2005; Schell et al., 1995; Wolosker et al., 1999). However, recent immunohistochemical studies have demonstrated that SR is more prominently expressed in neurons, rather than in astrocytes (Balu, Takagi, Puhl, Benneyworth, & Coyle, 2014; Ding, Ma, Nagahama, Yamada, & Semba, 2011; Ehmsen et al., 2013; Miya et al., 2008). Furthermore, using mice with conditional deletions of SR either in excitatory forebrain neurons or GFAP-expressing astrocytes, it was shown that the majority of SR was expressed in forebrain excitatory neurons, particularly in the neocortex and hippocampus, and that approximately 15 percent was expressed in astrocytes (Benneyworth, Li, Basu, Bolshakov, & Coyle, 2012). This pattern of neuronal expression was also observed in human post-mortem neocortex, as SR was found in both excitatory and inhibitory neurons, but not astrocytes (Balu et al., 2014). It should be noted that L-serine derived from astrocytes is the precursor for D-serine in the brain, as mice that lack the astrocyte-enriched enzyme phosphoglycerate 3-dehydrogenase, which catalyzes the first step in L-serine biosynthesis (Figure 2), display marked reductions in brain D-serine (Yang et al., 2010).
D-serine was also originally believed to be stored in, and released from astrocytes (Fossat et al., 2012; Martineau et al., 2013; Schell et al., 1995). More recent studies found that D-serine was located primarily in neurons (Curcio et al., 2013; Kartvelishvily, Shleper, Balan, Dumin, & Wolosker, 2006). However, those studies did not validate the specificity of D-serine immunostaining in brain tissue. Utilizing SR−/− mice as a negative control to optimize staining conditions (Balu et al., 2014; Ehmsen et al., 2013), D-serine was almost exclusively stored in neurons, particularly GABAergic neurons in the neocortex and hippocampus (Balu et al., 2014). Interestingly, SR and D-serine were only co-localized in approximately ~50% of neurons, depending on the brain region (Balu et al., 2014). This separation of SR and D-serine might be related to the enzymatic profile of SR, as the αβ-elimination reaction that metabolizes D-serine is more efficient than the racemization of L-serine to D-serine.
3.2. Glycine and Kynurenic Acid
Glycine acts as an NMDAR co-agonist in the forebrain. The affinity of GluN1 for glycine is allosterically regulated by the GluN2 subunit, with GluN2B containing NMDARs displaying higher affinity for glycine (Priestley et al., 1995). There are two types of sodium-dependent glycine transporters (GlyT), GlyT1 and GlyT2, which are considered the primary regulators of intra- and extracellular glycine levels (Betz, Gomeza, Armsen, Scholze, & Eulenburg, 2006). Although glycine concentration in mammalian CSF is high relative to its dissociation constant (KD) for the GMS, local glycine levels are functionally regulated at the synapse. While GlyT1 and GlyT2 are both expressed in the cerebellum and spinal cord, where inhibitory glycinergic neurotransmission is concentrated, GlyT1 is also found in the forebrain (Zafra, Gomeza, Olivares, Aragon, & Gimenez, 1995). GlyT1 is widely expressed in glial cells, but is also found in neurons at glutamatergic synapses (Cubelos, Gimenez, & Zafra, 2005) and is thought to regulate NMDAR activity by affecting glycine availability (Bergeron et al., 1998). The activity of GlyT1 is itself regulated by the endogenous inhibitor, sarcosine (N-methylglycine), an intermediate and byproduct in glycine synthesis and degradation.
Kynurenic acid is a competitive antagonist at the GMS of the NMDAR (Kessler, Terramani, Lynch, & Baudry, 1989). It is derived from the metabolism of tryphtophan by several unique kynurenine aminotransferases (KATs) that catalyze the irreversible transamination of kynurenine to kynurenic acid, which is present in the human brain at high nanomolar concentrations. In addition, kynurenic acid is a non-competitive antagonist at the α7 nicotinic acetylcholine receptor (α7nAChR) (Hilmas et al., 2001) and has antioxidant properties (Lugo-Huitron et al., 2011).
4. NMDA RECEPTOR HYPOFUNCTION AND SCHIZOPHRENIA
There is substantial pharmacologic, genetic, and biochemical evidence to support NMDAR hypofunction as a key etiological component of schizophrenia. Dissociative anesthetics such as ketamine and phencyclidine (PCP), which are NMDAR channel blockers, were known since their introduction to produce the full range of schizophrenia symptoms and cognitive deficits in healthy subjects (Javitt & Zukin, 1991; Krystal et al., 1994). In healthy volunteers, low doses of ketamine that do not cause delirium or dementia produce the physiological abnormalities associated with schizophrenia, including abnormal evoked related potentials (ERPs) (Umbricht et al., 2000), eye-tracking abnormalities (Radant, Bowdle, Cowley, Kharasch, & Roy-Byrne, 1998), and enhanced subcortical dopamine release (Kegeles et al., 2000). Meanwhile, stabilized schizophrenia patients exhibit an increased sensitivity to ketamine (Lahti, Weiler, Tamara Michaelidis, Parwani, & Tamminga, 2001).
Although there have been mixed results in showing NMDAR expression abnormalities in human post-mortem brain tissue, depending on the brain region examined and methodology used, there is accumulating biochemical evidence suggesting reduced NMDAR function in schizophrenia. Recent meta-analysis found significant decreases in the expression of GluN1 mRNA (medium to large effect size) and GluN1 protein (medium effect size) in the prefrontal cortex (PFC) of subjects with schizophrenia (Catts, Lai, Weickert, Weickert, & Catts, 2015). This same meta-analysis found no consistent statistically significant changes in cortical mRNA and protein expression of GluN2 (A, B, D) or GluN3A subunits in schizophrenia, with the exception of reduced GluN2C mRNA in the PFC (Catts et al., 2015; Weickert et al., 2013). In the postsynaptic density fraction obtained from human post-mortem dorsolateral PFC tissue, there is a marked reduction in the activity of signaling cascades downstream of the NMDAR in schizophrenia, despite an apparent increase in NMDAR density and GluN1 expression (Banerjee et al., 2015). In the hippocampus, there have been several reports of reduced GluN1 levels selectively in the dentate gyrus of patients with schizophrenia (X. M. Gao et al., 2000; Law & Deakin, 2001; Stan et al., 2015).
There are also numerous abnormalities of NMDAR GMS modulators in the brain and the periphery of patients with schizophrenia. SR and D-serine are reduced in schizophrenia (Bendikov et al., 2007; Goltsov et al., 2006; Hashimoto et al., 2005; Labrie, Wang, Barger, Baker, & Roder, 2009; Morita et al., 2007). Kynurenic acid, an endogenous GMS antagonist, is elevated in the cerebral spinal fluid and post-mortem brain tissue of patients with schizophrenia (Erhardt et al., 2001; Schwarcz et al., 2001). The levels of N-acetylaspartylglutamate (NAAG), an endogenous NMDAR antagonist and mGluR3 agonist, are regulated by the enzyme glutamate carboxypeptidase II (GCP-II). The decreased activity of GCP-II and increased NAAG levels in the brains of patients with schizophrenia supports the hypothesis that NAAG-mediated signaling is disrupted in schizophrenia (Bergeron & Coyle, 2012).
Furthermore, there is evidence suggesting that altered glutamate levels could be contributing to NMDAR hypofunction in schizophrenia. A meta-analysis examining in vivo glutamate concentrations in schizophrenia measured by proton magnetic resonance spectroscopy found lower glutamate concentrations that progressively decreased with age in frontal brain regions, but did not find significant glutamate differences between schizophrenia and control groups in the hippocampus (Marsman et al., 2013). Although there have been inconsistent findings regarding hippocampal glutamate levels in schizophrenia, one recent proton magnetic resonance spectroscopy study found that glutamate levels were reduced in the hippocampus of patients with schizophrenia, regardless of whether they were medicated (Stan et al., 2015).
Individuals with NMDAR autoimmune encephalitis, which is associated with antibodies against the extracellular epitopes of the GluN1 subunit (IgG GluN1), initially present with psychiatric symptoms similar to schizophrenia (Dalmau et al., 2007). It is believed these antibodies cause NMDAR internalization (Dalmau, Lancaster, Martinez-Hernandez, Rosenfeld, & Balice-Gordon, 2011). Most research now suggests that IgG GluN1 antibodies detected in cases of anti-NMDAR encephalitis are not found in patients with schizophrenia (de Witte et al., 2015; Kayser & Dalmau, 2014). Although (Steiner et al., 2013) detected serum NMDAR antibodies (IgA and/or IgM GluN) in acutely ill schizophrenic patients, reanalysis of control sera from this cohort by the same group showed that the frequency of IgA and IgM antibodies was similar to that of the patients (Steiner et al., 2014). On the other hand, a recent meta-analysis showed significantly higher odds of NMDAR antibody seropositivity in individuals with schizophrenia or schizoaffective, major depressive, or bipolar, disorders compared with healthy controls, based on high-specificity, but not low specificity, thresholds (Pearlman & Najjar, 2014). In order to fully understand the clinical and pathophysiological implications of NMDAR antibodies in psychiatric disorders, it will be important to have adequately powered longitudinal studies that employ standardized assay methods and seropositivity thresholds, and quantify NMDAR antibodies in both sera and cerebrospinal fluid (Pearlman & Najjar, 2014).
As mentioned earlier, recent large-scale studies have identified both common and rare genetic variants important for glutamatergic signaling and the postsynaptic density, with increased risk for schizophrenia. Copy number variant (CNV) analyses have implicated de novo mutations in genes that encode the NMDAR and proteins associated with the postsynaptic density with increased risk of schizophrenia (Fromer et al., 2014; Kirov et al., 2012; Purcell et al., 2014). Although these rare variants are highly penetrant and etiologically relevant to schizophrenia, common genetic variation seems to account for much more of the variance in liability for developing this disorder (Purcell et al., 2014). The largest schizophrenia GWAS to date (~37,000 cases and ~113,000 controls) identified 108 genetic loci, including ~600 brain-enriched genes (Schizophrenia Working Group of the Psychiatric Genomics, 2014), that are involved in dopaminergic (DRD2) and glutamatergic transmission including: SRR (serine racemase), the metabotropic 3 glutamate receptor (GRM3), glutamate receptor 1 (GRIA1), and the GRIN2A (GluN 2A). Interestingly, most of these identified schizophrenia genetic risk loci identified in that study were located within non-coding regions of the genome, and thus are of unknown function. However, recent work has shown that a portion of the schizophrenia GWAS risk SNPs is enriched for alleles that regulate gene expression (eSNPs) and are located within cis-regulatory elements (i.e., enhancer and promoter regions), suggesting a functional link between risk-associated non-coding SNPs and 3D genomic architecture (Roussos et al., 2014). Biological pathway analyses of GWAS data (~60,000 subjects from the Psychiatric Genetics Consortium) revealed that genetic variants associated with schizophrenia are enriched in pathways related to the postsynaptic density, the postsynaptic membrane, dendritic spines, and histone methylation (Network & Pathway Analysis Subgroup of Psychiatric Genomics, 2015). It should be noted that most of the loci identified by GWAS contain more than one gene. Thus, much work is needed to determine which variant(s), in which gene(s) of a particular locus, are contributing to the increased risk for schizophrenia.
Finally, there is an abundance of data from pharmacologic and genetic animal models that support the hypothesis of NMDAR hypofunction contributing to the pathophysiology of schizophrenia (Balu & Coyle, 2011; Inta, Monyer, Sprengel, Meyer-Lindenberg, & Gass, 2010; Meltzer et al., 2013). In pharmacologically induced models, administration of NMDAR antagonists (phencyclidine (PCP), dizocilpine (MK-801), or ketamine) or kynurenic acid to animals leads to neurochemical, morphological, and cognitive deficits similar to what is observed in schizophrenia (Meltzer et al., 2013; Pershing et al., 2015). Genetic animal models have also provided a wealth of data suggesting that reduced NMDAR activity can lead to changes in the brain and behavior that is similar to what is observed in schizophrenia. For example, mice lacking the enzyme SR (SR−/−), have reduced D-serine and display NMDAR hypofunction (Basu et al., 2009). Similar to what is observed in schizophrenia, SR−/− mice have excitatory neurons in the cortex and hippocampus with reduced dendritic spines, reduced cortico-hippocampal volume, enlarged lateral ventricles, and increased GABA concentrations in the prefrontal cortex (Balu et al., 2012; Balu & Coyle, 2012; Balu et al., 2013; Basu et al., 2009; DeVito et al., 2011; Puhl et al., 2015). SR−/− mice also show reduced brain derived neurotrophic factor (BDNF) expression, Akt signaling, and microRNA-132 levels, which is accompanied by learning and cognitive deficits dependent on the hippocampus and prefrontal cortex (Balu et al., 2013; DeVito et al., 2011). Other genetic models have found that deleting the GluN1 subunit of the NMDAR, either constitutively, in corticolimbic inhibitory neurons, in parvalbumin inhibitory neurons, or in forebrain pyramidal neurons leads to cognitive, social, and electrophysiological (i.e., gamma band oscillations) changes reminiscent of schizophrenia (Belforte et al., 2009; Jadi, Margarita Behrens, & Sejnowski, 2015; Tatard-Leitman et al., 2015). Mice lacking the protein dybsindin, (an original candidate risk gene), which is reduced in schizophrenia, display NMDAR hypofunction, disrupted inhibitory transmission, hyperexcitability in the PFC, as well as deficits in working memory and learning (Carlson et al., 2011; Glen et al., 2014; Karlsgodt et al., 2011; Yuan et al., 2015).
4.1. Circuit based framework for reduced NMDAR function in schizophrenia
There are several neurocircuitry hypotheses for how reduced NMDAR activity contributes to the symptoms of schizophrenia. One hypothesis is that the cognitive impairments are due to hypofunctional NMDARs on cortical γ-amino- butyric acid (GABA) interneurons, particularly fast-spiking parvalbumin interneurons, leading to changes in cortical network oscillations (Jadi et al., 2015; Lisman et al., 2008). This hypothesis is supported by the fact that patients with schizophrenia have reduced parvalbumin expression in the dorsolateral PFC and abnormal gamma band oscillations (Gonzalez-Burgos, Cho, & Lewis, 2015; Jadi et al., 2015), which have been implicated in the synchronization of neural ensembles during working memory and attention. NMDAR hypofunction on cortical interneurons could also lead to an increased tone of glutamatergic projection neurons, resulting in the overstimulation of GABAergic interneurons in the ventral tegmental area. These activated GABA neurons in turn blunt the mesocortical dopamine pathway, preventing adequate dopamine release in the PFC, and thus could contribute to the negative and cognitive symptoms (Ellaithy, Younkin, Gonzalez-Maeso, & Logothetis, 2015). Serotonin (5-HT) receptor subtypes 5-HT1A and 5-HT2A, which are important for emotion and cognition, are elevated and reduced, respectively, in the PFC of patients with schizophrenia (Selvaraj, Arnone, Cappai, & Howes, 2014),. These receptors are highly expressed in pyramidal neurons of the PFC (Kia et al., 1996) and regulate NMDAR activity (Yuen, Jiang, Chen, Feng, & Yan, 2008; Yuen et al., 2005). Thus, changes in PFC 5-HT signaling could contribute to the cognitive and negative symptoms by altering NMDAR activity in the PFC. Another potential consequence of cortical interneuron disinhibition is that the increased firing of cortical glutamatergic projection neurons leads to hyperactivation of the dopamine mesolimbic pathway and ultimately the positive symptoms (Ellaithy et al., 2015). It is also possible that over activation of the hippocampal-ventral tegmental area loop in schizophrenia can lead to increased dopamine release and onset of psychosis (Lisman et al., 2008).
5. AUGMENTING NMDA RECEPTOR FUNCTION TO TREAT SCHIZOPHRENIA
To this day, the major pharmacological treatment strategy for schizophrenia is based on a serendipitous discovery over 50 years ago of the antipsychotic effects of chlorpromazine (Delay & Deniker, 1955), which was initially developed as an antihistamine to reduce intraoperative autonomic stress. The discovery that these antipsychotic drugs increased dopamine turnover and that their clinical potency correlated with D2 receptor affinity, led to the development of 10 “first-generation” dopamine D2 blocker antipsychotic medications. It was not until 20 years later when another serendipitous finding that clozapine, a dibenzodiazepine derivative of the tricyclic antidepressant imipramine, was found in clinical trials to possess antipsychotic efficacy superior to that of existing agents (Kane, Honigfeld, Singer, & Meltzer, 1988). This result started an era of intensive research by the pharmaceutical industry to develop additional “second-generation” antipsychotics with a high 5-HT2/D2 ratio, as this was believed to be the therapeutic mechanism of action for clozapine (Meltzer, 1989). Unfortunately, a series of second-generation drugs developed on the basis of favorable 5-HT2/D2 ratios failed to outperform first-generation drugs in large publicly funded multicenter trials (P. B. Jones et al., 2006; Lieberman et al., 2005; Sikich et al., 2008). Although these second-generation antipsychotics produced fewer extrapyramidal side effects than first-generation compounds, they produced other serious side effects including hyperlipidemia, glucose intolerance, and weight gain. In light of these findings, there has been a shift to focus on other components (i.e. cognitive and negative symptoms) of schizophrenia rather than just the antipsychotic responsive positive symptoms that primarily engage the dopaminergic system. This section will highlight some of the various strategies employed to augment NMDAR function in schizophrenia.
5.1. The glycine modulatory site
Since the GMS of the NMDAR is not saturated in vivo (Bergeron et al., 1998), it is therefore a potential therapeutic target, supporting the hypothesis that administration of GMS agonists could benefit patients by enhancing activation of NMDARs. There are several strategies by which the availability or concentration of GMS co-agonists and antagonists can be altered as a means to augment NMDAR function. Thus, targeting the GMS has been a very active area of research in both the pharmaceutical industry and academia.
There have been more than 70 placebo-controlled clinical trials of GMS agonists in schizophrenia, including D-serine, glycine, D-cycloserine (DCS), and D-alanine. Taken as a whole, the results have been mixed. Many studies reported significant improvements over multiple symptom domains while others did not. Intrinsic differences in efficacy between GMS co-agonists (Goff, Tsai, Manoach, & Coyle, 1995; Quartermain, Mower, Rafferty, Herting, & Lanthorn, 1994), and methodological factors, most notably small sample sizes, subject compliance, and differences in concurrent antipsychotic use, likely contributed to the variability among these trials. Unfortunately, an NIMH funded large, multi-center, placebo controlled clinical trial of glycine and DCS added on to antipsychotic drugs in schizophrenic patients reported no effects of either drug on negative symptoms or cognition (Buchanan et al., 2007). Recent meta-analysis of 17 double-blind randomized placebo-controlled trials testing whether glutamate positive modulators (GMS modulators: D-serine, D-cycloserine, benzoate, Org25935; AMPA receptor: Minocyclin, CX516; NMDAR: N-acetyl cysteine, pregnenelone, L-carnosine) improve cognitive deficits (overall cognitive function and eight specific cognitive domains) in schizophrenia found that these types of drugs were not superior to placebo as an adjunctive therapy to antipsychotics (Iwata et al., 2015). It should be noted that earlier meta-analyses found that GMS agonists were able to improve multiple symptom domains in conjunction with antipsychotic treatment (Choi, Wykes, & Kurtz, 2013; Tsai & Lin, 2010). However, this most recent meta-analysis by Iwata et al., 2015 included more studies, subjects, and drugs than the previous studies (Choi et al., 2013; Tsai & Lin, 2010).
All of the aforementioned studies tested whether augmenting NMDAR function could improve symptoms in patients with established schizophrenia. It is possible that pharmacologic intervention in the earlier stages of the disorder might produce more robust effects. A recent double-blind, placebo-controlled, parallel-group randomized pilot trial in the United States found that D-serine improved negative symptoms in clinical high-risk teenage patients (Kantrowitz et al., 2015). Even though a larger sample size and replication is needed, this study suggests that D-serine could be effective in treating the prodromal symptoms of schizophrenia.
An alternative approach to directly targeting the GMS is to block reuptake of co-agonist. Research has focused on inhibiting glycine reuptake via GlyT1 blockade because the uptake and release mechanisms for D-serine are poorly understood. Sarcosine (N-methylglycine), an intermediate and byproduct in glycine synthesis and degradation, is an endogenous and competitive GlyT1 antagonist. The (Tsai & Lin, 2010) meta-analysis found it effective on total psychopathology, negative symptoms, and general psychopathology. However, sarcosine-derived GlyT1 inhibitors produce undesirable side effects, such as hypoactivity and ataxia, and have therefore prompted the development of non-sarcosine-based GlyT1inhibitors. Unfortunately, the noncompetitive GlyT-1 antagonist, bitopertin, which significantly reduced negative symptoms in a Phase-II trial in schizophrenia, failed to reach its endpoints to improve negative symptoms in multicenter Phase II/III trials (Bugarski-Kirola, Wang, Abi-Saab, & Blattler, 2014). One reason for the poor clinical outcomes with this class of drugs could be that inhibiting GlyT1 augments NMDAR function primarily at extra-synaptic receptors as suggested by recent electrophysiological studies (Li et al., 2013; Papouin et al., 2012), thereby undercutting therapeutic effects.
GMS agonist availability can also be increased by inhibiting DAAO, the enzyme that catabolizes D-amino acids, like D-serine and D-alanine. The expression and activity of DAAO are increased in patients with schizophrenia, and early candidate gene association studies suggested that DAAO was a schizophrenia risk gene (Verrall, Burnet, Betts, & Harrison, 2010). Benzoate is a naturally occurring DAAO inhibitor that is considered a generally safe food preservative by the FDA. In a small randomized, double-blind, placebo-controlled trial, sodium benzoate significantly improved positive, negative, and cognitive symptoms in patients with schizophrenia stabilized on antipsychotics (Lane et al., 2013). However, this study did not measure plasma D-serine levels following benzoate treatment. There is also an effort to determine whether more potent DAAO inhibitors would increase the bioavailability of D-serine for uptake into the brain. Interestingly, co-administration of novel DAAO inhibitors (much more potent than benzoate at inhibiting enzyme activity) with D-serine does not significantly increase peripheral D-serine levels in dogs and baboons compared to D-serine alone, even though these drugs were effective in doing so in rodents (Rojas et al., 2015). This study raises concerns about the viability of inhibiting peripheral DAAO in humans and suggests that the therapeutic effects seen with benzoate in patients with schizophrenia (Lane et al., 2013) were due to mechanisms other than DAAO inhibition.
Another strategy to restore the NMDAR hypofunction observed in schizophrenia could be to reduce the levels of the endogenous GMS antagonist, kynurenic acid. Preclinical studies in normal rodents demonstrate that cognition is impaired by the administration of either kynurenic acid or kynurenine, the immediate precursor to kynurenic acid (Herman, Bubser, Conn, & Jones, 2012). Kynurenine aminotransferase II inhibitors, which block the formation of kynurenic acid, prevent the cognitive and auditory-gating deficits in rodents and nonhuman primates caused by drugs such as, ketamine and amphetamine (Kozak et al., 2014; Schwarcz, Bruno, Muchowski, & Wu, 2012). These findings suggest that modulation of the kynurenine pathway could be a novel mechanism by which to attenuate the cognitive symptoms in schizophrenia. It should be noted that kynurenic acid is also a potent non-competitive antagonist of the α-7-nicotinic acetylcholine receptor (α7nAChR) and that the cognitive impairments produced by kyurenic acid could be mediated in part by its interaction with the α7nAChR (Albuquerque & Schwarcz, 2013).
In sum, there has been a great effort made to design novel compounds aimed at enhancing NMDAR activity, particularly by targeting the GMS. An optimistic interpretation of the studies conducted so far that directly or indirectly target the NMDAR GMS, is that they provide a proof of principle for the therapeutic effects of augmenting NMDAR function in schizophrenia, but not strong evidence for a feasible long-term treatment. For example, D-serine and glycine have poor pharmacokinetics and brain penetrance, exogenous glycine may preferentially act at extra-synaptic NMDARs, and chronic DCS results in receptor desensitization.
5.2. Allosteric modulation of the NMDA receptor
Although neuroactive steroids can directly regulate NMDAR activity, certain neurosteroids have low potency and poor selectivity, thus making them poor candidates for therapeutic agents. It was recently found that the major brain-derived cholesterol metabolite, 24(S)-hydroxycholesterol, is a very potent and selective NMDAR positive allosteric modulator (PAM) with a distinct mechanism that does not overlap with other allosteric modulators (Paul et al., 2013). Furthermore, synthetic oxysterol derivatives (i.e., SGE-201 and SGE-301) had desirable in vivo drug-like properties and reversed NMDAR antagonist-induced cognitive and social deficits (Paul et al., 2013). Although the exact mechanism by which activating this novel oxysterol site augments NMDAR function is unknown, these findings suggest that it could serve as a therapeutic drug target.
5.3. Metabotropic glutamate receptors 2 and 3
Group II metabotropic glutamate receptors (mGlu) are widely expressed throughout the rodent and human brain. The mGlu subtype 2 (mGlu2) is expressed in many of the brain regions implicated in schizophrenia, including the hippocampus, striatum, prefrontal cortex, and amygdala (Herman et al., 2012). These receptors are found presynaptically, functioning as autoreceptors to inhibit glutamate release, and are also found postsynaptically. There is evidence for crosstalk between mGlu2 and 5-HT2A and for the formation of mGlu2-5-HT2A receptor heterocomplexes (Ellaithy et al., 2015). The mGlu subtype 3 (mGlu3) shows a more diffuse expression pattern than mGlu2,, being expressed presynaptically and in astrocytes (Herman et al., 2012). Although preclinical evidence and initial clinical trials showed promise for mGlu2/3 agonists (orthosteric agonists lack subtype specificity) as novel treatments for schizophrenia, subsequent and larger clinical trials with the Lily mGlu2/3 prodrug were inconclusive (Kinon et al., 2011) and failed to demonstrate improvements compared to placebo (Hopkins, 2013). Positive allosteric modulators (PAMs) of type II mGlu receptors might hold more promise than traditional agonists. PAMs are unlike traditional agonists because they do not directly activate the receptor, but rather enhance signaling in the presence of endogenous agonist. These types of compounds may be better than traditional agonists because they are less likely to cause receptor internalization and have a safer side effect profile. These types of compounds are better than traditional agonists because they do not cause receptor internalization and have a safer side effect profile. Two mGlu2 PAMs have so far reached clinical trials. A Phase IIa study with ADX71149 (Addex and Janssen) identified patients with residual negative symptoms as those most likely to benefit from adjunctive treatment with ADX71149 (Ellaithy et al., 2015). Potentiation of mGlu3 might also be a therapeutic target to treat the cognitive symptoms of schizophrenia, as mGlu3 negative allosteric modulators impair synaptic plasticity and learning in mice (Walker et al., 2015).
5.4. Metabotropic glutamate receptor 5
The mGlu subtype 5 (mGlu5) is expressed throughout the brain, but is enriched in cortico-limbic regions that are important for affect and memory (Herman et al., 2012). It is primarily located postsynaptically in both excitatory and inhibitory neurons (Biber et al., 1999; Herman et al., 2012). In particular, mGlu5, via its physical interaction with the NMDAR in the postsynaptic density, can enhance NMDAR function. This enhancement is independent of Gαq signaling and is facilitated by adaptor proteins, such as Shank and Homer (C. Gao, Tronson, & Radulovic, 2013). mGlu5 is a leading target for novel therapeutics to treat schizophrenia and cognitive disorders, especially since the development of subtype specific PAMs. One newly developed PAM, VU0409551, was found to reverse the deficits in NMDAR function, synaptic plasticity, and memory in SR−/− mice, a genetic model relevant to schizophrenia (Balu et al., 2013). VU0409551 also produced robust antipsychotic-like and cognitive enhancing effects in pharmacologically induced animal models of schizophrenia (Rook et al., 2015). These results support positive allosteric modulation of mGlu5 as a promising mechanism by which to treat schizophrenia.
5.5. Cholinergic receptors
Muscarinic acetylcholine receptors (mAChRs) are G-protein coupled metabotropic receptors that are widely distributed throughout the neocortex and five isoforms (M1–M5) (Caulfield & Birdsall, 1998). The M1 mAChR is the predominant subtype in the brain, being expressed throughout the striatum, cortex, and postsynaptically on the cell bodies and dendrites of hippocampal pyramidal neurons and granule cells (Marino, Rouse, Levey, Potter, & Conn, 1998). Importantly, M1 mAChR activation in the hippocampus and other forebrain regions potentiates NMDAR currents (Marino et al., 1998). Thus, there has been interest in determining whether M1 mAChRs can alleviate some of the psychotic and cognitive deficits observed in schizophrenia through enhancement of NMDAR neurotransmission. Consistent with this hypothesis, was a small clinical trial in schizophrenia that demonstrated the antipsychotic efficacy of the putative M1/M4 selective mAChR agonist xanomaline (Shekhar et al., 2008). However, current cholinergic therapeutics are limited in their applicability because of aversive side-effect profiles that are attributed to peripheral activation of M2 and M3 mAChRs (Wess, Eglen, & Gautam, 2007). Thus, there has been an effort to generate novel and selective M1 mAChR PAMs (C. K. Jones, Byun, & Bubser, 2012). One such example is VU0453595, which was recently found to reverse the electrophysiological and behavioral deficits in PCP-treated mice (Ghoshal et al., 2015).
nAChRs, a family of receptors that is comprised of 16 different subunits, are widely expressed in the brain, mediate fast synaptic signaling, and modulate the release of other neurotransmitters. Although most nAChRs subunits assemble only into heteropentameric receptor ion channel combinations, the α7 subunits are able to generate functional homomeric nAChRs (Dani & Bertrand, 2007). These receptors have distinct characteristics due to their homopentameric composition that distinguishes them from the other nAChR subtypes including: rapid desensitization, less sensitivity to nicotine as an agonist, and a higher permeability to calcium (Dani & Bertrand, 2007). The majority of the nAChRs in the brain are composed of either α4β2 or α7 subunits. α7 nAChRs are abundantly expressed in the hippocampus and cortex, and play an important role in cognitive functions in both rodents and humans (Dani & Bertrand, 2007). Activation of α7 nAChRs increases cholinergic neurotransmission, the release of glutamate and dopamine, and is pro-cognitive. For these reasons, the α7 nAChR has been a therapeutic target for cognitive impairments in schizophrenia and Alzheimer’s disease. One new promising drug that engages the α7 nAChR is encenicline (EVP-6124; [(R)-7-chloro-N-quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide]), a high-affinity partial agonist that potentiates the response to the natural agonist acetylcholine (Prickaerts et al., 2012). Encenicline appears to work as a neuromodulator, with its pro-cognitive effects mediated in part by modulating multiple neurotransmitter systems including glutamate, dopamine, and acetylcholine in the prefrontal cortex and other brain regions (Huang et al., 2014). A recent phase 2, study found that encenicline was generally well tolerated and significantly improved cognition and function in patients with schizophrenia (Keefe et al., 2015). Efforts are also being made to design α7 nAChR PAMs and α4β2 agonists in hopes of ameliorating the cognitive symptoms in schizophrenia (C. K. Jones et al., 2012).
Schizophrenia is a highly heterogeneous disorder that is clinically defined based only on phenomenology, and not yet based on biology. Thus, it is possible that some of the variability in pharmacological responses reported in the above clinical trials could be due to variations in the pathology across schizophrenia patients. As we begin to better understand the neurobiological and genetic underpinnings of schizophrenia, mechanistically novel drugs could be potentially be tested in selected populations that will most likely benefit from that type of intervention.
6. CONCLUSION
Data from clinical, genetic, and animal model studies strongly implicate the NMDAR as a central hub for many of the pathological brain processes that occur in schizophrenia. It has been long known that the dissociative anesthetics and NMDAR antagonists, PCP and ketamine, produce the full range of schizophrenia symptoms and cognitive deficits in healthy subjects (Javitt & Zukin, 1991; Krystal et al., 1994), while ketamine exacerbates symptoms in stabilized schizophrenia patients (Lahti, Weiler, Tamara Michaelidis, Parwani, & Tamminga, 2001). Patients with NMDAR autoimmune encephalitis that produce antibodies against the GluN1 subunit, initially present with psychiatric symptoms similar to schizophrenia. Furthermore, evidence suggests that a certain population of patients with schizophrenia produce antibodies against the NMDAR. Data from patients and human post-mortem brains also suggests that the NMDAR is hypofunctional in schizophrenia. The recent explosion of large-scale GWAS and sequencing studies has provided significant evidence for disturbances in genes related to glutamatergic signaling, the postsynaptic density, and dendritic spines. Finally, there is a vast literature on pharmacologic and genetic animal models of NMDAR hypofunction that recapitulate the various neuropathological, electrophysiological, and behavioral abnormalities observed in schizophrenia. It should be noted, however, that this review is an oversimplification of an extremely complex disease. In schizophrenia, there are also disturbances to the GABAergic, cholinergic, and dopaminergic neurotransmitter systems (Coyle, Balu, Benneyworth, Basu, & Roseman, 2010; Lisman et al., 2008), as well as disruptions to astrocytes and white matter integrity (Mighdoll, Tao, Kleinman, & Hyde, 2015). Furthermore, there is convincing evidence that the immune system and oxidative stress play key roles in the pathophysiology of schizophrenia (Leza et al., 2015). Thus, schizophrenia likely results from various combinations of environmental disruptions in brain development and numerous genetic vulnerabilities. These etiological complexities imply that no single pharmacological approach will treat all aspects of the illness nor be effective for all patients with schizophrenia.
Acknowledgments
The National Institute of Mental Health supported this work (4R00MH099252-03). I would like to personally thank Dr. Coyle for being a great mentor during my postdoctoral fellowship and continuing to be a staunch advocate of my career.
ABBREVIATIONS
- DAAO
D-amino acid oxidase
- DCS
D-cycloserine
- ERP
evoked related potential
- GABA
γ-amino-butyric acid
- GCP-II
glutamate carboxypeptidase II
- GlyT
glycine transporter
- GMS
glycine modulatory site
- mAChRs
muscarinic acetylcholine receptors
- mGlu5
metabotropic glutamate receptor 5
- NAAG
N-acetylaspartylglutamate
- nAChR
Nicotinic acetylcholine receptors
- NMDAR
NMDA receptor
- PAM
positive allosteric modulator
- PCP
phencyclidine
- PFC
prefrontal cortex
- PLP
fold II pyridoxal 5′ phosphate
- SR
serine racemase
Footnotes
CONFLICT OF INTEREST
D.T.B has no conflicts of interest to declare.
References
- Albuquerque EX, Schwarcz R. Kynurenic acid as an antagonist of alpha7 nicotinic acetylcholine receptors in the brain: facts and challenges. Biochem Pharmacol. 2013;85(8):1027–1032. doi: 10.1016/j.bcp.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Carlson GC, Talbot K, Kazi H, Hill-Smith TE, Easton RM, … Lucki I. Akt1 deficiency in schizophrenia and impairment of hippocampal plasticity and function. Hippocampus. 2012;22(2):230–240. doi: 10.1002/hipo.20887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Coyle JT. Neuroplasticity signaling pathways linked to the pathophysiology of schizophrenia. Neurosci Biobehav Rev. 2011;35(3):848–870. doi: 10.1016/j.neubiorev.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Coyle JT. Neuronal d-serine regulates dendritic architecture in the somatosensory cortex. Neuroscience letters. 2012;517(2):77–81. doi: 10.1016/j.neulet.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Li Y, Puhl MD, Benneyworth MA, Basu AC, Takagi S, … Coyle JT. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc Natl Acad Sci U S A. 2013;110(26):E2400–2409. doi: 10.1073/pnas.1304308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Takagi S, Puhl MD, Benneyworth MA, Coyle JT. D-serine and serine racemase are localized to neurons in the adult mouse and human forebrain. Cell Mol Neurobiol. 2014;34(3):419–435. doi: 10.1007/s10571-014-0027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Wang HY, Borgmann-Winter KE, MacDonald ML, Kaprielian H, Stucky A, … Hahn CG. Src kinase as a mediator of convergent molecular abnormalities leading to NMDAR hypoactivity in schizophrenia. Mol Psychiatry. 2015;20(9):1091–1100. doi: 10.1038/mp.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu AC, Tsai GE, Ma CL, Ehmsen JT, Mustafa AK, Han L, … Coyle JT. Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol Psychiatry. 2009;14(7):719–727. doi: 10.1038/mp.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, … Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2009 doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendikov I, Nadri C, Amar S, Panizzutti R, De Miranda J, Wolosker H, Agam G. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr Res. 2007;90(1–3):41–51. doi: 10.1016/j.schres.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Benneyworth MA, Li Y, Basu AC, Bolshakov VY, Coyle JT. Cell Selective Conditional Null Mutations of Serine Racemase Demonstrate a Predominate Localization in Cortical Glutamatergic Neurons. Cellular and molecular neurobiology. 2012 doi: 10.1007/s10571-012-9808-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron R, Coyle JT. NAAG, NMDA receptor and psychosis. Curr Med Chem. 2012;19(9):1360–1364. doi: 10.2174/092986712799462685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron R, Meyer TM, Coyle JT, Greene RW. Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc Natl Acad Sci U S A. 1998;95(26):15730–15734. doi: 10.1073/pnas.95.26.15730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz H, Gomeza J, Armsen W, Scholze P, Eulenburg V. Glycine transporters: essential regulators of synaptic transmission. Biochem Soc Trans. 2006;34(Pt 1):55–58. doi: 10.1042/BST0340055. [DOI] [PubMed] [Google Scholar]
- Biber K, Laurie DJ, Berthele A, Sommer B, Tolle TR, Gebicke-Harter PJ, … Boddeke HW. Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. J Neurochem. 1999;72(4):1671–1680. doi: 10.1046/j.1471-4159.1999.721671.x. [DOI] [PubMed] [Google Scholar]
- Boehning D, Snyder SH. Novel neural modulators. Annu Rev Neurosci. 2003;26:105–131. doi: 10.1146/annurev.neuro.26.041002.131047. [DOI] [PubMed] [Google Scholar]
- Buchanan RW, Javitt DC, Marder SR, Schooler NR, Gold JM, McMahon RP, … Carpenter WT. The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psychiatry. 2007;164(10):1593–1602. doi: 10.1176/appi.ajp.2007.06081358. [DOI] [PubMed] [Google Scholar]
- Bugarski-Kirola D, Wang A, Abi-Saab D, Blattler T. A phase II/III trial of bitopertin monotherapy compared with placebo in patients with an acute exacerbation of schizophrenia - results from the CandleLyte study. Eur Neuropsychopharmacol. 2014;24(7):1024–1036. doi: 10.1016/j.euroneuro.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Canu E, Agosta F, Filippi M. A selective review of structural connectivity abnormalities of schizophrenic patients at different stages of the disease. Schizophr Res. 2015;161(1):19–28. doi: 10.1016/j.schres.2014.05.020. [DOI] [PubMed] [Google Scholar]
- Cardno AG, Marshall EJ, Coid B, Macdonald AM, Ribchester TR, Davies NJ, … Murray RM. Heritability estimates for psychotic disorders: the Maudsley twin psychosis series. Arch Gen Psychiatry. 1999;56(2):162–168. doi: 10.1001/archpsyc.56.2.162. [DOI] [PubMed] [Google Scholar]
- Carlson GC, Talbot K, Halene TB, Gandal MJ, Kazi HA, Schlosser L, … Siegel SJ. Dysbindin-1 mutant mice implicate reduced fast-phasic inhibition as a final common disease mechanism in schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(43):E962–970. doi: 10.1073/pnas.1109625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catts VS, Lai YL, Weickert CS, Weickert TW, Catts SV. A quantitative review of the postmortem evidence for decreased cortical N-methyl-d-aspartate receptor expression levels in schizophrenia: How can we link molecular abnormalities to mismatch negativity deficits? Biol Psychol. 2015 doi: 10.1016/j.biopsycho.2015.10.013. [DOI] [PubMed] [Google Scholar]
- Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50(2):279–290. [PubMed] [Google Scholar]
- Choi KH, Wykes T, Kurtz MM. Adjunctive pharmacotherapy for cognitive deficits in schizophrenia: meta-analytical investigation of efficacy. Br J Psychiatry. 2013;203(3):172–178. doi: 10.1192/bjp.bp.111.107359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan JJ, Srinivasan NG. The occurrence of certain D-amino acids in insects. Biochemistry. 1966;5(4):1185–1190. doi: 10.1021/bi00868a010. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Balu D, Benneyworth M, Basu A, Roseman A. Beyond the dopamine receptor: novel therapeutic targets for treating schizophrenia. Dialogues Clin Neurosci. 2010;12(3):359–382. doi: 10.31887/DCNS.2010.12.3/jcoyle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubelos B, Gimenez C, Zafra F. Localization of the GLYT1 glycine transporter at glutamatergic synapses in the rat brain. Cereb Cortex. 2005;15(4):448–459. doi: 10.1093/cercor/bhh147. [DOI] [PubMed] [Google Scholar]
- Curcio L, Podda MV, Leone L, Piacentini R, Mastrodonato A, Cappelletti P, … D’Ascenzo M. Reduced D-serine levels in the nucleus accumbens of cocaine-treated rats hinder the induction of NMDA receptor-dependent synaptic plasticity. Brain. 2013 doi: 10.1093/brain/awt036. [DOI] [PubMed] [Google Scholar]
- Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63–74. doi: 10.1016/S1474-4422(10)70253-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE, Voloschin A, … Lynch DR. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Annals of neurology. 2007;61(1):25–36. doi: 10.1002/ana.21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- De Miranda J, Panizzutti R, Foltyn VN, Wolosker H. Cofactors of serine racemase that physiologically stimulate the synthesis of the N-methyl-D-aspartate (NMDA) receptor coagonist D-serine. Proc Natl Acad Sci U S A. 2002;99(22):14542–14547. doi: 10.1073/pnas.222421299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miranda J, Santoro A, Engelender S, Wolosker H. Human serine racemase: moleular cloning, genomic organization and functional analysis. Gene. 2000;256(1–2):183–188. doi: 10.1016/s0378-1119(00)00356-5. [DOI] [PubMed] [Google Scholar]
- de Witte LD, Hoffmann C, van Mierlo HC, Titulaer MJ, Kahn RS, Martinez-Martinez P European Consortium of Autoimmune Mental D. Absence of N-Methyl-D-Aspartate Receptor IgG Autoantibodies in Schizophrenia: The Importance of Cross-Validation Studies. JAMA Psychiatry. 2015;72(7):731–733. doi: 10.1001/jamapsychiatry.2015.0526. [DOI] [PubMed] [Google Scholar]
- Delay J, Deniker P. Neuroleptic effects of chlorpromazine in therapeutics of neuropsychiatry. J Clin Exp Psychopathol. 1955;16(2):104–112. [PubMed] [Google Scholar]
- DeVito LM, Balu DT, Kanter BR, Lykken C, Basu AC, Coyle JT, Eichenbaum H. Serine racemase deletion disrupts memory for order and alters cortical dendritic morphology. Genes Brain Behav. 2011;10(2):210–222. doi: 10.1111/j.1601-183X.2010.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Ma N, Nagahama M, Yamada K, Semba R. Localization of D-serine and serine racemase in neurons and neuroglias in mouse brain. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2011;32(2):263–267. doi: 10.1007/s10072-010-0422-2. [DOI] [PubMed] [Google Scholar]
- Ehmsen JT, Ma TM, Sason H, Rosenberg D, Ogo T, Furuya S, … Wolosker H. D-serine in glia and neurons derives from 3-phosphoglycerate dehydrogenase. J Neurosci. 2013;33(30):12464–12469. doi: 10.1523/JNEUROSCI.4914-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellaithy A, Younkin J, Gonzalez-Maeso J, Logothetis DE. Positive allosteric modulators of metabotropic glutamate 2 receptors in schizophrenia treatment. Trends Neurosci. 2015;38(8):506–516. doi: 10.1016/j.tins.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt S, Blennow K, Nordin C, Skogh E, Lindstrom LH, Engberg G. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci Lett. 2001;313(1–2):96–98. doi: 10.1016/s0304-3940(01)02242-x. [DOI] [PubMed] [Google Scholar]
- Farrell MS, Werge T, Sklar P, Owen MJ, Ophoff RA, O’Donovan MC, … Sullivan PF. Evaluating historical candidate genes for schizophrenia. Mol Psychiatry. 2015;20(5):555–562. doi: 10.1038/mp.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltyn VN, Bendikov I, De Miranda J, Panizzutti R, Dumin E, Shleper M, … Wolosker H. Serine racemase modulates intracellular D-serine levels through an alpha, beta-elimination activity. J Biol Chem. 2005;280(3):1754–1763. doi: 10.1074/jbc.M405726200. [DOI] [PubMed] [Google Scholar]
- Fossat P, Turpin FR, Sacchi S, Dulong J, Shi T, Rivet JM, … Mothet JP. Glial D-serine gates NMDA receptors at excitatory synapses in prefrontal cortex. Cerebral cortex. 2012;22(3):595–606. doi: 10.1093/cercor/bhr130. [DOI] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, … O’Donovan MC. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014 doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusar-Poli P, Borgwardt S, Crescini A, Deste G, Kempton MJ, Lawrie S, … Sacchetti E. Neuroanatomy of vulnerability to psychosis: a voxel-based meta-analysis. Neurosci Biobehav Rev. 2011;35(5):1175–1185. doi: 10.1016/j.neubiorev.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Fusar-Poli P, Smieskova R, Kempton MJ, Ho BC, Andreasen NC, Borgwardt S. Progressive brain changes in schizophrenia related to antipsychotic treatment? A meta-analysis of longitudinal MRI studies. Neurosci Biobehav Rev. 2013;37(8):1680–1691. doi: 10.1016/j.neubiorev.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Tronson NC, Radulovic J. Modulation of behavior by scaffolding proteins of the post-synaptic density. Neurobiol Learn Mem. 2013;105:3–12. doi: 10.1016/j.nlm.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XM, Sakai K, Roberts RC, Conley RR, Dean B, Tamminga CA. Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am J Psychiatry. 2000;157(7):1141–1149. doi: 10.1176/appi.ajp.157.7.1141. [DOI] [PubMed] [Google Scholar]
- Ghoshal A, Rook JM, Dickerson JW, Roop GN, Morrison RD, Jalan-Sakrikar N, … Conn PJ. Potentiation of M Muscarinic Receptor Reverses Plasticity Deficits and Negative and Cognitive Symptoms in a Schizophrenia Mouse Model. Neuropsychopharmacology. 2015 doi: 10.1038/npp.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glausier JR, Lewis DA. Dendritic spine pathology in schizophrenia. Neuroscience. 2013;251:90–107. doi: 10.1016/j.neuroscience.2012.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glen WB, Jr, Horowitz B, Carlson GC, Cannon TD, Talbot K, Jentsch JD, Lavin A. Dysbindin-1 loss compromises NMDAR-dependent synaptic plasticity and contextual fear conditioning. Hippocampus. 2014;24(2):204–213. doi: 10.1002/hipo.22215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DC, Tsai G, Manoach DS, Coyle JT. Dose-finding trial of D-cycloserine added to neuroleptics for negative symptoms in schizophrenia. Am J Psychiatry. 1995;152(8):1213–1215. doi: 10.1176/ajp.152.8.1213. [DOI] [PubMed] [Google Scholar]
- Goltsov AY, Loseva JG, Andreeva TV, Grigorenko AP, Abramova LI, Kaleda VG, … Rogaev EI. Polymorphism in the 5′-promoter region of serine racemase gene in schizophrenia. Mol Psychiatry. 2006;11(4):325–326. doi: 10.1038/sj.mp.4001801. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Cho RY, Lewis DA. Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol Psychiatry. 2015;77(12):1031–1040. doi: 10.1016/j.biopsych.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M, Yamauchi T, Kamiya N, Miyahara I, Yoshimura T, Mihara H, … Esaki N. Crystal structure of a homolog of mammalian serine racemase from Schizosaccharomyces pombe. J Biol Chem. 2009;284(38):25944–25952. doi: 10.1074/jbc.M109.010470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59(6):846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Hamase K, Konno R, Morikawa A, Zaitsu K. Sensitive determination of D-amino acids in mammals and the effect of D-amino-acid oxidase activity on their amounts. Biol Pharm Bull. 2005;28(9):1578–1584. doi: 10.1248/bpb.28.1578. [DOI] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H, Traynelis SF. Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron. 2014;81(5):1084–1096. doi: 10.1016/j.neuron.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Engberg G, Shimizu E, Nordin C, Lindstrom LH, Iyo M. Reduced D-serine to total serine ratio in the cerebrospinal fluid of drug naive schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(5):767–769. doi: 10.1016/j.pnpbp.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Henson MA, Roberts AC, Salimi K, Vadlamudi S, Hamer RM, Gilmore JH, … Philpot BD. Developmental regulation of the NMDA receptor subunits, NR3A and NR1, in human prefrontal cortex. Cereb Cortex. 2008;18(11):2560–2573. doi: 10.1093/cercor/bhn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman EJ, Bubser M, Conn PJ, Jones CK. Metabotropic glutamate receptors for new treatments in schizophrenia. Handb Exp Pharmacol. 2012;(213):297–365. doi: 10.1007/978-3-642-25758-2_11. [DOI] [PubMed] [Google Scholar]
- Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci. 2001;21(19):7463–7473. doi: 10.1523/JNEUROSCI.21-19-07463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins CR. Is there a path forward for mGlu(2) positive allosteric modulators for the treatment of schizophrenia? ACS Chem Neurosci. 2013;4(2):211–213. doi: 10.1021/cn400023y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Felix AR, Flood DG, Bhuvaneswaran C, Hilt D, Koenig G, Meltzer HY. The novel alpha7 nicotinic acetylcholine receptor agonist EVP-6124 enhances dopamine, acetylcholine, and glutamate efflux in rat cortex and nucleus accumbens. Psychopharmacology (Berl) 2014;231(23):4541–4551. doi: 10.1007/s00213-014-3596-0. [DOI] [PubMed] [Google Scholar]
- Hulshoff Pol HE, Kahn RS. What happens after the first episode? A review of progressive brain changes in chronically ill patients with schizophrenia. Schizophr Bull. 2008;34(2):354–366. doi: 10.1093/schbul/sbm168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inta D, Monyer H, Sprengel R, Meyer-Lindenberg A, Gass P. Mice with genetically altered glutamate receptors as models of schizophrenia: a comprehensive review. Neurosci Biobehav Rev. 2010;34(3):285–294. doi: 10.1016/j.neubiorev.2009.07.010. [DOI] [PubMed] [Google Scholar]
- Iwata Y, Nakajima S, Suzuki T, Keefe RS, Plitman E, Chung JK, … Uchida H. Effects of glutamate positive modulators on cognitive deficits in schizophrenia: a systematic review and meta-analysis of double-blind randomized controlled trials. Mol Psychiatry. 2015;20(10):1151–1160. doi: 10.1038/mp.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadi MP, Margarita Behrens M, Sejnowski TJ. Abnormal Gamma Oscillations in N-Methyl-D-Aspartate Receptor Hypofunction Models of Schizophrenia. Biol Psychiatry. 2015 doi: 10.1016/j.biopsych.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325(6104):529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- Jones CK, Byun N, Bubser M. Muscarinic and nicotinic acetylcholine receptor agonists and allosteric modulators for the treatment of schizophrenia. Neuropsychopharmacology. 2012;37(1):16–42. doi: 10.1038/npp.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PB, Barnes TR, Davies L, Dunn G, Lloyd H, Hayhurst KP, … Lewis SW. Randomized controlled trial of the effect on Quality of Life of second- vs first-generation antipsychotic drugs in schizophrenia: Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1) Arch Gen Psychiatry. 2006;63(10):1079–1087. doi: 10.1001/archpsyc.63.10.1079. [DOI] [PubMed] [Google Scholar]
- Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789–796. doi: 10.1001/archpsyc.1988.01800330013001. [DOI] [PubMed] [Google Scholar]
- Kantrowitz JT, Woods SW, Petkova E, Cornblatt B, Corcoran CM, Chen H, … Javitt DC. D-serine for the treatment of negative symptoms in individuals at clinical high risk of schizophrenia: a pilot, double-blind, placebo-controlled, randomised parallel group mechanistic proof-of-concept trial. Lancet Psychiatry. 2015;2(5):403–412. doi: 10.1016/S2215-0366(15)00098-X. [DOI] [PubMed] [Google Scholar]
- Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344(6187):992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsgodt KH, Robleto K, Trantham-Davidson H, Jairl C, Cannon TD, Lavin A, Jentsch JD. Reduced dysbindin expression mediates N-methyl-D-aspartate receptor hypofunction and impaired working memory performance. Biological psychiatry. 2011;69(1):28–34. doi: 10.1016/j.biopsych.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartvelishvily E, Shleper M, Balan L, Dumin E, Wolosker H. Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors. The Journal of biological chemistry. 2006;281(20):14151–14162. doi: 10.1074/jbc.M512927200. [DOI] [PubMed] [Google Scholar]
- Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr Res. 2014 doi: 10.1016/j.schres.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe RS, Meltzer HA, Dgetluck N, Gawryl M, Koenig G, Moebius HJ, … Hilt DC. Randomized, Double-Blind, Placebo-Controlled Study of Encenicline, an alpha7 Nicotinic Acetylcholine Receptor Agonist, as a Treatment for Cognitive Impairment in Schizophrenia. Neuropsychopharmacology. 2015 doi: 10.1038/npp.2015.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegeles LS, Abi-Dargham A, Zea-Ponce Y, Rodenhiser-Hill J, Mann JJ, Van Heertum RL, … Laruelle M. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biol Psychiatry. 2000;48(7):627–640. doi: 10.1016/s0006-3223(00)00976-8. [DOI] [PubMed] [Google Scholar]
- Kehoe LA, Bernardinelli Y, Muller D. GluN3A: an NMDA receptor subunit with exquisite properties and functions. Neural Plast. 2013;2013:145387. doi: 10.1155/2013/145387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempton MJ, Stahl D, Williams SC, DeLisi LE. Progressive lateral ventricular enlargement in schizophrenia: a meta-analysis of longitudinal MRI studies. Schizophr Res. 2010;120(1–3):54–62. doi: 10.1016/j.schres.2010.03.036. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Giedd J, Lau JY, Lewis DA, Paus T. Changes in the adolescent brain and the pathophysiology of psychotic disorders. Lancet Psychiatry. 2014;1(7):549–558. doi: 10.1016/S2215-0366(14)00081-9. [DOI] [PubMed] [Google Scholar]
- Kessler M, Terramani T, Lynch G, Baudry M. A glycine site associated with N-methyl-D-aspartic acid receptors: characterization and identification of a new class of antagonists. J Neurochem. 1989;52(4):1319–1328. doi: 10.1111/j.1471-4159.1989.tb01881.x. [DOI] [PubMed] [Google Scholar]
- Kia HK, Miquel MC, Brisorgueil MJ, Daval G, Riad M, El Mestikawy S, … Verge D. Immunocytochemical localization of serotonin1A receptors in the rat central nervous system. J Comp Neurol. 1996;365(2):289–305. doi: 10.1002/(SICI)1096-9861(19960205)365:2<289::AID-CNE7>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Kinon BJ, Zhang L, Millen BA, Osuntokun OO, Williams JE, Kollack-Walker S, … Group HS. A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J Clin Psychopharmacol. 2011;31(3):349–355. doi: 10.1097/JCP.0b013e318218dcd5. [DOI] [PubMed] [Google Scholar]
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, … Owen MJ. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17(2):142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science. 1988;241(4867):835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- Konopaske GT, Lange N, Coyle JT, Benes FM. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry. 2014;71(12):1323–1331. doi: 10.1001/jamapsychiatry.2014.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak R, Campbell BM, Strick CA, Horner W, Hoffmann WE, Kiss T, … Castner SA. Reduction of brain kynurenic Acid improves cognitive function. J Neurosci. 2014;34(32):10592–10602. doi: 10.1523/JNEUROSCI.1107-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, … Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Labrie V, Wang W, Barger SW, Baker GB, Roder JC. Genetic loss of D-amino acid oxidase activity reverses schizophrenia-like phenotypes in mice. Genes Brain Behav. 2009 doi: 10.1111/j.1601-183X.2009.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology. 2001;25(4):455–467. doi: 10.1016/S0893-133X(01)00243-3. [DOI] [PubMed] [Google Scholar]
- Lane HY, Lin CH, Green MF, Hellemann G, Huang CC, Chen PW, … Tsai GE. Add-on treatment of benzoate for schizophrenia: a randomized, double-blind, placebo-controlled trial of D-amino acid oxidase inhibitor. JAMA Psychiatry. 2013;70(12):1267–1275. doi: 10.1001/jamapsychiatry.2013.2159. [DOI] [PubMed] [Google Scholar]
- Law AJ, Deakin JF. Asymmetrical reductions of hippocampal NMDAR1 glutamate receptor mRNA in the psychoses. Neuroreport. 2001;12(13):2971–2974. doi: 10.1097/00001756-200109170-00043. [DOI] [PubMed] [Google Scholar]
- Lesh TA, Niendam TA, Minzenberg MJ, Carter CS. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36(1):316–338. doi: 10.1038/npp.2010.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt JJ, Bobrow L, Lucia D, Srinivasan P. A selective review of volumetric and morphometric imaging in schizophrenia. Curr Top Behav Neurosci. 2010;4:243–281. doi: 10.1007/7854_2010_53. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28(2):325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- Leza JC, Garcia-Bueno B, Bioque M, Arango C, Parellada M, Do K, … Bernardo M. Inflammation in schizophrenia: A question of balance. Neurosci Biobehav Rev. 2015;55:612–626. doi: 10.1016/j.neubiorev.2015.05.014. [DOI] [PubMed] [Google Scholar]
- Li Y, Sacchi S, Pollegioni L, Basu AC, Coyle JT, Bolshakov VY. Identity of endogenous NMDAR glycine site agonist in amygdala is determined by synaptic activity level. Nat Commun. 2013;4:1760. doi: 10.1038/ncomms2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO … Clinical Antipsychotic Trials of Intervention Effectiveness, I. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31(5):234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo-Huitron R, Blanco-Ayala T, Ugalde-Muniz P, Carrillo-Mora P, Pedraza-Chaverri J, Silva-Adaya D, … La Cruz VP. On the antioxidant properties of kynurenic acid: free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol Teratol. 2011;33(5):538–547. doi: 10.1016/j.ntt.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 1998;95(19):11465–11470. doi: 10.1073/pnas.95.19.11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in schizophrenia: a focused review and meta-analysis of (1)H-MRS studies. Schizophr Bull. 2013;39(1):120–129. doi: 10.1093/schbul/sbr069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau M, Shi T, Puyal J, Knolhoff AM, Dulong J, Gasnier B, … Mothet JP. Storage and uptake of D-serine into astrocytic synaptic-like vesicles specify gliotransmission. J Neurosci. 2013;33(8):3413–3423. doi: 10.1523/JNEUROSCI.3497-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer HY. Clinical studies on the mechanism of action of clozapine: the dopamine-serotonin hypothesis of schizophrenia. Psychopharmacology (Berl) 1989;99(Suppl):S18–27. doi: 10.1007/BF00442554. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Rajagopal L, Huang M, Oyamada Y, Kwon S, Horiguchi M. Translating the N-methyl-D-aspartate receptor antagonist model of schizophrenia to treatments for cognitive impairment in schizophrenia. Int J Neuropsychopharmacol. 2013;16(10):2181–2194. doi: 10.1017/S1461145713000928. [DOI] [PubMed] [Google Scholar]
- Mighdoll MI, Tao R, Kleinman JE, Hyde TM. Myelin, myelin-related disorders, and psychosis. Schizophr Res. 2015;161(1):85–93. doi: 10.1016/j.schres.2014.09.040. [DOI] [PubMed] [Google Scholar]
- Miya K, Inoue R, Takata Y, Abe M, Natsume R, Sakimura K, … Mori H. Serine racemase is predominantly localized in neurons in mouse brain. J Comp Neurol. 2008;510(6):641–654. doi: 10.1002/cne.21822. [DOI] [PubMed] [Google Scholar]
- Morita Y, Ujike H, Tanaka Y, Otani K, Kishimoto M, Morio A, … Kuroda S. A genetic variant of the serine racemase gene is associated with schizophrenia. Biol Psychiatry. 2007;61(10):1200–1203. doi: 10.1016/j.biopsych.2006.07.025. [DOI] [PubMed] [Google Scholar]
- Mothet JP, Pollegioni L, Ouanounou G, Martineau M, Fossier P, Baux G. Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter D-serine. Proc Natl Acad Sci U S A. 2005;102(15):5606–5611. doi: 10.1073/pnas.0408483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network & Pathway Analysis Subgroup of Psychiatric Genomics C. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015;18(2):199–209. doi: 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14(6):383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladepeche L, Ruel J, Sacchi S, Labasque M, Hanini M, … Oliet SH. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150(3):633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- Paul SM, Doherty JJ, Robichaud AJ, Belfort GM, Chow BY, Hammond RS, … Zorumski CF. The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J Neurosci. 2013;33(44):17290–17300. doi: 10.1523/JNEUROSCI.2619-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman DM, Najjar S. Meta-analysis of the association between N-methyl-d-aspartate receptor antibodies and schizophrenia, schizoaffective disorder, bipolar disorder, and major depressive disorder. Schizophr Res. 2014;157(1–3):249–258. doi: 10.1016/j.schres.2014.05.001. [DOI] [PubMed] [Google Scholar]
- Perala J, Suvisaari J, Saarni SI, Kuoppasalmi K, Isometsa E, Pirkola S, … Lonnqvist J. Lifetime prevalence of psychotic and bipolar I disorders in a general population. Arch Gen Psychiatry. 2007;64(1):19–28. doi: 10.1001/archpsyc.64.1.19. [DOI] [PubMed] [Google Scholar]
- Pershing ML, Bortz DM, Pocivavsek A, Fredericks PJ, Jorgensen CV, Vunck SA, … Bruno JP. Elevated levels of kynurenic acid during gestation produce neurochemical, morphological, and cognitive deficits in adulthood: implications for schizophrenia. Neuropharmacology. 2015;90:33–41. doi: 10.1016/j.neuropharm.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prickaerts J, van Goethem NP, Chesworth R, Shapiro G, Boess FG, Methfessel C, … Konig G. EVP-6124, a novel and selective alpha7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of alpha7 nicotinic acetylcholine receptors. Neuropharmacology. 2012;62(2):1099–1110. doi: 10.1016/j.neuropharm.2011.10.024. [DOI] [PubMed] [Google Scholar]
- Priestley T, Laughton P, Myers J, Le Bourdelles B, Kerby J, Whiting PJ. Pharmacological properties of recombinant human N-methyl-D-aspartate receptors comprising NR1a/NR2A and NR1a/NR2B subunit assemblies expressed in permanently transfected mouse fibroblast cells. Mol Pharmacol. 1995;48(5):841–848. [PubMed] [Google Scholar]
- Puhl MD, Mintzopoulos D, Jensen JE, Gillis TE, Konopaske GT, Kaufman MJ, Coyle JT. In vivo magnetic resonance studies reveal neuroanatomical and neurochemical abnormalities in the serine racemase knockout mouse model of schizophrenia. Neurobiol Dis. 2015;73:269–274. doi: 10.1016/j.nbd.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, … Sklar P. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506(7487):185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartermain D, Mower J, Rafferty MF, Herting RL, Lanthorn TH. Acute but not chronic activation of the NMDA-coupled glycine receptor with D-cycloserine facilitates learning and retention. Eur J Pharmacol. 1994;257(1–2):7–12. doi: 10.1016/0014-2999(94)90687-4. [DOI] [PubMed] [Google Scholar]
- Radant AD, Bowdle TA, Cowley DS, Kharasch ED, Roy-Byrne PP. Does ketamine-mediated N-methyl-D-aspartate receptor antagonism cause schizophrenia-like oculomotor abnormalities? Neuropsychopharmacology. 1998;19(5):434–444. doi: 10.1016/S0893-133X(98)00030-X. [DOI] [PubMed] [Google Scholar]
- Rapoport JL, Giedd JN, Gogtay N. Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatry. 2012;17(12):1228–1238. doi: 10.1038/mp.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas C, Alt J, Ator NA, Thomas AG, Wu Y, Hin N, … Slusher BS. D-Amino Acid Oxidase Inhibition Increases D-Serine Plasma Levels in Mouse But not Monkey or Dog. Neuropsychopharmacology. 2015 doi: 10.1038/npp.2015.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook JM, Xiang Z, Lv X, Ghoshal A, Dickerson JW, Bridges TM, … Conn PJ. Biased mGlu5-Positive Allosteric Modulators Provide In Vivo Efficacy without Potentiating mGlu5 Modulation of NMDAR Currents. Neuron. 2015;86(4):1029–1040. doi: 10.1016/j.neuron.2015.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos P, Mitchell AC, Voloudakis G, Fullard JF, Pothula VM, Tsang J, … Sklar P. A role for noncoding variation in schizophrenia. Cell Rep. 2014;9(4):1417–1429. doi: 10.1016/j.celrep.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell MJ, Molliver ME, Snyder SH. D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci U S A. 1995;92(9):3948–3952. doi: 10.1073/pnas.92.9.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. 2012;13(7):465–477. doi: 10.1038/nrn3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Rassoulpour A, Wu HQ, Medoff D, Tamminga CA, Roberts RC. Increased cortical kynurenate content in schizophrenia. Biol Psychiatry. 2001;50(7):521–530. doi: 10.1016/s0006-3223(01)01078-2. [DOI] [PubMed] [Google Scholar]
- Selvaraj S, Arnone D, Cappai A, Howes O. Alterations in the serotonin system in schizophrenia: a systematic review and meta-analysis of postmortem and molecular imaging studies. Neurosci Biobehav Rev. 2014;45:233–245. doi: 10.1016/j.neubiorev.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, … Felder CC. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165(8):1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Sikich L, Frazier JA, McClellan J, Findling RL, Vitiello B, Ritz L, … Lieberman JA. Double-blind comparison of first- and second-generation antipsychotics in early-onset schizophrenia and schizo-affective disorder: findings from the treatment of early-onset schizophrenia spectrum disorders (TEOSS) study. Am J Psychiatry. 2008;165(11):1420–1431. doi: 10.1176/appi.ajp.2008.08050756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan AD, Ghose S, Zhao C, Hulsey K, Mihalakos P, Yanagi M, … Tamminga CA. Magnetic resonance spectroscopy and tissue protein concentrations together suggest lower glutamate signaling in dentate gyrus in schizophrenia. Mol Psychiatry. 2015;20(4):433–439. doi: 10.1038/mp.2014.54. [DOI] [PubMed] [Google Scholar]
- Steiner J, Teegen B, Schiltz K, Bernstein HG, Stoecker W, Bogerts B. Prevalence of N-methyl-D-aspartate receptor autoantibodies in the peripheral blood: healthy control samples revisited. JAMA Psychiatry. 2014;71(7):838–839. doi: 10.1001/jamapsychiatry.2014.469. [DOI] [PubMed] [Google Scholar]
- Steiner J, Walter M, Glanz W, Sarnyai Z, Bernstein HG, Vielhaber S, … Stoecker W. Increased prevalence of diverse N-methyl-D-aspartate glutamate receptor antibodies in patients with an initial diagnosis of schizophrenia: specific relevance of IgG NR1a antibodies for distinction from N-methyl-D-aspartate glutamate receptor encephalitis. JAMA Psychiatry. 2013;70(3):271–278. doi: 10.1001/2013.jamapsychiatry.86. [DOI] [PubMed] [Google Scholar]
- Tamminga CA, Stan AD, Wagner AD. The hippocampal formation in schizophrenia. Am J Psychiatry. 2010;167(10):1178–1193. doi: 10.1176/appi.ajp.2010.09081187. [DOI] [PubMed] [Google Scholar]
- Tatard-Leitman VM, Jutzeler CR, Suh J, Saunders JA, Billingslea EN, Morita S, … Siegel SJ. Pyramidal cell selective ablation of N-methyl-D-aspartate receptor 1 causes increase in cellular and network excitability. Biol Psychiatry. 2015;77(6):556–568. doi: 10.1016/j.biopsych.2014.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522–537. doi: 10.2174/138161210790361452. [DOI] [PubMed] [Google Scholar]
- Umbricht D, Schmid L, Koller R, Vollenweider FX, Hell D, Javitt DC. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry. 2000;57(12):1139–1147. doi: 10.1001/archpsyc.57.12.1139. [DOI] [PubMed] [Google Scholar]
- Verrall L, Burnet PW, Betts JF, Harrison PJ. The neurobiology of D-amino acid oxidase and its involvement in schizophrenia. Mol Psychiatry. 2010;15(2):122–137. doi: 10.1038/mp.2009.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AG, Wenthur CJ, Xiang Z, Rook JM, Emmitte KA, Niswender CM, … Conn PJ. Metabotropic glutamate receptor 3 activation is required for long-term depression in medial prefrontal cortex and fear extinction. Proc Natl Acad Sci U S A. 2015;112(4):1196–1201. doi: 10.1073/pnas.1416196112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, … Weickert TW. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol Psychiatry. 2013;18(11):1185–1192. doi: 10.1038/mp.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nature reviews Drug discovery. 2007;6(9):721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- Wolosker H. Serine racemase and the serine shuttle between neurons and astrocytes. Biochim Biophys Acta. 2011;1814(11):1558–1566. doi: 10.1016/j.bbapap.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Wolosker H, Blackshaw S, Snyder SH. Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(23):13409–13414. doi: 10.1073/pnas.96.23.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolosker H, Dumin E, Balan L, Foltyn VN. D-amino acids in the brain: D-serine in neurotransmission and neurodegeneration. FEBS J. 2008;275(14):3514–3526. doi: 10.1111/j.1742-4658.2008.06515.x. [DOI] [PubMed] [Google Scholar]
- Wolosker H, Mori H. Serine racemase: an unconventional enzyme for an unconventional transmitter. Amino Acids. 2012;43(5):1895–1904. doi: 10.1007/s00726-012-1370-3. [DOI] [PubMed] [Google Scholar]
- Yang JH, Wada A, Yoshida K, Miyoshi Y, Sayano T, Esaki K, … Furuya S. Brain-specific Phgdh deletion reveals a pivotal role for L-serine biosynthesis in controlling the level of D-serine, an N-methyl-D-aspartate receptor co-agonist, in adult brain. The Journal of biological chemistry. 2010;285(53):41380–41390. doi: 10.1074/jbc.M110.187443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, Yang F, Xiao Y, Tan S, Husain N, Ren M, … Je HS. Regulation of Brain-Derived Neurotrophic Factor Exocytosis and Gamma-Aminobutyric Acidergic Interneuron Synapse by the Schizophrenia Susceptibility Gene Dysbindin-1. Biol Psychiatry. 2015 doi: 10.1016/j.biopsych.2015.08.019. [DOI] [PubMed] [Google Scholar]
- Yuen EY, Jiang Q, Chen P, Feng J, Yan Z. Activation of 5-HT2A/C receptors counteracts 5-HT1A regulation of n-methyl-D-aspartate receptor channels in pyramidal neurons of prefrontal cortex. J Biol Chem. 2008;283(25):17194–17204. doi: 10.1074/jbc.M801713200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Jiang Q, Chen P, Gu Z, Feng J, Yan Z. Serotonin 5-HT1A receptors regulate NMDA receptor channels through a microtubule-dependent mechanism. J Neurosci. 2005;25(23):5488–5501. doi: 10.1523/JNEUROSCI.1187-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Gomeza J, Olivares L, Aragon C, Gimenez C. Regional distribution and developmental variation of the glycine transporters GLYT1 and GLYT2 in the rat CNS. Eur J Neurosci. 1995;7(6):1342–1352. doi: 10.1111/j.1460-9568.1995.tb01125.x. [DOI] [PubMed] [Google Scholar]