

Abstract
While selective estrogen receptor modulators, such as tamoxifen, have contributed to increased survival in patients with hormone receptor-positive breast cancer, the development of resistance to these therapies has led to the need to investigate other targetable pathways involved in oncogenic signaling. Approval of the mTOR inhibitor everolimus in the therapy of secondary endocrine resistance demonstrates the validity of this approach. Importantly, mTOR activation regulates eukaryotic messenger RNA translation. Eukaryotic translation initiation factor 4E (eIF4E), a component of the cap-dependent translation complex eIF4F, confers resistance to drug-induced apoptosis when overexpressed in multiple cell types. The eIF4F complex is downstream of multiple oncogenic pathways, including mTOR, making it an appealing drug target. Here, we show that the eIF4F translation pathway was hyperactive in tamoxifen-resistant (TamR) MCF-7L breast cancer cells. While overexpression of eIF4E was not sufficient to confer resistance to tamoxifen in MCF-7L cells, its function was necessary to maintain resistance in TamR cells. Targeting the eIF4E subunit of the eIF4F complex through its degradation using an antisense oligonucleotide (ASO) or via sequestration using a mutant 4E-BP1 inhibited the proliferation and colony formation of TamR cells and partially restored sensitivity to tamoxifen. Further, the use of these agents also resulted in cell cycle arrest and induction of apoptosis in TamR cells. Finally, the use of a pharmacologic agent which inhibited the eIF4E-eIF4G interaction also decreased the proliferation and anchorage-dependent colony formation in TamR cells. These results highlight the eIF4F complex as a promising target for patients with acquired resistance to tamoxifen and, potentially, other endocrine therapies.
Electronic supplementary material
The online version of this article (doi:10.1007/s12672-017-0296-3) contains supplementary material, which is available to authorized users.
Keywords: Endocrine-resistant Breast Cancer, Tamoxifen Resistance, TamR Cells, Anchorage-dependent Colony Formation, eIF4F Complex
Introduction
The therapeutic targeting of the estrogen receptor has significantly contributed to the decline in breast cancer deaths in recent years. Tamoxifen, a selective estrogen receptor modulator (SERM), has been shown to increase the 15-year survival rate of hormone receptor-positive breast cancer patients when used as an adjuvant systemic therapy [1]. Recently, the benefit of continuing tamoxifen for 10 years after definitive resection of estrogen receptor (ER)-positive breast cancer has been reported [2], demonstrating that breast cancer cells can remain responsive to ER inhibition for extended periods of time. However, not all ER-positive breast cancers respond to SERMs and the overall benefit of tamoxifen is hindered by both primary (de novo) and secondary (acquired) resistance to the drug, occurring in nearly half of all patients treated with tamoxifen. The development of resistance to established therapies has led researchers to investigate alternative signaling pathways to target. Most solid tumors have multiple signaling pathways altered, making single-agent targeted therapies ineffective. Targeting common downstream signaling nodes or hubs, however, would, in theory, be effective, provided that hub is active in a given cancer.
One common signaling hub found to be upregulated in several solid tumors is the cap-dependent translation pathway. Translation consists of four steps: initiation, elongation, termination, and recycling of ribosomes for continued use. Regulation of translation is controlled throughout the process; however, it is most tightly regulated in the initiation step. Initiation begins with the 43S ribosome subunit associating with the eIF4F translational complex and scanning the messenger RNA (mRNA) in search of the start codon [3]. The eIF4F translation-initiation complex consists of an RNA helicase (eIF4A), a scaffolding protein (eIF4G), and the cap-binding protein eukaryotic translation initiation factor 4E (eIF4E), which is the rate-limiting component of the complex. Mitogenic stimulation positively influences cap-dependent translation through intracellular signaling pathways. Convergence of these pathways occurs through activation of ribosomal S6 kinase and mTORC1, leading to phosphorylation of the translation-repressing 4E-binding proteins (4E-BPs). The primary source of regulation of this pathway occurs through the PI3K/Akt signaling pathway, ultimately relieving translational repression (through release of 4E-BP1 from eIF4E) and via Ras phosphorylation and activation of eIF4E [4]. Recently, the mTOR inhibitor everolimus in combination with tamoxifen has been shown to have a clinical benefit in advanced breast cancer [5] with the suggestion that patients with secondary endocrine resistance received the most benefit.
Because the eIF4F scaffold is downstream of multiple oncogenic pathways, it is not surprising that the cap-dependent translation pathway is often deregulated in human malignancy. Overexpression of eIF4E has been shown to transform mouse cells [6], induce tumor formation in a genetic mouse model with constitutive germ line expression of eIF4E [7], and lead to an aberrant self-renewal of mammary stem/progenitor cells resulting in preneoplastic mammary gland lesions in a mouse model [8]. Conversely, inhibiting cap-dependent translation in cancer cells with hyperactivation of the pathway using either pharmacologic [9] or genetic [10] manipulation leads to a decrease in xenograft tumor growth. Methods that increase eIF4E phosphorylation result in enhanced nuclear export of mRNAs and can contribute to cell transformation. Evidence from multiple experiments suggests that malignancies driven by different oncogenic pathways converge on and are dependent on hyperactivation of the eIF4F translational machinery.
Cap-dependent translation may be inhibited indirectly via targeting upstream signaling pathways or directly by targeting the eIF4F complex. Indirect targeting may be accomplished through inhibiting pathways that phosphorylate 4E-BPs, such as the PI3K/Akt/mTOR axis or by inhibiting the phosphorylation of eIF4E via the Ras/MAPK/ERK pathway. One disadvantage to indirect targeting is interruption of feedback loops in the case of the PI3K/Akt/mTOR pathway [11]. Direct inhibition of the eIF4F complex may be accomplished through disrupting the formation of the eIF4F complex or inhibiting the binding of eIF4E to the mRNA cap. Several pharmacologic compounds have been developed, such as 4EGI-1 [12] and 4E1RCat [13], which inhibit cap-dependent translation by preventing the eIF4E-eIF4G interaction. Multiple strategies designed to antagonize the eIF4E-cap interaction have also been developed. Decreasing eIF4E expression through the use of an antisense oligonucleotide (ASO) [14] and inhibiting cap binding through the use of chemical cap analogues, such as 4Ei-1 [15] have both demonstrated the ability to inhibit cap-dependent translation. The development of these agents has led to their exploration in multiple cancers, especially those with hyperactivation of the eIF4F pathway.
In this study, we sought to determine the role of the eIF4F translation pathway in an in vitro model of tamoxifen resistance. We found tamoxifen-resistant (TamR) cells had hyperactivation of the eIF4F pathway and enhanced cap-dependent translation. Further, inhibition of the pathway through either genetic or pharmacologic methods significantly inhibited the growth of TamR cells and partially restored their sensitivity to tamoxifen. Our findings suggest inhibition of the eIF4F pathway may be effective in endocrine-resistant breast cancer.
Materials and Methods
Cell Lines and Culture
Cells were grown in a humidified atmosphere containing 5% CO2 at 37 °C and supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin. MCF-7L cells were obtained from C. Kent Osborne (Baylor College of Medicine) and cultured in improved MEM Richter’s modification medium (zinc option) containing 11.25 nmol/L insulin and 5% FBS. TamR cells were generated and cultured as previously described in media containing 100 nM 4-hydroxytamoxifen (Sigma) [16]. T47D cells were obtained from ATCC and cultured in a MEM medium (with Earle’s salts and l-glutamine) containing 6 ng/L insulin, 100× non-essential amino acids, and 5% FBS. All cell lines are examined annually for mycoplasma infection and authentication by short tandem repeat analysis.
Plasmids, Retroviral Infection, and Transfection
Retroviral infection protocol and generation of the MSCV-M1GR1 constructs containing a green fluorescent protein (GFP) reporter and expressing 3HA-tagged 4E-BP1A37/A46 and eIF4E have been previously described [10]. The bicistronic reporter system used (pcDNA-rLuc-polIRES-fLuc) was generated as previously described [17]. GFP-positive cells were isolated using a Becton Dickinson fluorescence-activated cell sorting (FACS) DiVa (BD) with a 530/528 optical filter. For transfection, 2 × 105 cells/well were plated on a six-well plate in media without antibiotic. The next day, cells were transfected with 50 or 100 nM ASO using Oligofectamine (Invitrogen) according to the manufacturer’s instructions. Cells were incubated with Oligofectamine in Opti-MEM media (Invitrogen) for 6 h. Media were then removed and replaced with normal growth media without antibiotics. RNA for polyribosome preparations was collected the next day.
Immunoblot and Cap-Binding Analysis
Experimental procedures used have been previously described [17]. Lysates were clarified, and then protein concentration was determined using the bicinchoninic acid protein assay reagent kit (Pierce). Even amounts of protein (40 or 50 μg) were resolved on an SDS-PAGE gel and transferred to a nitrocellulose membrane. Blots were probed with the following primary antibodies: anti-actin (Sigma), anti-eIF4E (Transduction Laboratories), anti-eIF4E (Cell Signaling), and anti-eIF4G from Dr. Sonenberg (McGill University). ImageJ software was utilized to determine the relative density of protein bands.
Cell Proliferation Assay
Cells were cultured in media containing dextran-coated charcoal (DCC) FBS for 24 h before plating. Cells were plated in a six-well plate at a density of 1 × 104 cells/well and allowed to equilibrate overnight. Full DCC FBS-containing media were replaced with phenol red-free IMEM supplemented with 5% DCC FBS and tamoxifen added at the concentrations indicated in the figures. Cell were cultured for 5 or 7 days and then trypsinized and counted on a Coulter Counter (Beckman Coulter) or a hemocytometer.
Anchorage-Dependent Colony Formation
Cells were plated at a density of 1000 cells/well in triplicate in a six-well plate. Cultures were continued for 10 days and then fixed with 4% buffered formalin and stained with Coomassie blue, and pictures were taken. Colonies >3 pixels were quantified using GeneTools image analysis software (Syngene).
RNA, Polyribosome Preparations, and RNASeq Analysis
Total RNA was collected using TRI Reagent (Sigma) according to the manufacturer’s directions. Polyribosome-stratified RNA was fractionated and prepared as previously described [18]. Ten fractions were collected into tubes containing 10% SDS. RNA from these fractions was purified using TRI Reagent (Invitrogen). We obtained raw sequence reads from four samples of polyribosome-stratified RNA (two from parental tamoxifen-sensitive cell line and two from TamR cell line) and four corresponding raw sequence reads from total RNA. The raw sequence (fastq) was mapped to the human genome (NCBI build 36) using bowtie2 in TopHat (version 2.0.1) [19, 20] to generate BAM files. Then, the read counts for each transcript were calculated using the BAM files through bedtools (version 2.17.0) [21]. A negative binomial model has been considered a suitable model to analyze read count data generated from RNASeq experiments in the literature [22]. In addition, we use the total RNA read counts as offset in the negative binomial regression model to adjust for variation in RNA abundance across samples as implemented by Larsson et al. [23]. The empirical Bayesian method was applied to calculate tag-wise dispersions towards [24]. Differentially expressed genes between TamR and parental MCF-7L cells with p values <0.05 were considered in gene set enrichment analysis using the curated KEGG database.
Cell Cycle and Apoptosis Analysis
Cells were treated with either 100 nM tamoxifen or vehicle for 24 h and then fixed with ethanol and stained with propidium iodide, and DNA content was analyzed on a FACSCalibur flow cytometer (BD) as previously described [18].
Statistical Analysis
Statistical analysis was performed using either a one-way ANOVA with Tukey’s post hoc test or an unpaired t test. A p value <0.05 was considered significant unless otherwise noted.
Results
Acquired Tamoxifen Resistance in MCF-7L Breast Cancer Cells Requires Hyperactivation of eIF4F-Mediated Translation
Prior to examining the influence of tamoxifen resistance on translation, TamR MCF-7L cells were generated and characterized [16]. We next compared the protein expression levels of translational machinery components in the parental and TamR cells in both the presence and absence of tamoxifen (Fig. 1a). TamR cells displayed an enrichment of the hyperphosphorylated (inactive) isoform of 4E-BP1 (isoform γ), an alteration favoring eIF4F assembly, and increased cap-dependent translation through the release of eIF4E from sequestration by 4E-BP1. Cell lysates derived from tamoxifen-resistant MCF-7 and T47D cells were also subjected to cap analogue captured in order to examine the integrity of the eIF4F complex. eIF4E bound to the cap analogue is associated with its binding partners. Immunoblot analysis of the pull down shows that, while the integrity of the cap-dependent initiation complex decreased with tamoxifen treatment in tamoxifen-sensitive cells, the integrity of eIF4F increased with tamoxifen treatment in TamR cells, reflecting an increase in translational potential in both tamoxifen-resistant cell lines (Fig. 1b, c).
Fig. 1.

Analysis of the eIF4F translational complex in tamoxifen-sensitive and tamoxifen-resistant (TamR) MCF-7L breast carcinoma cells. a Cells were treated for 24 h with 100 nM tamoxifen or vehicle and then lysed using three freeze-thaw cycles. Equal amounts of protein were separated on an SDS-PAGE gel and immunoblotted with specific antibodies for the translational factors eIF4GI and eIF4E and the translational inhibitors 4E-BP2 and 4E-BP1. b MCF-7L (left) and T47D (right) cell lysates treated with either 100 nM tamoxifen or vehicle were incubated with 7-Me GTP Sepharose resin to capture the cap-binding protein eIF4E and its partners. The cap-bound material was eluted, subjected to SDS-PAGE, and immunoblotted. c Changes in integrity of the eIF4F complex in response to tamoxifen were quantified as differences between vehicle and tamoxifen treatments in eIF4GI/4E-BP1,2 relative density (R.D.) of the blots shown in b. d Plates of actively proliferating cells were treated for 24 h with 100 nM tamoxifen and then with cycloheximide (100 μg/mL) for 5 min followed by trypsinization and lysis. Cytoplasmic extract purified from the lysate was layered onto a sucrose gradient and then fractionated into ten, 1 mL fractions. Fractionated RNA was normalized to total RNA. e Cells transfected with a dual luciferase reporter construct (pcDNA3-rLuc-polIRES-fLuc) overnight. Transfection media were replaced with media containing 5% FBS and grown for another 24 h. Cells were then lysed, and levels of luciferase were read using a Dual-Luciferase Reporter Assay System (Promega)
Since MCF-7 cells have served as a model for endocrine-sensitive breast cancer [25], we focused our work on this cell line. We examined the effect of tamoxifen on translation by isolating ribosome-bound mRNA. MCF-7L and TamR cells were treated with tamoxifen for 24 h, and then total RNA was stratified into ten fractions by sucrose gradient centrifugation. Translationally active transcripts, with more bound ribosomes, migrate more rapidly through the gradient and are found in the heavy fractions (fractions 7–10). With tamoxifen treatment, a clear difference can be seen between the tamoxifen-sensitive MCF-7L cells and the TamR cells, with TamR cells having a larger percentage of mRNA in the heavy fractions (Fig. 1d). Additionally, we examined the relative levels of both cap-dependent and cap-independent translations in MCF-7L and TamR cells using a dual-luciferase bicistronic reporter system (Fig. 1e). While there was no significant difference in the cap-independent translation between the two cell lines, TamR cells have a 1.8-fold increase in cap-dependent translation compared to wild-type MCF-7L cells.
Genome-Wide Analysis of Polysome Preparations Revealed Enrichment of Translation-Related Pathways in TamR Cells
To examine the genome-wide effect of enhanced cap-dependent translation in TamR cells, we performed the RNASeq analysis on polyribosome preparations isolated from parental MCF-7L and TamR cells. mRNA was isolated and separated based on the number of bound ribosomes. For each cell line, both polyribosome-bound RNA (bound to greater than or equal to four ribosomes) and total cellular RNA were isolated and submitted for RNASeq analysis. Heavy ribosomal read counts (those RNAs bound to greater than or equal to four ribosomes) were compared between TamR and parental MCF-7L cells while adjusting total mRNA read counts as offset. To detect basal gene changes in the presence of tamoxifen between the cell lines, cells were starved for 24 h prior to being treated with tamoxifen (in the presence of charcoal-stripped serum) for 24 additional hours. We identify 21 transcripts with a false discovery rate of less than 0.05. A heatmap of the ratio of heavy ribosomal read counts and total mRNA read counts is shown in supplemental Figure 1. A list of differentially expressed genes with p < 0.05 is provided in supplemental Table 1. Among the top 2000 differentially regulated genes listed, 182 genes were reported to be upregulated by Lin et al. [26]. Among the top 2000 downregulated genes, 199 were reported to be downregulated by Lin et al. This common set of upregulated and downregulated genes is shown in supplemental Tables 2 and 3.
Using a p value <0.05, we performed gene set enrichment analysis using the curated KEGG pathway database. The KEGG-enriched pathways for TamR versus parental MCF-7L cells are shown in Table 1. Several translation-related pathways are highly significant, including ribosome (KEGGID: 03010), ribosome biogenesis in eukaryotes (KEGGID: 03008), and RNA polymerase (KEGGID: 03020). In addition, the phosphatidylinositol signaling pathway (KEGGID: 04070), which lies upstream of the mTOR pathway and leads to the activation of cap-dependent translation, was also found to be enriched. Thus, tamoxifen resistance is associated with increased translation of pathways associated with cap-dependent translation.
Table 1.
Enriched KEGG pathway analysis in TamR versus parental MCF-7L cells. Using a p value <0.05, gene set enrichment analysis was performed using the curated KEGG database
| KEGG ID | p value | Odds ratio | Exp count | Pathway |
|---|---|---|---|---|
| 03010 | 0.00 | 4.24 | 28.20 | Ribosome |
| 00230 | 0.00 | 2.29 | 41.98 | Purine metabolism |
| 04120 | 0.00 | 2.18 | 42.30 | Ubiquitin-mediated proteolysis |
| 00240 | 0.00 | 2.37 | 28.84 | Pyrimidine metabolism |
| 05016 | 0.00 | 1.79 | 50.32 | Huntington’s disease |
| 04114 | 0.00 | 1.95 | 31.09 | Oocyte meiosis |
| 04070 | 0.00 | 2.15 | 21.15 | Phosphatidylinositol signaling |
| 03020 | 0.00 | 3.03 | 9.29 | RNA polymerase |
| 05010 | 0.00 | 1.60 | 45.19 | Alzheimer’s disease |
| 04130 | 0.01 | 2.42 | 10.26 | SNARE interactions in vesicular transport |
| 05012 | 0.02 | 1.52 | 33.97 | Parkinson’s disease |
| 04260 | 0.02 | 1.94 | 13.46 | Cardiac muscle contraction |
| 01040 | 0.03 | 2.61 | 6.41 | Biosynthesis of unsaturated fatty acids |
| 04710 | 0.03 | 2.61 | 6.41 | Circadian rhythm in mammals |
| 03420 | 0.03 | 1.86 | 13.78 | Nucleotide excision repair |
| 00190 | 0.04 | 1.45 | 34.93 | Oxidative phosphorylation |
| 03008 | 0.04 | 1.56 | 22.75 | Ribosome biogenesis in eukaryotes |
Selected KEGG-enriched pathways in TamR versus parental MCF-7L cells with p < 0.05 are shown
eIF4E Overexpression Was Not Sufficient to Confer Resistance to Tamoxifen, but Its Function Was Necessary to Maintain Resistance in MCF-7L Cells
As we showed that tamoxifen resistance is associated with an increase in levels of hyperphosphorylated 4E-BP1, leading to the release of eIF4E from sequestration, we investigated if overexpression of eIF4E would confer partial or complete resistance of MCF-7L cells to tamoxifen treatment. MCF-7L cells were transduced with lentiviral particles containing either a control plasmid (vector) or a plasmid containing HA-tagged eIF4E (HA-eIF4E). After cells were GFP sorted, the level of eIF4E expression was determined via immunoblotting (Fig. 2a) and the relative level of cap-dependent translation was characterized (data not shown). While the relative level of cap-dependent translation increased in HA-eIF4E cells, there was no significant effect seen on monolayer growth in an MTT assay (Fig. 2b) or anchorage-dependent colony formation in a clonogenic efficiency assay (Fig. 2c, d) compared to vector control cells when treated with tamoxifen. Thus, overexpression of eIF4E alone does not confer resistance to tamoxifen treatment in proliferation or anchorage-dependent colony formation.
Fig. 2.

Effects of modulation of eIF4E on tamoxifen sensitivity in MCF-7L and TamR cells. a Western blot analysis demonstrating the lentivirus-mediated overexpression of HA-tagged eIF4E in tamoxifen-sensitive MCF-7L cells. Cells were transduced with a lentiviral eIF4E/GFP expression vector and flow sorted by GFP expression. b Cell growth assay to evaluate the responsiveness of MCF-7L tamoxifen-sensitive cells and eIF4E-overexpressing cells to tamoxifen. Cells were serum starved overnight before plating. Cells were grown in 5% dextran-coated charcoal (DCC)-treated FBS + treatment for 7 days and then trypsinized, stained with trypan blue, and counted. No significant difference was seen when each treatment was compared across cell lines by an unpaired t test. c Clonogenic efficiency assay examining the effect of eIF4E overexpression in response to tamoxifen in MCF-7 cells. Cells were plated and treated in full media. Colonies were allowed to grow for 10 days and then fixed and stained. Percent area was determined using GeneTools (Syngene) software and graphed in d. e Western blot showing the expression level of lentivirus-delivered HA-4E-BP1A37/A46 in TamR cells. Cells were harvested from full media and then lysed using three freeze-thaw cycles. Equal amounts of protein were resolved on an SDS-PAGE gel and immunoblotted to determine the expression of HA-4E-BP1A37/A46. f To evaluate cell growth, cells were plated in triplicate in a six-well plate and grown in 5% DCC FBS and either tamoxifen or vehicle for 5 days and then trypsinized and counted. g Stable cells were plated in triplicate in a six-well plate. After 24 h, media were replaced with media containing either tamoxifen or vehicle. Cells were allowed to grow for 10 days and then fixed and stained. Percent area was quantitated and graphed in h. A one-way ANOVA with Tukey’s post hoc test was used to compare the difference between vehicle and tamoxifen-treated groups
We next transduced TamR cells with lentiviral particles containing either a control plasmid or a plasmid expressing a 4E-BP1 mutant that cannot be inactivated (4E-BP1A37/A46) by phosphorylation, causing irreversible inhibition of eIF4E. 4E-BP1A37/A46 levels were evaluated post sorting via immunoblotting (Fig. 2e). In contrast to the wild-type cells, 4E-BP1A37/A46 expression in TamR cells resulted in a significant reduction in cell number in response to tamoxifen, while vector control cells remained resistant to tamoxifen (Fig. 2f). A similar response to tamoxifen in 4E-BP1A37/A46-transduced TamR cells was seen when anchorage-dependent colony formation was assessed (Fig. 2g, h). Previous work (data not shown) showed that this construct reduced the sensitivity of cells to estradiol and IGF-I. In these tamoxifen-resistant cells, the data suggest that irreversible inhibition of eIF4E in TamR cells partially restores sensitivity to tamoxifen.
Downregulation of eIF4E with Antisense Oligonucleotides Cooperated with Tamoxifen to Suppress Growth and Viability of TamR Cells
In order to further investigate the role of eIF4E in tamoxifen resistance, an eIF4E-targeted antisense oligonucleotide (4E-ASO) was used to knock down levels of eIF4E in TamR cells (Fig. 3a). Mock and mismatch ASO (MM ASO)-transfected cells were used as controls. To elucidate the effect of eIF4E knockdown on translation, a polyribosome preparation was performed on the MM ASO- and 4E-ASO-treated cells (Fig. 3b). 4E-ASO treatment resulted in a decrease in the heavy fraction of total RNA compared to the MM ASO-transfected cells, indicating a decrease in mRNA translation. Similar to overexpression of the mutant 4E-BP1A37/A46, 4E-ASO-treated TamR cells have a significant reduction in proliferation (Fig. 3c) and anchorage-dependent colony formation (Fig. 3d, e) with tamoxifen treatment. Thus, decreasing eIF4E expression also partially restores tamoxifen sensitivity in TamR cells.
Fig. 3.
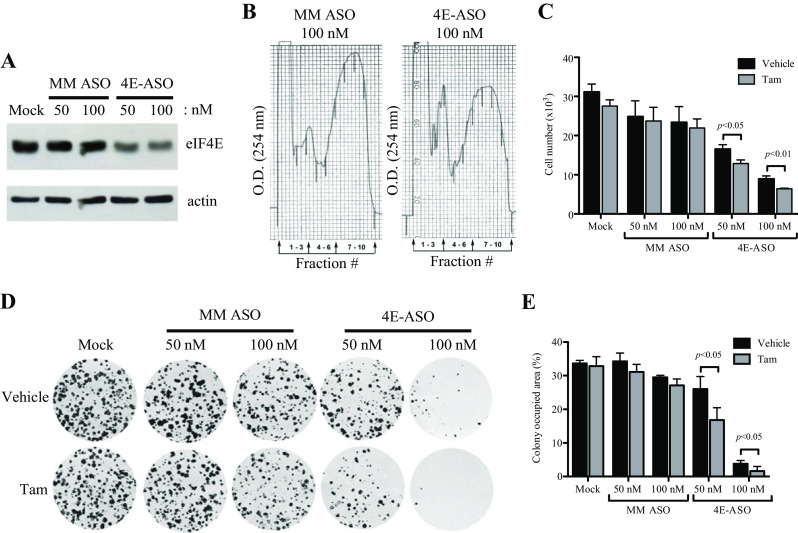
Reduction in levels of eIF4E alters responsiveness of TamR cells to tamoxifen treatment. a Western blot showing relative levels of eIF4E. Cells were plated and transfected overnight, and lysates were collected. Lysates were subjected to SDS-PAGE gel electrophoresis and then immunoblotted with specific antibodies for eIF4E and actin. b Polyribosome analysis. Polyribosomes were collected from MCF-7L cells treated with either mismatch or eIF4E ASO and resolved into ten discrete fractions on a sucrose gradient. Shown is the optical density (O.D.); the heavier polyribosomes correspond to the higher numbered fractions. c Cell proliferation assay. Cells were plated, transfected, and then grown in media supplemented with 5% DCC FBS and 100 nM tamoxifen or vehicle for 5 days. They were then trypsinized and counted. d Cells were plated in triplicate in a six-well plate and transfected overnight. Media were then replaced with media containing either tamoxifen or vehicle, and cells were allowed to grow for 10 days and then fixed and stained, and percent area was quantitated and graphed in e. One-way ANOVA was used to compare differences in vehicle and tamoxifen treatments
Inhibition of eIF4E in TamR Cells Suppressed Actively Proliferating Cells and Increased Levels of Apoptosis with Tamoxifen Treatment
In order to determine the mechanism of eIF4F inhibition of TamR cells, we examined cell cycle progression and apoptosis by flow cytometry. Tamoxifen treatment had no effect on cell cycle progression of the vector-treated cells and a minor effect on MM ASO-treated cells. However, tamoxifen significantly decreased the percentage of actively proliferating cells in the 4E-BP1A37/A46 cell line (Fig. 4a) as well as the 50 nM 4E-ASO-transfected cells (Fig. 4b). Additionally, TamR 4E-BP1A37/A46 cells showed a significant increase in apoptosis as measured by sub-G1 peak when treated with tamoxifen (Fig. 4c), an observation supported by a decrease in colony number in anchorage-dependent colony formation (data not shown). There was also a significant increase in the percent of apoptotic cells with tamoxifen treatment in 4E-ASO-transfected cells (Fig. 4d). These data suggest that inhibiting the eIF4F pathway in conjunction with tamoxifen treatment results in both decreased cell cycle progression and apoptosis in TamR cells.
Fig. 4.

Effect of eIF4E inhibition coupled with tamoxifen treatment on actively proliferating (a, b) and apoptotic (c, d) cells. TamR cells stably expressing a non-repressible 4E-BP1 mutant (HA-4E-BP1A37/A46) (a, c) or being transfected with eIF4E-targeted ASO (4E-ASO) or mismatched ASO (MM ASO) (b, d) were plated and then treated with either tamoxifen or vehicle for 5 days. Cells were then fixed with ethanol and stained with propidium iodine. DNA content was determined on a FACSCalibur flow cytometer (BD). Either Student’s t test or one-way ANOVA was used to compare differences in vehicle and tamoxifen treatments; *p < 0.05, **p < 0.01, ***p < 0.001
TamR Cells Display Increased Sensitivity to Inhibition of cap-Dependent Translation Using 4E1RCat Compared to Tamoxifen-Sensitive Cells
4E1RCat is a small molecule inhibitor of eIF4E that blocks both the eIF4E:4E-BP1 and eIF4E:eIF4G interactions, thereby preventing cap-dependent but not cap-independent translation. Treatment of MCF-7L and TamR cells with 3 or 5 μM 4E1RCat resulted in a significant decrease in cap-dependent translation without significantly altering cap-independent translation (Fig. 5a) measured by the reporter assay. To determine the effect of 4E1RCat on proliferation in MCF-7L and TamR cell lines, cells were treated with vehicle or 4E1RCat for 5 days. Compared to MCF-7L cells, 3 μM 4E1RCat treatment suppressed TamR monolayer growth (Fig. 5b), indicating TamR cells were more sensitive to 4E1RCat treatment. Both MCF-7L and TamR cells were suppressed by 5 μM 4E1RCat. Additionally, there was also a significant decrease in anchorage-dependent colony formation in both cell lines; however, the difference when MCF-7L cells were compared to TamR cells was not significant (Fig. 5c, d; data not shown). In all assays, treatment with 4E1RCat resulted in decreasing cells to basal (untreated) status; therefore, adding tamoxifen to 4E1RCat treatment did not result in a further decrease in translation, proliferation, or anchorage-dependent growth (data not shown). Thus, TamR cells are responsive to pharmacologic inhibition of eIF4F-mediated translation and may be more sensitive to treatment than tamoxifen-sensitive cells.
Fig. 5.

Treatment with the cap-dependent translation inhibitor 4E1RCat successfully inhibits proliferation and colony formation. a To evaluate the effectiveness and specificity of 4E1RCat, a dual-luciferase reporter construct (pcDNA3-rLuc-polIRES-fLuc) was transfected overnight into MCF-7L and TamR cells. Cells were then treated with 4E1RCat at the concentrations indicated for 24 h, and levels of luciferase were read. b To examine the effect of 4E1RCat on cell proliferation, 1 × 104 cells/well were seeded into a six-well plate and grown in media containing 5% FBS plus either vehicle or treatment at the concentrations indicated. After 5 days, cells were trypsinized, stained with trypan blue, and counted. c The effect of eIF4E inhibition on anchorage-dependent colony formation was addressed using a clonogenic efficiency assay. Cells were plated in a six-well plate in triplicate in full growth media. The next day, media were replaced with media containing the treatments indicated. After 10 days, cells were fixed and stained with Coomassie blue, and the area was assessed as described in Fig. 2. Results were graphed in d. Significance was assessed using a one-way ANOVA with Tukey’s post hoc test
Discussion
Resistance to endocrine therapy is a major clinical problem in breast cancer. Typically, primary resistance is defined as a lack of benefit from endocrine therapy documented by the progression of disease within 6 months of treatment. Secondary resistance occurs when progression occurs after 6 months of clinical benefit of endocrine therapy. Most of the preclinical data model secondary resistance as most ER-positive cell lines display a growth requirement for estradiol. Analysis of cells selected for secondary endocrine resistance has shown the activation of the PI3K pathway [27]. Indeed, the approval of the mTOR antagonist everolimus has validated the importance of this signaling. In addition, multiple other clinical trials are underway to study other pathway inhibitors such as Akt and PI3K itself. Since the PI3K pathway results in the activation of the mRNA cap-dependent translational machinery, it is plausible that enhanced translation is a mechanism for endocrine resistance.
To evaluate this possibility, we studied the cells we previously created to be resistant to tamoxifen. These cells demonstrated upregulation of cap-dependent mRNA translation as shown by hyperphosphorylation of 4E-BP1, increased eIF4G binding to eIF4E, and elevated levels of mRNA bound to the heavy ribosome fraction. While forced expression of eIF4E alone did not result in tamoxifen resistance, cells selected for resistance to tamoxifen were sensitive to inhibition of this pathway. Notably, mTORC1 activation increases ribosomal biogenesis [28] which was able to be shown in evaluating the ribosome-bound mRNA species. These results are consistent with the therapeutic benefit of mTOR inhibitors in tamoxifen-resistant breast cancer. The clinical results suggest that mTOR inhibition is most effective in the treatment of patients with secondary resistance to tamoxifen [5]. This clinical finding is consistent with our results, and enhanced activity of the cap-dependent translational machinery is not sufficient to cause primary resistance.
While mTORC1 inhibition is now approved for the treatment of endocrine-resistant breast cancer, one problem with drugs of this class is the disruption of the negative feedback pathway to insulin and insulin-like growth factor signaling. By inhibiting mTORC1, the key adaptor protein insulin receptor substrate 1 is not downregulated [11, 29]. This disruption of negative feedback leads to increased activation of pathways upstream of mTORC1 including Akt. While the clinical implications for this upregulation are not clear, it is notable that insulin-like growth factor and insulin signaling have been implicated in endocrine resistance [30, 31]. We have also shown the insulin-like growth factors regulate gene transcription and are sufficient to active transcriptional activity of ER [32]. Thus, inhibition of transcriptional and translational activities mediated by growth factors and their downstream effectors should be effective in treating endocrine-responsive and endocrine-resistant breast cancers. Further, identifying targets that do not further enhance PI3K activity would be valuable. Our data show that inhibition of cap-dependent translational does not affect upstream signaling (data not shown).
We used several genetic constructs, antisense oligonucleotides, and a pharmacologic inhibitor to show that disruption of the cap-dependent translation reduces tamoxifen resistance. Thus, inhibition of this process, alone or in combination with upstream signal transduction inhibitors, might be clinically exploited. Further development of drugs specifically designed to suppress cap-dependent translation should be developed to validate the importance of this pathway.
Electronic supplementary material
(PPTX 13166 kb)
(CSV 1305 kb)
(CSV 21 kb)
(CSV 17 kb)
Compliance with Ethical Standards
Financial Support
This study received grants from the Department of Defense Breast Cancer Research Program Pre-Doctoral Fellowship (grant BC093938; DHF). Public Health Service (grant CA74285; DY), Komen for the Cure (grant SAC 11039; DY), Mayo Clinic SPORE in Breast Cancer (P50CA116201), and Cancer Center Support (grant P30 CA77398).
Conflict of Interest
The authors declare that they have no conflict of interests.
Footnotes
Dedra H. Fagan and Lynsey M. Fettig contributed equally to this work.
References
- 1.Early Breast Cancer Trialists’ Collaborative Group (2005) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 365:1687–1717 [DOI] [PubMed]
- 2.Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, Abraham M, Medeiros Alencar VH, Badran A, Bonfill X, Bradbury J, Clarke M, Collins R, Davis SR, Delmestri A, Forbes JF, Haddad P, Hou MF, Inbar M, Khaled H, Kielanowska J, Kwan WH, Mathew BS, Mittra I, Muller B, Nicolucci A, Peralta O, Pernas F, Petruzelka L, Pienkowski T, Radhika R, Rajan B, Rubach MT, Tort S, Urrutia G, Valentini M, Wang Y, Peto R, Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) Collaborative Group Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381:805–816. doi: 10.1016/S0140-6736(12)61963-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joshi B, Cai AL, Keiper BD, Minich WB, Mendez R, Beach CM, Stepinski J, Stolarski R, Darzynkiewicz E, Rhoads RE. Phosphorylation of eukaryotic protein synthesis initiation factor 4E at Ser-209. J Biol Chem. 1995;270:14597–14603. doi: 10.1074/jbc.270.24.14597. [DOI] [PubMed] [Google Scholar]
- 5.Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G, Abadie-Lacourtoisie S, Eymard JC, Debled M, Spaeth D, Legouffe E, Allouache D, El Kouri C, Pujade-Lauraine E. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30:2718–2724. doi: 10.1200/JCO.2011.39.0708. [DOI] [PubMed] [Google Scholar]
- 6.Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature. 1990;345:544–547. doi: 10.1038/345544a0. [DOI] [PubMed] [Google Scholar]
- 7.Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, Pandolfi PP. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–486. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 8.Avdulov S, Herrera J, Smith K, Peterson M, Gomez-Garcia JR, Beadnell TC, Schwertfeger KL, Benyumov AO, Manivel JC, Li S, Bielinsky AK, Yee D, Bitterman PB, Polunovsky VA. EIF4E threshold levels differ in governing normal and neoplastic expansion of mammary stem and luminal progenitor cells. Cancer Res. 2015;75:687–697. doi: 10.1158/0008-5472.CAN-14-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, Halperin JA, Wagner G. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 10.Avdulov S, Li S, Michalek V, Burrichter D, Peterson M, Perlman DM, Manivel JC, Sonenberg N, Yee D, Bitterman PB, Polunovsky VA. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell. 2004;5:553–563. doi: 10.1016/j.ccr.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 11.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan S, Li Y, Yue P, Khuri FR, Sun SY. The eIF4E/eIF4G interaction inhibitor 4EGI-1 augments trail-mediated apoptosis through c-FLIP down-regulation and DR5 induction independent of inhibition of cap-dependent protein translation. Neoplasia. 2010;12:346–356. doi: 10.1593/neo.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cencic R, Hall DR, Robert F, Du Y, Min J, Li L, Qui M, Lewis I, Kurtkaya S, Dingledine R, Fu H, Kozakov D, Vajda S, Pelletier J. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc Natl Acad Sci U S A. 2011;108:1046–1051. doi: 10.1073/pnas.1011477108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graff JR, Konicek BW, Vincent TM, Lynch RL, Monteith D, Weir SN, Schwier P, Capen A, Goode RL, Dowless MS, Chen Y, Zhang H, Sissons S, Cox K, McNulty AM, Parsons SH, Wang T, Sams L, Geeganage S, Douglass LE, Neubauer BL, Dean NM, Blanchard K, Shou J, Stancato LF, Carter JH, Marcusson EG. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest. 2007;117:2638–2648. doi: 10.1172/JCI32044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia Y, Chiu TL, Amin EA, Polunovsky V, Bitterman PB, Wagner CR. Design, synthesis and evaluation of analogs of initiation factor 4E (eIF4E) cap-binding antagonist Bn7-GMP. Eur J Med Chem. 2010;45:1304–1313. doi: 10.1016/j.ejmech.2009.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fagan DH, Uselman RR, Sachdev D, Yee D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: implications for breast cancer treatment. Cancer Res. 2012;72:3372–3380. doi: 10.1158/0008-5472.CAN-12-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li S, Sonenberg N, Gingras AC, Peterson M, Avdulov S, Polunovsky VA, Bitterman PB. Translational control of cell fate: availability of phosphorylation sites on translational repressor 4E-BP1 governs its proapoptotic potency. Mol Cell Biol. 2002;22:2853–2861. doi: 10.1128/MCB.22.8.2853-2861.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larsson O, Li S, Issaenko OA, Avdulov S, Peterson M, Smith K, Bitterman PB, Polunovsky VA. Eukaryotic translation initiation factor 4E induced progression of primary human mammary epithelial cells along the cancer pathway is associated with targeted translational deregulation of oncogenic drivers and inhibitors. Cancer Res. 2007;67:6814–6824. doi: 10.1158/0008-5472.CAN-07-0752. [DOI] [PubMed] [Google Scholar]
- 19.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with tophat and cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larsson O, Sonenberg N, Nadon R. Anota: analysis of differential translation in genome-wide studies. Bioinformatics. 2011;27:1440–1441. doi: 10.1093/bioinformatics/btr146. [DOI] [PubMed] [Google Scholar]
- 24.Robinson MD, Smyth GK. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2008;9:321–332. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- 25.Lee AV, Oesterreich S, Davidson NE (2015) MCF-7 cells—changing the course of breast cancer research and care for 45 years. J Natl Cancer Inst 107(7). doi:10.1093/jnci/djv073 [DOI] [PubMed]
- 26.Lin X, Li J, Yin G, Zhao Q, Elias D, Lykkesfeldt AE, Stenvang J, Brunner N, Wang J, Yang H, Bolund L, Ditzel HJ. Integrative analyses of gene expression and DNA methylation profiles in breast cancer cell line models of tamoxifen-resistance indicate a potential role of cells with stem-like properties. Breast Cancer Res. 2013;15:R119. doi: 10.1186/bcr3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H, Higham C, Garcia-Echeverria C, Shyr Y, Arteaga CL. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120:2406–2413. doi: 10.1172/JCI41680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006;25:6384–6391. doi: 10.1038/sj.onc.1209883. [DOI] [PubMed] [Google Scholar]
- 29.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. Akt inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fox EM, Miller TW, Balko JM, Kuba MG, Sanchez V, Smith RA, Liu S, Gonzalez-Angulo AM, Mills GB, Ye F, Shyr Y, Manning HC, Buck E, Arteaga CL. A kinome-wide screen identifies the insulin/IGF-1 receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res. 2011;71:6773–6784. doi: 10.1158/0008-5472.CAN-11-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fox EM, Kuba MG, Miller TW, Davies BR, Arteaga CL. Autocrine IGF-I/insulin receptor axis compensates for inhibition of AKT in ER-positive breast cancer cells with acquired resistance to estrogen deprivation. Breast Cancer Res. 2013;15:R55. doi: 10.1186/bcr3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Becker MA, Ibrahim YH, Oh AS, Fagan DH, Byron SA, Sarver AL, Lee AV, Shaw LM, Fan C, Perou CM, Yee D. Insulin receptor substrate adaptor proteins mediate prognostic gene expression profiles in breast cancer. PLoS One. 2016;11:e0150564. doi: 10.1371/journal.pone.0150564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PPTX 13166 kb)
(CSV 1305 kb)
(CSV 21 kb)
(CSV 17 kb)