

Abstract
Background
Cerebral palsy (CP) is the most common motor disability in childhood with a worldwide prevalence of ranging between 1.5 to >4 per 1000 live births. Hypoxic ischemic encephalopathy (HIE) contributes to the burden of CP, but the long-term neuropathological findings of this association remain limited.
Methodology
Thirty-four term Macaca nemestrina were included in this long-term neuropathologic study: 9 control animals delivered by Cesarean section, and 25 animals with perinatal asphyxia who delivered by cesarean section after 15–18 minutes of umbilical cord occlusion (UCO). UCO animals were randomized to saline (n = 11), therapeutic hypothermia (TH) only (n = 6), or TH+erythropoietin (Epo; n = 8). Epo was given on days 1, 2, 3, and 7. Animals had serial developmental assessments and underwent MRI with diffusion tensor imaging at 9 months of age followed by necropsy. Histology and immunohistochemical staining of brain and brainstem sections was performed.
Results
All UCO animals demonstrated and met standard diagnostic criteria for human neonates with moderate-to-severe HIE. Four animals developed moderate-to-severe CP (3 UCO, 1 UCO+TH), 9 had mild CP (2 UCO, 3 UCO+TH, 3 UCO+TH+Epo, 1 control) and 2 UCO animals died. None of the animals treated with TH+Epo died, had moderate-to-severe CP, or demonstrated signs of long-term neuropathological toxicity. Compared to animals grouped together as non-CP (controls and mild CP only), animals with CP (moderate & severe) demonstrated decreased fractional anisotropy of multiple white matter tracks including corpus callosum and internal capsule on using track based special statistics (TBSS). Animals with CP had decreased staining for cortical neurons, and increased brainstem glial scarring compared to animals without CP. Cerebellar cell density of the internal granular layer and white matter was decreased in CP animals compared to control animals without CP.
Conclusions/Significance
In this nonhuman primate HIE model, animals treated with TH+Epo had less brain pathology noted by TBSS and with immunohistochemical staining supporting the long-term safety of TH+Epo in the setting of HIE. Animals who developed CP showed white matter changes noted by TBSS, subtle histopathologic changes in both white and gray matter and brainstem injury that correlated with CP severity. This HIE model may lend itself to further study of the relationship between brainstem injury and CP.
Keywords: Brain injury, Central nervous system, Cerebellum, Developing brain, Erythropoietin, Hypothermia therapy, Hypoxic-ischemic encephalopathy, Immunohistochemistry, Monkey
Introduction
Intrapartum hypoxia is one important cause of hypoxic ischemic encephalopathy (HIE), a problem that contributes to 22% of neonatal deaths worldwide.[1,2] Common causes of intrapartum hypoxia are intrauterine asphyxia brought on by circulatory problems, such as clotting of placental arteries, placental abruption, or inflammatory processes.[3] In developed countries, therapeutic hypothermia (TH) initiated within 6 hours of birth is the standard of care for term and near term neonates with moderate or severe HIE,[4–7] and results in less death and better neurodevelopmental outcomes up to at least 18 months of age for survivors compared to untreated neonates.[8] Despite TH, 46% of treated neonates with HIE still die or sustain major neurodevelopmental disability, with approximately 20% developing cerebral palsy (CP).[4–12] Since TH provides only partial treatment benefit, adjunctive therapies are needed. Erythropoietin (Epo), a hematopoietic cytokine with neuroprotective effects, is a promising treatment for neonates with HIE[13,14] that could be used concurrently with TH.[15,16]
To better understand treatment strategies that may improve outcomes in neonates with HIE (e.g., TH+Epo), we developed a nonhuman primate model of acute perinatal asphyxia using umbilical cord occlusion (UCO) prior to delivery to produce moderate-to-severe HIE.[17,18] In our model, UCO for 15–18 minutes results in clinical and laboratory manifestations of HIE comparable to human newborns entered in the major TH clinical trials.[4,5] In addition to early physical exam and MRS findings consistent with moderate-to-severe HIE, animals demonstrate transient kidney and liver biochemical dysfunction, abnormal amplitude-integrated electroencephalogram (aEEG), feeding intolerance, progressive spasticity, and cognitive delay.[17,18] We found that UCO for 15 to 18 minutes resulted in death or moderate-to-severe CP in 43% of saline treated and 44% of TH treated animals, but in none of the animals treated with TH+Epo.[18] Compared to UCO saline, UCO+TH+Epo treated animals had improved motor and cognitive responses, cerebellar growth, and diffusion tensor imaging measures on MRI. These findings suggest Epo treatment may play a key role in preventing neurodevelopmental impairment following HIE.
Long-term neuropathologic findings following perinatal asphyxia are limited. While we previously reported on 4 animals exposed to UCO for 15 minutes who had evidence of neuronal degeneration with gliosis, extensive immunohistochemistry of normal vs. injured or treated animals was not available.[17] Here we present long-term neuropathologic findings from a proof-of-concept, open-label, randomized controlled study to evaluate the incidence of death or CP by treatment group. We also provide a secondary analysis of the neuropathology of animals with and without CP to improve our understanding of the underlying neuropathological substrates of this injury.
Methods and Animals
Animals
The parent study testing the safety and efficacy of TH+Epo at improving survival and preventing the development of moderate-to-severe CP after HIE in a term nonhuman primate model of perinatal asphyxia included 56 Macaca nemestrina (pigtailed macaques) randomized to different treatment groups.[18] This current study focused on brain histopathology at 9 months of age, which was available in 34 animals (25 with HIE and 9 non-UCO cesarean delivered control animals). An overview of the experimental UCO model used to produce HIE in Macaca nemestrina is summarized in Figure 1. The Animal Care and Use Committees at the University of Washington in accordance with US National Institutes of Health (NIH) guidelines approved all experimental protocols.
Figure 1.
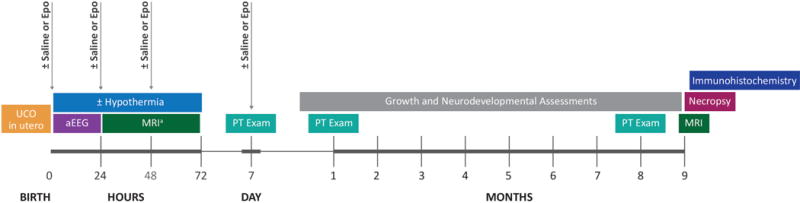
An overview of the procedures and assessments for the nonhuman primate model of perinatal asphyxia. UCO = umbilical cord occlusion, Epo = erythropoietin, aEEG = amplitude-integrated Electroencephalography, MRI = Magnetic resonance and diffusion tensor imaging; PT = physical therapy. aThe first MRI scan was performed at either 24 or 72 hours of age.
Delivery and Resuscitation
Animals were delivered 1–8 days prior to term (168±2 days) by hysterotomy under maternal general anesthesia with sevoflurane. Following uterine incision, the umbilical cord was exteriorized (while keeping amniotic fluid and the fetus in the uterine cavity) and clamped (UCO group; n=25) for either 15 (n=9) or 18 minutes (n=16) to induce perinatal asphyxia. Control animals (n=9) were also delivered by hysterotomy, but did not undergo UCO. A 2.5-Fr Vygon™ umbilical arterial catheter was inserted prior to delivery. Fetuses were delivered, weighed and stabilized by a team of neonatologists using standardized American Academy of Pediatrics neonatal resuscitation principles.[19] Resuscitations included endotracheal intubation, positive pressure ventilation, chest compressions, and bolus epinephrine as indicated. Apgar scores were assigned at 1, 5, 10, and 20 minutes. A covered heating pad, radiant warmer, and polyethylene sheet were used to provide initial thermal support during stabilization, followed by placement into a thermal-neutral incubator. Monitoring included pulse oximetry, rectal thermometry, and aEEG (BrainZ BRM3, Natus Medical Incorp., San Carlos, CA, USA).
Treatment Groups
To focus on the long-term neuropathologic effects of perinatal asphyxia, data from the UCO animals were compared with data from cesarean section control animals. The 25 animals delivered after UCO for either 15 or 18 minutes were assigned to 1 of 3 treatment groups: saline (n=11: 3 UCOx15 min, 8 UCOx18 min), TH only (33.5°C for 72 h; n=6: 2 UCOx15 min, 4 UCOx18 min), or TH+Epo (n=8, 4 UCOx15 min, 4 UCOx18 min). The Epo dose was 3,500 U/kg × 1 dose followed by 3 doses of 2,500 U/kg, or Epo 1,000 U/kg/day × 4 doses given intravenously. Nine animals of similar gestational age were delivered by cesarean section to serve as control animals. Primary outcome measures were made using the allocated treatment group as the variable. For the secondary analysis, as previously described,[18] animals were designated as non-CP (normal or mild CP only) or as CP (moderate to severe CP).
Animal Care
Post-resuscitation care, which closely mimicked care practices in human newborns requiring intensive care, was conducted as previously reported.[20] Briefly, study animals were maintained for a minimum of 3 days on parenteral fluids with adjustments made to maintain euglycemia and hydration. For infants treated with TH, enteral feedings were started after rewarming once the animals had a normal abdominal exam and stooling was established (typically on postnatal day 4). Routine labs included serial electrolytes, arterial blood gas, and lactate level measurements (iSTAT ®; HESKA Corp., Loveland, CO, USA). Animals with clinical seizure activity were treated with phenobarbital (5 mg/kg) with repeated doses given (dose range: 5–30 mg/kg) until clinical seizure activity ceased. Data for all of these parameters has been previously reported.[18,20]
Psychological and environmental enrichment activities were performed based on the Infant Primate Research Laboratory care protocol.[21] A physical therapist skilled in neonatal care and blinded to treatment group performed sequential exams at 1 week, 1 month, and 8 months to document any evidence of motor abnormalities and contractures consistent with CP. Animals were evaluated on their ability to control active movement and muscle tone at each joint was graded on the Ashford scale of 0 (normal) to 4 (affected parts rigid in flexion or extension).[19] Based on serial evaluations, animals at the end of the study were characterized as: normal (no CP), mild CP, moderate CP, or severe CP. Animals who died were placed in the severe category.
MRI acquisition and analyses
All surviving UCO animals, non-UCO cesarean-section delivered control animals, and five colony animals underwent sedated MR imaging as previously described at 24 or 72 hours [20], and again at 9 months. Brain MRI from surviving animals diagnosed with CP (n=6; 1 severe, 2 moderate, and 3 mild) were compared to control animals (n=5). Total scan time was approximately 2 hours and consisted of magnetization prepared rapid gradient echo (MPRAGE), high resolution T1 weighted imaging, diffusion tensor imaging (DTI), and MR spectroscopy acquired on a Philips Achieva 3.0 Tesla magnet with X-series Quasar Dual gradient system. Two 8-channel array head coils were custom-made to fit neonatal and juvenile macaques. Details of the sequence acquisition were performed as previously reported.[20]
DTI
Voxelwise statistical analysis of the fractional anisotropy (FA) data was carried out using TBSS (Tract-Based Spatial Statistics[22]), part of FSL.[23] First, FA images were created by fitting a tensor model to the raw diffusion data using FDT, and then brain-extracted using BET.[24] All subjects’ FA data were then aligned into a common space using the nonlinear registration tool FNIRT,[25,26] which uses a b-spline representation of the registration warp field.[27] Scans at 3 days and 9 months of age from representative control animals were used to align animals instead of the human normal space. Next, the mean FA image was created and thinned to create a mean FA skeleton, which represents the centers of all tracts common to the group. Each subject’s aligned FA data was then projected onto this skeleton, and the resulting data fed into voxelwise cross-subject statistics. Voxelwise cross-subject statistics were performed on non-FA DTI data (first eigenvalue, second eigenvalue, third eigenvalue, mean diffusivity, mode of anisotropy, volume ratio, and raw T2 signal without diffusion weighting) using the tbss_non_FA command. Threshold-Free Cluster Enhancement results with correction for multiple comparisons were used to determine significance.[28]
Histopathological analysis
At 9 months of age, animals were sedated with ketamine and then euthanized with an overdose of intravenous sodium pentobarbital. After terminal perfusion with 4% paraformaldehyde, the animal’s brains were immediately removed, and immersion fixed with 4% paraformaldehyde, and paraffin embedded. Hematoxylin and eosin (H&E) stains from a subgroup of animals with CP (n=3; 2 UCO animals, 1 UCO+TH animal) were compared to cesarean section control animals (n=4). Four μm thick sections from the level of the thalamus with associated cerebral cortex, corpus callosum, and hippocampus were routinely processed, embedded in paraffin, and H&E stained. These slides were qualitatively evaluated in a blinded manner by a board certified veterinary pathologist (JS).
Immunohistochemical (IHC) staining
Six μm-thick paraffin sections were cut and IHC staining was performed as previously described [29,30], including citric acid antigen retrieval, blocking with 2% serum (respective to secondary antibody species), and overnight primary and one hour secondary antibody incubation. BrdU staining was performed as previously described including 2 mol/L HCl treatment for 30 minutes followed by 0.1 mol/L sodium borate for 5 min. Antibodies used and the concentrations are listed in Table 1.
Table 1.
List of antibodies and suppliers
| Antibody | Supplier | Reference | Dilution |
|---|---|---|---|
| GFAP | DAKO | Z0334 | 1/500 |
| Iba1 | Wako | 019-19741 | 1/200 |
| CD68 | DAKO | m0814 | 1/500 |
| CD45 | DAKO | m0701 | 1/500 |
| Ki67 | DAKO | M7240 | 1/500 |
| MBP | Chemicon | MAB 382 | 1/500 |
| Olig2 | IBL | OD803 | 1/200 |
| NeuN | Chemicon | MAB377 | 1/2000 |
| Calbindin | Swant | 300 | 1/2000 |
| MAP2 | Millipore | MAB 378 | 1/200 |
| BrdU | BD | 347580 | 1:100 |
| Goat anti-rabbit | Vector Labs. | BA-9200 | 1/500 |
| Goat anti-mouse | Vector Labs. | BA-1000 | 1/500 |
Analysis was made for glial fibrillary acidic protein (GFAP), ionized calcium binding adaptor molecule 1 (Iba1), myelin basic protein (MBP), and microtubule-associated protein 2 (MAP2) using densitometric analysis, as previously described [31,32] using Image J (NIH, USA). Counts were made of CD68, CD45, Ki67, Olig2, NeuN, Calbindin and BrdU. Figure 2 demonstrates brain regions evaluated with IHC staining. Analyses were made in 3 fields of view (FOV) per brain region for densitometric analysis and 2 FOV per brain region for counts as previously described [31,32]. IHC data across fields of view were averaged and the mean used in statistical analyses in Graphpad prism (version 5 for Mac). IHC analyses of the effects of UCO were made with a Mann-Whitney U test, and the effects of treatment on UCO injury were analyzed using a one-way ANOVA with a Dunnet’s post-hoc (comparing UCO to UCO+TH and UCO to UCO+TH+Epo) which included an adjustment for multiple comparisons. Comparisons of CP versus control for all brain regions were made using a Mann Whitney U test. Significance for all analysis was accepted at a p<0.05.
Figure 2.
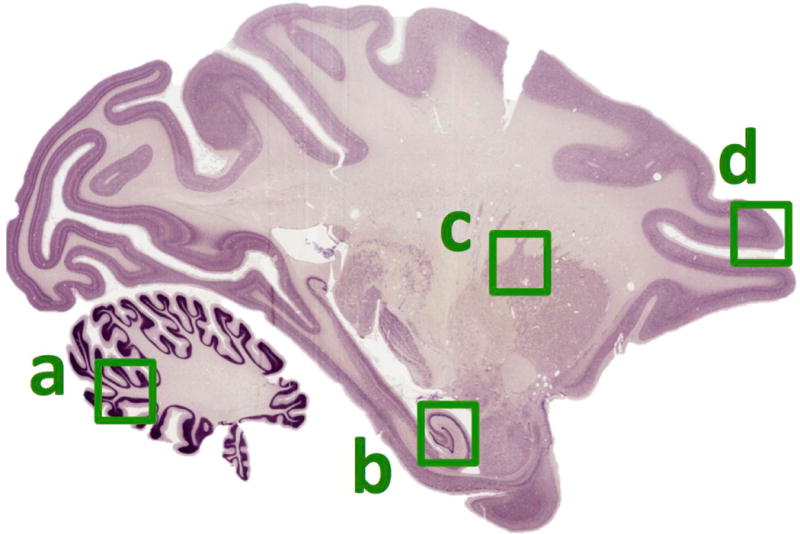
Representative outline of brain areas analyzed via immunohistochemistry shown on a illustrative sagittal section of H&E stained macaque brain. a, cerebellum in which the white matter and/or granule cell layer were analyzed. b, hippocampus in which cell counts of the whole cornu amnios layer (CA) or densitometry with the CA1 central to the images were made. c, corona radiata white matter tract. d, prefrontal cortex, where analysis were centered on layer 4.
Brainstem analysis
Brainstem tissues were embedded in paraffin and 4 μm-thick sections from the pons and medulla were H&E stained or immunostained using the Ventana Bench Mark Ultra automated immunostainer (Ventana Medical Systems, Tucson, AZ) according to the manufacturer’s instructions. Sections were subjected to an antigen retrieval process of 8 minutes using Ventana’s Cell Conditioner 1 (Ventana Medical Systems, Tucson, AZ) followed by GFAP detection using an anti-GFAP antibody (Ventana Medical Systems, predilute Ready to Use).
Brainstem scoring
Brainstems were analyzed in 12 animals (5 controls, 5 UCO, and 2 UCO+TH). The samples in each case included various levels of medulla, pons, and/or spinal cord. The brainstem was hemisected in most cases. H&E and GFAP slides from each case were accessed. In assessing H&E sections, the extent of neuronal loss and the presence or absence of gliosis as defined by the presence of gemistocytic cells with hyper-eosinophilic cytoplasm (reactive astrocytes) were noted. In assessing GFAP sections, the presence of glial fiber staining with a “feltwork” appearance in the tissue with and without gliosis as defined by the presence of GFAP-positive astrocytic cell body staining (reactive astrocytes) was noted. After assessing H&E and GFAP stains, composite scores were given as follows: 0, no pathology by H&E and GFAP (no neuronal loss by H&E; mainly feltwork pattern of GFAP staining); 1, mild gliosis with no obvious neuronal loss; 2, moderate gliosis with possible neuronal loss but mild; 3, severe injury with widespread gliosis and neuronal loss. To assess the strength and direction of association between brainstem severity scores and CP severity scores, a Spearman’s rank correlation coefficient was performed (Graphpad Prism).
Morphometric measurements of cerebellum
After brain MRI, cerebellar tissue was available for 4 CP and 4 non-CP (control, no UCO) animals for morphometric cerebellar measurements. The cerebellum was imbedded in paraffin and sagittal sections 4 μm thick were made. Slides were stained for H&E, calbindin, GFAP, and BrdU on the Leica Bond Automated Immunostainer with Leica bond kits (Leica Microsystems Inc., Buffalo Grove, IL). Slides were baked for 30 minutes at 60°C and deparaffinized. Antigen retrieval comprised of citrate or EDTA for 20m at 100°C. Slides were blocked in normal donkey serum (10% in TBS) for 10 minutes at room temperature. Primary antibodies were applied at room temperature for 30 min. Leica goat anti-mouse HRP polymer was applied for 30 minutes at room temperature and blocked with peroxide for 10 min. Leica bond mixed refine (DAB) detection applied twice for 10 minutes at room temperature. Slides were counterstained with hematoxylin.
Slides were scanned using a Nanozoomer Digital Pathology slide scanner (Olympus America; Center Valley, Pennsylvania). The digital images were then imported into Visiopharm software (Hoersholm, Denmark) for analysis. Measures included thickness of the molecular layer, Purkinje number, Bergman glia density, density of the internal granular layer, density of cells in the white matter, and BrdU+ cell counts. Cerebellar analyses were performed using Image J software (NIH, USA). Statistical analysis (2-sided t-tests with a p<0.05) was performed using IBM SPSS Statistics version 19.0 (SPSS Inc., IBM Company, Chicago, IL).
Results
Animal characteristics
After delivery, all animals exposed to UCO were flaccid without spontaneous respirations or movement, required intubation and mechanical ventilation, and met standard diagnostic criteria for human neonates with moderate-to-severe HIE. These animals had Apgar scores less than 5 at 10 minutes of age, needed positive pressure ventilation at 10 minutes of age, had severe metabolic acidosis within the first 60 postnatal minutes, and depressed voltages on aEEG. Slightly more females (19/34; 55.9%) than males (15/34; 44.1%) were included in this study. Body weights were not significantly different at necropsy whether assessed by treatment group, outcome (CP), or sex.
Primary Outcome Measures: Effect of treatment on the diagnosis of CP
Of the 34 animals in this analysis, 4 animals had moderate-to-severe CP (3 UCO, 1 UCO+TH) and 9 had mild CP based on serial behavioral assessments by a neonatal physical therapist who was blinded to animal treatment groups. Two female UCO animals died, one at 3 days and one at 270 days of age. The early death was due to severe neurologic compromise. The late death occurred in an UCO+TH animal with a history of persistent tachypnea who unexpectedly collapsed and was noted to have a combination of endo- and myocarditis with cardiac compromise and chronic pulmonary atelectasis on autopsy. None of the animals treated with TH+Epo died or had moderate-to-severe CP. Analysis with two-way ANOVA demonstrated that males had a greater burden of CP (53%, 8 total: 4 mild, 3 moderate, 1 severe) than females (26%, 6 mild) across treatment groups (p=0.029), and this analysis also confirmed our previous reports on the efficacy of our treatments to reduce the burden of CP (p=0.021; Figure 3a).
Figure 3.
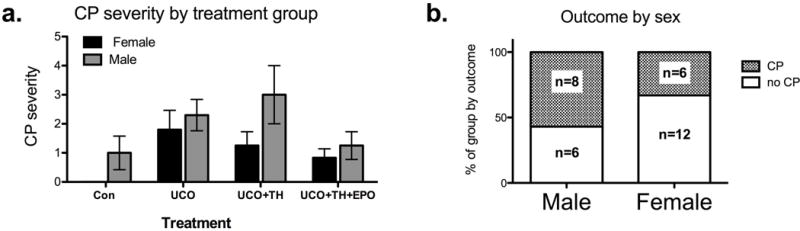
Sex specific analysis by treatment and outcome. a. Males suffered from more CP (p=0.029) and CP overall was reduced by treatment (p=0.021, 2-way ANOVA), and b. in an assessment of the proportion of animals suffering from CP, male sex was highly associated with a diagnosis of CP (p<0.01, Fisher’s exact test).
Primary Outcome Measures: Effects of UCO and treatments on neuropathology
We initially assessed the effects of UCO on the brain and the effects of TH with or without Epo using immunohistochemistry for neurons (NeuN), astrocytes (GFAP), microglia (Iba1), oligodendrocytes (Olig2), microglia/macrophage (CD68, CD45), and proliferation (Ki67 & BrdU), (Table 2). Significant effects of UCO were: reduced NeuN positive cell number in the cortex (Figure 4a) and cerebellum, reduced MBP density in the hippocampus and the corona radiata, decreased GFAP density in the cerebellum (Figure 4b), decreased CD68 positive cells in the hippocampus and increased Ki67 positive cells in the cortex. The effects of either TH or TH+Epo were minimal. Namely, TH+Epo prevented the loss of MBP density in the hippocampus, but exacerbated the loss in the corona radiata and TH and TH+Epo prevented the decrease in GFAP staining associated with UCO in the cerebellum. Very sparse staining (1–4 positive cells per entire brain section) was noted for BrdU and therefore, analysis was not conducted. TH+Epo (versus TH alone) as a therapy was not associated with long-term changes in the brain for these measures despite decreasing the group-wide incidence of CP (Figure 3a). The size of the groups precluded any meaningful sex specific analysis of the neuropathology data by treatment group.
Table 2.
Immunohistochemical analysis by treatment group
| Target | Ab | Treatment | Hippocampus | Corona Radiata | Cortex | Cerebellum (White & Grey matter) | |
|---|---|---|---|---|---|---|---|
| Neurons | NeuN | Control | 78.34±6.01 | 27.73±5.19 | 146.1±18.14* | 94.93±4.94** | |
| UCO | 73.06±4.52 | 23.17±8.54 | 107.3±5.92 | 63.48±6.22 | |||
| UCO+TH | 72.52±7.43 | 31.40±6.07 | 109.1±13.93 | 82.47±8.04 | |||
| UCO+TH+Epo | 79.06±4.72 | 24.57±4.26 | 110.5±9.19 | 73.24±7.29 | |||
| Oligoden-drocytes | Olig2 | Control | 148.5±32.50 | 300.4±38.02 | 355.4±27.77 | 356.40±42.88* | |
| UCO | 118.0±36.29 | 246.2±24.07 | 352.8±41.52 | 243.5±27.80 | |||
| UCO+TH | 213.0±35.68 | 242.8±34.26 | 320.6±37.11 | 304.0±27.10 | |||
| UCO+TH+Epo | 188.5±32.50 | 272.6±19.95 | 314.8±41.30 | 282.4±37.64 | |||
| Myelin | MBP | Control | 17.25±2.49* | 61.80±36.08* | 17.07±2.24 | 69.67±6.03 | |
| UCO | 10.40±0.99 | 34.92±3.38 | 15.92±2.42 | 58.85±6.06 | |||
| UCO+TH | 12.42±1.22 | 30.00±0.87 | 18.33±3.15 | 68.93±6.08 | |||
| UCO+TH+Epo | 27.53±3.23### | 23.81±2.71# | 16.06±0.97 | 64.95±7.89 | |||
| Astrocytes | GFAP | Control | 39.94±6.43 | 55.27±1.64 | 31.47±4.55 | 66.20±5.86*** | |
| UCO | 33.22±3.83 | 50.63±2.42 | 29.17±3.57 | 34.70±2.01 | |||
| UCO+TH | 39.67±2.51 | 58.10±2.45 | 28.90±4.57 | 68.86±3.14§§§ | |||
| UCO+TH+Epo | 34.83±2.12 | 57.62±4.24 | 31.33±2.52 | 54.95±3.89### | |||
| Microglia | Iba1 | Control | 25.78±4.74 | 18.00±2.09 | 20.80±3.61 | 11.47±1.58 | |
| UCO | 16.61±2.75 | 19.52±0.60 | 19.33±1.96 | 11.91±1.38 | |||
| UCO+TH | 26.83±3.55 | 19.71±0.68 | 19.67±0.43 | 11.91–1.38 | |||
| UCO+TH+Epo | 19.45±4.10 | 18.28±3.18 | 20.97±1.29 | 8.05±0.77 | |||
| Macrophage/Microglia | CD68 | Control | 19.09±2.79* | 15.60±2.34 | 12.27±2.86 | 20.41±0.99 | 5.10±1.03 |
| UCO | 10.19±2.96 | 15.09±2.69 | 11.72±1.25 | 21.65±3.08 | 8.16±2.13 | ||
| UCO+TH | 14.47±4.04 | 22.13±3.74 | 10.73±1.19 | 33.20±7.68 | 6.20±1.21 | ||
| UCO+TH+Epo | 16.36±1.38 | 12.12±1.79 | 13.95±2.22 | 20.41±2.03 | 4.64±0.50 | ||
| Proliferation | Ki67 | Control | 3.00±0.86 | 6.87±2.11 | 0.33±0.11* | 0.00±0.00 | 0.50±0.50 |
| UCO | 9.91±5.83 | 12.26±2.96 | 2.15±0.39 | 1.79±0.74 | 2.67±1.84 | ||
| UCO+TH | 3.87±1.08 | 6.40±2.07 | 1.86±0.34 | 1.40±0.60 | 0.40±0.40 | ||
| UCO+TH+Epo | 2.16±0.65 | 4.99±1.76 | 1.55±0.32 | 1.14±0.60 | 1.00±0.65 | ||
Ab=antibody. UCO=umbilical cord occlusion. TH=therapeutic hypothermia. Epo=erythropoietin. Cerebella analysis made across the white and gray unless specified.
; significance in Mann-Whitney U test of UCO vs Control. Where p<0.05; ANOVA comparing UCO, UCO+TH & UCO+TH+ Epo then §,§§,§§§ designates the level of significance in a Dunnets post-test of UCO vs UCO+TH and #,##,### designates the level of significance in post-test of UCO vs UCO+TH+Epo.
Figure 4.
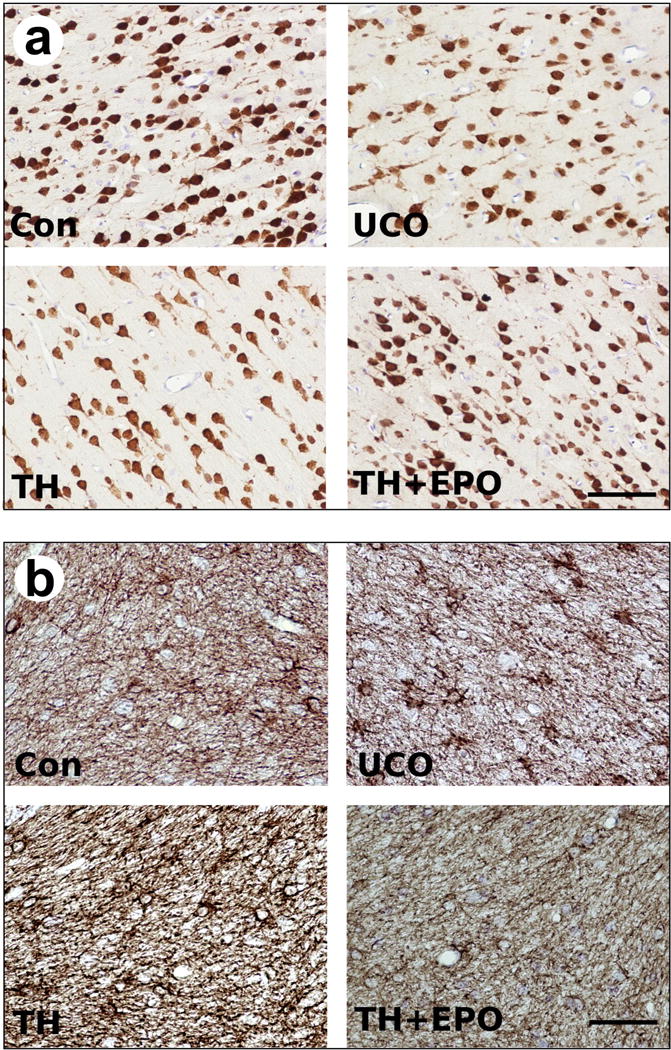
Representative images of immunohistological staining for a) NeuN and b) GFAP in the cerebellum for control cesarean delivered animals (Con), asphyxiated animals (UCO), asphyxiated animals treated with therapeutic hypothermia (TH) and asphyxiated animals treated with therapeutic hypothermia plus Epo (TH+Epo). Scale bar = 70um.
Secondary Outcome Measures: Effects of sex on CP diagnosis
When adjusting our data to proportions of animals suffering from CP in line with our secondary outcomes measures, we observed a strong association between a diagnosis of CP and male sex (Fisher’s exact test, p<0.0001; Figure 3b). As above, for body weight, no differences by sex were observed for CP versus no-CP animals.
Secondary Outcome Measures: Changes associated with CP via Magnetic Resonance Imaging
Fiber density, axonal diameter, and myelination were evaluated using FA measurements. FMRIB Software Library tract-based spatial statistics revealed changes in FA, first, second, and third eigenvalues, and mean diffusivity (MD) of CP and control animals (Figure 5) in the first 72 hours after injury.[33] Decreased FA was demonstrated in the corpus callosum, anterior and posterior limbs of the internal capsule, as well as multiple other white matter tracks in the animals with CP (6 CP and 4 control animals). To examine differences of brain structural integrity between control and CP animals, MD measures were obtained, which demonstrated increased signal in CP animals compared to controls. In animals with CP, both the MRI obtained within the first 72 hours of age and the MRI obtained at 9 months of age, demonstrated similar regions of decreased FA (Figure 5). No differences in brain volume between groups were present.
Figure 5.
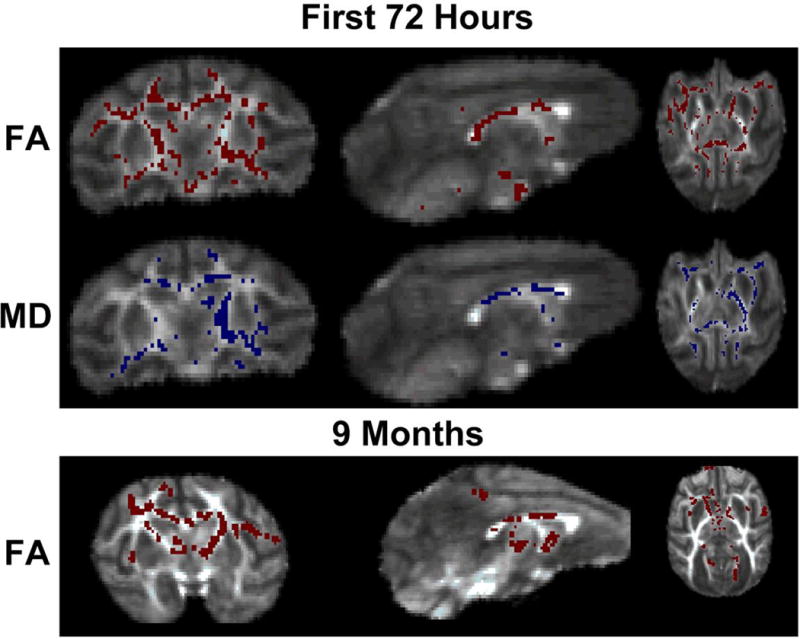
Tract-Based Spatial Statistics images show the average fractional anisotropy (FA) image of the subjects with red areas representing increased signal in control animals compared to animals with CP (p<0.05). Scans performed in the first 72 hours demonstrated decreased FA in the corpus callosum, anterior and posterior limbs of the internal capsule, as well as multiple other white matter tracks in the animals with CP (6 CP and 4 control). Blue areas represent regions of increased mean diffusivity (MD) in CP animals compared to controls (p<0.05). At nine months, animals with CP continued to have decreased FA in the same areas as found during the MRI taken within the first 72 hours of age (6 CP and 5 control).
Secondary Outcome Measures: Neuropathology associated with CP
No consistent qualitative abnormalities were observed on examination of H&E stained slides of the thalamic region examined from animals with CP (n=3: 1 mild, 1 moderate, and 1 severe) compared to cesarean section controls (n=4, 3 without CP, 1 mild CP). One UCO animal diagnosed with moderate CP had mild focally extensive white matter vacuolation and mild multifocal expansion of the arteriolar tunica media, of unknown significance (Figure 6).
Figure 6.
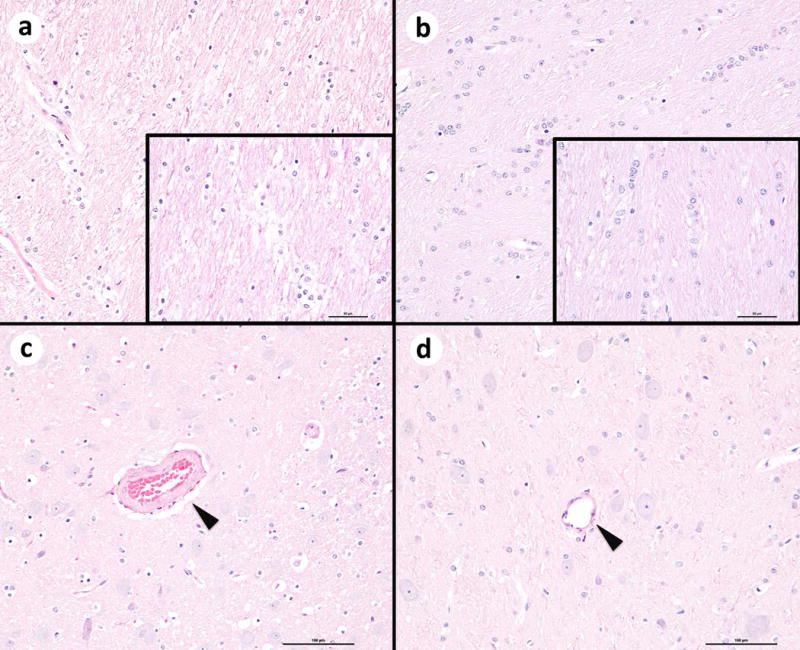
Hematoxylin and eosin (H&E) images of the brain at the level of the thalamus from two animals. Left side, umbilical cord occlusion (UCO) group animal with moderate CP; Right side, control animal. a White matter tracts show mild vacuolation and disorganization compared to the control animal (b). Original magnification 20x. Insets: Higher magnification image, (bar = 50 microns). c Within the gray matter, the tunica media of the vessel wall (arrow head) is mildly expanded in the UCO animal. Bar = 100 microns. d Representative vessel (arrow head) from the control animal. Bar = 100 microns.
In the cortex, compared to the no-CP classified animals, those with CP had decreased numbers of NeuN positive cells (p=0.0430), but increased numbers of Ki67 positive cell (p=0.001) as shown in Figure 7. Numbers of KI67 positive cells were also increased in the CP group in the hippocampus (p=0.0326). Also in the hippocampus, numbers of CD68 positive cells was decreased in CP animals compared to animals classified into no-CP (p=0.009). No other differences in the white matter (corona radiata), cortex, cerebellum, or hippocampus, including staining for Iba1 and MBP, were noted. A sex specific analysis of the neuropathological data revealed that in females there were no significant differences between CP and no-CP groups (supplementary figure 1). Analysis specifically of the males revealed that the effects from the whole group analysis remained for neurons in the cortex (p=0.0879), and additionally in the cerebellum (p=0.0173; supplementary figure 2). Further, in male CP animals, the observations of increased numbers of Ki67 positive cells in the cortex (p=0.120) and decreased numbers of CD68 positive cells in the hippocampus (p=0.0159) was larger than from the mix sex analysis. However, there was no longer any significant increase in hippocampal Ki67 expression (p=0.662).
Figure 7.
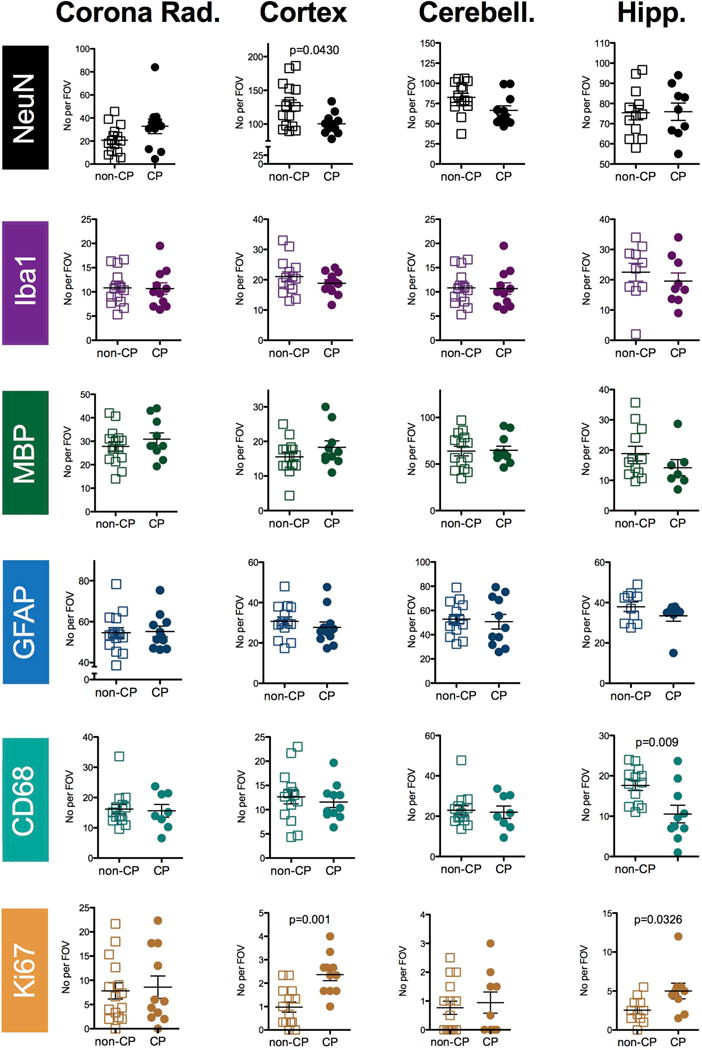
Scatter plots of the data from the immunohistochemical staining separated by outcome. Data from animals diagnosed with no CP or mild CP (‘non-CP’) versus animals diagnosed with moderate or severe CP (CP) for the corona radiate (Corona Rad.), the Cortex, the cerebellum (cerebell.) and the hippocampus (Hipp.) as outlined in Figure 2. Groups were compared with a Mann-Whitney U test and p values <0.05 are indicated on the graphs.
Secondary Outcome Measures: Changes in the brainstem associated with CP Brainstem scoring
Brainstems were analyzed in 12 animals (5 controls, 5 UCO, and 2 UCO+TH). Abnormalities were noted in all 5 animals with CP (3 UCO and 2 UCO+TH), which included one UCO animal who died at 270 days of age. One control animal without CP had a brainstem score of 1 (mild gliosis with no obvious neuronal loss). Animals with CP demonstrated neuronal loss and reactive astrocytes with increased cytoplasmic GFAP-positive staining compared to the control animals, as demonstrated in Figure 8g. A strong association between brainstem severity scores and CP severity scores was demonstrated by Spearman’s rank correlation coefficient (R = 0.973), as seen in Figure 8h.
Figure 8.
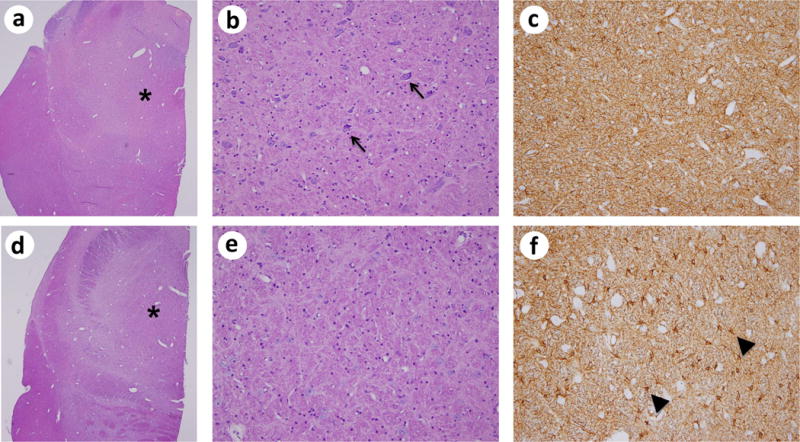
Brainstem Histology. Images a–c show the brainstem of a control animal taken at the level of the pons. Images d–f show the brainstem of a CP animal taken at the level of the pons. a and d are images of H&E stains taken at 2x magnification with an asterisks indicating the area of the pontine tegmentum from which images b and e (H&E, 20x) and c and f (GFAP, 20x) were taken. The high power H&E image of the CP animal (e) shows neuronal loss compared to the non-CP non-UCO animal (b)[examples of neurons in b are marked with arrows]. The GFAP staining in the control animal (c) shows the normal appearance of astrocytes with predominate staining in the fibers [feltwork pattern] with scattered detectable cell bodies. The GFAP staining in the CP animal (f) shows a substantial increase in reactive astrocytes with increased cytoplasmic GFAP-positive staining [examples of reactive astrocytes are marked with arrowheads]. In (g) brainstem histology severity scores (Y-axis) were assessed using H&E and GFAP stains with composite scores given as follows: 0, no pathology by H&E and GFAP (no neuronal loss by H&E; mainly feltwork pattern of GFAP staining); 1, mild gliosis with no obvious neuronal loss; 2, moderate gliosis with possible neuronal loss but mild; 3, severe injury with widespread gliosis and neuronal loss. CP severity scores (x axis) were characterized as: 1, normal (no CP); 2, mild; 3, moderate or; 4, severe CP or death. In (h), analysis of the brain stem and CP severity scores was demonstrated by Spearman’s rank correlation coefficient (R = 0.973).
Secondary Outcome Measures: Morphometric measurements of the cerebellum associated with CP
Cerebellar tissue was available from 4 CP animals and 4 animals without CP (no-CP; no-UCO control only). At 9 months of age, the external granular layer is not present. There were no differences in thickness of the molecular layer (CP 334 ± 56 μm vs no-CP controls 263 ± 26 μm), Purkinje cell density (CP 6.3 ± 0.8 cells/500 μm vs no-CP control 6.8 ± 1.0 cells/500 μm), or Bergman glial fiber density (CP 20.2± 1.5 fibers/100 μm vs no-CP control 18.2± 2.6 fibers/100 μm). The cell density of the internal granular layer and white matter was significantly decreased in CP animals (2-sided t-tests, p<0.05; Figure 9). No differences in cerebellar volume were present between groups.
Figure 9.
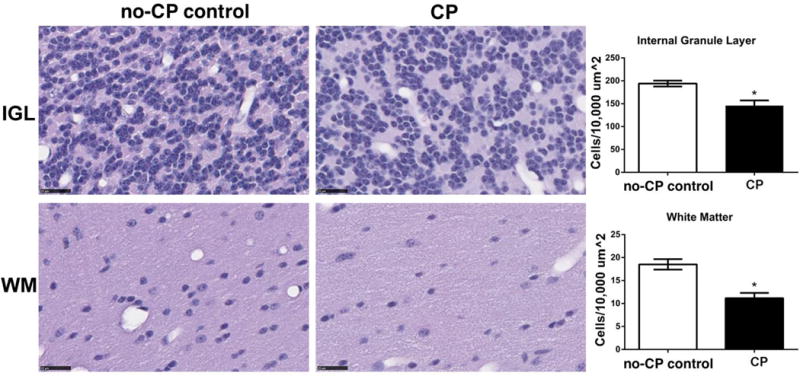
Cerebellar internal granule layer (IGL) and white matter (WM) tract cell density was compared between control (n=5) and animals that had CP (n=4). CP animals had decreased cell density in the IGL and WM as compared to control animals (*2-sided t-test, p<0.05,) Black bars are 25 μm long.
Discussion
The nonhuman primate brain shares a similar complexity and development to that of humans, which facilitates neurocognitive testing, analogous to testing done in humans, to be conducted over time [21]. These attributes make this UCO model attractive for studying the long-term neuropathology of CP related to HIE and the safety and efficacy of neurotherapeutics. We previously reported that compared to UCO saline, animals treated with TH+Epo had less death or CP at 9 months of age, with a relative risk reduction of 0.911 (95% CI –0.429 to 0.994), an absolute risk reduction of 0.395 (95% CI 0.072–0.635).[18] The primary purpose of the current study was to confirm the long-term safety and efficacy of TH+Epo, including neuropathological assessment. We found that indeed TH+Epo reduced the long-term risk of CP or death and that TH+Epo treated animals (compared to TH only) were not negatively impacted (somatic growth, head circumference, neuropathology scores). Based on IHC analysis there were no signs of long-term toxicity of TH+Epo, which supports further research of this combined treatment strategy to promote improved long-term outcomes in neonates with HIE.
Our secondary aim was to assess the neuropathology of animals who developed CP by 9 months of age, which is comparable to 3 years of human development, after exposure to UCO compared to animals without CP (i.e., control animals ± UCO animals with only mild CP). Animals with CP demonstrated decreased FA and increased mean diffusivity values across multiple white matter tracks including corpus callosum and internal capsule on MRI. In children with congenital hemiparesis and sensorimotor deficits, reduced FA and increased MD have been observed for precentral gyrus and paracentral lobule connections within the PLIC on the hemisphere contralateral to the impaired side.[34] The significance of the decrease FA in the anterior limb of the internal (ALIC) capsule, a region which contains thalamo-cortical, cortico-pontine fibers, and caudate/pallidum fibers, is not clear. Bilateral disruptions in the thalamo-cortical connections of the ALIC have been demonstrated in schizophrenia and have been associated with poor memory performance.[35] Decreased white matter FA is observed in infants diagnosed with HIE and the severity of the loss in FA is associated with long-term outcome [36].
In neonates with HIE, the correlation of DTI with brain pathology findings is not established, which prevents us from drawing significant conclusions regarding radiological–pathological correlations in our animals with CP. We did not observe changes in either oligodendrocyte cell number (Olig2 positive cell counts, data not shown) or myelin-staining intensity to directly support that decreased FA might relate to hypomyelination. However, important ultrastructural changes such as altered myelin-wrapping efficiency or altered G ratios (axon diameter/total fiber diameter) can affect FA values and are associated with perinatal brain injury [29,37]. Gliosis is considered an important contributor to perinatal injury following hypoxia-ischemia [38,39] and it is thought that this gliosis might persist, preventing repair and precipitating further neurodegeneration (see, [40]). However, there were no changes in the density of staining for astrocytes (GFAP) or the number of microglia (Iba1+). Given the potential relevance of activation state rather than morphology or number in an in vivo context our analysis might not have had the sensitivity to detect long-term (tertiary) changes in glia. In regards to the decreased macrophage/microglia number (CD68) in the hippocampus, in isolation the relevance of this change is purely speculative such as possibly mediating effects on memory by changing the local inflammatory milieu [41]. Similarly, there appeared to be a long term, but isolated effect of increased proliferation in the CP group. This occurred in the cortex, which suffered a reduction in mature neuronal numbers, and so it is tempting to simply suggest this reflects a long-term (failed) regenerative response, but this requires additional work to specifically characterize the cell type proliferating.
Animals with CP had decreased staining for cortical neurons and increased brainstem glial scarring compared to animals in the control group. We reported previously that UCO in this model was associated with reduced cerebellar volumes and that this was corrected with TH+Epo [18]. Providing neuropathological evidence to support our previous work, cerebellar cell density of the internal granular layer and white matter was decreased in CP animals compared to control animals.
There was a strong correlation between brainstem injury severity (neuronal loss and glial scarring) and CP severity. A possible explanation for this neuronal loss could be damaged neural pathways (e.g., motor cortex to brainstem) that led to demise of remote brainstem neurons following UCO-induced HIE. In a neonatal rat model, Reinebrant et al demonstrated that disrupted serotonergic inputs to the motor cortex resulted in brainstem serotonergic neuronal loss after hypoxic-ischemic injury.[42] Disruption of the brainstem and spinal cord serotonergic system has also been proposed to account for the motor defects observed in neonatal mice using the Rice– Vanucci hypoxic-ischemic injury model.[43] Future studies in our UCO model investigating damaged neural pathways, such as serotonergic system pathway, after neonatal HIE may reveal cellular mechanisms of axonal degeneration relevant to the development of CP.
An important observation from this study was the greater likelihood of males to suffer from CP following exposure to UCO. This observation is in agreement with clinical findings, including from the Surveillance of Cerebral Palsy in Europe (SCPE) that males are more likely to suffer CP following perinatal events such as HIE.[44,45] We noted (despite low power in this analysis) that effects specific to males drove the majority of the neuropathological changes in animals suffering CP in this model. Sex specific differences in basic cellular responses to insult may underpin some of the vulnerability to perinatal events [46] as may differences in the maturation of various regions altering susceptibility.[47] Unfortunately, only the overall outcome and IHC analysis were sufficiently powered to approach the question of sex specific effects.
Limited information on the long-term neuropathology of HIE-related brain injury in relation to cognitive impairments and CP is available to guide strategies to improve childhood outcomes. The inability to accurately determine the cause of HIE in human newborns remains a substantial shortcoming of clinical medicine. Not only is the timing and mechanism of brain injury based on speculation, but the prevention of HIE has been unattainable. Retrospective analyses of HIE in human newborns is typically hindered by conjecture about underlying causation, lack of any accurate information on duration and degree of insult(s), and nebulous explanations about key factors that predict, trigger, and perpetuate the pathologic processes related to HIE. While an imperfect model, the nonhuman primate UCO model of HIE has a clearly defined injury mechanism (UCO) and duration of initial insult (15 or 18 minutes of UCO), which allows the study of treatments initiated at uniform times across groups. Similar to humans, not all animals with HIE developed CP or had apparent long-term neurodevelopment deficits. Why some animals and human newborns tolerate severe hypoxic insults associated with HIE and others die or are permanently devastated remains a conundrum. While most CP cases are not secondary to HIE, the cases that are related have been associated with severe HIE based on Sarnat criteria.[48] Consistent with this relationship, animals subjected to UCO in our current study demonstrated findings consistent with severe HIE and went on to develop CP in 35% (12/34) cases. In this study,[48] TH did not reduce the long-term outcome of death or CP. Whether this discrepancy with human studies is due to the UCO model mechanism of injury, limited sample size, or other unrecognized factors remains unclear. One factor that must be considered is the duration of cooling, since 72 hours in a neonatal macaque is not equivalent to 72 hours in a human neonate, given that the rate of aging in these animals is approximately three times as fast as humans.[49] A recent phase III trial studying the effect of depth and duration of cooling on outcomes of moderate to severe HIE, demonstrated a higher adjusted risk ratio for NICU deaths in babies cooled for 120 hours compared to 72 hours (1.37; 95%CI, 0.92–2.04)[50]. Thus, the 72-hour duration of TH in the UCO+TH animals may have exceeded the window of benefit. While combined treatment with TH+Epo looks promising,[15,16] we suspect that additional strategies will be needed to mitigate long-term neurodevelopment injury in neonates with moderate-to-severe HIE.
Neonates with HIE typically demonstrate two main brain MRI injury patterns, a watershed predominant or a basal ganglia/thalamus pattern, as demonstrated by Miller et al who studied a cohort study of 173 term neonates with HIE in order to determine antenatal and perinatal risk factors and neurodevelopmental deficits associated with these patterns.[51] In their study, newborns delivered by emergent cesarean section who required intensive resuscitation at birth with more severe encephalopathy and seizures, had basal ganglia/thalamus injury patterns detected on MRI that were associated with the most impaired motor and cognitive outcome at 30 months. In our nonhuman primate study, UCO induced an acute, abrupt hypoxic insult associated with intensive resuscitation at birth and severe encephalopathy and seizures; however no pattern of gross brain MRI abnormalities was detected either early (days 1–3) or late (6 or 9 months).[18] Although these animals did not demonstrate basal ganglia/thalamus injury patterns typically associated with severe neuromotor and cognitive outcomes in human neonates, 35% (12/34) still developed CP (4 animals had moderate-to-severe CP and 9 had mild CP). The explanation for this apparent discrepancy between neuroimaging and clinical presentation is not clear. While many children with CP have abnormalities detectable on MRI, this is not always the case, as children with all clinical subtypes of CP, including severe functional severity, can have normal conventional MRIs.[52] A retrospective study by Numata et al investigating associations between neuroimaging findings, motor function, epileptic episodes, and cognitive function in 86 CP patients with spastic diplegia who were born at term found no abnormalities on conventional MRI in 41.9% of patients.[53] Using a population-based CP registry (Quebec, Canada), Benini et al studied 213 patients to identify distinctive clinical features characterizing children with CP and normal-appearing MRI findings.[54] They demonstrated that 42% (15 of 36) of the children with normal-appearing MRI displayed a high degree of functional disability (Gross Motor Functional Classification System IV–V), compared with 33% (30 of 90) with abnormal MRI and that normal-appearing MRI was more common in children with dyskinetic CP and less common in children spastic hemiplegic CP, compared with other CP variants. Abnormal connectivity may be important in the development of CP,[55] and unfortunately, MRI structural connectivity measurements to determine if animals with CP had disrupted sensorimotor pathways compared animals without CP was not conducted in our study.
This study has many limitations, including a small number of animals in each group due to the substantial expense of the model, differences in UCO durations (15 minutes in 9 animals, 18 minutes in 18 animals), and neuropathological analysis of only 34 of 56 animals (60.7%) from the parent study (only animals that survived to 9 months were examined). This later point is important as it means that we may not have detected the most severe neuropathological injuries as these lead to early death. During the course of the study, the UCO duration was changed from 15 to 18 minutes to promote more consistent and significant long-term neurodevelopmental disability important for testing neuroprotective strategies. However, the distribution of animals exposed to UCO for 15 or 18 minutes was consistent between the groups and across sexes. Since all animals received serial behavioral testing and physical therapy, long-term neurodevelopmental outcomes may have been modified by this therapy, a phenomenon that could affect interpretation of the long-term impact of early treatment (TH±Epo) vs serial physical therapy for newborns with HIE. This issue of an apparent enriched environment is important as physical therapy improves outcomes across preclinical models [56,57], and a varied home environment is standard for babies. As such, any therapy must be able to be effective in the context of an enriched environment. Another criticism of our study is that we included animals with mild CP in the ‘non-CP’ when we analyzed scatter plot data from immunohistochemical staining based on outcomes. Due to limited animal numbers, this ‘non CP’ group was compared to animals diagnosed with moderate or severe CP (CP). We recognize that humans with mild CP can have significant disabilities, including cognitive impairment, behavioral problems, and epilepsy, and therefore future, larger studies would benefit from not combining different categories of CP for analysis.
Another study limitation is the lack of IHC staining on thalamic tissue. Thalamic abnormalities were not seen on MRI or with H&E staining (except for mild changes in one animal with moderate CP). These findings likely exclude severe/frank thalamic lesions from being involved in the observed outcomes, but as IHC staining was not performed we cannot exclude subtle deep gray matter pathology. Brainstem histopathological analysis was limited to 12 animals and did not include any TH+Epo animals (8 total TH+Epo animals; 5 without CP, 3 with mild CP), which prevents assessment of this treatment effect. In addition, MRI analysis of the brainstem was not performed, which prevents radiologic correlation with histopathology findings. In addition, the project is important given the need for a robust model of HIE, although this study is limited in statistical power due to the ethical and practical limitations of a primate model. Part of the limitations of the statistics is that multiple regions and cell types were analyzed, increasing the chance of false positive results. However, our neuropathological assessments supplement strong observations on behavioral deficits with robust clinical applicability. Therefore, despite these limitations, this study offers valuable insight into the long-term brain neuropathology associated with CP secondary to HIE, findings that are not ascertainable in humans due to obvious ethical reasons and secondary to limitations of post-mortem human studies (including variability in postmortem delay and in underlying etiology).
In conclusion, animals with CP associated with severe HIE secondary to UCO demonstrated decreased FA of multiple white matter tracks including corpus callosum and internal capsule, decreased IHC staining for cortical neurons, and increased brainstem glial scarring compared to animals without CP. In addition, cerebellar cell density of the internal granular layer and white matter was decreased in CP animals compared to control animals without CP. However, our UCO model was not associated with a basal ganglia/thalamus injury pattern typical of neonates with HIE who develop CP, which suggests possible structural connectivity abnormalities. Future studies using this model should include MRI brainstem imaging, including structural connectivity measurements to evaluate for disrupted sensorimotor pathways that may play a key role in animals with CP.
Supplementary Material
Supplementary figure 1. Scatter plots of the data from the immunohistochemical staining of females only separated by outcome. Data from animals diagnosed with no CP or mild CP (‘non-CP’) versus animals diagnosed with moderate or severe CP (CP) for the corona radiate (Corona Rad.), the Cortex, the cerebellum (cerebell.) and the hippocampus (Hipp.) as outlined in Figure 2. Groups were compared with a Mann-Whitney U test and p values <0.05 are indicated on the graphs.
Supplementary figure 2. Scatter plots of the data from the immunohistochemical staining of males only separated by outcome. Data from animals diagnosed with no CP or mild CP (‘non-CP’) versus animals diagnosed with moderate or severe CP (CP) for the corona radiate (Corona Rad.), the Cortex, the cerebellum (cerebell.) and the hippocampus (Hipp.) as outlined in Figure 2. Groups were compared with a Mann-Whitney U test and p values <0.05 are indicated on the graphs.
Acknowledgments
We thank for Kylie Cory and Tom Gerbert for their animal care work, and Olivia Janson and Henry Smelser for technical assistance. This study was supported by the US NIH/Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01-HD-52820-01A2 and R01-HD-52820-01A3).
This study was supported by grants from NICHD: R01-HD-52820, and HD083091; Inserm, Université Paris Diderot, Université Sorbonne-Paris-Cité, DHU PROTECT, PremUP, Fondation de France, Fondation pour la Recherche sur le Cerveau, Roger de Spoelberch Foundation, Leducq Foundation, and the Wellcome Trust (WSCR P32674). In addition, the authors acknowledge financial support from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy’s & St Thomas' NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust. The supporting bodies played no role in any aspect of study design, analysis, interpretation or decision to publish this data.
References
- 1.Mortality GBD, Causes of Death C Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117–171. doi: 10.1016/S0140-6736(14)61682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurinczuk JJ, White-Koning M, Badawi N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev. 2010;86:329–338. doi: 10.1016/j.earlhumdev.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Locatelli A, Incerti M, Ghidini A, Greco M, Villa E, Paterlini G. Factors associated with umbilical artery acidemia in term infants with low Apgar scores at 5 min. Eur J Obstet Gynecol Reprod Biol. 2008;139:146–150. doi: 10.1016/j.ejogrb.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, Fanaroff AA, Poole WK, Wright LL, Higgins RD, Finer NN, Carlo WA, Duara S, Oh W, Cotten CM, Stevenson DK, Stoll BJ, Lemons JA, Guillet R, Jobe AH, National Institute of Child H, Human Development Neonatal Research N Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353:1574–1584. doi: 10.1056/NEJMcps050929. [DOI] [PubMed] [Google Scholar]
- 5.Gluckman PD, Wyatt JS, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, Polin RA, Robertson CM, Thoresen M, Whitelaw A, Gunn AJ. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365:663–670. doi: 10.1016/S0140-6736(05)17946-X. [DOI] [PubMed] [Google Scholar]
- 6.Azzopardi DV, Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, Kapellou O, Levene M, Marlow N, Porter E, Thoresen M, Whitelaw A, Brocklehurst P, Group TS Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med. 2009;361:1349–1358. doi: 10.1056/NEJMoa0900854. [DOI] [PubMed] [Google Scholar]
- 7.Jacobs SE, Morley CJ, Inder TE, Stewart MJ, Smith KR, McNamara PJ, Wright IM, Kirpalani HM, Darlow BA, Doyle LW, Infant Cooling Evaluation C Whole-body hypothermia for term and near-term newborns with hypoxic-ischemic encephalopathy: a randomized controlled trial. Arch Pediatr Adolesc Med. 2011;165:692–700. doi: 10.1001/archpediatrics.2011.43. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG. Cooling for newborns with hypoxic ischaemic encephalopathy. The Cochrane database of systematic reviews. 2013;1:CD003311. doi: 10.1002/14651858.CD003311.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunn AJ, Gluckman PD, Gunn TR. Selective head cooling in newborn infants after perinatal asphyxia: a safety study. Pediatrics. 1998;102:885–892. doi: 10.1542/peds.102.4.885. [DOI] [PubMed] [Google Scholar]
- 10.Eicher DJ, Wagner CL, Katikaneni LP, Hulsey TC, Bass WT, Kaufman DA, Horgan MJ, Languani S, Bhatia JJ, Givelichian LM, Sankaran K, Yager JY. Moderate hypothermia in neonatal encephalopathy: safety outcomes. Pediatr Neurol. 2005;32:18–24. doi: 10.1016/j.pediatrneurol.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 11.Simbruner G, Mittal RA, Rohlmann F, Muche R. neo.n EnTP: Systemic hypothermia after neonatal encephalopathy: outcomes of neo. nEURO.network RCT Pediatrics. 2010;126:e771–778. doi: 10.1542/peds.2009-2441. [DOI] [PubMed] [Google Scholar]
- 12.Zhou WH, Cheng GQ, Shao XM, Liu XZ, Shan RB, Zhuang DY, Zhou CL, Du LZ, Cao Y, Yang Q, Wang LS, China Study G Selective head cooling with mild systemic hypothermia after neonatal hypoxic-ischemic encephalopathy: a multicenter randomized controlled trial in China. The Journal of pediatrics. 2010;157:367–372. 372, e361–363. doi: 10.1016/j.jpeds.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 13.Zhu C, Kang W, Xu F, Cheng X, Zhang Z, Jia L, Ji L, Guo X, Xiong H, Simbruner G, Blomgren K, Wang X. Erythropoietin improved neurologic outcomes in newborns with hypoxic-ischemic encephalopathy. Pediatrics. 2009;124:e218–226. doi: 10.1542/peds.2008-3553. [DOI] [PubMed] [Google Scholar]
- 14.Elmahdy H, El-Mashad AR, El-Bahrawy H, El-Gohary T, El-Barbary A, Aly H. Human recombinant erythropoietin in asphyxia neonatorum: pilot trial. Pediatrics. 2010;125:e1135–1142. doi: 10.1542/peds.2009-2268. [DOI] [PubMed] [Google Scholar]
- 15.Wu YW, Bauer LA, Ballard RA, Ferriero DM, Glidden DV, Mayock DE, Chang T, Durand DJ, Song D, Bonifacio SL, Gonzalez FF, Glass HC, Juul SE. Erythropoietin for neuroprotection in neonatal encephalopathy: safety and pharmacokinetics. Pediatrics. 2012;130:683–691. doi: 10.1542/peds.2012-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu YW, Mathur AM, Chang T, McKinstry RC, Mulkey SB, Mayock DE, Van Meurs KP, Rogers EE, Gonzalez FF, Comstock BA, Juul SE, Msall ME, Bonifacio SL, Glass HC, Massaro AN, Dong L, Tan KW, Heagerty PJ, Ballard RA. High-Dose Erythropoietin and Hypothermia for Hypoxic-Ischemic Encephalopathy: A Phase II Trial. Pediatrics. 2016;137 doi: 10.1542/peds.2016-0191. [DOI] [PubMed] [Google Scholar]
- 17.Juul SE, Aylward E, Richards T, McPherson RJ, Kuratani J, Burbacher TM. Prenatal cord clamping in newborn Macaca nemestrina: a model of perinatal asphyxia. Dev Neurosci. 2007;29:311–320. doi: 10.1159/000105472. [DOI] [PubMed] [Google Scholar]
- 18.Traudt CM, McPherson RJ, Bauer LA, Richards TL, Burbacher TM, McAdams RM, Juul SE. Concurrent erythropoietin and hypothermia treatment improve outcomes in a term nonhuman primate model of perinatal asphyxia. Dev Neurosci. 2013;35:491–503. doi: 10.1159/000355460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kattwinkel JE. Textbook of Neonatal Resuscitation. 6th. Elk Grove Village, IL/ Dallas, TX: American Heart Association/American Academy of Pediatrics; 2011. [Google Scholar]
- 20.Jacobson Misbe EN, Richards TL, McPherson RJ, Burbacher TM, Juul SE. Perinatal asphyxia in a nonhuman primate model. Dev Neurosci. 2011;33:210–221. doi: 10.1159/000327246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burbacher TM, Grant KS. Methods for studying nonhuman primates in neurobehavioral toxicology and teratology. Neurotoxicol Teratol. 2000;22:475–486. doi: 10.1016/s0892-0362(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 22.Smith SM, Jenkinson M, Johansen-Berg H, Rueckert D, Nichols TE, Mackay CE, Watkins KE, Ciccarelli O, Cader MZ, Matthews PM, Behrens TEJ. Tract-based spatial statistics: Voxelwise analysis of multi-subject diffusion data. Neuroimage. 2006;31:1487–1505. doi: 10.1016/j.neuroimage.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 23.Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TEJ, Johansen-Berg H, Bannister PR, De Luca M, Drobnjak I, Flitney DE, Niazy RK, Saunders J, Vickers J, Zhang Y, De Stefano N, Brady JM, Matthews PM. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23(Supplement 1):S208–S219. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 24.Smith SM. Fast robust automated brain extraction. Human Brain Mapping. 2002;17:143–155. doi: 10.1002/hbm.10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersson JLR, Jenkinson M, Smith S. Non-linear optimisation. 2007 FMRIB technical report TR07JA1 from www.fmriboxacuk/analysis/techrep.
- 26.Andersson JLR, Jenkinson M, Smith S. Non-linear registration, aka Spatial normalisation. 2007 FMRIB technical report TR07JA2 from www.fmriboxacuk/analysis/techrep.
- 27.Rueckert D, Sonoda LI, Hayes C, Hill DL, Leach MO, Hawkes DJ. Nonrigid registration using free-form deformations: application to breast MR images. IEEE Trans Med Imaging. 1999;18:712–721. doi: 10.1109/42.796284. [DOI] [PubMed] [Google Scholar]
- 28.Smith SM, Jenkinson M, Johansen-Berg H, Rueckert D, Nichols TE, Mackay CE, Watkins KE, Ciccarelli O, Cader MZ, Matthews PM, Behrens TE. Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data. Neuroimage. 2006;31:1487–1505. doi: 10.1016/j.neuroimage.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 29.Favrais G, van de Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, Lacaud A, Saliba E, Dammann O, Gallego J, Sizonenko S, Hagberg H, Lelievre V, Gressens P. Systemic inflammation disrupts the developmental program of white matter. Ann Neurol. 2011;70:550–565. doi: 10.1002/ana.22489. [DOI] [PubMed] [Google Scholar]
- 30.Ezzati M, Bainbridge A, Broad KD, Kawano G, Oliver-Taylor A, Rocha-Ferreira E, Alonso-Alconada D, Fierens I, Rostami J, Hassell KJ, Tachtsidis I, Gressens P, Hristova M, Bennett K, Lebon S, Fleiss B, Yellon D, Hausenloy DJ, Golay X, Robertson1 NJ. Immediate remote ischemic postconditioning after hypoxia ischemia in piglets protects cerebral white matter but not grey matter. J Cereb Blood Flow Metab. 2015 doi: 10.1177/0271678X15608862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fleiss B, Coleman HA, Castillo-Melendez M, Ireland Z, Walker DW, Parkington HC. Effects of birth asphyxia on neonatal hippocampal structure and function in the spiny mouse. Int J Dev Neurosci. 2011;29:757–766. doi: 10.1016/j.ijdevneu.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Gressens P, Dingley J, Plaisant F, Porter H, Schwendimann L, Verney C, Tooley J, Thoresen M. Analysis of neuronal, glial, endothelial, axonal and apoptotic markers following moderate therapeutic hypothermia and anesthesia in the developing piglet brain. Brain Pathol. 2008;18:10–20. doi: 10.1111/j.1750-3639.2007.00095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Douaud G, Jbabdi S, Behrens TEJ, Menke RA, Gass A, Monsch AU, Rao A, Whitcher B, Kindlmann G, Matthews PM, Smith S. DTI measures in crossing-fibre areas: Increased diffusion anisotropy reveals early white matter alteration in MCI and mild Alzheimer’s disease. Neuroimage. 2011;55:880–890. doi: 10.1016/j.neuroimage.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsao H, Pannek K, Fiori S, Boyd RN, Rose S. Reduced integrity of sensorimotor projections traversing the posterior limb of the internal capsule in children with congenital hemiparesis. Res Dev Disabil. 2014;35:250–260. doi: 10.1016/j.ridd.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 35.Rosenberger G, Nestor PG, Oh JS, Levitt JJ, Kindleman G, Bouix S, Fitzsimmons J, Niznikiewicz M, Westin CF, Kikinis R, McCarley RW, Shenton ME, Kubicki M. Anterior limb of the internal capsule in schizophrenia: a diffusion tensor tractography study. Brain Imaging Behav. 2012;6:417–425. doi: 10.1007/s11682-012-9152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tusor N, Wusthoff C, Smee N, Merchant N, Arichi T, Allsop JM, Cowan FM, Azzopardi D, Edwards AD, Counsell SJ. Prediction of neurodevelopmental outcome after hypoxic-ischemic encephalopathy treated with hypothermia by diffusion tensor imaging analyzed using tract-based spatial statistics. Pediatr Res. 2012;72:63–69. doi: 10.1038/pr.2012.40. [DOI] [PubMed] [Google Scholar]
- 37.Billiards SS, Haynes RL, Folkerth RD, Borenstein NS, Trachtenberg FL, Rowitch DH, Ligon KL, Volpe JJ, Kinney HC. Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathol. 2008;18:153–163. doi: 10.1111/j.1750-3639.2007.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delcour M, Olivier P, Chambon C, Pansiot J, Russier M, Liberge M, Xin D, Gestreau C, Alescio-Lautier B, Gressens P, Verney C, Barbe MF, Baud O, Coq JO. Neuroanatomical, Sensorimotor and Cognitive Deficits in Adult Rats with White Matter Injury Following Prenatal Ischemia. Brain Pathol. 2012;22:1–16. doi: 10.1111/j.1750-3639.2011.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shrivastava K, Chertoff M, Llovera G, Recasens M, Acarin L. Short and long-term analysis and comparison of neurodegeneration and inflammatory cell response in the ipsilateral and contralateral hemisphere of the neonatal mouse brain after hypoxia/ischemia. Neurology research international. 2012;2012:781512. doi: 10.1155/2012/781512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fleiss B, Gressens P. Tertiary mechanisms of brain damage: a new hope for treatment of cerebral palsy? The Lancet Neurology. 2012;11:556–566. doi: 10.1016/S1474-4422(12)70058-3. [DOI] [PubMed] [Google Scholar]
- 41.Bilbo SD, Barrientos RM, Eads AS, Northcutt A, Watkins LR, Rudy JW, Maier SF. Early-life infection leads to altered BDNF and IL-1beta mRNA expression in rat hippocampus following learning in adulthood. Brain Behav Immun. 2008;22:451–455. doi: 10.1016/j.bbi.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 42.Reinebrant HE, Wixey JA, Buller KM. Disruption of raphe serotonergic neural projections to the cortex: a potential pathway contributing to remote loss of brainstem neurons following neonatal hypoxic-ischemic brain injury. Eur J Neurosci. 2012;36:3483–3491. doi: 10.1111/j.1460-9568.2012.08276.x. [DOI] [PubMed] [Google Scholar]
- 43.Bellot B, Peyronnet-Roux J, Gire C, Simeoni U, Vinay L, Viemari JC. Deficits of brainstem and spinal cord functions after neonatal hypoxia-ischemia in mice. Pediatr Res. 2014;75:723–730. doi: 10.1038/pr.2014.42. [DOI] [PubMed] [Google Scholar]
- 44.Surveillance of Cerebral Palsy in E. Surveillance of cerebral palsy in Europe: a collaboration of cerebral palsy surveys and registers. Surveillance of Cerebral Palsy in Europe (SCPE) Dev Med Child Neurol. 2000;42:816–824. doi: 10.1017/s0012162200001511. [DOI] [PubMed] [Google Scholar]
- 45.Johnston MV, Hagberg H. Sex and the pathogenesis of cerebral palsy. Dev Med Child Neurol. 2007;49:74–78. doi: 10.1017/s0012162207000199.x. [DOI] [PubMed] [Google Scholar]
- 46.Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, Graham SH, Clark RS. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem. 2004;279:38563–38570. doi: 10.1074/jbc.M405461200. [DOI] [PubMed] [Google Scholar]
- 47.Cerghet M, Skoff RP, Swamydas M, Bessert D. Sexual dimorphism in the white matter of rodents. J Neurol Sci. 2009;286:76–80. doi: 10.1016/j.jns.2009.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pappas A, Korzeniewski SJ. Long-Term Cognitive Outcomes of Birth Asphyxia and the Contribution of Identified Perinatal Asphyxia to Cerebral Palsy. Clin Perinatol. 2016;43:559–572. doi: 10.1016/j.clp.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 49.Roth GS, Mattison JA, Ottinger MA, Chachich ME, Lane MA, Ingram DK. Aging in rhesus monkeys: relevance to human health interventions. Science. 2004;305:1423–1426. doi: 10.1126/science.1102541. [DOI] [PubMed] [Google Scholar]
- 50.Shankaran S, Laptook AR, Pappas A, McDonald SA, Das A, Tyson JE, Poindexter BB, Schibler K, Bell EF, Heyne RJ, Pedroza C, Bara R, Van Meurs KP, Grisby C, Huitema CM, Garg M, Ehrenkranz RA, Shepherd EG, Chalak LF, Hamrick SE, Khan AM, Reynolds AM, Laughon MM, Truog WE, Dysart KC, Carlo WA, Walsh MC, Watterberg KL, Higgins RD, Eunice Kennedy Shriver National Institute of Child H, Human Development Neonatal Research N Effect of depth and duration of cooling on deaths in the NICU among neonates with hypoxic ischemic encephalopathy: a randomized clinical trial. JAMA. 2014;312:2629–2639. doi: 10.1001/jama.2014.16058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller SP, Ramaswamy V, Michelson D, Barkovich AJ, Holshouser B, Wycliffe N, Glidden DV, Deming D, Partridge JC, Wu YW, Ashwal S, Ferriero DM. Patterns of brain injury in term neonatal encephalopathy. The Journal of pediatrics. 2005;146:453–460. doi: 10.1016/j.jpeds.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 52.Bax M, Tydeman C, Flodmark O. Clinical and MRI correlates of cerebral palsy: the European Cerebral Palsy Study. JAMA. 2006;296:1602–1608. doi: 10.1001/jama.296.13.1602. [DOI] [PubMed] [Google Scholar]
- 53.Numata Y, Onuma A, Kobayashi Y, Sato-Shirai I, Tanaka S, Kobayashi S, Wakusawa K, Inui T, Kure S, Haginoya K. Brain magnetic resonance imaging and motor and intellectual functioning in 86 patients born at term with spastic diplegia. Developmental medicine and child neurology. 2013;55:167–172. doi: 10.1111/dmcn.12013. [DOI] [PubMed] [Google Scholar]
- 54.Benini R, Dagenais L, Shevell MI, Registre de la Paralysie Cerebrale au Quebec C Normal imaging in patients with cerebral palsy: what does it tell us? The Journal of pediatrics. 2013;162:369–374. e361. doi: 10.1016/j.jpeds.2012.07.044. [DOI] [PubMed] [Google Scholar]
- 55.Rose S, Guzzetta A, Pannek K, Boyd R. MRI structural connectivity, disruption of primary sensorimotor pathways, and hand function in cerebral palsy. Brain Connect. 2011;1:309–316. doi: 10.1089/brain.2011.0034. [DOI] [PubMed] [Google Scholar]
- 56.Johansson BB. Functional outcome in rats transferred to an enriched environment 15 days after focal brain ischemia. Stroke. 1996;27:324–326. doi: 10.1161/01.str.27.2.324. [DOI] [PubMed] [Google Scholar]
- 57.Will B, Galani R, Kelche C, Rosenzweig MR. Recovery from brain injury in animals: relative efficacy of environmental enrichment, physical exercise or formal training (1990–2002) Prog Neurobiol. 2004;72:167–182. doi: 10.1016/j.pneurobio.2004.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. Scatter plots of the data from the immunohistochemical staining of females only separated by outcome. Data from animals diagnosed with no CP or mild CP (‘non-CP’) versus animals diagnosed with moderate or severe CP (CP) for the corona radiate (Corona Rad.), the Cortex, the cerebellum (cerebell.) and the hippocampus (Hipp.) as outlined in Figure 2. Groups were compared with a Mann-Whitney U test and p values <0.05 are indicated on the graphs.
Supplementary figure 2. Scatter plots of the data from the immunohistochemical staining of males only separated by outcome. Data from animals diagnosed with no CP or mild CP (‘non-CP’) versus animals diagnosed with moderate or severe CP (CP) for the corona radiate (Corona Rad.), the Cortex, the cerebellum (cerebell.) and the hippocampus (Hipp.) as outlined in Figure 2. Groups were compared with a Mann-Whitney U test and p values <0.05 are indicated on the graphs.