

Abstract
Continuous‐flow multistep synthesis is combined with quasi‐continuous final‐product purification to produce pure products from crude reaction mixtures. In the nucleophilic aromatic substitution of 2,4‐difluoronitrobenzene with morpholine followed by a heterogeneous catalytic hydrogenation, the desired monosubstituted product can be continuously separated from the co‐ and by‐products in a purity of over 99 % by coupling a flow reactor sequence to a multiple dual‐mode (MDM) centrifugal partition chromatography (CPC) device. This purification technique has many advantages over HPLC, such as higher resolution and no need for column replacement or silica recycling, and it does not suffer from irreversible adsorption.
Keywords: centrifugal partition chromatography, continuous flow chemistry, multistep synthesis, purification
The continuous‐flow synthesis of active pharmaceutical ingredients (APIs) and their intermediates1, 2, 3, 4, 5, 6, 7, 8, 9 is actively encouraged by regulatory agencies,10 and has many advantages over batch processing.11, 12 However, the continuous manufacturing of the final dosage form of drugs by coupling the synthesis with formulation demands highly pure APIs. Consequently, continuous‐flow purification is inevitable in most cases. Nonetheless, continuous synthesis is usually followed by “discontinuous” purification because the number of available options for continuous purification is limited.1, 13 The existing methods6, 14 can be classified as in‐line work‐up and final‐product purification depending on their primary place of application within a multistep sequence.1 In‐line work‐up can remove co‐products while it cannot eliminate by‐products that are structurally related to the desired product. High purity can be achieved by final‐product purification by multicolumn chromatography,15 simulated moving bed (SMB) chromatography,16, 17, 18, 19 catch‐and‐release chromatography,20, 21, 22, 23 crystallization,15, 19, 24, 25, 26 or recrystallization,2, 27 although these methods have their drawbacks. Crystallization usually requires semi‐batch processing, and catch‐and‐release chromatography can only be categorized as a truly continuous purification method when automated switching between multiple columns is employed.28 The operation of SMB chromatography is technically complex; furthermore, it utilizes expensive solid adsorbents, and challenging separations may require additional crystallization.15, 18
Centrifugal partition chromatography (CPC) is a countercurrent separation technique29, 30, 31, 32, 33 that is widely used for the purification of natural products, small molecules, and biological compounds. CPC does not require a solid stationary phase as two non‐miscible phases are applied instead; one of them is used as the mobile phase and the other one as the stationary phase, which is maintained inside the rotating column by centrifugal forces. In the ascending mode (AM), the upper (lighter) phase is the mobile phase and the lower (denser) phase is the stationary phase, whereas in the descending mode (DM), the opposite holds (Figure 1). Batchwise separations can be converted into semi‐continuous purification by using the multiple dual‐mode (MDM) approach,30, 34, 35, 36, 37 which means that the liquid nature of the stationary phase is used to regenerate it by inversing the stationary and mobile phases multiple times and re‐injecting the sample solution in between. Choosing the most suitable biphasic liquid system (BLS) is like choosing the column and eluent in high‐performance liquid chromatography (HPLC). As a rule of thumb, the partition coefficients should be around 0.5–1.5 for the compound of interest, and the settling time of the phases should not exceed 20 s.38
Figure 1.
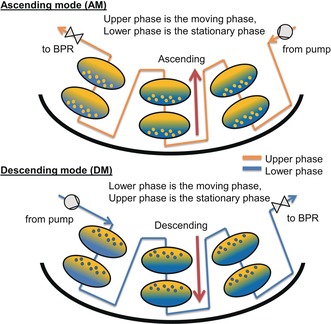
Working principle of AM and DM CPC devices.
CPC has many advantages over HPLC, such as higher resolution, no need for column replacement or silica recycling, and it is absolutely free of irreversible adsorption.38
To address the purification issues currently faced in flow synthesis, we decided to develop a new continuous final‐product purification method based on CPC. Herein, we report the first successful coupling of a multistep flow synthesis and MDM CPC to accomplish the quasi‐continuous purification and production of a pure product. This paper is organized into five sections as follows: flow reaction, finding the proper BLS, CPC method development, automation, and coupling the reaction to the purification.
The target molecule, 4‐fluoro‐2‐(morpholin‐4‐yl)aniline (5 a), which is a key intermediate in the synthesis of bioactive carbazoles,39 was synthesized in a nucleophilic aromatic substitution (SNAr) reaction16 of 2,4‐difluoronitrobenzene (1) with morpholine (2) followed by heterogeneous catalytic hydrogenation (Figure 2).
Figure 2.
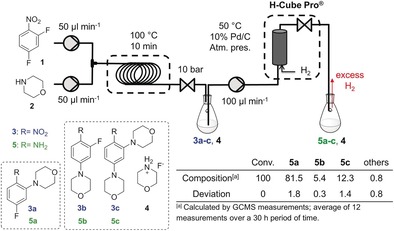
Continuous‐flow SNAr reaction of 2,4‐difluoronitrobenzene (1) with morpholine (2) followed by a heterogeneous continuous hydrogenation of the nitro compounds 3 a–3 c to the corresponding anilines 5 a–5 c using a loop reactor and the H‐Cube Pro™ device.
The first reaction step was performed in ethanol (EtOH) at 100 °C with a residence time of 10 min in a loop reactor connected to a Zaiput® back pressure regulator adjusted to 10 bar. The resulting crude reaction mixture of compounds 3 a–3 c and the morpholine hydrofluoride salt (4) was introduced into an H‐Cube Pro™ reactor containing a 10 % Pd/C cartridge at 50 °C and atmospheric pressure. The product (5 a) and all of the intermediates (3 a–3 c) and by‐products (5 b and 5 c) were isolated and characterized (see the Supporting Information). The average 5 a content of the reaction mixture was about 81 % along with 5 % of 5 b and 12 % of 5 c.
First, an adequate BLS was developed through extensive experimentation to differentiate between the regioisomers 5 a and 5 b, which are similar in every physicochemical property, including the pK a value (Table 1). A mixture of n‐hexane (n‐Hex), methyl tert‐butyl ether (MTBE), EtOH, and water in a ratio of 1:1:1:1 (v/v) gave ideal partition coefficients (K U/L) for anilines 5 a–5 c (Table 1) and exhibited a short settling time of 16 s (see the Supporting Information).
Table 1.
Measured physicochemical parameters of anilines 5 a–5 c.
| Entry | Parameter | 5 a | 5 b | 5 c |
|---|---|---|---|---|
| 1 | K U/L [a] | 1.86 | 0.49 | 0.24 |
| 2 | pK a [b] | 4.08±0.015 | 4.06±0.029 | 4.76±0.023 |
[a] The partition coefficients (K U/L) were determined by GCMS measurements in the biphasic solvent system n‐Hex/MTBE/EtOH/H2O (1:1:1:1, v/v); K U/L=peak area of the compound in the upper phase divided by the peak area of the compound in the lower phase. [b] The pK a values were determined by UV spectrophotometric titrations (see the Supporting Information).
Employing the chosen BLS in the initial batchwise CPC experiments performed on a 100 mL capacity column (Armen SCPC‐100+1000‐B apparatus with a SpotPrepII system) showed practically baseline separation for the product both in AM and DM. The operating conditions on the equilibrated column were 5–10 mL sample injection, a mobile‐phase flow rate of 5 mL min−1, and a rotation speed of 2000 rpm. Owing to the distinctively higher partition coefficient of the desired product (5 a) as compared to the by‐products 5 b and 5 c, the product was eluted first in AM (the upper phase is the mobile phase), and eluted last in DM (the lower phase is the mobile phase), which is ideal for our purification purposes.
Using these optimized conditions without modification, an MDM method was developed for quasi‐continuous purification. After the column had been equilibrated and the first sample injection had taken place, the by‐products 5 b and 5 c were simply washed out from the column in DM. Next, the sample solution was injected into the column again, and finally, the product from both injections was eluted and collected in AM. This process could be repeatedly reversed several times, without post‐washing and equilibration of the column between cycles. The efficiency and recovery were not affected as compared to the single AM and DM separation approach. In this way, stable, uninterrupted MDM CPC separation was conducted for more than 5 h. The purity of the product was more than 99.9 % (GCMS), and the recovery of 5 a was 91 %.
To connect the reaction stream with the purification unit, it was essential to automate the sample intake, which was enabled by the programmable magnetic valves of the SpotPrepII device. Owing to the increased dead volume before the column, a prolonged elution time was necessary in AM to achieve the same recovery.
To match the composition of the sample intake of the CPC separation with the output of the continuous‐flow reactor, the EtOH solution of the product was mixed with the other components of the chosen BLS. After phase separation using a separating funnel, the upper and lower phases of the resulting biphasic mixture were separately introduced into the CPC device (Figure 3 A) according to the program (see the Supporting Information). The phase separation unit served as a buffer flask, and also allowed the escape of excess hydrogen from the reduction step (which would otherwise be forcing out the liquid from the CPC column). To achieve overall continuous operation, the total inlet throughput of the reaction stream and the other components of the sample solution into the buffer flask must be equal or greater than the throughput of the outlet over a certain period of time. For this purpose, the elution times (both in AM and DM), the time and flow rate of the sample intakes, the flow rates of the reaction stream and the other components of the sample solution, and their volume contraction factor were considered.
Figure 3.
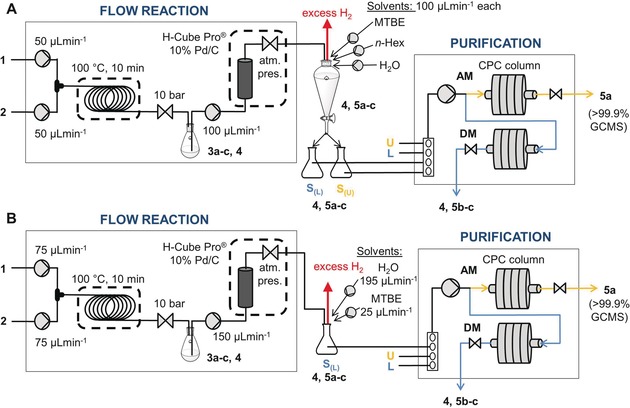
Flow chart of the two‐step synthesis followed by a quasi‐continuous MDM CPC purification using A) a two‐phase sample intake or B) a one‐phase sample intake. U=upper phase of the chosen BLS, L=lower phase of the chosen BLS, S(L)=sample solution in the lower phase, S(u)=sample solution in the upper phase.
The whole system (two‐step reaction and purification) could be continuously operated, and the yield of isolated product, its purity, and the productivity values were satisfactory (Table 2, entry 1).
Table 2.
Results obtained with the system combining the two‐step synthesis with purification.
| Entry | Sample intake method | Yield[a] [%] | Purity[b] [%] | Productivity[c] [g h−1 L−1] |
|---|---|---|---|---|
| 1 | two‐phase[d] | 57 | >99.9 | 1.44 |
| 2 | one‐phase[e] | 59 | >99.9 | 2.27 |
To increase the productivity by increasing the sample solution concentration and its throughput, a one‐phase sample intake method was developed (Figure 3 B). The sample solution was prepared as a single‐phase mixture of the reaction stream in EtOH and combined with MTBE and water using two additional pumps (flow rates of 195, 25, and 150 μL min−1 for H2O, MTBE, and EtOH, respectively) in a composition that corresponds to the lower phase of the BLS (the ratios were determined by GC‐FID or 1H NMR spectroscopy for the organic compounds and Karl Fischer titration for the water content; see the Supporting Information). The more concentrated sample solution and the higher throughput of the reaction stream gave a productivity that was 60 % higher (Table 2, entry 2) than that of the two‐phase sample intake method.
In summary, we have developed a system for the multistep continuous‐flow synthesis and purification of a complex reaction mixture, utilizing quasi‐continuous multiple dual‐mode centrifugal partition chromatography, which can be operated in a truly continuous manner by using buffer flasks and a few pumps (see SI) and by synchronizing the flow reaction with the purification. The productivity was increased significantly by the one‐phase intake of the sample solution.
The throughput could be easily increased by scaling up the column capacity40, 41, 42, 43 or by converting it into a true moving‐bed system44, 45, 46, 47 by introducing the sample solution continuously into the intermediate point of the column (e.g., between two columns). This system is the first continuous‐flow adsorbent‐free final‐product purification technique, and should find wide applicability in the synthesis of APIs or intermediates thereof.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
R.Ö. thanks Gedeon Richter Talentum and the Pro Progressio Foundation for financial support. Special thanks go to the RotaChrom Technológiai Kft. for consultation.
R. Örkényi, J. Éles, F. Faigl, P. Vincze, A. Prechl, Z. Szakács, J. Kóti, I. Greiner, Angew. Chem. Int. Ed. 2017, 56, 8742.
Contributor Information
Róbert Örkényi, Email: orkenyi.robert@gmail.com.
Dr. István Greiner, Email: i.greiner@richter.hu.
References
- 1.P. Bana, R. Örkényi, K. Lövei, Á. Lakó, G. I. Túrós, J. Éles, F. Faigl, I. Greiner, Bioorg. Med. Chem 2016, https://doi.org/10.1016/j.bmc.2016.12.046. [DOI] [PubMed]
- 2. Adamo A. et al., Science 2016, 352, 61–67. [DOI] [PubMed] [Google Scholar]
- 3. Baumann M., Baxendale I. R., Beilstein J. Org. Chem. 2015, 11, 1194–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsubogo T., Oyamada H., Kobayashi S., Nature 2015, 520, 329–332. [DOI] [PubMed] [Google Scholar]
- 5. Kobayashi S. S., Chem. Asian J. 2016, 11, 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ley S. V., Fitzpatrick D. E., Ingham R. J., Myers R. M., Angew. Chem. Int. Ed. 2015, 54, 3449–3464; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3514–3530. [Google Scholar]
- 7. Ley S. V., Fitzpatrick D. E., Myers R. M., Battilocchio C., Ingham R. J., Angew. Chem. Int. Ed. 2015, 54, 10122–10136; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10260–10275. [Google Scholar]
- 8. Webb D., Jamison T. F., Chem. Sci. 2010, 1, 675. [Google Scholar]
- 9. Gutmann B., Cantillo D., Kappe C. O., Angew. Chem. Int. Ed. 2015, 54, 6688–6728; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6788–6832. [Google Scholar]
- 10.Z. Brennan, “http://www.in-pharmatechnologist.com/Processing/FDA-calls-on-manufacturers-to-begin-switch-from-batch-to-continuous-production”, 2015.
- 11. Darvas F., Hessel V., Dormán G., Flow Chemistry, Vol. 1, De Gruyter, Berlin, 2014. [Google Scholar]
- 12. Darvas F., Dormán G., Fekete M., Flow Chemistry, Vol. 2, De Gryter, Berlin, 2014. [Google Scholar]
- 13. Agostino F. J., Krylov S. N., TrAC Trends Anal. Chem. 2015, 72, 68–79. [Google Scholar]
- 14. Darvas F., Dormán G., Fekete M., Flow Chemistry, Vol. 2, De Gryter, Berlin, 2014, pp. 213–252. [Google Scholar]
- 15. Gilmore K., Kopetzki D., Lee J. W., Horváth Z., McQuade D. T., Seidel-Morgenstern A., Seeberger P. H., Chem. Commun. 2014, 50, 12652–12655. [DOI] [PubMed] [Google Scholar]
- 16. O'Brien A. G. et al., Angew. Chem. Int. Ed. 2012, 51, 7028–7030; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7134–7137. [Google Scholar]
- 17. Lee J. W., Horváth Z., O'Brien A. G., Seeberger P. H., Seidel-Morgenstern A., Chem. Eng. J. 2014, 251, 355–370. [Google Scholar]
- 18. Horváth Z., Horosanskaia E., Lee J. W., Lorenz H., Gilmore K., Seeberger P. H., Seidel-Morgenstern A., Org. Process Res. Dev. 2015, 19, 624–634. [Google Scholar]
- 19. Sreedhar B., Shen B., Li H., Rousseau R., Kawajiri Y., Org. Process Res. Dev. 2017, 21, 31–43. [Google Scholar]
- 20. Baxendale I. R., Deeley J., Griffiths-Jones C. M., Ley S. V, Saaby S., Tranmer G. K., Chem. Commun. 2006, 2566. [DOI] [PubMed] [Google Scholar]
- 21. Hartwig J., Kirschning A., Chem. Eur. J. 2016, 22, 3044–3052. [DOI] [PubMed] [Google Scholar]
- 22. Tamborini L., Romano D., Pinto A., Bertolani A., Molinari F., Conti P., J. Mol. Catal. B 2012, 84, 78–82. [Google Scholar]
- 23. Tamborini L., Romano D., Pinto A., Contente M., Iannuzzi M. C., Conti P., Molinari F., Tetrahedron Lett. 2013, 54, 6090–6093. [Google Scholar]
- 24. Borukhova S., Noël T., Metten B., de Vos E., Hessel V., ChemSusChem 2013, 6, 2220–2225. [DOI] [PubMed] [Google Scholar]
- 25. Mascia S. et al., Angew. Chem. Int. Ed. 2013, 52, 12359–12363; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12585–12589. [Google Scholar]
- 26. Heider P. L. et al., Org. Process Res. Dev. 2014, 18, 402–409. [Google Scholar]
- 27. Monbaliu J.-C. C. M., Stelzer T., Revalor E., Weeranoppanant N., Jensen K. F., Myerson A. S., Org. Process Res. Dev. 2016, 20, 1347–1353. [Google Scholar]
- 28. Zak J., Ron D., Riva E., Harding H. P., Cross B. C. S., Baxendale I. R., Chem. Eur. J. 2012, 18, 9901–9910. [DOI] [PubMed] [Google Scholar]
- 29. Berthod A. in Comprehensive Analytical Chemistry, Vol. 38 (Ed.: A. Berthod), 2002, pp. 1–397. [Google Scholar]
- 30. Friesen J. B., McAlpine J. B., Chen S. N., Pauli G. F., J. Nat. Prod. 2015, 78, 1765–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oelmeier S. A., Ladd-Effio C., Hubbuch J., J. Chromatogr. A 2013, 1319, 118–126. [DOI] [PubMed] [Google Scholar]
- 32.S. Ignatova in 7th Int. Conf. Countercurrent Chromatogr. Hangzhou, August 6–8, 2012, Session 1, K-2, Hangzhou, China.
- 33. Ignatova S., Sutherland I., J. Chromatogr. A 2015, 1425, 1–7. [DOI] [PubMed] [Google Scholar]
- 34. Mekaoui N., Berthod A., J. Chromatogr. A 2011, 1218, 6061–6071. [DOI] [PubMed] [Google Scholar]
- 35. Delannay E., Toribio A., Boudesocque L., Nuzillard J.-M., Zèches-Hanrot M., Dardennes E., Le Dour G., Sapi J., Renault J.-H., J. Chromatogr. A 2006, 1127, 45–51. [DOI] [PubMed] [Google Scholar]
- 36. Hu R., Pan Y., TrAC Trends Anal. Chem. 2012, 40, 15–27. [Google Scholar]
- 37. Roullier C., Chollet-Krugler M., Bernard A., Boustie J., J. Chromatogr. B 2009, 877, 2067–2073. [DOI] [PubMed] [Google Scholar]
- 38. Ito Y., J. Chromatogr. A 2005, 1065, 145–168. [DOI] [PubMed] [Google Scholar]
- 39. Kroth H., Nampally S., Molette J., Gabellieri E., Benderitter P. A. R., Froestl W., Schieferstein H., Mueller A., Schmitt-Willich H., Berndt M., WO2015110263, 2015.
- 40. Sutherland I. A., Audo G., Bourton E., Couillard F., Fisher D., Garrard I., Hewitson P., Intes O., J. Chromatogr. A 2008, 1190, 57–62. [DOI] [PubMed] [Google Scholar]
- 41. Bouju E., Berthod A., Faure K., J. Chromatogr. A 2015, 1409, 70–78. [DOI] [PubMed] [Google Scholar]
- 42. Bouju E., Berthod A., Faure K., J. Chromatogr. A 2016, 1466, 59–66. [DOI] [PubMed] [Google Scholar]
- 43. Kotland A., Chollet S., Diard C., Autret J.-M., Meucci J., Renault J.-H., Marchal L., J. Chromatogr. A 2016, 1474, 59–70. [DOI] [PubMed] [Google Scholar]
- 44. Hewitson P., Ignatova S., Ye H., Chen L., Sutherland I., J. Chromatogr. A 2009, 1216, 4187–4192. [DOI] [PubMed] [Google Scholar]
- 45. Ignatova S., Hewitson P., Mathews B., Sutherland I., J. Chromatogr. A 2011, 1218, 6102–6106. [DOI] [PubMed] [Google Scholar]
- 46. Goll J., Frey A., Minceva M., J. Chromatogr. A 2013, 1284, 59–68. [DOI] [PubMed] [Google Scholar]
- 47. Hopmann E., Goll J., Minceva M., Chem. Eng. Technol. 2012, 35, 72–82. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary