

ABSTRACT
Salmonella enterica serovar Typhimurium can inject effector proteins into host cells via type III secretion systems (T3SSs). These effector proteins modulate a variety of host transcriptional responses to facilitate bacterial growth and survival. Here we show that infection of host cells with S. Typhimurium specifically induces the ubiquitination of tumor necrosis factor receptor-associated factor 6 (TRAF6). This TRAF6 ubiquitination is triggered by the Salmonella pathogenicity island 1 (SPI-1) T3SS effectors SopB and SopE2. We also demonstrate that TRAF6 is involved in the SopB/SopE2-induced phosphorylation of signal transducer and activator of transcription 3 (STAT3), a signaling event conducive to the intracellular growth of S. Typhimurium. Specifically, TRAF6 mediates lysine-63 ubiquitination within the Src homology 2 (SH2) domain of STAT3, which is an essential step for STAT3 membrane recruitment and subsequent phosphorylation in response to S. Typhimurium infection. TRAF6 ubiquitination participates in STAT3 phosphorylation rather than serving as only a hallmark of E3 ubiquitin ligase activation. Our results reveal a novel strategy in which S. Typhimurium T3SS effectors broaden their functions through the activation of host proteins in a ubiquitination-dependent manner to manipulate host cells into becoming a Salmonella-friendly zone.
KEYWORDS: Salmonella enterica serovar Typhimurium, TRAF6, ubiquitination, STAT3, Salmonella effectors
INTRODUCTION
Infection with bacterial pathogens often induces host inflammatory responses (1), which are initiated by the recognition of microbial products, collectively known as pathogen-associated molecular patterns (PAMPs). PAMPs are recognized by Toll-like receptors (TLRs) or cytosolic NOD-like receptors (NLRs) (1, 2), which activate nuclear factor κB (NF-κB) and mitogen-activated protein kinases (MAPKs) and induce the production of proinflammatory cytokines critical for host defenses (3, 4). To overcome host defenses, some pathogens have developed strategies to dampen the host innate immune response by inactivating MAPKs or NF-κB signaling. For instance, the Shigella flexneri phosphothreonine lyase OspF, which is injected into host cells by a type III secretion system (T3SS), inactivates the innate immune response by dephosphorylating MAPKs (5). The Yersinia protein YopJ/YopP, a T3SS effector protein containing both deubiquitinating and acetyltransferase activities, inhibits the activation of NF-κB and prevents MAPK phosphorylation (6–8).
Bacterial pathogens also trigger host inflammatory responses so as to gain access to essential nutrients and compete with the intestinal microbial flora during infection. Salmonella enterica serovar Typhimurium (9), a causative agent of food poisoning, stimulates innate immune responses in cultured epithelial cells with the T3SS effector proteins SopE, SopE2, and SopB (10). Our previous observations of tumor necrosis factor receptor-associated factor 6 (TRAF6)-mediated ubiquitination of SopB point to the importance of TRAF6 during S. Typhimurium infection (11, 12). TRAF6 is a unique member of the TRAF family that contains E3 ubiquitin ligase activity (13). TRAF6 plays a crucial role in mediating the signals from the tumor necrosis factor (TNF) receptor superfamily and the interleukin-1 receptor (IL-1R)/TLR superfamily (14). Upon bacterial infection, TRAF6 has been shown to mediate the activation of MAPKs and NF-κB downstream of MyD88 in IL-1/TLR signaling (15, 16). In addition to NF-κB and MAPKs, signal transducer and activator of transcription 3 (STAT3) plays an important role in host inflammatory responses. STAT3 belongs to a family of transcription factors that transduce cellular signals from a number of cytokines and soluble growth factors, such as IL-6 family cytokines, epidermal growth factor (EGF), and platelet-derived growth factor (PDGF) (17). Following activation, STAT3 is phosphorylated before its dimerization in the cytoplasm. Then the dimerized protein moves from the cytosol to the nucleus to initiate target gene transcription (18). STAT3 is found to be activated by certain pathogens. For example, Helicobacter pylori activates STAT3, which plays an important role in gastric carcinogenesis (19). In the case of S. Typhimurium, STAT3 is activated, subsequently reprogramming gene expression in epithelial cells, converting host cells to a metabolic state that is conducive to the intracellular replication of Salmonella (20). However, the mechanisms of STAT3 activation by S. Typhimurium remain unclear. Here we report a previously unidentified ubiquitination-dependent activation of STAT3 that is mediated by TRAF6. This function of TRAF6 is initiated by the Salmonella pathogenicity island 1 (SPI-1) T3SS effectors SopB and SopE2 from S. Typhimurium.
RESULTS
S. Typhimurium SPI-1 T3SS effectors trigger TRAF6 ubiquitination.
Since TRAF6 plays a critical role in integrating multiple upstream receptor signals to induce downstream activation of transcription factors during bacterial infection (21), we examined the responses of TRAF6 to bacterial pathogens. First, we infected cultured wild-type (WT) mouse embryonic fibroblasts (MEFs) with various pathogens, including S. Typhimurium, Shigella flexneri, Listeria monocytogenes, and Staphylococcus aureus. Significant portions of modified TRAF6 were observed in cells infected with S. Typhimurium at a multiplicity of infection (MOI) of 10 at 8 and 16 h postinfection, whereas the mobility of TRAF6 remained unchanged for the other three pathogens during the same periods (Fig. 1A). As a consequence, the total amount of TRAF6, including both modified and unmodified TRAF6, was found to be significantly increased by S. Typhimurium at 8 and 16 h postinfection when measured by ImageJ and normalized to values for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Fig. 1B). Simultaneously, modification of TRAF6 was found to be a general response to S. Typhimurium, since analogous observations were made in intestinal epithelial Henle-407 cells and mouse RAW 264.7 macrophages, although the chains of TRAF6 modification seemed to differ in different cell types (Fig. 1C). As in the MEFs for which results are shown in Fig. 1B, the total amounts of TRAF6 in both Henle-407 cells and mouse RAW 264.7 macrophages were increased by S. Typhimurium (Fig. 1D).
FIG 1.
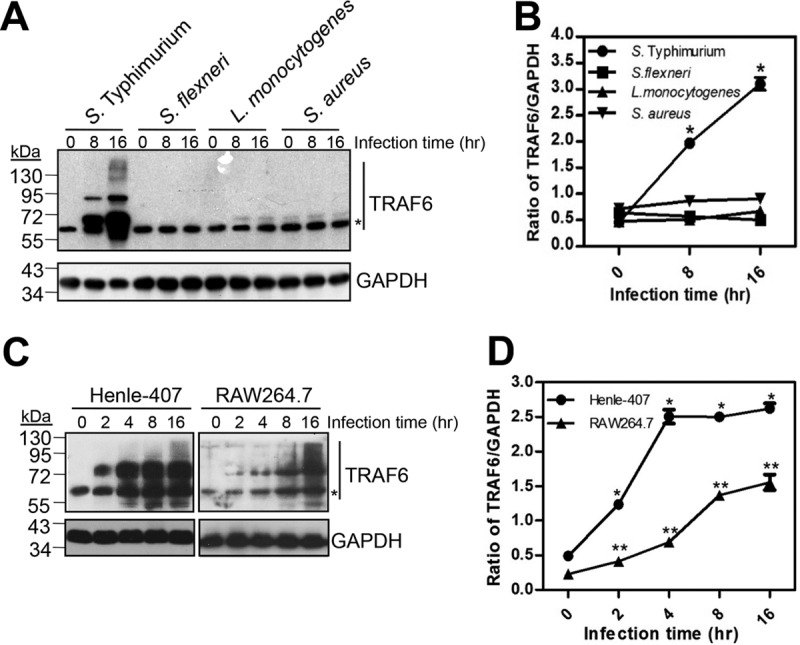
S. Typhimurium induces the modification of TRAF6. (A) Induction of TRAF6 modification in MEFs by S. Typhimurium, Shigella flexneri, Listeria monocytogenes, and Staphylococcus aureus. Cultured MEFs were infected with equal amounts of bacteria at an MOI of 10 for 1 h. The infected cells were then chased in the presence of gentamicin and were lysed at the indicated times. The proteins of the infected cells were then separated and subjected to Western blot analysis with rabbit anti-TRAF6, using anti-GAPDH as a loading control. The asterisk indicates the predicted mobility of unmodified TRAF6. (B) Quantified ratio of total TRAF6 to GAPDH (from panel A). Each band was measured with ImageJ, and measurements were normalized to GAPDH values. Values are means (± SD) for three independent experiments. Single and double asterisks indicate statistically significant differences (P < 0.05) in the ratios of TRAF6 to GAPDH within Henle-407 cells and RAW 264.7 cells, respectively, from the values for uninfected cells as determined by Student’s t test.
To further exploit this TRAF6-involved infection strategy by S. Typhimurium, we employed MEFs as the host cells for further investigation, since we had our own line of Traf6−/− MEFs. It has been shown previously that TRAF6 undergoes autoubiquitination (22). To determine whether the changes in TRAF6 mobility caused by S. Typhimurium were due to ubiquitination, we infected Traf6+/+ MEFs with S. Typhimurium for 8 h. TRAF6 was immunoprecipitated and was detected using anti-ubiquitin via Western blotting. To better separate unmodified and modified TRAF6, we ran immunoprecipitated TRAF6 on a 7.5% SDS-PAGE gel for a longer time than that used for whole-cell lysate (WCE) separations (for which a 10% SDS-PAGE gel was used). As shown in Fig. 2A, ubiquitinated TRAF6 was readily detected in S. Typhimurium-infected Traf6+/+MEFs, confirming that the observed change in TRAF6 mobility was due to ubiquitination. Moreover, we infected Traf6+/+ and Traf6−/− MEFs with S. Typhimurium. As shown in Fig. S1 in the supplemental material, both ubiquitinated TRAF6 and unmodified TRAF6 were robustly visualized in the anti-TRAF6 immunoblot from Traf6+/+ MEFs, while no signals were detected from the Traf6−/− MEFs, demonstrating the specific responses of TRAF6 upon S. Typhimurium infection.
FIG 2.

S. Typhimurium T3SS effector-dependent ubiquitination of TRAF6. (A) TRAF6 is ubiquitinated upon S. Typhimurium infection. Traf6+/+ MEFs were infected with S. Typhimurium for 8 h. (Left) The presence of ubiquitinated TRAF6 [TRAF6-(Ub)n] in the infected cells was analyzed by immunoprecipitation (IP) with rabbit anti-TRAF6 and Western blotting (7.5% SDS-PAGE gels) (IB) with mouse anti-TRAF6 or mouse anti-ubiquitin. (Right) Whole-cell lysates (WCE) were probed on Western blots from 10% SDS-PAGE gels with rabbit anti-TRAF6, mouse anti-ubiquitin, and anti-GAPDH (as a loading control). (B) Dependence of TRAF6 ubiquitination on the S. Typhimurium effectors SopB and SopE2. Cultured Traf6+/+ MEFs were infected with wild-type S. Typhimurium or the ΔsopB, ΔsopE2, ΔsopB ΔsopE2, or ΔinvA mutant at an MOI of 10 for 1 h, followed by Western blotting with rabbit anti-TRAF6. (C) Quantification of the ratio of total TRAF6 to GAPDH (from panel B). Values are means (± SD) for three independent experiments. Single or double asterisks indicate statistically significant differences (P < 0.05) between the wild type (WT) and the ΔsopB mutant or between the WT and the ΔsopE2 mutant, respectively, using two-way analysis of variance (ANOVA) with Prism software. (D) Traf6+/+ MEFs either were not infected (n.i.) or were infected with the indicated S. Typhimurium mutant expressing Yersinia pseudotuberculosis invasin protein at an MOI of 50 or with wild-type S. Typhimurium at an MOI of 10 for 1 h, followed by Western blotting with rabbit anti-TRAF6, using anti-GAPDH as a loading control.
We next investigated whether S. Typhimurium T3SS effectors are involved in TRAF6 ubiquitination. As shown in Fig. 2B, TRAF6 ubiquitination was strictly dependent on the T3SS, since TRAF6 ubiquitination was abolished in S. Typhimurium ΔinvA, a T3SS-defective mutant (23), in contrast to WT S. Typhimurium. Since we showed previously that SopB interacts directly with TRAF6 (11), we reasoned that SopB may be responsible for TRAF6 ubiquitination. We found that deletion of SopB decreased TRAF6 ubiquitination but did not eliminate it (Fig. 2B), suggesting that SopB is not the only factor involved in TRAF6 ubiquitination upon S. Typhimurium infection. Due to the well-known cooperation of SPI-1 T3SS effectors SopE and SopE2 with SopB (10, 20), we also examined the effects of SopE and SopE2 on TRAF6 ubiquitination. However, upon screening the genome, we found that SopE is totally absent in S. Typhimurium strain LT2 (GenBank accession no. NC_003197.1). Thus, we constructed a ΔsopE2 mutant and a ΔsopB ΔsopE2 double mutant in S. Typhimurium strain LT2. We found that TRAF6 ubiquitination was decreased upon infection with the ΔsopE2 mutant but was completely abolished upon infection with the ΔsopB ΔsopE2 mutant (Fig. 2B), suggesting that both SopB and SopE2 are required for TRAF6 ubiquitination. In accordance with a significant decrease in TRAF6 ubiquitination, the total amount of TRAF6 upon infection with the ΔsopB or ΔsopE2 mutant was smaller than that with wild-type S. Typhimurium (Fig. 2C). Deletion of invA or dual deletion of sopB and sopE2 drastically reduced the total amount of TRAF6 from that in wild-type S. Typhimurium-infected cells. However, it is hard to ascertain on the basis of our present results whether the effectors SopB and SopE2 induce the expression of TRAF6 during S. Typhimurium infection, since protein levels are determined by multiple factors, such as mRNA expression and protein degradation. Even the subcellular shift of posttranslationally modified protein from the primary position of synthesis might stimulate the production of protein. Although our present results indicate that SopB and SopE2 significantly stimulated the increase in TRAF6 ubiquitination during S. Typhimurium infection, much further work is necessary to clarify how the expression of TRAF6 is regulated by S. Typhimurium infection.
To test whether TRAF6 ubiquitination was due to extracellular or intracellular bacteria, we expressed the Yersinia pseudotuberculosis invasin protein in the invasion-defective S. Typhimurium ΔinvA and ΔsopB ΔsopE2 mutants. The Yersinia invasin protein is known to mediate the invasion of bacteria via the α4β1 integrin receptors (24). Meanwhile, to ensure comparable intracellular bacterial numbers for the invasin-expressing mutants and wild-type S. Typhimurium, the MOI of these mutants were elevated nearly 5-fold over that of wild-type S. Typhimurium during infection. This manipulation is due to a lower level of invasion by the invasin-expressing strains than by wild-type S. Typhimurium (data not shown). Despite the presence of equivalent numbers of intracellular bacteria in the ΔinvA or ΔsopB ΔsopE2 mutant (see Fig. S2 in the supplemental material), the expression of invasin did not rescue the defect in TRAF6 ubiquitination observed in wild-type MEFs (Fig. 2D). Taken together, these results suggest that S. Typhimurium triggers TRAF6 ubiquitination through the SPI-1 T3SS effectors SopE2 and SopB.
TRAF6 mediates ubiquitination-dependent STAT3 phosphorylation induced by S. Typhimurium.
To determine the biological function of TRAF6, we examined the activation of three signaling pathways known to be responsible for the regulation of host transcriptional responses upon S. Typhimurium infection: MAPKs, NF-κB, and STAT3. We found that deletion of TRAF6 in Traf6−/− MEFs abolished the constitutive phosphorylation of extracellular signal-regulated kinase (ERK) and p38 and the activation of NF-κB as measured by IκBα degradation, which were observed in Traf6+/+ MEFs (Fig. 3A). Moreover, the activation of nearly all of the MAPKs and NF-κB was decreased by various degrees in Traf6−/− MEFs in the early phase of infection. However, the impact of the absence of TRAF6 on MAPKs and NF-κB gradually disappeared with prolonged infection times, though at various time points for different MAPKs. For instance, analogous levels of phosphorylated ERK, phosphorylated p38, and phosphorylated Jun N-terminal protein kinase (JNK) were reached for wild-type Traf6+/+ and Traf6−/− MEFs at 4 h, 8 h, and 16 h postinfection, respectively. In addition, analogous NF-κB activation was observed at 8 h postinfection. In summary, these results indicate that the absence of TRAF6 led to delays in the activation of MAPKs and NF-κB but did not abolish their activation. Ultimately, all the MAPKs and NF-κB reached a plateau independently of the function of TRAF6.
FIG 3.
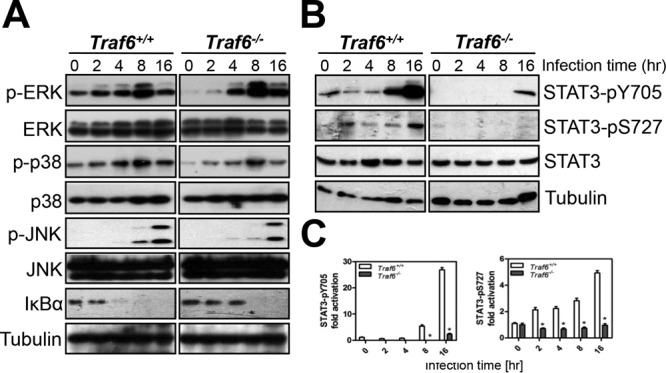
The absence of TRAF6 inhibits the phosphorylation of MAPKs, the activation of NF-κB, and the phosphorylation of STAT3 upon S. Typhimurium infection. (A and B) Infected cells were analyzed by Western blotting with antibodies against MAPKs and IκBα (A) or against STAT3 and the phosphorylated forms of STAT3 (pY705 and pS727) (B). Tubulin was detected as a loading control. (C) Quantification of the fold activation of STAT3-pY705 and STAT3-pS727 (relative to levels in uninfected cells). Values are means (± SD) for three independent experiments. Asterisks indicate statistically significant differences (P < 0.05) from the values for Traf6+/+ cells as determined by Student's t test.
Distinct from the effects on MAPKs and NF-κB, TRAF6 deletion substantially diminished STAT3 phosphorylation in Traf6−/− cells relative to the robust induction of STAT3 phosphorylation at Y705 at 8 to 16 h postinfection and at S727 at 2 h postinfection in Traf6+/+ cells (Fig. 3B and C). As has been well established, the phosphorylation of STAT3 at position S727 and its phosphorylation at position Y705 are induced and regulated independently (25). Therefore, the decreases in ERK and JNK phosphorylation that occur in the absence of TRAF6 may explain the inhibition of STAT3 S727 phosphorylation in Traf6−/− MEFs infected with S. Typhimurium. However, in contrast to STAT3 S727 phosphorylation, STAT3 Y705 phosphorylation coincided with TRAF6 ubiquitination, which is initiated by SPI-1 T3SS effectors SopB and SopE2 (as shown in Fig. 2). Moreover, the phosphorylation of STAT3 was previously shown to be induced by SopB and SopE2 (20), suggesting an underlying correlation between TRAF6 and STAT3 phosphorylation at Y705.
STAT3 is phosphorylated before its entrance into the nucleus to regulate target gene transcription (18). To confirm TRAF6-dependent STAT3 phosphorylation during S. Typhimurium infection, we performed immunofluorescence assays to visualize STAT3-pY705 in cells. We found that nearly 70% of the Salmonella-infected Traf6+/+ MEFs stained positive for STAT3-pY705 (green), which colocalized with the nucleus (blue) at 8 h postinfection; in contrast, only approximately 17% of Traf6−/− MEFs were positive for STAT3-pY705, though with only small amounts of green staining (see Fig. S3A and B in the supplemental material). It was shown previously that effector-driven STAT3 phosphorylation plays a central role in the modulation of host gene expression to promote Salmonella-induced filament (SIF) formation and Salmonella-containing vacuole (SCV) maturation in the late stages of infection (20); thus, we measured S. Typhimurium replication within Traf6+/+ and Traf6−/− MEFs. We found that S. Typhimurium replication was significantly lower in Traf6−/− cells than in Traf6+/+ cells (Fig. S3C), confirming the involvement of TRAF6 in STAT3 phosphorylation during S. Typhimurium infection. To examine whether the differences in bacterial burden between Traf6+/+ and Traf6−/− cells led to different STAT3 phosphorylation by S. Typhimurium, we used chloramphenicol, a bacterial protein synthesis inhibitor, to impede the intracellular replication of the bacteria. Since equal numbers of intracellular bacteria were observed in Traf6+/+ and Traf6−/− MEFs after 1 h of infection (Fig. S3C), we chose to add chloramphenicol to the medium at this point. We found that robust STAT3 phosphorylation was readily detected in Traf6+/+ cells after 8 and 16 h of infection (see Fig. S4 in the supplemental material), a finding consistent with the results seen in Fig. 3B, where no chloramphenicol was present. These results indicate that the SopB and SopE2 effectors delivered early in infection play a vital role in STAT3 phosphorylation, although a proportion of newly synthesized effector proteins could be translocated during the late stage of infection. These results are consistent with the manner of STAT3 activation revealed by Hannemann et al. (20). Meanwhile, significantly weaker STAT3 phosphorylation was observed in Traf6−/− cells than in Traf6+/+ cells in the presence of chloramphenicol (Fig. S4), taking into account the TRAF6-dependent STAT3 phosphorylation achieved through more-extensive replication of S. Typhimurium in Traf6+/+ MEFs than in Traf6−/− MEFs. Taken together, these results suggest that TRAF6 mediates STAT3 phosphorylation during S. Typhimurium infection.
Next, we investigated whether STAT3 is ubiquitinated by TRAF6 because TRAF6 possesses E3-ubiquitin ligase activity (13). Polyubiquitinated STAT3 was readily detected 2 h after S. Typhimurium infection (Fig. 4A, left). The amount of ubiquitinated STAT3 gradually increased and reached its highest level at 8 h postinfection. In comparison, little, if any, STAT3 ubiquitination was detected in Salmonella-infected Traf6−/− MEFs (Fig. 4A, right), indicating a requirement for TRAF6 in the ubiquitination of STAT3 during S. Typhimurium infection. To examine whether the E3-ubiquitin ligase activity of TRAF6 is required for STAT3 ubiquitination and phosphorylation, we complemented Traf6−/− MEFs with either wild-type TRAF6 or TRAF6 C70A, a RING mutant lacking E3-ubiquitin ligase activity. We found that in contrast to wild-type TRAF6, the C70A mutant failed to restore either STAT3 ubiquitination or STAT3 phosphorylation after 8 h of infection (Fig. 4B). Meanwhile, the number of intracellular bacteria in C70A-expressing Traf6−/− MEFs was significantly lower than that in cells expressing wild-type TRAF6 (Fig. S3D). These data suggest that STAT3 ubiquitination and phosphorylation depend on the E3-ubiquitin ligase activity of TRAF6. To test the formation of a TRAF6-mediated polyubiquitin chain on STAT3, we first cotransfected HEK293T cells with TRAF6 as well as K63-only or K48-only mutants of ubiquitin and then infected these cells with S. Typhimurium for 8 h. Robust STAT3 polyubiquitination was visualized in cells expressing both TRAF6 and K63-only ubiquitin at 8 h after infection by S. Typhimurium, whereas relatively weak STAT3 polyubiquitination was detected in cells expressing both TRAF6 and K48-only ubiquitin (see Fig. S5 in the supplemental material). These data suggest that during S. Typhimurium infection, STAT3 is ubiquitinated with lysine-63-linked polyubiquitin chains.
FIG 4.
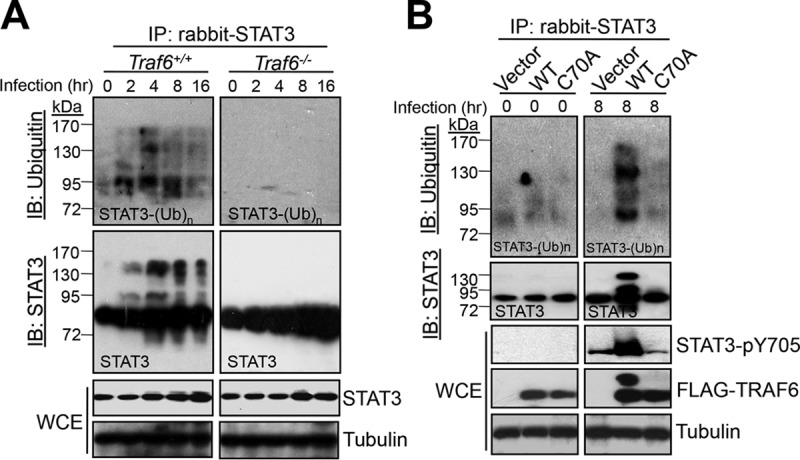
TRAF6 catalyzes STAT3 ubiquitination upon S. Typhimurium infection. (A) Traf6+/+ MEFs were first serum starved and then infected with S. Typhimurium at an MOI of 10 for the indicated times. WCEs were collected for immunoprecipitation (IP) with rabbit anti-STAT3, followed by Western blotting (IB) with mouse anti-STAT3 and mouse anti-ubiquitin. WCEs were probed with rabbit anti-STAT3 and rabbit anti-tubulin (as a loading control). (B) Importance of the TRAF6 C70 residue for S. Typhimurium-induced STAT3 ubiquitination and phosphorylation. Traf6−/− MEFs were transfected with either mock plasmids (Vector), pcDNA4-FLAG-TRAF6 (WT), or pcDNA4-FLAG-TRAF6 C70A (C70A) for 24 h before infection with S. Typhimurium for 8 h. STAT3 was immunoprecipitated as described for panel A, and the phosphorylation of STAT3 was detected as for Fig. 3B.
STATs share a highly conserved structure with five functional domains: an NH2-terminal coiled-coil domain, a DNA-binding domain, a linker domain, an Src homology 2 (SH2) domain, and a trans-activation domain (26). Among these, the SH2 domain is necessary for STAT3 binding to membrane receptors and subsequent phosphorylation (27). To clarify the dependence of STAT3 ubiquitination on the SH2 domain, we coexpressed TRAF6 with wild-type STAT3 or a STAT3 ΔSH2 mutant (devoid of the SH2 domain) in HEK293T cells, followed by S. Typhimurium infection. As shown in Fig. 5A, TRAF6 expression failed to promote the ubiquitination of the STAT3 ΔSH2 mutant, in contrast to wild-type STAT3. To analyze the lysine residues within the SH2 domain for possible ubiquitination sites (Fig. 5B), we mutated all six lysine residues (Lys548, Lys551, Lys557, Lys573, Lys574, and Lys591) within the SH2 domain to Ala [the K(1-6)A mutant]. We found that STAT3 ubiquitination and phosphorylation were substantially abolished in the STAT3 K(1-6)A mutant (Fig. 5C), suggesting that ubiquitination within the SH2 domain of STAT3 is essential for STAT3 phosphorylation. Since the first step of STAT3 phosphorylation is its recruitment to the membrane, we investigated whether STAT3 ubiquitination is vital for membrane recruitment. As shown in Fig. 5D and in Fig. S6 in the supplemental material, only 0.08% of the STAT3 K(1-6)A mutant was recruited to the membrane even when coexpressed with TRAF6, in contrast to 68.2% of wild-type STAT3. Simultaneously, the phosphorylation of STAT3 in the membrane was greatly diminished, demonstrating the role of STAT3 ubiquitination in membrane recruitment and phosphorylation. Moreover, we compared the subcellular localizations of STAT3 in Traf6+/+ and Traf6−/− MEFs. The results in Fig. S7 in the supplemental material indicated that both STAT3 and phosphorylated STAT3 were acquired from the plasma membrane fractions in the presence of TRAF6 in wild-type MEFs. However, neither STAT3 nor phosphorylated STAT3 was detected in Traf6−/− MEFs, further confirming the dependence of STAT3 membrane recruitment and subsequent phosphorylation on TRAF6-catalyzed STAT3 ubiquitination. Thus, we concluded that TRAF6 ubiquitinates STAT3 within its SH2 domain and that this step is required for STAT3 recruitment and phosphorylation upon S. Typhimurium infection.
FIG 5.

Mutation of lysines within the SH2 domain abolishes STAT3 ubiquitination. (A) Ubiquitination and phosphorylation of FLAG-STAT3 or FLAG-STAT3 ΔSH2 when cotransfected with HA-TRAF6 into HEK293T cells for 24 h before infection with S. Typhimurium for 8 h. FLAG-STAT3 or FLAG-STAT3 ΔSH2 was pulled down using anti-FLAG M2 magnetic beads and was then analyzed with rabbit anti-STAT3 and rabbit anti-STAT3-pY705. WCEs were probed with mouse anti-FLAG and rabbit anti-tubulin (as a loading control). (B) Diagram indicating the locations of lysine residues within the SH2 domain of STAT3. (C) Effects of mutations in the STAT3 SH2 domain lysine residues on STAT3 ubiquitination and phosphorylation. FLAG-STAT3 or FLAG-STAT3 K(1-6)A was cotransfected with HA-TRAF6 into HEK293T cells for 24 h before infection with S. Typhimurium for the indicated times. FLAG-STAT3 was pulled down and detected as described for panel A. (D) Effects of mutations in the STAT3 SH2 domain lysine residues on STAT3 membrane localization and phosphorylation. FLAG-STAT3 or FLAG-STAT3 K(1-6)A was cotransfected with HA-TRAF6 into HEK293T cells for 24 h before infection with S. Typhimurium for 8 h. WCE and plasma membrane (Mem) fractions were analyzed by Western blotting with the indicated antibodies.
TRAF6 ubiquitination is required for STAT3 phosphorylation upon S. Typhimurium infection.
Previously, the SPI-1 T3SS effectors SopE, SopE2, and SopB were shown to be required for STAT3 phosphorylation during S. Typhimurium infection (20). In agreement with these findings, deletion of sopB decreased, and double deletion of sopB and sopE2 completely abolished, the activation of STAT3 observed for wild-type S. Typhimurium LT2 at 8 and 16 h postinfection (see Fig. S8A and B in the supplemental material). However, until now, it was not clear whether TRAF6 ubiquitination induced by SopB and SopE2 was important for STAT3 ubiquitination and phosphorylation or if it served only as a hallmark of E3 ubiquitin ligase activation. Thus, we mutated TRAF6 Lys124, an essential site for TRAF6 autoubiquitination (28), to Ala and found that ectopic expression of the K124A mutant in Traf6−/− MEFs not only dramatically impaired TRAF6 ubiquitination but simultaneously decreased STAT3 ubiquitination and phosphorylation from that with wild-type TRAF6 in response to S. Typhimurium infection (Fig. 6A and B), confirming the involvement of TRAF6 ubiquitination in STAT3 ubiquitination and phosphorylation induced by the SPI-1 T3SS effectors SopB and SopE2.
FIG 6.
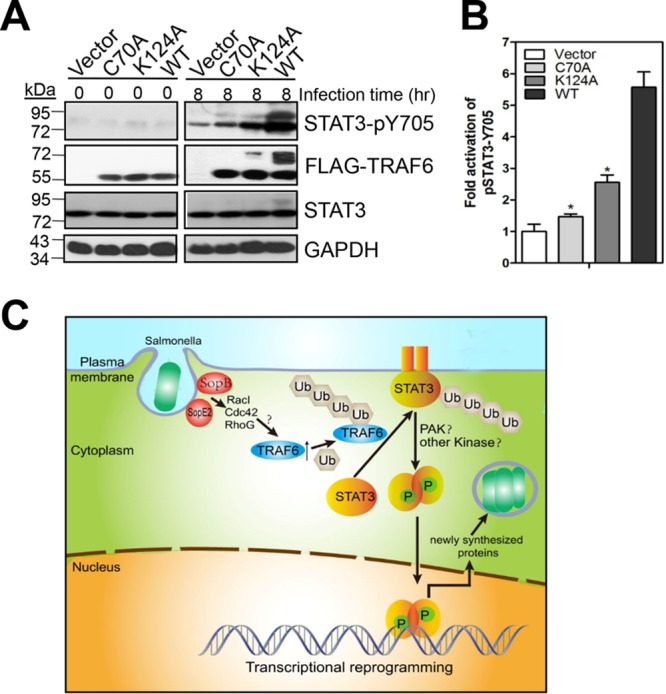
TRAF6 ubiquitination is important for the induction of STAT3 phosphorylation by S. Typhimurium. (A) Importance of the TRAF6 K124 residue in STAT3 phosphorylation. Traf6−/− MEFs were transfected with mock plasmids, pcDNA4-FLAG-TRAF6, pcDNA4-FLAG-TRAF6 C70A, or pcDNA4-FLAG-TRAF6 K124A for 24 h before infection with S. Typhimurium for 8 h. WCEs were probed with mouse anti-FLAG, rabbit anti-pSTAT3 (Y705), rabbit anti-STAT3, or anti-GAPDH (as a loading control). (B) Quantification of the fold activation of STAT3-pY705 (relative to that for Traf6−/− MEFs expressing a mock plasmid). Values are means (± SD) for three independent experiments. Asterisks indicate statistically significant (P < 0.05) differences from the values for Traf6−/− MEFs expressing wild-type TRAF6 as determined by Student's t test. (C) Schematic representation. SPI-1 T3SS effectors SopB and SopE2 initiate the ubiquitination of TRAF6 via the activation of RacI, Cdc42, and RhoG. TRAF6 catalyzes STAT3 ubiquitination, which is essential for STAT3 membrane recruitment and subsequent phosphorylation. TRAF6 ubiquitination not only acts as a hallmark of activation of ubiquitin ligase activity but also is involved in TRAF6-catalyzed STAT3 ubiquitination. STAT3 phosphorylation modulates host gene expression to promote intracellular bacterial replication.
DISCUSSION
It is well established that S. Typhimurium manipulates host cell gene expression either by stimulating innate immune receptors with conserved bacterial products or through SPI-1 T3SS effectors, which stimulate transcriptional responses independently of innate immune receptor activation. In this study, we have defined an SPI-1 T3SS effector-driven TRAF6-dependent regulation of STAT3 phosphorylation, which modulates host transcriptional responses to adapt to invading bacteria. We found that unlike Shigella flexneri, Listeria monocytogenes, and Staphylococcus aureus, S. Typhimurium induces the ubiquitination of TRAF6 within host cells specifically through the T3SS SPI-1 effectors SopB and SopE2, suggesting that a distinct TRAF6-dependent strategy is used by Salmonella.
SopB is a phosphatidylinositol 4-phosphatase and 5-phosphatase that promotes Salmonella invasion by redundantly activating Rac I, Cdc42, and RhoG (29). SopE2 is a known guanine nucleotide exchange factor (GEF) capable of activating RacI and Cdc42 (30). It is also known that S. Typhimurium SopB and SopE2 exert their function on STAT3 phosphorylation by redundantly activating RacI, Cdc42, and RhoG (20), and we have demonstrated here that SopB and SopE2 initiate TRAF6 ubiquitination (Fig. 2). We have also shown that TRAF6 ubiquitination is required for STAT3 phosphorylation (Fig. 6A). Therefore, we hypothesize that SopB and SopE2 initiate TRAF6 ubiquitination via the activation of RacI, Cdc42, and RhoG (Fig. 6C). In support of our hypothesis, TRAF6 is found to be a ubiquitin ligase responsible for ubiquitination-dependent Akt phosphorylation (13), and SopB promotes Akt phosphorylation, which is induced by S. Typhimurium via the RacI–phosphatidylinositol 3-kinase (PI3K) signaling pathway (31). Additionally, SopE2 is reported to enhance the activity of TRAF6 in activating NF-κB-dependent transcription during S. Typhimurium infection (32). TRAF6 has also been found to act downstream of the RacI/PI3K/Akt signaling cascade in TLR2 signaling for nitric oxide production in chondrocytes (33), and RacI knockdown inhibits both the redox-dependent formation of a TRAF6–NF-κB-inducing kinase (NIK) complex and the recruitment of both TRAF6 and NIK to IL-1R during NF-κB activation (34). RacI is frequently found to interact with Myd88 and TRAF6 in IL-1R signaling, reactive oxygen species (ROS) production in osteoclastogenesis, and TRANCE-mediated NF-κB activation (35). All these findings supply evidence for an interaction between RacI and TRAF6, further suggesting the possible involvement of RacI, Cdc42, and RhoG in SopB- and SopE2-dependent TRAF6 ubiquitination.
Since TRAF6 is known to act downstream of the TLR superfamily (36), the loss of TRAF6 might cause major decreases in TLR signaling. Therefore, STAT3 phosphorylation could be reduced later in the infection, when the TLR-driven expression of IL-6 and IL-10 (37), which activate STAT3 by autocrine/paracrine signaling (38), would be blocked. However, this possibility has been excluded by Hannemann et al. (20); when they treated uninfected cultured epithelial cells with supernatants obtained from S. Typhimurium-infected cells at different times after infection, no detectable STAT3 phosphorylation was observed in these cells, showing that STAT3 activation is a consequence of Salmonella infection that is independent of the autocrine or paracrine pathways (20). Our examination of the function of TRAF6 with regard to STAT3 in S. Typhimurium-infected cells has revealed that TRAF6 catalyzes the lysine-63-linked polyubiquitination of STAT3 within the SH2 domain. Wei et al. argued previously that TRAF6 catalyzes STAT3 ubiquitination upon alpha interferon (IFN-α) stimulation (39), and they concluded that TRAF6 overexpression ubiquitinates STAT3 and depresses the transcriptional activity of the STAT3-responsive promoter in response to IFN-α. However, they did not supply any details with regard to TRAF6-dependent STAT3 ubiquitination. In contrast to their conclusion, we regard TRAF6 as essential for STAT3 phosphorylation in response to S. Typhimurium infection. In our speculation, STAT3 ubiquitination might serve as a signal for STAT3 membrane recruitment and subsequent phosphorylation or as a platform for the binding of kinases or other factors vital for STAT3 phosphorylation (Fig. 6C). With regard to kinases for STAT3 phosphorylation, although Hannemann et al. (20) proved that PAK (p21-activated kinase) and Abl tyrosine kinases are involved in a noncanonical phosphorylation of STAT3, it is not clear whether there are other kinases that phosphorylate STAT3 aside from these enzymes or whether ubiquitination-dependent STAT3 phosphorylation requires the participation of PAK and Abl tyrosine kinases. Besides STAT3, a variety of proteins, such as the mammalian target of rapamycin (mTOR) (40), IκB kinase (IKK) (41), SM22α K21 (42), glycogen synthase kinase 3β (GSK-3β) (43), Akt (13), and transforming growth factor β (TGF-β) type I receptor (TβRI) (44), have been reported to be ubiquitinated by TRAF6, generating a nondegradative lysine-63-linked polyubiquitin chain prior to phosphorylation and activation. However, it is difficult to clarify whether STAT3 is a substrate of TRAF6, due to the absence of direct evidence, such as an in vitro reconstitution study.
Concerted TRAF6 ubiquitination and the kinetics of STAT3 phosphorylation, both of which are activated by the SPI-1 T3SS effectors SopB and SopE2, suggest that TRAF6 might serve as an infection-associated protein, providing a potential therapeutic target against Salmonella. In support of our finding, TRAF6 was found to be translocated into pathogen-containing vacuoles (PVs) (45) and was involved in the ubiquitin-dependent labeling of PVs, which provide a safe haven for many intracellular bacterial pathogens (46). In conclusion, we have described here a mechanism by which S. Typhimurium T3SS effectors broaden their functions through the activation of host proteins in a ubiquitination-dependent manner to modulate host transcriptional responses so as to adapt host cells to the intracellular replication of bacteria.
MATERIALS AND METHODS
Bacterial strains, cells, growth conditions, cDNA constructs, and other reagents.
Wild-type (WT) S. Typhimurium LT2, as described in reference 5, was used in this study. ΔsopB, ΔinvA, ΔsopE2, and ΔsopB ΔsopE2 mutants were constructed using the λRed recombination system as described in reference 47. Shigella flexneri 2a strain 301, Listeria monocytogenes 10403S, and Staphylococcus aureus 0485 were purchased from the China General Microbiological Culture Collection Center (CGMCC). The human intestinal epithelial cell line (Henle-407), the RAW 264.7 cell line, and the HEK293T cell line were purchased from the American Type Culture Collection (ATCC). Wild-type Traf6+/+ and Traf6−/− mouse embryonic fibroblasts (MEFs) were generously donated by Jun-ichiro Inoue and Jin Gohda of the University of Tokyo. All cells were cultured in antibiotic-free Dulbecco's modified Eagle medium (DMEM; Gibco) supplemented with 10% bovine fetal serum (FBS) at 37°C under a 5% CO2 atmosphere.
DNA encoding the Yersinia pseudotuberculosis invasin (GenBank accession no. CNI21406.1) was synthesized and subcloned into the pBAD24 vector. The S. Typhimurium ΔinvA and ΔsopB ΔsopE2 strains were each transformed with pBAD24-invasin. The expression of invasin, which was under the control of the arabinose-inducible araABC promoter in these S. Typhimurium strains, was induced by adding 100 mmol/liter arabinose to the medium. cDNA for TRAF6 or STAT3 was inserted into the pcDNA4-FLAG vector for the expression of FLAG-tagged proteins as described in reference 48. cDNA for TRAF6 was inserted into the pcDNA3-HA vector for the expression of hemagglutinin (HA)-tagged TRAF6 as described in reference 48. The TRAF6 C70A, TRAF6 K124A, and STAT3 K(1-6)A point mutants were generated using a QuikChange site-directed mutagenesis kit (Stratagene). The STAT3 ΔSH2 mutant was constructed in our lab by two-step PCR using KOD-Plus high-fidelity DNA polymerase (Koyobo, Japan). The pcDNA3-K63-only ubiquitin plasmid and the pcDNA3-K48-only ubiquitin plasmid were donated by H. Li from Xinan University, as described in reference 49. All plasmids were verified by DNA sequencing.
Rabbit anti-phospho-ERK, rabbit anti-phospho-p38, rabbit anti-phospho-JNK, rabbit anti-ERK, rabbit anti-p38, mouse anti-JNK, mouse anti-IκBα, rabbit anti-phospho-STAT3 (Y705), mouse anti-STAT3, and rabbit anti-phospho-STAT3 (S727) antibodies, as well as Alexa Fluor 488-conjugated secondary rabbit antibodies, were purchased from Cell Signaling Technology. Rabbit anti-TRAF6, chicken anti-GAPDH, mouse anti-ubiquitin (K63 specific), mouse anti-ubiquitin (K48 specific), a secondary rabbit antibody, a secondary mouse antibody, and a secondary chicken antibody were purchased from Millipore. Mouse anti-FLAG and anti-FLAG M2 magnetic beads were purchased from Sigma. Rabbit anti-N-cadherin, rabbit anti-STAT3, mouse anti-TRAF6, mouse-anti-ubiquitin, and rabbit anti-tubulin were purchased from Santa Cruz Biotechnology. DAPI (4′,6-diamidino-2-phenylindole) and gentamicin were purchased from Roche. Pierce Sepharose A/G beads were purchased from Thermo Scientific.
Bacterial infections, transfection, and Western blotting.
Cells at a confluence of 80% were starved in FBS-deficient DMEM for 2 h. Starved cells were then infected with S. Typhimurium or with Shigella flexneri, Listeria monocytogenes, or Staphylococcus aureus at an MOI of 10 for 1 h. The infected cells were subsequently incubated with DMEM and 100 μg/ml gentamicin for 30 min. Finally, the infected cells were washed once with prewarmed phosphate-buffered saline (PBS) and then incubated with DMEM and 10 μg/ml gentamicin until the harvest times. Transfection and Western blotting were performed as described previously (11). Protein bands in images of Western blots were quantified with ImageJ software.
Immunofluorescence microscopy.
Traf6+/+ and Traf6−/− MEFs grown on glass coverslips were infected with S. Typhimurium, washed once with prewarmed PBS, and fixed in 4% paraformaldehyde in PBS for 15 min at 37°C before being permeabilized for 15 min with 0.3% Triton X-100 in PBS. Cells were then washed five times with PBS and blocked with 2% normal goat serum in PBS for 1 h at room temperature, followed by incubation at 4°C overnight with rabbit-anti-phospho-STAT3 (Y705). The next day, after five washes with PBS, the cells were incubated with Alexa Fluor 488 (green)-conjugated anti-rabbit secondary antibodies and DAPI for 2 h at room temperature before being washed twice with PBS. Finally, the glass coverslips were mounted on glass slides and were examined using a Nikon Eclipse Ti fluorescence microscope, a digital sight camera, and NIS-Elements Basic Research software.
Immunoprecipitation.
For immunoprecipitation (50), the infected cells were lysed in lysis buffer (50 mM Tris [pH 7.6], 1 M NaCl, 1% NP-40, 0.5% Triton X-100, 5% sodium deoxycholate, 1 mM phenylmethane sulfonyl fluoride [PMSF], and a protease inhibitor cocktail). The cells were disrupted by mechanical lysis with a Dounce homogenizer. After centrifugation (at 15,000 × g for 30 min at 4°C), the cell extracts were precleared using Sepharose A/G beads for 2 h with continuous shaking at 4°C. The beads were removed by centrifugation, and the supernatants were then incubated with a primary antibody at 4°C overnight. The immune complex was captured by incubation with new Sepharose A/G beads for 1 h at 37°C. The beads were then washed three times with lysis buffer using centrifugation. The pellets containing the Sepharose A/G immune complexes were suspended in 1× SDS loading buffer and were analyzed by immunoblotting.
Isolation of plasma membranes.
Plasma membranes were isolated as described previously (50). After HEK293T cells (on a 10-cm plate) were infected (MOI, 10) for 8 h, the harvested cells were incubated in a buffer (20 mM HEPES [pH 7.2], 0.5 mM EGTA, 1 mM PMSF, and a protease inhibitor cocktail) on ice for 30 min. The cells were disrupted by mechanical lysis with a Dounce homogenizer, and the lysates were centrifuged twice at 15,000 × g and 4°C for 30 min to remove bacteria and debris. The resulting supernatants were centrifuged at 100,000 × g and 4°C to separate the membrane (pellet) from the cytoplasmic (supernatant) fractions. The presence of STAT3 and phosphorylated STAT3 in the membrane fractions was detected by Western blotting.
Intracellular S. Typhimurium numbering assays.
Cells were infected, washed, and treated with 100 μg/ml gentamicin to kill extracellular bacteria. After infection, the infected cells were lysed with 0.3% Triton X-100, and 10-fold serial dilutions of the homogenates were plated on LB agar. CFU, representing the numbers of intracellular bacteria, are reported as the means ± standard deviations (SD) from three independent experiments.
Statistical analyses.
Unless otherwise noted, results are presented as the means ± SD for three independent experiments. Statistical significance was calculated by a one-tailed distributed paired Student t test or by two-way analysis of variance (ANOVA) with Prism software (GraphPad Software, Inc.). P values of <0.05 were considered to indicate significant differences.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sunny Shin for critical review of the manuscript. We are grateful to Jun-ichiro Inoue and Jin Gohda of the University of Tokyo for the gift of the Traf6+/+ and Traf6−/− MEFs. We thank Hongtao Li of Xinan University for giving us a variety of plasmids and strains used in this study.
This work was supported by the National Natural Science Foundation of China (grants 81101220 and 31540066 [to H.-H.R.] and grant 31270050 [to S.-Y.W.]), the Tianjin Innovation Team Building Project (grant TD12-5049), and the NIH (R01AI120489 [to J.Z.]).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00081-17.
REFERENCES
- 1.Akira S. 2009. Pathogen recognition by innate immunity and its signaling. Proc Jpn Acad Ser B Phys Biol Sci 85:143–156. doi: 10.2183/pjab.85.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barton GM. 2008. A calculated response: control of inflammation by the innate immune system. J Clin Invest 118:413–420. doi: 10.1172/JCI34431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leber JH, Crimmins GT, Raghavan S, Meyer-Morse NP, Cox JS, Portnoy DA. 2008. Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog 4:e6. doi: 10.1371/journal.ppat.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sethi S, Chakraborty T. 2011. Role of TLR-/ NLR-signaling and the associated cytokines involved in recruitment of neutrophils in murine models of Staphylococcus aureus infection. Virulence 2:316–328. doi: 10.4161/viru.2.4.16142. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Xu H, Zhou Y, Zhang J, Long C, Li S, Chen S, Zhou JM, Shao F. 2007. The phosphothreonine lyase activity of a bacterial type III effector family. Science 315:1000–1003. doi: 10.1126/science.1138960. [DOI] [PubMed] [Google Scholar]
- 6.Bliska JB. 2006. Yersinia inhibits host signaling by acetylating MAPK kinases. ACS Chem Biol 1:349–351. doi: 10.1021/cb600261k. [DOI] [PubMed] [Google Scholar]
- 7.Mukherjee S, Hao YH, Orth K. 2007. A newly discovered post-translational modification—the acetylation of serine and threonine residues. Trends Biochem Sci 32:210–216. doi: 10.1016/j.tibs.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Zhou H, Monack DM, Kayagaki N, Wertz I, Yin J, Wolf B, Dixit VM. 2005. Yersinia virulence factor YopJ acts as a deubiquitinase to inhibit NF-κB activation. J Exp Med 202:1327–1332. doi: 10.1084/jem.20051194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roppenser B, Grinstein S, Brumell JH. 2012. Modulation of host phosphoinositide metabolism during Salmonella invasion by the type III secreted effector SopB. Methods Cell Biol 108:173–186. doi: 10.1016/B978-0-12-386487-1.00009-2. [DOI] [PubMed] [Google Scholar]
- 10.Bruno VM, Hannemann S, Lara-Tejero M, Flavell RA, Kleinstein SH, Galan JE. 2009. Salmonella Typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog 5:e1000538. doi: 10.1371/journal.ppat.1000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruan HH, Li Y, Zhang XX, Liu Q, Ren H, Zhang KS, Zhao H. 2014. Identification of TRAF6 as a ubiquitin ligase engaged in the ubiquitination of SopB, a virulence effector protein secreted by Salmonella typhimurium. Biochem Biophys Res Commun 447:172–177. doi: 10.1016/j.bbrc.2014.03.126. [DOI] [PubMed] [Google Scholar]
- 12.Ruan H, Zhang Z, Tian L, Wang S, Hu S, Qiao JJ. 2016. The Salmonella effector SopB prevents ROS-induced apoptosis of epithelial cells by retarding TRAF6 recruitment to mitochondria. Biochem Biophys Res Commun 478:618–623. doi: 10.1016/j.bbrc.2016.07.116. [DOI] [PubMed] [Google Scholar]
- 13.Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG, Lin HK. 2009. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325:1134–1138. doi: 10.1126/science.1175065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollet I, Opina CJ, Zimmerman C, Leong KG, Wong F, Karsan A. 2003. Bacterial lipopolysaccharide directly induces angiogenesis through TRAF6-mediated activation of NF-κB and c-Jun N-terminal kinase. Blood 102:1740–1742. doi: 10.1182/blood-2003-01-0288. [DOI] [PubMed] [Google Scholar]
- 15.Stockhammer OW, Rauwerda H, Wittink FR, Breit TM, Meijer AH, Spaink HP. 2010. Transcriptome analysis of Traf6 function in the innate immune response of zebrafish embryos. Mol Immunol 48:179–190. doi: 10.1016/j.molimm.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Verstak B, Nagpal K, Bottomley SP, Golenbock DT, Hertzog PJ, Mansell A. 2009. MyD88 adapter-like (Mal)/TIRAP interaction with TRAF6 is critical for TLR2- and TLR4-mediated NF-κB proinflammatory responses. J Biol Chem 284:24192–24203. doi: 10.1074/jbc.M109.023044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang HF, Lai R. 2014. STAT3 in cancer—friend or foe? Cancers (Basel) 6:1408–1440. doi: 10.3390/cancers6031408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kortylewski M, Feld F, Kruger KD, Bahrenberg G, Roth RA, Joost HG, Heinrich PC, Behrmann I, Barthel A. 2003. Akt modulates STAT3-mediated gene expression through a FKHR (FOXO1a)-dependent mechanism. J Biol Chem 278:5242–5249. doi: 10.1074/jbc.M205403200. [DOI] [PubMed] [Google Scholar]
- 19.Piao JY, Lee HG, Kim SJ, Kim DH, Han HJ, Ngo HK, Park SA, Woo JH, Lee JS, Na HK, Cha YN, Surh YJ. 23 February 2016. Helicobacter pylori activates IL-6-STAT3 signaling in human gastric cancer cells: potential roles for reactive oxygen species. Helicobacter doi: 10.1111/hel.12298. [DOI] [PubMed] [Google Scholar]
- 20.Hannemann S, Gao B, Galan JE. 2013. Salmonella modulation of host cell gene expression promotes its intracellular growth. PLoS Pathog 9:e1003668. doi: 10.1371/journal.ppat.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y. 2008. TRAF6 autoubiquitination-independent activation of the NFκB and MAPK pathways in response to IL-1 and RANKL. PLoS One 3:e4064. doi: 10.1371/journal.pone.0004064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. 2007. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of IκB kinase activation. J Biol Chem 282:4102–4112. doi: 10.1074/jbc.M609503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Worrall LJ, Vuckovic M, Strynadka NC. 2010. Crystal structure of the C-terminal domain of the Salmonella type III secretion system export apparatus protein InvA. Protein Sci 19:1091–1096. doi: 10.1002/pro.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isberg RR, Falkow S. 1985. A single genetic locus encoded by Yersinia pseudotuberculosis permits invasion of cultured animal cells by Escherichia coli K-12. Nature 317:262–264. doi: 10.1038/317262a0. [DOI] [PubMed] [Google Scholar]
- 25.Chung J, Uchida E, Grammer TC, Blenis J. 1997. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol 17:6508–6516. doi: 10.1128/MCB.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becker S, Groner B, Muller CW. 1998. Three-dimensional structure of the Stat3β homodimer bound to DNA. Nature 394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- 27.Zhang T, Kee WH, Seow KT, Fung W, Cao X. 2000. The coiled-coil domain of Stat3 is essential for its SH2 domain-mediated receptor binding and subsequent activation induced by epidermal growth factor and interleukin-6. Mol Cell Biol 20:7132–7139. doi: 10.1128/MCB.20.19.7132-7139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parks EE, Ceresa BP. 2014. Cell surface epidermal growth factor receptors increase Src and c-Cbl activity and receptor ubiquitylation. J Biol Chem 289:25537–25545. doi: 10.1074/jbc.M114.579581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piscatelli HL, Li M, Zhou D. 2016. Dual 4- and 5-phosphatase activities regulate SopB-dependent phosphoinositide dynamics to promote bacterial entry. Cell Microbiol 18:705–719. doi: 10.1111/cmi.12542. [DOI] [PubMed] [Google Scholar]
- 30.Stender S, Friebel A, Linder S, Rohde M, Mirold S, Hardt WD. 2000. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol Microbiol 36:1206–1221. doi: 10.1046/j.1365-2958.2000.01933.x. [DOI] [PubMed] [Google Scholar]
- 31.Knodler LA, Finlay BB, Steele-Mortimer O. 2005. The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J Biol Chem 280:9058–9064. doi: 10.1074/jbc.M412588200. [DOI] [PubMed] [Google Scholar]
- 32.Huang FC, Werne A, Li Q, Galyov EE, Walker WA, Cherayil BJ. 2004. Cooperative interactions between flagellin and SopE2 in the epithelial interleukin-8 response to Salmonella enterica serovar Typhimurium infection. Infect Immun 72:5052–5062. doi: 10.1128/IAI.72.9.5052-5062.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu-Bryan R, Pritzker K, Firestein GS, Terkeltaub R. 2005. TLR2 signaling in chondrocytes drives calcium pyrophosphate dihydrate and monosodium urate crystal-induced nitric oxide generation. J Immunol 174:5016–5023. doi: 10.4049/jimmunol.174.8.5016. [DOI] [PubMed] [Google Scholar]
- 34.Li Q, Engelhardt JF. 2006. Interleukin-1β induction of NFκB is partially regulated by H2O2-mediated activation of NFκB-inducing kinase. J Biol Chem 281:1495–1505. doi: 10.1074/jbc.M511153200. [DOI] [PubMed] [Google Scholar]
- 35.Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, Kim N, Lee SY. 2005. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 106:852–859. doi: 10.1182/blood-2004-09-3662. [DOI] [PubMed] [Google Scholar]
- 36.Bezbradica JS, Schroder K. 2014. TRAF6 is a nexus for TLR-STAT1 crosstalk. Immunol Cell Biol 92:737–738. doi: 10.1038/icb.2014.71. [DOI] [PubMed] [Google Scholar]
- 37.Walsh MC, Lee J, Choi Y. 2015. Tumor necrosis factor receptor-associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol Rev 266:72–92. doi: 10.1111/imr.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caldenhoven E, van Dijk T, Raaijmakers JA, Lammers JW, Koenderman L, De Groot RP. 1995. Activation of the STAT3/acute phase response factor transcription factor by interleukin-5. J Biol Chem 270:25778–25784. doi: 10.1074/jbc.270.43.25778. [DOI] [PubMed] [Google Scholar]
- 39.Wei J, Yuan Y, Jin C, Chen H, Leng L, He F, Wang J. 2012. The ubiquitin ligase TRAF6 negatively regulates the JAK-STAT signaling pathway by binding to STAT3 and mediating its ubiquitination. PLoS One 7:e49567. doi: 10.1371/journal.pone.0049567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Owen KA, Meyer CB, Bouton AH, Casanova JE. 2014. Activation of focal adhesion kinase by Salmonella suppresses autophagy via an Akt/mTOR signaling pathway and promotes bacterial survival in macrophages. PLoS Pathog 10:e1004159. doi: 10.1371/journal.ppat.1004159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen ZJ. 2012. Ubiquitination in signaling to and activation of IKK. Immunol Rev 246:95–106. doi: 10.1111/j.1600-065X.2012.01108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dong LH, Li L, Song Y, Duan ZL, Sun SG, Lin YL, Miao SB, Yin YJ, Shu YN, Li H, Chen P, Zhao LL, Han M. 2015. TRAF6-mediated SM22α K21 ubiquitination promotes G6PD activation and NADPH production, contributing to GSH homeostasis and VSMC survival in vitro and in vivo. Circ Res 117:684–694. doi: 10.1161/CIRCRESAHA.115.306233. [DOI] [PubMed] [Google Scholar]
- 43.Ko R, Park JH, Ha H, Choi Y, Lee SY. 2015. Glycogen synthase kinase 3β ubiquitination by TRAF6 regulates TLR3-mediated pro-inflammatory cytokine production. Nat Commun 6:6765. doi: 10.1038/ncomms7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sundar R, Gudey SK, Heldin CH, Landstrom M. 2015. TRAF6 promotes TGFβ-induced invasion and cell-cycle regulation via Lys63-linked polyubiquitination of Lys178 in TGFβ type I receptor. Cell Cycle 14:554–565. doi: 10.4161/15384101.2014.990302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haldar AK, Foltz C, Finethy R, Piro AS, Feeley EM, Pilla-Moffett DM, Komatsu M, Frickel EM, Coers J. 2015. Ubiquitin systems mark pathogen-containing vacuoles as targets for host defense by guanylate binding proteins. Proc Natl Acad Sci U S A 112:E5628–E5637. doi: 10.1073/pnas.1515966112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar Y, Valdivia RH. 2009. Leading a sheltered life: intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe 5:593–601. doi: 10.1016/j.chom.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Ding X, Cui J, Xu H, Chen J, Gong YN, Hu L, Zhou Y, Ge J, Lu Q, Liu L, Chen S, Shao F. 2011. Cysteine methylation disrupts ubiquitin-chain sensing in NF-κB activation. Nature 481:204–208. doi: 10.1038/nature10690. [DOI] [PubMed] [Google Scholar]
- 49.Zhu Y, Li H, Hu L, Wang J, Zhou Y, Pang Z, Liu L, Shao F. 2008. Structure of a Shigella effector reveals a new class of ubiquitin ligases. Nat Struct Mol Biol 15:1302–1308. doi: 10.1038/nsmb.1517. [DOI] [PubMed] [Google Scholar]
- 50.Patel JC, Hueffer K, Lam TT, Galan JE. 2009. Diversification of a Salmonella virulence protein function by ubiquitin-dependent differential localization. Cell 137:283–294. doi: 10.1016/j.cell.2009.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.