

Abstract
Three‐dimensional structure determination of integral membrane proteins has advanced in unprecedented detail our understanding of mechanistic events of how ion channels, transporters, receptors, and enzymes function. This exciting progress required a tremendous amount of methods development, as exemplified here with G protein‐coupled receptors (GPCRs): Optimizing the production of GPCRs in recombinant hosts; increasing the probability of crystal formation using high‐affinity ligands, nanobodies, and minimal G proteins for co‐crystallization, thus stabilizing receptors into one conformation; using the T4 lysozyme technology and other fusion partners to promote crystal contacts; advancing crystallization methods including the development of novel detergents, and miniaturization and automation of the lipidic cubic phase crystallization method; the concept of conformational thermostabilization of GPCRs; and developing microfocus X‐ray synchrotron technologies to analyze small GPCR crystals. However, despite immense progress to explain how GPCRs function, many receptors pose intractable hurdles to structure determination at this time. Three emerging methods, serial femtosecond crystallography, micro electron diffraction, and single particle electron cryo‐microscopy, hold promise to overcome current limitations in structural membrane biology.
Keywords: integral membrane protein, G protein‐coupled receptor, three‐dimensional structure determination, lipidic cubic phase (LCP), serial femtosecond crystallography (SFX), micro electron diffraction (MicroED), single particle electron cryo‐microscopy (cryo‐EM)
Introduction
Determining three‐dimensional structures of integral membrane proteins was largely deemed impossible until the 1970s. In 1975, a seminal paper by Richard Henderson and Nigel Unwin1 presented the first structural information for an integral membrane protein, a 7 Å resolution structural model of bacteriorhodopsin, a light‐driven proton pump from the archaebacterium Halobacterium salinarium. This structure was obtained from two‐dimensional crystals imaged by room temperature electron microscopy, but it took another 15 years of methods development before an atomic model of bacteriorhodopsin was published in 1990, which was the first structure of any protein determined by electron microscopy.2 In the intervening period, the first structure of an integral membrane protein, a bacterial photosynthetic reaction center, was determined by X‐ray crystallography,3 and in 1988, Hartmut Michel, Johann Deisenhofer, and Robert Huber were awarded the Nobel Prize in Chemistry for this work. These accomplishments launched the era of membrane protein structural biology.
During the last three decades, breathtaking progress has been made in elucidating how membrane proteins function through determining their three‐dimensional structures and utilizing information from nonstructural experimental approaches. For this to happen, strategies, reagents, and tools for recombinant expression, purification, stabilization, crystallization and data collection needed to be developed4 because many membrane protein targets, especially eukaryotic membrane proteins, are not abundant naturally and are unstable in detergent solution owing to their conformational flexibility. This review summarizes concepts that have advanced the field of membrane protein structure determination, using GPCRs as an example, and discusses emerging avenues of investigation and new technological developments with the potential to overcome current limitations in membrane protein structural biology.
G protein‐coupled receptors
GPCRs are integral membrane proteins that reside in the plasma membrane of eukaryotic cells. They are central to transmitting signals from the extracellular milieu to the inside of the cell. Ligand binding on the extracellular surface of GPCRs leads to conformational changes within the receptor, triggering the activation of G proteins and the interaction with kinases and arrestin molecules in the cytosol.5 With more than 800 members in the human genome, GPCRs constitute the largest protein family involved in signal transduction, and are major targets for drug development.
GPCRs exist in an equilibrium between inactive, nonsignaling states, and active states that can interact with signaling partners within the cell. In contrast to rhodopsin, many GPCRs exhibit basal activity that has been correlated with structural instability.6 Ligands range from inverse agonists, which inhibit basal activity, to full agonists, which maximally activate the receptor. The conformational dynamics of GPCRs provide the basis for the complexity and fine‐tuning of hormone signaling, but also render GPCRs difficult to crystallize because of their conformational heterogeneity and instability.
In the year 2000, the first crystal structure was determined of a GPCR, the visual pigment rhodopsin.7 The second crystal structure, the β2‐adrenergic receptor (β2AR), was published only in 2007.8, 9, 10 10 years later, crystal structures of 38 unique GPCRs have been reported.11 Extraordinary progress towards understanding the activation mechanism of GPCRs was achieved by the crystallization of receptors in an active state, coupled to a cytoplasmic heterotrimeric G protein,12 an engineered mini G protein,13 G protein mimicking nanobodies (Nbs),14, 15, 16, 17 or arrestin18 (Fig. 1). This exciting progress required a tremendous amount of methods development, with the most notable achievements described below. The foundation of today's successes was the decades of research by groups throughout the world who were convinced that structures of GPCRs will be solved through sufficient effort and the development of novel approaches. Indeed, it took two decades of hard work after the cloning of the respective cDNAs19, 20, 21, 22, 23 before the structures of the β2AR,8, 9, 10 the β1‐adrenergic receptor (β1AR),24 the adenosine A2a receptor (A2aR),25, 26, 27, 28 and the neurotensin receptor (NTSR1)29, 30, 31, 32 were determined.
Figure 1.
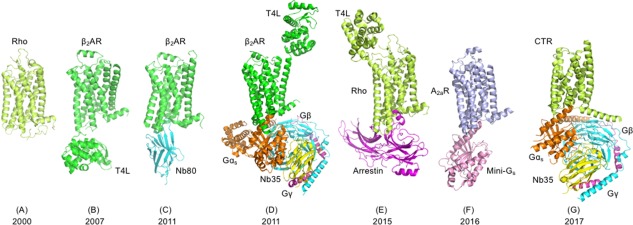
Milestones of GPCR structure determination: PDB codes are given in parentheses. (A) Rhodopsin (1F88), (B) β2‐adrenergic receptor (2RH1), (C) β2AR with nanobody (3P0G), (D) β2AR with heterotrimeric G protein (3SN6), (E) Rhodopsin with arrestin (4ZWJ), (F) Adenosine A2a receptor with mini G protein (5G53), (G) Calcitonin receptor (CTR) with heterotrimeric G protein (5UZ7). Structures shown in panels A–F were solved by X‐ray crystallography, whereas the structure shown in panel G was solved by single particle cryo‐EM. The T4 lysozyme fusion partner facilitating crystal formation is indicated as T4L. Individual protein chains of receptor complexes are labeled and shown in different colors. Publication years are given at bottom of figure.
What are the challenges in solving membrane protein structures?
This section addresses key considerations for membrane protein crystallization and serves as a guide to navigate through the different aspects discussed in this review. Hurdles for obtaining high‐resolution structures may present themselves at virtually every step, from recombinant expression to crystal formation and diffraction data acquisition.
Despite the miniaturization of the actual crystallization process by robotics, the target GPCR needs to be produced in yeast, insect cells, or mammalian cells at milligram scale. These production methods are preferred because only very few GPCRs can be expressed in functional form in bacteria.29, 33, 34, 35 Owing to space limitations, the reader is referred to reviews that cover the topic of recombinant expression of membrane proteins in detail [e.g., Refs. 36, 37, 38].
GPCRs are highly dynamic molecules and their inherent flexibility is the underlying cause for their often‐observed biochemical instability. Successful structure determination requires that detergent‐solubilized GPCRs are maintained in a biologically relevant form, as aggregated or denatured protein will not form crystals. Bovine rhodopsin was the first GPCR to be crystallized,7, 39 reflecting its high stability in many detergents as long as the receptor was kept in its inactive, dark state. However, most other GPCRs are labile in the presence of detergent40 owing to their highly dynamic properties. Remedies to stabilize receptors include addition of a high‐affinity ligand to stabilize a particular signaling conformation thus reducing conformational flexibility and increasing receptor stability;41 conformational thermostabilization of GPCRs42 as a means to lock the receptor in a particular ligand‐dependent state; or complexation with signaling mimetics or partners such as nanobodies,43 mini G proteins44, 45 or G protein12 to improve stability. Continued efforts to develop novel mild detergents such as bicyclic alkylmaltosides,46 facial amphiphiles,47 and maltose‐neopentylglycols48, 49 have been instrumental in receptor stabilization and concomitant formation of well‐ordered crystals.12
Protein crystals are materials in which molecules are arranged in a highly ordered microscopic structure characterized by symmetry. Thus, any flexible entity of a receptor, such as poorly structured termini and loops, or dynamic features of the transmembrane core, counteract the growth of highly diffracting crystals. Unstructured receptor regions have been removed by protein engineering in many receptor constructs optimized for crystallization. Replacement of loops by crystallization modules such as T4 lysozyme (T4L) has restricted the movement of adjacent transmembrane helices.10 Conformational thermostabilization42 and co‐crystallization with conformation‐specific antibodies,9, 50, 51 nanobodies,14, 15, 16, 17 or G protein12, 13 have resulted in more homogenous protein preparations, and diffracting crystals.
Crystallogenesis requires that protein molecules engage in crystal lattice contacts. As for water‐soluble proteins, polar interactions contribute to the stabilization of crystals of membrane proteins.52 The polar surfaces of many GPCRs are small, and using stabilizing, large detergents such as dodecyl‐maltoside, results in occlusion of the hydrophilic receptor regions essential to form crystal contacts. Small detergents such as octyl‐glucoside are in theory better suited for crystallization52 but are often highly denaturing precluding their use for most GPCR targets. Two approaches for promoting receptor crystal formation have evolved: Increasing the hydrophilic area by engineered fusion proteins such as T4L,10, 53 antibodies,9, 50, 51 or nanobodies,43 so that mild detergents with a large micelle can be used; or thermostabilizing receptors to make them withstand harsh, short‐chain detergents.42
Fusion partners and antibody fragments
Obtaining high‐resolution structures of GPCRs other than rhodopsin has been challenging because of their inherent structural instability. In addition, many GPCRs contain relatively small hydrophilic loops, and the scarcity of hydrophilic regions available to form potential crystal contacts is a major impediment to obtaining diffraction quality GPCR crystals. Replacement of inner loop 3 (IL3) with T4L, a well‐folded protein that adds a polar surface and restricts the movement of the adjacent transmembrane helices, proved critical for obtaining the well‐ordered crystals of the β2AR.10 An expanded set of fusion partners for crystallizing GPCRs has subsequently been developed, most notably a thermostabilized apocytochrome b562 (BRIL),53 rubredoxin,53 and the catalytic domain of Pyrococcus abysii glycogen synthase (PGS).54 These fusion partners have been used in various ways to replace IL3 (β2AR‐T4L,10 chemokine receptor CCR5‐rubredoxin,55 orexin receptor OX2‐PGS54), or IL2 (metabotropic glutamate receptor mGlu5‐T4L),56 or positioned at the receptor N‐terminus (T4L‐β2AR,12, 57 BRIL‐nociceptin/orphanin FQ receptor58).
Antibody fragment‐mediated crystallization59 has also been successful through reducing the conformational dynamics of the receptor and extending the hydrophilic surface area available to make crystal contacts. Receptors crystallized in the presence of mouse monoclonal antibody fragments include β2AR bound to Fab5, which recognizes a three‐dimensional epitope on the intracellular side of the β2AR,9, 50 and A2aR bound to Fab2838, which binds to the intracellular surface of A2AR where it acts as an allosteric antagonist.51
Nanobodies and mini G proteins
The formation of highly ordered 3D crystals necessitates that the receptor is preferentially in one particular conformation. This is achieved by the use of high‐affinity inverse agonist or full agonist ligands, and in the case of active‐state receptors, by complexation with a signaling partner or mimetic. To date, only one structure of a GPCR‐arrestin complex (rhodopsin‐arrestin18) and only one structure of a GPCR‐G protein complex (β2AR bound to the adenylate cyclase stimulating G protein Gs 12) have been reported, providing the first insight into the arrangement of those complexes. In addition, receptors have been crystallized in an active state coupled to peptide fragments of the Gα subunit60, 61 or arrestin.62 Crystallizing GPCR‐arrestin and GPCR‐G protein complexes remains very challenging, but the development of G protein mimicking nanobodies63 and most recently minimal G proteins44 facilitated crystallization of several active‐state receptors (β2AR‐Nb80,16 β2AR‐Nb6B9,17 muscarinic receptor M2R‐Nb9–8,15 μ‐opioid receptor‐Nb39,14 A2AR‐mini Gs 13).
Nanobodies43 are the recombinant variable domains of camelid heavy‐chain antibodies with full antigen‐binding capability, and are selected from peripheral blood lymphocytes of an immunized llama through combinatorial methods such as phage display, yeast display, or ribosome display.43, 63 Nanobodies have evolved as alternatives to conventional antibodies because of their elongated shape and convex antigen binding site, allowing access to cavities on proteins that are difficult to access by conventional antibodies. Nanobodies have been instrumental in obtaining structures of conformationally stabilized active‐state GPCRs.
Mini G proteins are engineered, small soluble proteins composed of a truncated form of the GTPase domain of the G protein, including point mutations to stabilize the protein in the absence of Gβγ and in the presence of detergent. Three truncations eliminate the switch III region, part of the N terminus, and the α‐helical domain of the Gα protein.44 Mini G proteins efficiently couple to GPCRs in the absence of Gβγ subunits.44 A growing number of mini G proteins provide now the tools to study GPCRs in their active conformation and facilitate complex formation of GPCRs for crystallization experiments.45
In surfo and in meso crystallization
Common and successful approaches to membrane protein crystallization are methods that make use of detergent‐solubilized membrane protein preparations in crystallization trials (in surfo: vapor diffusion, dialysis, microbatch) [see Ref. 64]. While protein–protein interactions dominate the crystallization of soluble proteins, both detergent–detergent and protein–detergent interactions are additional considerations for the membrane protein crystallization, and choosing the right detergent has been key to successful membrane protein crystallization efforts. GPCRs (except rhodopsin) show poor stability in detergent solutions, especially in harsh short‐chain detergents, preferred for crystal contacts to occur. Conformational thermostabilization is one possible approach to make the protein more tolerant to short‐chain detergents, and this approach has been successfully used to determine GPCR structures in surfo.29, 42, 65 Alternatively, receptors have been prepared in mild, stabilizing detergents such as dodecylmaltoside or neopentylglycol detergents. However, the hydrophilic regions of the membrane protein, essential to form crystal contacts, are then often occluded by the detergent micelle, preventing crystal growth. Extending the hydrophilic surface area by using a monoclonal Fab antibody fragment51 or replacing IL3 with BRIL66 allowed the structures of antagonist‐bound A2AR to be determined from crystals grown by vapor diffusion and using large‐micelle detergent.
Lipidic cubic phase (LCP) is a membrane‐mimetic matrix suitable for stabilization and crystallization of membrane proteins in a lipidic environment (in meso crystallogenesis).64 This methodology was introduced by Ehud Landau and Jürg Rosenbusch back in 1996,67 and optimized for miniaturization and automation by Martin Caffrey and Vadim Cherezov.64 The success of using LCP for crystallization can be attributed to the stabilizing membrane‐like environment of LCP and type I crystal packing, where crystal contacts are formed between both polar and nonpolar parts of the protein, contributing to better order and diffraction. LCP crystallization accommodates membrane proteins prepared in mild long‐chain detergents. Almost all in meso GPCR structures have utilized receptors modified by fusion partners, although no fusion partner was used for the chemokine CCR9 receptor68 and the high‐resolution structure of β1AR.69 LCP crystallization is not limited to GPCRs, but structures of other membrane proteins, both α‐helical and β‐barrel, including single and multisubunit proteins, have been reported [see Ref. 70]. Crystals grown in meso are often small, and the development of microfocus X‐ray synchrotron technologies such as the minibeam71 combined with rasterization capabilities72 and the development of fast detector technologies have been indispensable for data collection.
Two alternative crystallization techniques, in which protein is crystallized from a lipid membrane environment, are the bicelle crystallization method9, 73 and crystallization by vesicle fusion.74 These methods have had some successes with a variety of membrane proteins.
Conformational thermostabilization
GPCRs are highly dynamic molecules with low thermal barriers between the inactive and active signaling states, displaying in many cases basal activity. Furthermore, GPCRs show poor stability in detergent solution. Therefore, conformational flexibility and instability, once extracted from the membrane, impede the growth of well‐ordered crystals.42 Two strategies have been used to address these shortcomings: Use of high‐affinity ligands, with slow off‐rates to allow full occupancy during the crystallization process, lock the wild‐type receptor into a single conformation, which increases the receptor stability in detergent solution and therefore the probability of crystal formation. Alternatively, improving the thermostability of the GPCR itself makes the protein more tolerant to short‐chain detergents, preferred in vapor diffusion crystallization experiments. The latter approach allows co‐crystallization with low‐affinity ligands, because the enhanced stability is encoded within the receptor protein itself.42 To isolate thermostabilizing mutations in a receptor, mutations are introduced throughout the receptors by either systematic scanning mutagenesis [see Refs. 65, 75] or random mutagenesis as part of an evolution‐based strategy.76, 77 In systematic scanning mutagenesis, the most stabilizing single mutations are then combined to generate a mutant receptor with an augmented melting temperature. The combination of mutagenesis with a specific selection strategy was used to stabilize agonist78, 79 and antagonist65, 78 conformations of GPCRs.
Crystal structures of agonist‐bound GPCRs have relied on the use of either exceptionally high‐affinity agonists or receptor stabilization by mutagenesis. However, many endogenous ligands bind with low affinity and are structurally not tractable in complex with a receptor. Using directed evolution, Ring and colleagues17 used yeast surface display together with a conformationally specific selection strategy to engineer a high‐affinity nanobody that minimizes receptor heterogeneity and stabilizes the active state of the β2AR to determine the crystal structure of the activated receptor bound to the endogenous agonist adrenaline.
In situ data collection
Membrane protein crystals are often tiny and fragile, and thus pose challenges for handling such as mounting the crystals or soaking in cryoprotectants. Recently published methods on in situ room temperature data collection have made a major contribution to address these issues. Axford et al.80 mounted an entire 96‐well sitting‐drop crystallization plate at a synchrotron beam line and successfully collected a data set from many membrane protein crystals. Huang and colleagues81 implemented an in situ approach with in meso crystallization, termed the “in meso in situ serial crystallography” (IMISX) method. Both methodologies pave new avenues to data collection from crystals that are too fragile and too small to mount without losing diffraction quality through mechanical shear‐induced stress.
Techniques that complement structural work
The focus of this review is on approaches yielding high resolution structural information. By nature, crystal structures provide static rather than dynamic views and typically capture the lowest energy state within an ensemble of conformations that may differ from other signaling conformations of membrane‐inserted receptors, not observed in crystal structures. Therefore, techniques that complement structural work are essential to understand how proteins function. For example, molecular dynamics simulations sample interactions of a membrane protein with lipid and partner proteins, and their internal dynamics;30, 82, 83 single molecule spectroscopy and super‐resolution imaging allows the study of macromolecular function;84 fluorescence spectroscopy probes conformational changes within a receptor,85, 86 and native mass spectrometry has been used to characterize noncovalent protein–protein and protein–ligand complexes and lipid interactions.87 Nuclear magnetic resonance (NMR) techniques and site‐directed electron paramagnetic resonance (EPR) spectroscopy88, 89, 90 are powerful tools for examining intramolecular dynamics and distance distributions between domains of a protein.91
Nuclear magnetic resonance (NMR)
Two main methods have been employed to study the structure and dynamics of membrane proteins, namely solution NMR and solid‐state NMR. Solution NMR methods require that the sample undergoes fast rotational diffusion and this therefore poses limitations on the size of the target membrane protein and its preparation (detergent‐solubilized and lipid micelles, small bicelles, nanodiscs).92 Solid‐state NMR methods have principally no size limitations because they are used on static samples, or samples rotated at high speed, and thus allow membrane protein samples to be prepared in lipid bilayers mimicking the protein's natural environment. However, solid‐state NMR methods still meet technical barriers, because solid‐state NMR spectra are complex, leading to spectral crowding of uniformly labeled samples of large proteins, restricting the size of structures that can be determined.92
Solving the complete structure of a membrane protein has been very challenging and has been impaired by substantial difficulties of sample preparation of sufficient quantities of isotopically 15N‐ and 13C‐labeled protein, limited thermal stability, and sample heterogeneity. Nevertheless, the backbone assignment of the archaeal seven‐helix transmembrane receptor sensory rhodopsin II93 has been accomplished by solution NMR. Furthermore, the backbone structure of a phospholipid bilayer embedded GPCR, the chemokine receptor CXCR1, has been reported by using solid‐state NMR techniques.94 For this, CXCR1 was expressed and isotopically labeled in E. coli as inclusion bodies, purified, and refolded into DMPC proteoliposomes.
In contrast, selective labeling of residues with 19F, 13C, or 15N has been used to provide evidence for ligand‐ and G protein‐dependent conformational states and energy landscapes of GPCRs, and their local dynamics by solution NMR methods.91 Chemical conjugation with isotope‐labeled compounds, either alkylation of cysteine residues or dimethylation of lysine residues, has been used to gather site‐specific information and to avoid presently intractable spectral overlap of a uniformly labeled sample. Chemical labeling is limited by solvent accessibility and the efficiency of the chemical reactions. Despite this limitation, chemical labeling has the advantage that highly sensitive nuclei, such as 19F, can be incorporated, thus leading to excellent signal/noise ratios. Selective isotopic labeling during protein biosynthesis accesses any labeling sites, even those in the hydrophobic environment of the membrane protein. Site‐specific labeling is an excellent tool for fingerprinting the range of potential states of a receptor, but a major challenge remains of relating these observations to the structural changes that give rise to these phenomena.
19F NMR probes have been introduced into detergent‐purified receptors91, 95, 96 at naturally occurring cysteine residues or at a selected position mutated to a cysteine96 and provide exquisite sensitivity to solvent exposure or side chain packing. To monitor a unique receptor environment associated with specific conformational changes requires use of minimal cysteine receptor versions,91 which have to be thoroughly tested for ligand binding and G protein activation to assure integrity of the mutated receptor. Specific labeling of methyl groups, such as 13CH3ε‐methionines, provide ideal NMR probes for high molecular weight proteins because the length and flexibility of the methionine side chain compensates for the slow tumbling, improving the resolution and sensitivity of the spectra. Methionine residues are dispersed throughout the receptor and many are found in the transmembrane region, not accessible for study by fluorine NMR, which requires the addition of small‐molecule probes to surface‐exposed cysteines.97 To study specific receptor regions, methionines that face the exterior of the receptor may be mutated thus simplifying the spectrum and eliminating signals from 13CH3ε‐methionine residues that do not undergo structural changes when comparing active and inactive structures.97 The labeling of methionine residues is achieved by growing for example insect cells in methionine‐deficient media in the presence of 13C methyl‐labeled methionine. Alternatively, 13C labels can be introduced into detergent‐purified GPCRs by reductive methylation of, for example lysine residues, with13C‐formaldehyde.98 The above NMR methods have provided data of functional relevance on selected side‐chains. A recent investigation by Isogai and colleagues99 studied receptor motions followed at backbone sites by selective labeling with 15N‐valine, accomplished by growing insect cells in medium devoid of unlabeled valine in the presence of 15N‐valine.99
In elegant work, Smith and colleagues100 have characterized the activation triggers of the visual pigment rhodopsin using solid‐state NMR approaches. The opsin is produced in HEK293S cells and reconstituted with retinal. The information is obtained through measurements of chemical shift and dipolar couplings. This does not yield complete 3D protein structural information because of the expense of uniform 13C‐, 15N‐labeling of proteins using eukaryotic cell lines. Rather, selective labeling of specific amino acid types or different ligands allows the structural features of the ligand or protein of interest to be studied in exquisite detail. For this, stable tetracycline‐inducible suspension HEK293S cell lines containing the opsin gene and its mutants were used to express rhodopsin101 in medium supplemented with specific 13C‐labeled amino acids.
The conformational and dynamic heterogeneity observed in these and other recent NMR studies provides new insights into membrane‐protein function that would have been impossible to obtain through crystallography alone.
New Approaches
Despite tremendous progress to elucidate the structural biology of GPCRs, many receptors pose unsurmountable obstacles to structure determination owing to a multitude of reasons such as very low expression levels, high instability even in mild detergents, extensive post‐translational modifications, formation of microcrystals not suitable for use with current synchrotron mini X‐ray beams, or resistance to crystallization. Three emerging methods, serial femtosecond crystallography, micro electron diffraction, and single particle electron cryo‐microscopy, hold promise in addressing some of these limitations. The first two approaches relate to micro‐crystals, whereas the latter does not require crystals at all.
Serial femtosecond crystallography (SFX)
SFX102 is an emerging technique that relies on extremely bright and ultrashort pulses produced by X‐ray free‐electron lasers (XFELs), permitting the collection of high‐resolution diffraction intensities from very small crystals at room temperature with minimal radiation damage, using the principle of “diffraction before destruction”.103 A single diffraction pattern is collected before each crystal is destroyed by the powerful XFEL pulse. To obtain a single data set, diffraction patterns from thousands of randomly oriented crystals need to be collected. The crystals are therefore delivered into the beam in a continuous hydrated stream, and diffraction images are recorded by a detector operating at the XFEL pulse repetition rate.103 Technical improvements related to the delivery, detector technology, data acquisition, and analysis104 have transformed macromolecular crystallography. It is now possible to determine structures using XFEL using less than a milligram of protein.105 In 2011, a low‐resolution electron density map from crystals of photosystem I demonstrated proof‐of‐principle103 and now structures have been determined at sub‐2 Å resolution.106 Several structures of GPCRs at high resolution have also been determined.18, 107, 108, 109, 110, 111 Most excitingly, time‐resolved serial femtosecond crystallography at an X‐ray free electron laser has allowed the visualization of conformational changes in bacteriorhodopsin from nanoseconds to milliseconds following photoactivation112 and the light‐induced structural changes for oxygen evolution by photosystem II.113 However, access to XFELs for SFX is currently very limited and not a routine approach for most membrane protein crystallographers.
Micro electron diffraction (MicroED)
MicroED114 is a newly developed technique that records electron diffraction patterns from very small crystals in the electron microscope. Extremely tiny 3D crystals are required as electrons cannot penetrate thick samples. Samples are vitrified, as hydration is critical. The specimen is rotated and multiple diffraction patters are taken from a single crystal, resulting in a series of diffraction patterns that can be indexed and processed with available X‐ray crystallography software. The continuous rotation of the sample requires direct electron detectors. The extremely low electron dose rate results in greatly reduced radiation damage of biological samples that are dose sensitive, but this still produces quality diffraction patterns of high resolution. Proof of principle of the MicroED approach was achieved using lysozyme, yielding a structure to 2.9 Å resolution from 3D microcrystals in cryogenic conditions.115 Subsequently, several protein structures have been solved using improved techniques,114 but the structure of a membrane protein by MicroED is yet to be determined.
Single particle electron cryo‐microscopy (Cryo‐EM)
To date, the majority of membrane protein structures has been determined by X‐ray crystallography and by electron crystallography. These approaches necessitate the availability of well‐ordered, three‐dimensional, or two‐dimensional crystals that are a major road block for many membrane protein targets.116 In contrast, single particle cryo‐EM sidesteps the requirement of well‐ordered crystals for structure determination, thus providing an attractive alternative to crystallography. Major technological advances in the last few years have improved detection (direct electron detection camera), classification of images of heterogeneous sample conformations, correction for beam‐induced motion blurring of images, and dose‐fractionation to use the best subframes for data processing.117, 118 This has resulted in the ‘resolution revolution’119 and an increasing rate of high‐resolution structures deposited in the EM Database.120 Indeed, single particle cryo‐EM has already been used extensively to characterize soluble proteins in multiple conformations at high resolution121 and is being used increasingly on membrane proteins.122, 123, 124, 125, 126, 127, 128 The recent structure determination of hemoglobin (molecular weight 64 kDa) at 3.2 Å resolution is a milestone in cryo‐EM,129 which has had an immediate impact in the GPCR field; the identical methodology was recently used to determine the structure of the calcitonin receptor coupled to heterotrimeric G s.130 Further structures of GPCRs in complex with other binding partners will no doubt follow swiftly, which will result in a revolution in our understanding of the active states of GPCRs.
Conclusions
Membrane protein structure determination has advanced beyond all expectations our insight into mechanistic aspects of how they function. New arising concepts and technological developments hold promise to overcome some of the remaining experimental limitations, propelling us into an exciting time, where structures of integral membrane proteins are no longer regarded as insurmountable challenges.
ACKNOWLEDGMENTS
The research of R.G. is supported by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke. The author has no conflict of interest to declare. I thank Chris Tate (LMB‐MRC) and Anirban Banerjee (NICHD, NIH) for comments on the manuscript.
REFERENCES
- 1. Henderson R, Unwin PN (1975) Three‐dimensional model of purple membrane obtained by electron microscopy. Nature 257:28–32. [DOI] [PubMed] [Google Scholar]
- 2. Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH (1990) Model for the structure of bacteriorhodopsin based on high‐resolution electron cryo‐microscopy. J Mol Biol 213:899–929. [DOI] [PubMed] [Google Scholar]
- 3. Deisenhofer J, Epp O, Miki K, Huber R, Michel H (1985) Structure of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3A resolution. Nature 318:618–624. [DOI] [PubMed] [Google Scholar]
- 4. Grisshammer R (2013) Why we need many more G protein‐coupled receptor structures. Expert Rev Proteomics 10:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rosenbaum DM, Rasmussen SG, Kobilka BK (2009) The structure and function of G‐protein‐coupled receptors. Nature 459:356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gether U, Ballesteros JA, Seifert R, Sanders‐Bush E, Weinstein H, Kobilka BK (1997) Structural instability of a constitutively active G protein‐coupled receptor. Agonist‐independent activation due to conformational flexibility. J Biol Chem 272:2587–2590. [DOI] [PubMed] [Google Scholar]
- 7. Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M (2000) Crystal structure of rhodopsin: a G protein‐coupled receptor. Science 289:739–745. [DOI] [PubMed] [Google Scholar]
- 8. Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC (2007) High‐resolution crystal structure of an engineered human beta2‐adrenergic G protein‐coupled receptor. Science 318:1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK (2007) Crystal structure of the human beta2 adrenergic G‐protein‐coupled receptor. Nature 450:383–387. [DOI] [PubMed] [Google Scholar]
- 10. Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK (2007) GPCR engineering yields high‐resolution structural insights into beta2‐adrenergic receptor function. Science 318:1266–1273. [DOI] [PubMed] [Google Scholar]
- 11. http://blanco.biomol.uci.edu/mpstruc/. Accessed 30 May, 2017.
- 12. Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK (2011) Crystal structure of the beta2 adrenergic receptor–Gs protein complex. Nature 477:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carpenter B, Nehme R, Warne T, Leslie AG, Tate CG (2016) Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature 536:104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, Kobilka BK (2015) Structural insights into mu‐opioid receptor activation. Nature 524:315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504:101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK (2011) Structure of a nanobody‐stabilized active state of the beta(2) adrenoceptor. Nature 469:175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK (2013) Adrenaline‐activated structure of beta2‐adrenoceptor stabilized by an engineered nanobody. Nature 502:575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, Xu Q, de Waal PW, Ke J, Tan MH, Zhang C, Moeller A, West GM, Pascal BD, Van Eps N, Caro LN, Vishnivetskiy SA, Lee RJ, Suino‐Powell KM, Gu X, Pal K, Ma J, Zhi X, Boutet S, Williams GJ, Messerschmidt M, Gati C, Zatsepin NA, Wang D, James D, Basu S, Roy‐Chowdhury S, Conrad CE, Coe J, Liu H, Lisova S, Kupitz C, Grotjohann I, Fromme R, Jiang Y, Tan M, Yang H, Li J, Wang M, Zheng Z, Li D, Howe N, Zhao Y, Standfuss J, Diederichs K, Dong Y, Potter CS, Carragher B, Caffrey M, Jiang H, Chapman HN, Spence JC, Fromme P, Weierstall U, Ernst OP, Katritch V, Gurevich VV, Griffin PR, Hubbell WL, Stevens RC, Cherezov V, Melcher K, Xu HE (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X‐ray laser. Nature 523:561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dixon RA, Kobilka BK, Strader DJ, Benovic JL, Dohlman HG, Frielle T, Bolanowski MA, Bennett CD, Rands E, Diehl RE, Mumford RA, Slater EE, Sigal IS, Caron MG, Lefkowitz RJ, Strader CD (1986) Cloning of the gene and cDNA for mammalian beta‐adrenergic receptor and homology with rhodopsin. Nature 321:75–79. [DOI] [PubMed] [Google Scholar]
- 20. Furlong TJ, Pierce KD, Selbie LA, Shine J (1992) Molecular characterization of a human brain adenosine A2 receptor. Brain Res Mol Brain Res 15:62–66. [DOI] [PubMed] [Google Scholar]
- 21. Kobilka BK, Dixon RA, Frielle T, Dohlman HG, Bolanowski MA, Sigal IS, Yang‐Feng TL, Francke U, Caron MG, Lefkowitz RJ (1987) cDNA for the human beta 2‐adrenergic receptor: a protein with multiple membrane‐spanning domains and encoded by a gene whose chromosomal location is shared with that of the receptor for platelet‐derived growth factor. Proc Natl Acad Sci USA 84:46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tanaka K, Masu M, Nakanishi S (1990) Structure and functional expression of the cloned rat neurotensin receptor. Neuron 4:847–854. [DOI] [PubMed] [Google Scholar]
- 23. Yarden Y, Rodriguez H, Wong SK, Brandt DR, May DC, Burnier J, Harkins RN, Chen EY, Ramachandran J, Ullrich A, Ross EM (1986) The avian beta‐adrenergic receptor: primary structure and membrane topology. Proc Natl Acad Sci USA 83:6795–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Warne T, Serrano‐Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF (2008) Structure of a beta1‐adrenergic G‐protein‐coupled receptor. Nature 454:486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dore AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, Hurrell E, Bennett K, Congreve M, Magnani F, Tate CG, Weir M, Marshall FH (2011) Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 19:1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322:1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG (2011) Agonist‐bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474:521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC (2011) Structure of an agonist‐bound human A2A adenosine receptor. Science 332:322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Egloff P, Hillenbrand M, Klenk C, Batyuk A, Heine P, Balada S, Schlinkmann KM, Scott DJ, Schutz M, Pluckthun A (2014) Structure of signaling‐competent neurotensin receptor 1 obtained by directed evolution in Escherichia coli . Proc Natl Acad Sci USA 111:E655–E662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krumm BE, Lee S, Bhattacharya S, Botos I, White CF, Du H, Vaidehi N, Grisshammer R (2016) Structure and dynamics of a constitutively active neurotensin receptor. Sci Rep 6:38564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krumm BE, White JF, Shah P, Grisshammer R (2015) Structural prerequisites for G‐protein activation by the neurotensin receptor. Nat Commun 6:7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, Gvozdenovic‐Jeremic J, Shah P, Shiloach J, Tate CG, Grisshammer R (2012) Structure of the agonist‐bound neurotensin receptor. Nature 490:508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Calandra B, Tucker J, Shire D, Grisshammer R (1997) Expression in Escherichia coli and characterisation of the human central CB1 and peripheral CB2 cannabinoid receptors. Biotechnol Lett 19:425–428. [Google Scholar]
- 34. Grisshammer R, Duckworth R, Henderson R (1993) Expression of a rat neurotensin receptor in Escherichia coli . Biochem J 295:571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weiss HM, Grisshammer R (2002) Purification and characterization of the human adenosine A(2a) receptor functionally expressed in Escherichia coli . Eur J Biochem 269:82–92. [DOI] [PubMed] [Google Scholar]
- 36. Andrell J, Tate CG (2013) Overexpression of membrane proteins in mammalian cells for structural studies. Mol Membr Biol 30:52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grisshammer R, Tate CG (1995) Overexpression of integral membrane proteins for structural studies. Q Rev Biophys 28:315–422. [DOI] [PubMed] [Google Scholar]
- 38. Tate CG, Grisshammer R (1996) Heterologous expression of G‐protein‐coupled receptors. Trends Biotechnol 14:426–430. [DOI] [PubMed] [Google Scholar]
- 39. Li J, Edwards PC, Burghammer M, Villa C, Schertler GF (2004) Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol 343:1409–1438. [DOI] [PubMed] [Google Scholar]
- 40. Lee S, Mao A, Bhattacharya S, Robertson N, Grisshammer R, Tate CG, Vaidehi N (2016) How do short chain nonionic detergents destabilize G‐protein‐coupled receptors? J Am Chem Soc 138:15425–15433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shibata Y, White JF, Serrano‐Vega MJ, Magnani F, Aloia AL, Grisshammer R, Tate CG (2009) Thermostabilization of the neurotensin receptor NTS1. J Mol Biol 390:262–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tate CG (2012) A crystal clear solution for determining G‐protein‐coupled receptor structures. Trends Biochem Sci 37:343–352. [DOI] [PubMed] [Google Scholar]
- 43. Manglik A, Kobilka BK, Steyaert J (2017) Nanobodies to study G protein‐coupled receptor structure and function. Annu Rev Pharmacol Toxicol 57:19–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carpenter B, Tate CG (2016) Engineering a minimal G protein to facilitate crystallisation of G protein‐coupled receptors in their active conformation. Protein Eng Des Sel 29:583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nehme R, Carpenter B, Singhal A, Strege A, Edwards PC, White CF, Du H, Grisshammer R, Tate CG (2017) Mini‐G proteins: novel tools for studying GPCRs in their active conformation. PLoS One 12:e0175642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hovers J, Potschies M, Polidori A, Pucci B, Raynal S, Bonnete F, Serrano‐Vega MJ, Tate CG, Picot D, Pierre Y, Popot JL, Nehme R, Bidet M, Mus‐Veteau I, Busskamp H, Jung KH, Marx A, Timmins PA, Welte W (2011) A class of mild surfactants that keep integral membrane proteins water‐soluble for functional studies and crystallization. Mol Membr Biol 28:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee SC, Bennett BC, Hong WX, Fu Y, Baker KA, Marcoux J, Robinson CV, Ward AB, Halpert JR, Stevens RC, Stout CD, Yeager MJ, Zhang Q (2013) Steroid‐based facial amphiphiles for stabilization and crystallization of membrane proteins. Proc Natl Acad Sci USA 110:E1203–E1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bae HE, Mortensen JS, Ribeiro O, Du Y, Ehsan M, Kobilka BK, Loland CJ, Byrne B, Chae PS (2016) Tandem neopentyl glycol maltosides (TNMs) for membrane protein stabilisation. Chem Commun (Camb) 52:12104–12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chae PS, Rasmussen SG, Rana RR, Gotfryd K, Chandra R, Goren MA, Kruse AC, Nurva S, Loland CJ, Pierre Y, Drew D, Popot JL, Picot D, Fox BG, Guan L, Gether U, Byrne B, Kobilka B, Gellman SH (2010) Maltose‐neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat Methods 7:1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Day PW, Rasmussen SG, Parnot C, Fung JJ, Masood A, Kobilka TS, Yao XJ, Choi HJ, Weis WI, Rohrer DK, Kobilka BK (2007) A monoclonal antibody for G protein‐coupled receptor crystallography. Nat Methods 4:927–929. [DOI] [PubMed] [Google Scholar]
- 51. Hino T, Arakawa T, Iwanari H, Yurugi‐Kobayashi T, Ikeda‐Suno C, Nakada‐Nakura Y, Kusano‐Arai O, Weyand S, Shimamura T, Nomura N, Cameron AD, Kobayashi T, Hamakubo T, Iwata S, Murata T (2012) G‐protein‐coupled receptor inactivation by an allosteric inverse‐agonist antibody. Nature 482:237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Michel H (1983) Crystallization of membrane proteins. Trends Biochem Sci 8:56–59. [Google Scholar]
- 53. Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, Kunken J, Xu F, Cherezov V, Hanson MA, Stevens RC (2012) Fusion partner toolchest for the stabilization and crystallization of G protein‐coupled receptors. Structure 20:967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yin J, Mobarec JC, Kolb P, Rosenbaum DM (2015) Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature 519:247–250. [DOI] [PubMed] [Google Scholar]
- 55. Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, Li T, Ma L, Fenalti G, Li J, Zhang W, Xie X, Yang H, Jiang H, Cherezov V, Liu H, Stevens RC, Zhao Q, Wu B (2013) Structure of the CCR5 chemokine receptor‐HIV entry inhibitor maraviroc complex. Science 341:1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dore AS, Okrasa K, Patel JC, Serrano‐Vega M, Bennett K, Cooke RM, Errey JC, Jazayeri A, Khan S, Tehan B, Weir M, Wiggin GR, Marshall FH (2014) Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511:557–562. [DOI] [PubMed] [Google Scholar]
- 57. Zou Y, Weis WI, Kobilka BK (2012) N‐terminal T4 lysozyme fusion facilitates crystallization of a G protein coupled receptor. PLoS One 7:e46039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, Roth BL, Cherezov V, Stevens RC (2012) Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485:395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hunte C, Michel H (2002) Crystallisation of membrane proteins mediated by antibody fragments. Curr Opin Struct Biol 12:503–508. [DOI] [PubMed] [Google Scholar]
- 60. Deupi X, Edwards P, Singhal A, Nickle B, Oprian D, Schertler G, Standfuss J (2012) Stabilized G protein binding site in the structure of constitutively active metarhodopsin‐II. Proc Natl Acad Sci USA 109:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP (2008) Crystal structure of opsin in its G‐protein‐interacting conformation. Nature 455:497–502. [DOI] [PubMed] [Google Scholar]
- 62. Szczepek M, Beyriere F, Hofmann KP, Elgeti M, Kazmin R, Rose A, Bartl FJ, von Stetten D, Heck M, Sommer ME, Hildebrand PW, Scheerer P (2014) Crystal structure of a common GPCR‐binding interface for G protein and arrestin. Nat Commun 5:4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pardon E, Laeremans T, Triest S, Rasmussen SG, Wohlkonig A, Ruf A, Muyldermans S, Hol WG, Kobilka BK, Steyaert J (2014) A general protocol for the generation of Nanobodies for structural biology. Nat Protoc 9:674–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Caffrey M, Cherezov V (2009) Crystallizing membrane proteins using lipidic mesophases. Nat Protoc 4:706–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Serrano‐Vega MJ, Magnani F, Shibata Y, Tate CG (2008) Conformational thermostabilization of the beta1‐adrenergic receptor in a detergent‐resistant form. Proc Natl Acad Sci USA 105:877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sun B, Bachhawat P, Chu ML, Wood M, Ceska T, Sands ZA, Mercier J, Lebon F, Kobilka TS, Kobilka BK (2017) Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proc Natl Acad Sci USA 114:2066–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Landau EM, Rosenbusch JP (1996) Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci USA 93:14532–14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Oswald C, Rappas M, Kean J, Dore AS, Errey JC, Bennett K, Deflorian F, Christopher JA, Jazayeri A, Mason JS, Congreve M, Cooke RM, Marshall FH (2016) Intracellular allosteric antagonism of the CCR9 receptor. Nature 540:462–465. [DOI] [PubMed] [Google Scholar]
- 69. Miller‐Gallacher JL, Nehme R, Warne T, Edwards PC, Schertler GF, Leslie AG, Tate CG (2014) The 2.1 A resolution structure of cyanopindolol‐bound beta1‐adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand‐free receptor. PloS One 9:e92727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cherezov V (2011) Lipidic cubic phase technologies for membrane protein structural studies. Curr Opin Struct Biol 21:559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Riekel C, Burghammer M, Schertler G (2005) Protein crystallography microdiffraction. Curr Opin Struct Biol 15:556–562. [DOI] [PubMed] [Google Scholar]
- 72. Cherezov V, Hanson MA, Griffith MT, Hilgart MC, Sanishvili R, Nagarajan V, Stepanov S, Fischetti RF, Kuhn P, Stevens RC (2009) Rastering strategy for screening and centring of microcrystal samples of human membrane proteins with a sub‐10 microm size X‐ray synchrotron beam. J R Soc Interface 6:S587–S597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Faham S, Bowie JU (2002) Bicelle crystallization: a new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure. J Mol Biol 316:1–6. [DOI] [PubMed] [Google Scholar]
- 74. Takeda K, Sato H, Hino T, Kono M, Fukuda K, Sakurai I, Okada T, Kouyama T (1998) A novel three‐dimensional crystal of bacteriorhodopsin obtained by successive fusion of the vesicular assemblies. J Mol Biol 283:463–474. [DOI] [PubMed] [Google Scholar]
- 75. Magnani F, Serrano‐Vega MJ, Shibata Y, Abdul‐Hussein S, Lebon G, Miller‐Gallacher J, Singhal A, Strege A, Thomas JA, Tate CG (2016) A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat Protoc 11:1554–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dodevski I, Pluckthun A (2011) Evolution of three human GPCRs for higher expression and stability. J Mol Biol 408:599–615. [DOI] [PubMed] [Google Scholar]
- 77. Sarkar CA, Dodevski I, Kenig M, Dudli S, Mohr A, Hermans E, Pluckthun A (2008) Directed evolution of a G protein‐coupled receptor for expression, stability, and binding selectivity. Proc Natl Acad Sci USA 105:14808–14813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Magnani F, Shibata Y, Serrano‐Vega MJ, Tate CG (2008) Co‐evolving stability and conformational homogeneity of the human adenosine A2a receptor. Proc Natl Acad Sci USA 105:10744–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shibata Y, Gvozdenovic‐Jeremic J, Love J, Kloss B, White JF, Grisshammer R, Tate CG (2013) Optimising the combination of thermostabilising mutations in the neurotensin receptor for structure determination. Biochim Biophys Acta 1828:1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Axford D, Foadi J, Hu NJ, Choudhury HG, Iwata S, Beis K, Evans G, Alguel Y (2015) Structure determination of an integral membrane protein at room temperature from crystals in situ . Acta Crystallogr D Biol Crystallogr 71:1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Huang CY, Olieric V, Ma P, Panepucci E, Diederichs K, Wang M, Caffrey M (2015) In meso in situ serial X‐ray crystallography of soluble and membrane proteins. Acta Crystallogr D Biol Crystallogr 71:1238–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chavent M, Duncan AL, Sansom MS (2016) Molecular dynamics simulations of membrane proteins and their interactions: from nanoscale to mesoscale. Curr Opin Struct Biol 40:8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, Shaw DE (2011) Activation mechanism of the beta2‐adrenergic receptor. Proc Natl Acad Sci USA 108:18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Moerner WE (2015) Single‐molecule spectroscopy, imaging, and photocontrol: foundations for super‐resolution microscopy (nobel lecture). Angew Chem Int Ed Engl 54:8067–8093. [DOI] [PubMed] [Google Scholar]
- 85. Dawaliby R, Trubbia C, Delporte C, Masureel M, Van Antwerpen P, Kobilka BK, Govaerts C (2016) Allosteric regulation of G protein‐coupled receptor activity by phospholipids. Nat Chem Biol 12:35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yao XJ, Velez Ruiz G, Whorton MR, Rasmussen SG, DeVree BT, Deupi X, Sunahara RK, Kobilka B (2009) The effect of ligand efficacy on the formation and stability of a GPCR‐G protein complex. Proc Natl Acad Sci USA 106:9501–9506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Landreh M, Robinson CV (2015) A new window into the molecular physiology of membrane proteins. J Physiol 593:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hubbell WL, Lopez CJ, Altenbach C, Yang Z (2013) Technological advances in site‐directed spin labeling of proteins. Curr Opin Struct Biol 23:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Raghuraman H, Islam SM, Mukherjee S, Roux B, Perozo E (2014) Dynamics transitions at the outer vestibule of the KcsA potassium channel during gating. Proc Natl Acad Sci USA 111:1831–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Verhalen B, Dastvan R, Thangapandian S, Peskova Y, Koteiche HA, Nakamoto RK, Tajkhorshid E, McHaourab HS (2017) Energy transduction and alternating access of the mammalian ABC transporter P‐glycoprotein. Nature 543:738–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, Prosser RS, Kobilka BK (2015) Structural insights into the dynamic process of beta2‐adrenergic receptor signaling. Cell 161:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liang B, Tamm LK (2016) NMR as a tool to investigate the structure, dynamics and function of membrane proteins. Nat Struct Mol Biol 23:468–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gautier A, Mott HR, Bostock MJ, Kirkpatrick JP, Nietlispach D (2010) Structure determination of the seven‐helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat Struct Mol Biol 17:768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Park SH, Das BB, Casagrande F, Tian Y, Nothnagel HJ, Chu M, Kiefer H, Maier K, De Angelis AA, Marassi FM, Opella SJ (2012) Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature 491:779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K (2012) Biased signaling pathways in beta2‐adrenergic receptor characterized by 19F‐NMR. Science 335:1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ye L, Van Eps N, Zimmer M, Ernst OP, Prosser RS (2016) Activation of the A2A adenosine G‐protein‐coupled receptor by conformational selection. Nature 533:265–268. [DOI] [PubMed] [Google Scholar]
- 97. Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, Thian FS, Kobilka TS, Shaw DE, Mueller L, Prosser RS, Kobilka BK (2013) The dynamic process of beta(2)‐adrenergic receptor activation. Cell 152:532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, Huang W, Kobilka BK, Demene H, Granier S (2015) Propagation of conformational changes during mu‐opioid receptor activation. Nature 524:375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Isogai S, Deupi X, Opitz C, Heydenreich FM, Tsai CJ, Brueckner F, Schertler GF, Veprintsev DB, Grzesiek S (2016) Backbone NMR reveals allosteric signal transduction networks in the beta1‐adrenergic receptor. Nature 530:237–241. [DOI] [PubMed] [Google Scholar]
- 100. Kimata N, Reeves PJ, Smith SO (2015) Uncovering the triggers for GPCR activation using solid‐state NMR spectroscopy. J Magn Reson 253:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Reeves PJ, Thurmond RL, Khorana HG (1996) Structure and function in rhodopsin: high level expression of a synthetic bovine opsin gene and its mutants in stable mammalian cell lines. Proc Natl Acad Sci USA 93:11487–11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Schlichting I (2015) Serial femtosecond crystallography: The first five years. IUCrJ 2:246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chapman HN, Fromme P, Barty A, White TA, Kirian RA, Aquila A, Hunter MS, Schulz J, DePonte DP, Weierstall U, Doak RB, Maia FR, Martin AV, Schlichting I, Lomb L, Coppola N, Shoeman RL, Epp SW, Hartmann R, Rolles D, Rudenko A, Foucar L, Kimmel N, Weidenspointner G, Holl P, Liang M, Barthelmess M, Caleman C, Boutet S, Bogan MJ, Krzywinski J, Bostedt C, Bajt S, Gumprecht L, Rudek B, Erk B, Schmidt C, Homke A, Reich C, Pietschner D, Struder L, Hauser G, Gorke H, Ullrich J, Herrmann S, Schaller G, Schopper F, Soltau H, Kuhnel KU, Messerschmidt M, Bozek JD, Hau‐Riege SP, Frank M, Hampton CY, Sierra RG, Starodub D, Williams GJ, Hajdu J, Timneanu N, Seibert MM, Andreasson J, Rocker A, Jonsson O, Svenda M, Stern S, Nass K, Andritschke R, Schroter CD, Krasniqi F, Bott M, Schmidt KE, Wang X, Grotjohann I, Holton JM, Barends TR, Neutze R, Marchesini S, Fromme R, Schorb S, Rupp D, Adolph M, Gorkhover T, Andersson I, Hirsemann H, Potdevin G, Graafsma H, Nilsson B, Spence JC (2011) Femtosecond X‐ray protein nanocrystallography. Nature 470:73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Feld GK, Frank M (2014) Enabling membrane protein structure and dynamics with X‐ray free electron lasers. Curr Opin Struct Biol 27:69–78. [DOI] [PubMed] [Google Scholar]
- 105. Liu W, Wacker D, Wang C, Abola E, Cherezov V (2014) Femtosecond crystallography of membrane proteins in the lipidic cubic phase. Philos Trans R Soc Lond B Biol Sci 369:20130314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Suga M, Akita F, Hirata K, Ueno G, Murakami H, Nakajima Y, Shimizu T, Yamashita K, Yamamoto M, Ago H, Shen JR (2015) Native structure of photosystem II at 1.95 A resolution viewed by femtosecond X‐ray pulses. Nature 517:99–103. [DOI] [PubMed] [Google Scholar]
- 107. Batyuk A, Galli L, Ishchenko A, Han GW, Gati C, Popov PA, Lee MY, Stauch B, White TA, Barty A, Aquila A, Hunter MS, Liang M, Boutet S, Pu M, Liu ZJ, Nelson G, James D, Li C, Zhao Y, Spence JC, Liu W, Fromme P, Katritch V, Weierstall U, Stevens RC, Cherezov V (2016) Native phasing of x‐ray free‐electron laser data for a G protein‐coupled receptor. Sci Adv 2:e1600292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Fenalti G, Zatsepin NA, Betti C, Giguere P, Han GW, Ishchenko A, Liu W, Guillemyn K, Zhang H, James D, Wang D, Weierstall U, Spence JC, Boutet S, Messerschmidt M, Williams GJ, Gati C, Yefanov OM, White TA, Oberthuer D, Metz M, Yoon CH, Barty A, Chapman HN, Basu S, Coe J, Conrad CE, Fromme R, Fromme P, Tourwe D, Schiller PW, Roth BL, Ballet S, Katritch V, Stevens RC, Cherezov V (2015) Structural basis for bifunctional peptide recognition at human delta‐opioid receptor. Nat Struct Mol Biol 22:265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liu W, Wacker D, Gati C, Han GW, James D, Wang D, Nelson G, Weierstall U, Katritch V, Barty A, Zatsepin NA, Li D, Messerschmidt M, Boutet S, Williams GJ, Koglin JE, Seibert MM, Wang C, Shah ST, Basu S, Fromme R, Kupitz C, Rendek KN, Grotjohann I, Fromme P, Kirian RA, Beyerlein KR, White TA, Chapman HN, Caffrey M, Spence JC, Stevens RC, Cherezov V (2013) Serial femtosecond crystallography of G protein‐coupled receptors. Science 342:1521–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhang H, Han GW, Batyuk A, Ishchenko A, White KL, Patel N, Sadybekov A, Zamlynny B, Rudd MT, Hollenstein K, Tolstikova A, White TA, Hunter MS, Weierstall U, Liu W, Babaoglu K, Moore EL, Katz RD, Shipman JM, Garcia‐Calvo M, Sharma S, Sheth P, Soisson SM, Stevens RC, Katritch V, Cherezov V (2017) Structural basis for selectivity and diversity in angiotensin II receptors. Nature 544:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA, James D, Wang D, Nelson G, Weierstall U, Sawaya MR, Xu Q, Messerschmidt M, Williams GJ, Boutet S, Yefanov OM, White TA, Wang C, Ishchenko A, Tirupula KC, Desnoyer R, Coe J, Conrad CE, Fromme P, Stevens RC, Katritch V, Karnik SS, Cherezov V (2015) Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell 161:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Nango E, Royant A, Kubo M, Nakane T, Wickstrand C, Kimura T, Tanaka T, Tono K, Song C, Tanaka R, Arima T, Yamashita A, Kobayashi J, Hosaka T, Mizohata E, Nogly P, Sugahara M, Nam D, Nomura T, Shimamura T, Im D, Fujiwara T, Yamanaka Y, Jeon B, Nishizawa T, Oda K, Fukuda M, Andersson R, Bath P, Dods R, Davidsson J, Matsuoka S, Kawatake S, Murata M, Nureki O, Owada S, Kameshima T, Hatsui T, Joti Y, Schertler G, Yabashi M, Bondar AN, Standfuss J, Neutze R, Iwata S (2016) A three‐dimensional movie of structural changes in bacteriorhodopsin. Science 354:1552–1557. [DOI] [PubMed] [Google Scholar]
- 113. Suga M, Akita F, Sugahara M, Kubo M, Nakajima Y, Nakane T, Yamashita K, Umena Y, Nakabayashi M, Yamane T, Nakano T, Suzuki M, Masuda T, Inoue S, Kimura T, Nomura T, Yonekura S, Yu LJ, Sakamoto T, Motomura T, Chen JH, Kato Y, Noguchi T, Tono K, Joti Y, Kameshima T, Hatsui T, Nango E, Tanaka R, Naitow H, Matsuura Y, Yamashita A, Yamamoto M, Nureki O, Yabashi M, Ishikawa T, Iwata S, Shen JR (2017) Light‐induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 543:131–135. [DOI] [PubMed] [Google Scholar]
- 114. Nannenga BL, Gonen T (2016) MicroED opens a new era for biological structure determination. Curr Opin Struct Biol 40:128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Shi D, Nannenga BL, Iadanza MG, Gonen T (2013) Three‐dimensional electron crystallography of protein microcrystals. Elife 2:e01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Liao M, Cao E, Julius D, Cheng Y (2014) Single particle electron cryo‐microscopy of a mammalian ion channel. Curr Opin Struct Biol 27:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Bai XC, McMullan G, Scheres SH (2015) How cryo‐EM is revolutionizing structural biology. Trends Biochem Sci 40:49–57. [DOI] [PubMed] [Google Scholar]
- 118. Fernandez‐Leiro R, Scheres SH (2016) Unravelling biological macromolecules with cryo‐electron microscopy. Nature 537:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kuhlbrandt W (2014) Biochemistry. The resolution revolution. Science 343:1443–1444. [DOI] [PubMed] [Google Scholar]
- 120. Patwardhan A (2017) Trends in the electron microscopy data bank (EMDB). Acta Crystallogr D 73. Doi:10.1107/S2059798317004181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JL, Subramaniam S (2016) Breaking cryo‐EM resolution barriers to facilitate drug discovery. Cell 165:1698–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Bai XC, Rajendra E, Yang G, Shi Y, Scheres SH (2015) Sampling the conformational space of the catalytic subunit of human gamma‐secretase. Elife 4:e11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Cao E, Liao M, Cheng Y, Julius D (2013) TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504:113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Gao Y, Cao E, Julius D, Cheng Y (2016) TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534:347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Liao M, Cao E, Julius D, Cheng Y (2013) Structure of the TRPV1 ion channel determined by electron cryo‐microscopy. Nature 504:107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Meyerson JR, Chittori S, Merk A, Rao P, Han TH, Serpe M, Mayer ML, Subramaniam S (2016) Structural basis of kainate subtype glutamate receptor desensitization. Nature 537:567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Meyerson JR, Kumar J, Chittori S, Rao P, Pierson J, Bartesaghi A, Mayer ML, Subramaniam S (2014) Structural mechanism of glutamate receptor activation and desensitization. Nature 514:328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Shen H, Zhou Q, Pan X, Li Z, Wu J, Yan N (2017) Structure of a eukaryotic voltage‐gated sodium channel at near‐atomic resolution. Science 355:eaal4326. [DOI] [PubMed] [Google Scholar]
- 129. Khoshouei M, Radjainia M, Baumeister W, Danev R (2017) Cryo‐EM structure of haemoglobin at 3.2 Å determined with the Volta phase plate. DOI: 10.1101/087841. [DOI] [PMC free article] [PubMed]
- 130. Liang YL, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, Tarrasch J, Thal DM, Furness SGB, Christopoulos G, Coudrat T, Danev R, Baumeister W, Miller LJ, Christopoulos A, Kobilka BK, Wootten D, Skiniotis G, Sexton PM (2017) Phase‐plate cryo‐EM structure of a class B GPCR‐G‐protein complex. Nature. DOI: 10.1038/nature22327. [DOI] [PMC free article] [PubMed] [Google Scholar]