

High-resolution three-dimensional structure of chromosomes in a human prophase nucleus from electron microscopy.
Abstract
The human genetic material is packaged into 46 chromosomes. The structure of chromosomes is known at the lowest level, where the DNA chain is wrapped around a core of eight histone proteins to form nucleosomes. Around a million of these nucleosomes, each about 11 nm in diameter and 6 nm in thickness, are wrapped up into the complex organelle of the chromosome, whose structure is mostly known at the level of visible light microscopy to form a characteristic cross shape in metaphase. However, the higher-order structure of human chromosomes, between a few tens and hundreds of nanometers, has not been well understood. We show a three-dimensional (3D) image of a human prophase nucleus obtained by serial block-face scanning electron microscopy, with 36 of the complete set of 46 chromosomes captured within it. The acquired image allows us to extract quantitative 3D structural information about the nucleus and the preserved, intact individual chromosomes within it, including their positioning and full spatial morphology at a resolution of around 50 nm in three dimensions. The chromosome positions were found, at least partially, to follow the pattern of chromosome territories previously observed only in interphase. The 3D conformation shows parallel, planar alignment of the chromatids, whose occupied volumes are almost fully accounted for by the DNA and known chromosomal proteins. We also propose a potential new method of identifying human chromosomes in three dimensions, on the basis of the measurements of their 3D morphology.
INTRODUCTION
Since the very beginning of optical microscopy, investigations have been made into the different levels of structure found in human chromosomes and nuclei. The interpretation of information about the higher-order structure of human chromosomes, at the level of fine structure introduced by the formation of nucleosomes (1–3), still remains controversial in the literature (4–8). The familiar image of chromosomes showing an X shape due to the two arms of the sister chromatids joining at the centromere is, at least partly, caused by the standard preparation method of “dropping” the nuclear extract onto a flat surface. However, if the arms were free to flex, unconstrained by the mounting surface, they would be expected to follow relatively random directions pointing away from the tethering point of the centromeres. When the cell is entering metaphase, the DNA condensation process is believed to occur by self-assembly of more loosely coiled DNA originating in interphase onto a protein scaffold structure (9–11). Without a scaffold, they might be expected to coil around each other (10), which would cause topological problems in the ability of the chromatids to separate at mitosis. The role of the scaffolding proteins avoids this undesirable coiling.
When chromosome “spreads” are generated, usually by dropping whole prophase or metaphase nuclei onto glass slides, the resulting chromosomes lie down flat after drying in almost all cases, with the two chromatids side by side. This suggests, but does not prove, that the mitotic nuclear state of the chromosomes has parallel alignment of the chromatids. To resolve the question, we need three-dimensional (3D) high-resolution images of the nuclei without flattening them for viewing under a microscope.
To answer these structural biological questions about human chromosomes and nuclei, we have developed new protocols for preserving the 3D structure of whole nuclei (with chromosomes) (12), so they can be imaged by powerful new 3D microscopy methods (13–15). Here, a human prophase nucleus was imaged in three dimensions by serial block-face scanning electron microscopy (SBFSEM), which uses a scanning electron microscope (SEM) with a built-in serial sectioning ultramicrotome for 3D registration (13, 16).
These 3D structural images provide unique and vital information, because they allow the relative positions of the chromosomes to be determined within the internal space of the nucleus. This raises the questions of how the chromosome territories (CTs), known to form in interphase, collapse upon condensation of the chromatin and how the chromosomes are positioned in the nucleus before the formation of the spindle structure and the kinetochore. It is not believed that there are any sorting mechanisms active within the nucleus before spindle formation; hence, it would be surprising if the CT positioning information was lost upon the chromatin condensation, leading to compact chromosomes just before prometaphase during the cell cycle (17–20).
It is also vital to identify the chromosomes themselves. We do not yet have multicolor fluorescence in situ hybridization (M-FISH) type probes (21) available for electron microscopy; therefore, we have to rely instead on the chromosome size and shape information to identify the chromosomes. This is analogous to flow cytometry karyotyping, where chromosomes are identified by the strength of the fluorescence signal from a dye that is quantitatively bound to the DNA. For complete identification, two dyes that bind differently to AT- and GC-rich sequences are needed (22, 23). However, from our 3D imaging results, the chromosomes can be ranked according to their volumes/sizes and can be grouped by this information fairly reliably. The centromere positions provide secondary identification information, which is also used in our classification.
RESULTS
SBFSEM micrographs of a human prophase nucleus
Figure 1 shows two slices from the series of 2D backscattered electron (BSE) micrographs acquired by the SBFSEM measurements. It presents the distribution of condensed individual chromosomes in the nucleus (24–26) with a nearly circular envelope. Two of the chromosomes are highlighted in the zoomed-in insets of Fig. 1. A distinct porous network structure has been revealed in the imaged chromosomes, and the cross sections of the chromatid pairs of some of the chromosomes are visible in the original BSE micrographs. These features are easily distinguishable in the 3D maps without any further processing because of the high contrast in the BSE micrographs. As the DNA chain wraps around the proteins in the chromatin, we believe that the black regions in the imaged chromosomes are protein-DNA complexes “dyed” with Pt stain because SBFSEM does not have sufficient resolution to distinguish the DNA from the chromosomal proteins, although the Pt stain is nominally DNA-specific (27).
Fig. 1. Two slices from the acquired SBFSEM stack of 300 BSE micrographs of the nucleus with zoomed-in insets of two selected chromosomes.
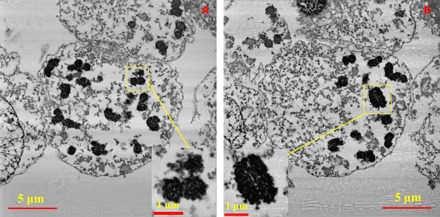
(A) Slice no. 36 of 300. (B) Slice no. 127 of 300. The insets in (A) and (B) are a broken chromosome and an identified intact chromosome A1 (assigned chromosome 1; see explanation in the text), respectively. The pixel size of the BSE micrographs is 11 nm × 11 nm, and the sectioning thickness is 20 nm.
3D spatial structure of the human prophase nucleus (with chromosomes)
Figure 2 (A and B) and movies S1 and S2 demonstrate individual chromosomes and their 3D spatial distribution in the human prophase nucleus, with its envelope revealed by SBFSEM. In total, 36 of the 46 chromosomes were captured; 19 of them were intact, and the other 17 were broken. The intact chromosomes are in yellow or green, and the two green ones are the chromosomes that have close contact with other chromosomes. The broken chromosomes in red are those chromosomes that are not fully visualized either because they were already partially cut away during the sample preparation (before the measurement) or because they were not completely sectioned by the SBFSEM system. In Fig. 2 (A and B) and movies S1 and S2, we can see that the distribution of the chromosomes in the nucleus is not random but grouped toward the two sides of the nucleus. Table 1 presents the volume statistical results of all the intact chromosomes identified within the nucleus, and the lengths and diameters of some of them are listed in Table 2. The volumes of the broken chromosomes are listed in table S1 as a reference. Compared with the known distribution of DNA sizes from the human genome (28), it allows the classification of the measured intact chromosomes into groups of similar sizes following the international convention (29) on the basis of their volumes/sizes (in three dimensions): group A for chromosomes 1 to 3, group BC for chromosomes 4 to 7 and X, group C for chromosomes 8 to 12, and group D for chromosomes 13 to 22 and Y. As expected, the distribution of volumes/sizes of the chromosomes indicates that the sampling (30) of the 36 chromosomes of the full set of 46 chromosomes is approximately random.
Fig. 2. 3D characterization and analysis of the imaged chromosomes within the human prophase nucleus.
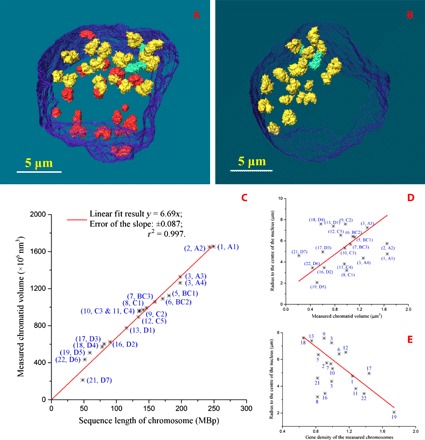
(A) 3D rendering of all the observed chromosomes in the nucleus, with its envelope in transparent blue, the intact chromosomes in yellow or green, and the broken ones in red. (B) 3D rendering of the intact chromosomes only in the nucleus. (C) Measured chromatid volumes versus the number of base pairs of the accordingly assigned chromosomes from the human genome sequence, with a linear fit. (D) Correlation between the measured chromatid volumes and the radii of the assigned chromosomes from the center of the nucleus. (E) Correlation between the gene density of measured chromosomes and their radii from the center of the nucleus. The chromosomes have been labeled by their assigned chromosome numbers in (E).
Table 1. Volume statistics of all the intact chromosomes in the nucleus.
|
Chromosome (number) |
Chromatid volume (V1) (μm3)* |
Cavity volume (V2) (μm3)† |
Ratio of V1/(V1 + V2) (%) |
Assigned chromosome (number) |
Calculated chromatid volume (μm3)‡ |
| A1 | 1.655 | 0.087 | 95.0 | 1 | 1.446 |
| A2 | 1.650 | 0.048 | 97.2 | 2 | 1.411 |
| A3 | 1.328 | 0.047 | 96.6 | 3 | 1.148 |
| A4 | 1.263 | 0.037 | 97.1 | 3 | 1.148 |
| BC1 | 1.124 | 0.083 | 93.1 | 5 | 1.049 |
| BC2 | 1.092 | 0.076 | 93.5 | 6 | 0.992 |
| BC3 | 1.056 | 0.036 | 96.7 | 7 (or X) | 0.923 |
| C1 | 0.993 | 0.102 | 90.7 | 8 | 0.849 |
| C2 | 0.972 | 0.053 | 94.8 | 9 | 0.819 |
| C3 | 0.963 | 0.074 | 92.9 | 10 | 0.786 |
| C4 | 0.955 | 0.054 | 94.7 | 11 | 0.783 |
| C5 | 0.895 | 0.049 | 94.8 | 12 | 0.777 |
| D1 | 0.776 | 0.054 | 93.5 | 13 | 0.668 |
| D2 | 0.625 | 0.066 | 90.4 | 16 | 0.524 |
| D3 | 0.603 | 0.037 | 94.2 | 17 | 0.471 |
| D4 | 0.572 | 0.045 | 92.7 | 18 | 0.453 |
| D5 | 0.509 | 0.029 | 94.6 | 19 | 0.343 |
| D6 | 0.435 | 0.035 | 92.6 | 22 | 0.298 |
| D7 | 0.213 | 0.014 | 93.8 | 21 | 0.279 |
*Chromatid volume is half of the measured volume of the chromosomes without cavities, that is, half of the volume of the black region only in the imaged chromosomes, with chromatid pairs measured by Avizo.
†Cavity volume is half of the volume of all the cavities in the chromosomes with chromatid pairs.
‡Calculated chromatid volumes were obtained by multiplying the sequence length of the assigned human chromosomes (in mega–base pairs) from the database by 5.80 nm3. The database is Archive Ensembl Release 68, July 2012 (28). The volume per base pair, 5.80 nm3, was obtained by theoretical calculation, which is explained in the main text.
Table 2. Statistical analysis of the chromatid pairs of selected intact chromosomes.
SSC1, single sister chromatid 1; SSC2, single sister chromatid 2; Ave., average value of SSC1 and SSC2.
|
Chromosome (number) |
Chromatid length (μm)* |
Ave. of cross-sectional area (μm2)† |
Ave. diameter of cross sections (μm)† |
Difference of the chromatid pairs in volume (%)‡ |
|
| A1 | SSC1 | 3.241 | 0.524 | 0.819 | 1.84 |
| SSC2 | 2.801 | 0.530 | |||
| Ave. | 3.021 | 0.527 | |||
| A2 | SSC1 | 2.869 | 0.512 | 0.799 | 6.54 |
| SSC2 | 3.007 | 0.492 | |||
| Ave. | 2.938 | 0.502 | |||
| A3 | SSC1 | 2.828 | 0.438 | 0.760 | 3.50 |
| SSC2 | 2.744 | 0.470 | |||
| Ave. | 2.786 | 0.454 | |||
| A4 | SSC1 | 2.766 | 0.421 | 0.742 | 0.31 |
| SSC2 | 2.768 | 0.443 | |||
| Ave. | 2.767 | 0.432 | |||
| BC1 | SSC1 | 2.236 | 0.492 | 0.776 | 9.00 |
| SSC2 | 2.531 | 0.453 | |||
| Ave. | 2.384 | 0.473 | |||
| BC3 | SSC1 | 2.085 | 0.439 | 0.760 | 4.61 |
| SSC2 | 1.914 | 0.468 | |||
| Ave. | 2.000 | 0.454 | |||
| D2 | SSC1 | 1.732 | 0.375 | 0.701 | 9.13 |
| SSC2 | 1.647 | 0.396 | |||
| Ave. | 1.690 | 0.386 |
*Chromatid lengths were obtained by measuring the separated two chromatids of the chromosomes using Avizo.
†“Ave. of cross-sectional area” represents the average of the areas of a few cross sections along the central axis of the chromatids, which were carried out using Avizo. “Ave. diameter of cross sections” represents the diameter calculated from the values of the average of the “Ave. of cross-sectional area” when we assume the cross sections are circles.
‡Difference was obtained by superposition calculation on the two chromatids by 3D volume registration using Avizo. The difference (in percentage) equals 2*abs(VC1 − VC2)/(VC1 + VC2)*100%, where VC1 is the volume of the SSC1 and VC2 is the volume of the SSC2.
Human prophase chromosome structure in three dimensions
We can see that the chromosomes imaged in our experiment are tightly packed without a great portion of “empty” space (Figs. 1 and 3), because, on average, only about 6% of the observed chromosome volume appears as cavities, in the form of less-dense regions in the imaged chromosomes, and the remaining 94% (Table 1) is the dyed chromosomal protein–DNA complex. In addition, the larger chromosomes, in groups A and B, appear to have slightly denser compaction than the smaller ones, in groups C and D, because the first seven chromosomes in Table 1 have on average 95.6% of their volume occupied by chromosomal protein–DNA complex and the later 12 chromosomes have about 93.3% of this value. As stated above, we believe that the black regions in the BSE micrographs of the chromosomes are chromosomal protein–DNA complexes. In chromosome A1 (Fig. 3, A to C), a typical example, there are a few cavities (in blue) connected to the external space surrounding the chromosome, and most of the cavities (in light green and green) are totally sealed inside the chromosome. The latter cavities need to be counted as part of the internal structure of the chromosomes, and we think they correspond to either unstained proteins or the cytoplasm-filled empty space within the chromosomes. The cavities are observed to distribute evenly along the two sister chromatids of the chromosome (Fig. 3C) and are positioned near the central axis of the chromatids. Those cavities, represented in green in 3D images or in white in the middle of Fig. 3A, which are at the interface region of the two sister chromatids (transition color region in the middle of Fig. 3, B and C), are generally larger than the cavities fully sealed inside the single chromatids. This is not hard to understand because the paired chromatids are structured for their ultimate separation into two single chromatids in metaphase, and the separation appears to be already starting to take place to generate more space in between.
Fig. 3. Rendering of the identified intact individual chromosomes.
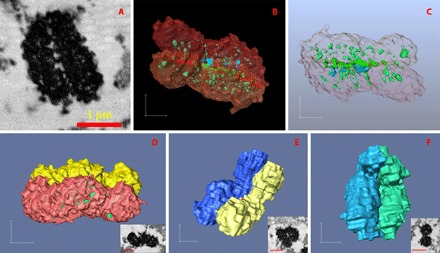
(A) An SBFSEM slice of the chromosome A1, assigned as chromosome 1. This is a different slice from the inset in Fig. 1B. (B) 3D rendering of the chromatid separated chromosome A1 showing cavities in solid light green, green, and blue. (C) 3D rendering of the cavity network in chromosome A1 viewed through a transparent chromosome surface. Bottom: 3D rendering of the surface views of the chromatid-separated chromosomes A2 (D), A4 (E), and BC1 (F), which are assigned as chromosomes 2, 3, and 6, respectively. The insets at the bottom-right corners of (D), (E), and (F) are the SBFSEM slices of the chromosomes A2, A4, and BC1, respectively. Scale bars, 500 nm [(B to F), in each direction] and 1 μm (insets in D to F).
In most cases, as shown in Figs. 3 and 4 (A to C), the chromosomes were observed to have two parallel chromatids with roughly the same (curved) cylindrical shape. In some cases, particularly for smaller chromosomes (fig. S1), the interface between the two chromatids is indistinct, making it hard to determine and to separate the chromatid pairs. However, in most of the examples, it was possible to reliably segment the images into pairs of chromatids (Figs. 3 and 4). To verify the similarity in conformation between the two sister chromatids, we performed a superposition calculation in three dimensions to overlay the pairs through volume registration using Avizo software (Fig. 4, D to F). As seen, the chromatid pairs in each chromosome match with each other in both shape and size, with an average volume difference of about 5%. Despite the fact that the segmentation of the sister chromatids is a semiautomatic operation with some subjective processing, the resulting volume differences between the sister chromatids are narrowly distributed from 0.31 to 9.13% (Table 2). Examples are provided in Table 2, with the lengths, cross-sectional areas, and diameters of the chromatids measured using image tools provided by Avizo. The diameters of the chromatid cross sections are notably narrowly distributed, with most of the variation in chromosome sizes/numbers accounted for by their lengths. On average, the diameter of the chromatids is about 765 nm. This suggests that a highly orchestrated folding of the sequence takes place, which is uniformly conserved across all the chromosomes. We can also see from Table 2 that whenever the two sister chromatids do not have identical sizes, the longer ones always have smaller cross sections. This indicates that the two sister chromatids with identical amounts of DNA appear to conserve volume but probably do not preserve their morphologies exactly during the chromatin condensation.
Fig. 4. Characterization and analysis of chromatid pairs of selected chromosomes.
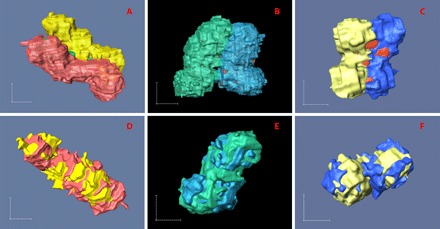
Left, middle, and right columns show chromosomes A3, BC3, and D2, respectively, which are assigned as chromosomes 3, 7, or X and 16 accordingly to the scheme of Table 1. (A to C) 3D rendering of the chromatid-separated chromosomes. (D to F) 3D superposition of the chromatid pairs through volume registration using Avizo. Scale bars, 500 nm (in each direction).
It was also striking that the 3D shapes of the chromosomes were far from “flat”; a selection of examples is shown in Figs. 3 and 4, with the chromatid pairs color-coded. The common shape for the larger chromosomes was not X-shaped but S-shaped (chromosomes A2 and A3) or C-shaped (chromosomes A1 and A4), whereas the “classical” X shape was seen in the smaller chromosomes (chromosomes BC3 and D2). It seems that the larger chromosomes more likely turned to show S shape or C shape, but the smaller chromosomes tend to be X-shaped. This is probably because, to form an S shape or C shape, the chromosomes need to be sufficiently long to bend a couple of times; however, to form an X shape, the chromosomes just need one knot point between the two chromatids (Fig. 4C). In all cases, the two sister chromatids follow a very similar shape, as if they were bonded together all along their length (Figs. 3 and 4). It is apparent that the overall structure is more flexible when transverse to the pairing direction than along it, with a flat plane of separation between the chromatids. It is also significant that no examples of twisted chromatid pairs were found either.
DISCUSSION
Looking further into all these findings, the volume information from Table 1 and the morphology details from the figures not only allow us to designate the chromosome groups as shown above but also allow us to roughly identify all the intact individual chromosomes and to accordingly obtain the volume per base pair of the imaged chromosomes. After a global comparison of the volumes of all the 36 measured chromosomes, both the intact and broken ones, we can assume that the biggest measured intact chromosome, A1, is very likely to be human chromosome 1 or 2. After checking the centromere positions, we assign the imaged chromosome A1 to be chromosome 1. We then assigned all the other imaged chromosomes to the chromosomes with closest agreement of relative volume to relative genome size. The full assignment (Fig. 2 and Table 1) was finished after a combined consideration of the centromere positions and shapes of the chromosomes. The known distribution of sizes among human chromosomes 1 to 23 from both flow cytometry (3) and the human genome sequence (28) is well represented by the 19 measured examples in Fig. 2C. The slope of the linear relationship of Fig. 2C, 6.69 ± 0.087 (with r2 = 0.997), provides the volume per base pair from our experiment as 6.69 nm3. To check for ambiguity in the assignment, if we choose the biggest chromosome A1 to be chromosome 2 instead and follow the same assignment process, the fitting gives a slope of 7.16 ± 0.15 (r2 = 0.991) (fig. S2 and table S2). This shows that the first assignment is a better solution.
We can also calculate the expected volume per base pair. The base pairs in the DNA double helix structure can be considered as a cylinder, which is 2 nm in diameter with a spacing of 0.34 nm, thus occupying a volume of 1.07 nm3/base pair (bp) (31, 32). The nucleosome, which contains about 146 bp (3, 33), can be considered also to have a cylindrical shape with a diameter of 11 nm and a thickness of 6 nm (3); hence, this histone-DNA complex has an aggregate volume of 3.91 nm3/bp. It is about four times the size of the DNA itself because of the solvent-filled spaces and the eight histone proteins making up its core particle (1). Since histones make up about 60% of the total protein mass of chromosomes in metaphase (34), we can estimate that if the histone protein complement takes up (3.91 − 1.07) nm3 = 2.84 nm3, then the remaining nonhistone chromosomal proteins would occupy (1 − 0.60)/0.60 × 2.84 nm3 = 1.89 nm3; thus, the total volume of chromosomal protein–DNA complex is (3.91 + 1.89) nm3 = 5.80 nm3/bp. According to this, the estimated volumes of a single chromatid of a human chromosome can be calculated, and the volumes of assigned ones are listed in Table 1. Our experimentally measured volume per base pair, 6.69 nm3, is 15% higher than the calculated value of 5.80 nm3. This suggests that our interpretation of the experimental results is reliable with respect to the chromosome size and shape measurement and confirms that the volumes of the 3D images we have obtained are largely accounted for by the complement of DNA and known proteins, with very little “empty” space. We can speculate that the measured volume is larger than the calculated one because the Pt stain (in the chromosomes) and additional cavities are too small to be resolved by SBFSEM. It is also known that the genome sequence length is less than the full DNA length.
The observed 3D shapes and tight parallel structure of the chromatid pairs without twisting (Figs. 3 and 4) can be understood by assuming that interchromatid interactions effectively bond each other along the entire length of the chromatids. If there were explicit contacts between the chromatids, these would impede any lateral flexing of the structure but still allow it to flex in the nonpaired direction. There are considerable implications for the nature of the scaffold structure if it is not allowed to twist; it may be that intertwining of the chromatids is prevented by anisotropies of the scaffold structure or that the interchromatid contacts are built in early in the condensation and serve to guide the construction of the scaffold. With further consideration of the porous network structure of chromosome A1 (Fig. 3, A to C), we hypothesize that the appearance of cavities distributed along the central axis of the chromatids might be due to unstained scaffolding protein structures.
Because the 3D structure of the partial nucleus was acquired (Figs. 1 and 2, A and B), the positions of all the imaged chromosomes inside the nucleus are retrievable from the 3D image. This presents us with the opportunity to follow the destiny of the CTs (35) established during interphase, earlier in the cell cycle. The correlation between the volumes of chromatids and the radii of the (assigned) chromosomes to the center of nucleus is presented in Fig. 2D. The radius here equals the distance between the center of mass of each chromosome and the center of mass of the measured partial nuclear envelope. The observed positive correlation demonstrates that the smaller chromosomes are generally closer to the center of the nucleus and that the larger chromosomes are nearer to the nuclear periphery, except for the biggest chromosomes. However, the preferred position of the chromosomes seems to have a stronger relation to their gene density (Fig. 2E). In agreement with a previous report (36), the assigned gene-rich chromosome 19 (chromosome D5) is found to be located closest to the nuclear center, whereas the assigned gene-poor chromosome 18 (chromosome D4) is one of the chromosomes staying furthest from the nuclear center (Fig. 2E). Because these nonrandom trends in the locations of chromosomes in the measured prophase nucleus agree with the CT information in interphase revealed by previous work (35–37), this provides evidence to suggest that the CT arrangement in the nucleus is at least partially preserved from interphase up to prophase of the cell cycle. This also suggests that the chromatin condenses locally within its interphase CTs before the chromosomes start to move to the equatorial plate of the cell. In a future study, the condensation process of the chromosomes could be investigated in more detail by comparing the 3D structure of chromosomes between the earlier and the later stages of prophase.
In summary, our analysis of the 3D spatial structure of a human prophase nucleus containing condensed chromosomes has revealed that the chromosomes have a porous network structure and that, in 3D space, they can be S-shaped and C-shaped, and not only X-shaped. The larger chromosomes were more likely found to show S shape or C shape, whereas the smaller chromosomes tend to be X-shaped. Experimentally, the measured volume per base pair is found to be 6.69 nm3, which is accounted for by the calculated volumes of DNA and known chromosomal proteins, with a 15% excess. The sister chromatids have curved cylindrical shapes with a well-conserved diameter of around 765 nm. The chromatids are about 2 to 3 μm long and remain in contact with each other all along their length without twisting, possibly because of interaction with or support from a scaffold “backbone.” The size analysis of the sister chromatid pairs indicates that although they are not exactly identical in morphology during the chromatin condensation process, the two sister chromatids have similar volume. The measured chromosomes can be roughly identified, even without the full set of 46 chromosomes, by the analysis of their volume/size distributions in combination with their centromere positions in space. This method could be used in the future to identify human chromosomes through 3D imaging approaches, avoiding the need for M-FISH type tools. The characteristic nonrandom positioning of the CTs in prophase resembles the pattern found in interphase (35–37): The smaller chromosomes are found closer to the center of the nucleus, whereas the larger ones are nearer the periphery. The radial position of the chromosomes within the nucleus also seems to correlate with their gene density, with the gene-rich chromosomes near the nuclear center and gene-poor ones near the periphery. This suggests that the chromatin condenses locally and that the CTs are at least partially maintained from interphase into prophase of the cell cycle.
MATERIALS AND METHODS
Chromosomes and nucleus embedding
The chromosomes and nuclei were obtained by bursting grown cells from a registered B lymphocyte Yoruba cell line (passage 4, male) cultured at 37°C in a 5% CO2 atmosphere. The cells were thymidine-synchronized for 16 hours using 2 mM thymidine followed by a Colcemid treatment (0.2 mg/ml) (Gibco Life Technologies) for 6 hours and a hypotonic treatment at 37°C using 0.075 M KCl for 5 min. Then, the sample was fixed in three changes of 3:1 methanol/acetic acid and then chemically fixed again by 2.5% (v/v) glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) for 1 hour. After fixation, the sample was stained with 5 mM platinum blue (self-synthesized in the laboratory) for 30 min at room temperature, then washed twice with Milli-Q water, and dehydrated by ethanol-water solutions (30, 50, 75, and 100%) for 15 min each. The sample was centrifuged after every washing or dehydration step to remove the treatment solution. After all the abovementioned procedures, we expected to obtain isolated nuclei and to remove the general contamination from other organelles as much as possible, which would minimize the consumption of the platinum blue stain and maximize our chance to gain homogeneously fully stained nuclear specimens containing chromosomes inside. The chromosomes and nuclei were finally embedded in a small volume of a four-component epoxy resin and subsequently left to cure overnight at 60°C. More fresh resin of the same recipe was added to fill the container to form a second part and left at 60°C again for about another 20 hours to be fully cured (12). The epoxy resin was made by following the standard recipe of hard resin based on Agar 100 epoxy from Agar Scientific, Elektron Technology UK Ltd.
SBFSEM measurement
The cured sample was trimmed to a pyramid-shaped block with a top face of about 500 μm × 500 μm using a conventional ultramicrotome (Leica Ultracut UCT) after mechanical polishing. Finally, the sample was serially sectioned and imaged by the SBFSEM system in an FEI Quanta 250 field-emission gun environmental SEM (FEGESEM) at 5 kV under a chamber pressure of 60 Pa of water vapor. The pixel size of generated BSE micrographs was set to 11 nm × 11 nm to cover the whole nucleus. The nominal sectioning thickness was 20 nm per slice with a diamond knife (horizontal) cutting speed of 0.3 mm/s; in total, 300 slices were collected for the measured sample.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Biotechnology and Biological Sciences Research Council through grant BB/H022597/1 and partially by the Engineering and Physical Sciences Research Council (EPSRC), UK, LATEST2 Programme through grant EP/H020047/1. The early work was partially supported by the EPSRC, UK, through a Dorothy Hodgkin Postgraduate Award to B.C. The work at the Tongji University was supported by the Tongji Talent Program “Materials Nanostructure” through grants 152221 and 152243. The work at the Brookhaven National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract DE-SC00112704. We thank Diamond Light Source, UK, for providing access to Avizo software. The SBFSEM measurements were performed on the (LATEST2 Programme–associated) FEI FEGESEM and Gatan 3View facilities at the School of Materials at the University of Manchester. Author contributions: B.C., M.Y., and I.R. conceived the project. A.K.E. synthesized the platinum blue stain. M.Y. performed the cell culture and sample purification, whereas B.C. performed the resin embedding and the remaining sample preparation. B.C., T.H., and M.Y. performed the SBFSEM measurements. B.C. analyzed the data. I.R. and G.T. supervised the project. B.C. and I.R. prepared the manuscript, incorporating contributions from all authors. Competing interests: I.R. is a paid consultant at Tongji University and declares that he has competing interests as Chair of Science Advisory Committee, Linac Coherent Light Source, Stanford Linear Accelerator Center, Stanford University, at the time of this research. I.R. also declares that he has competing interests as Chairman of the Royal Society Conference “Real and reciprocal space x-ray imaging” and as Organizer of the first UK-Japan Chromosome Imaging Workshop overlapping the time of the research. I.R. also declares that he has competing interests as Chair of the Science Advisory Committee of the ALBA Synchrotron, Barcelona, Spain, and as a member of the Science Advisory Committee of the European X-ray Free Electron Laser facility in Hamburg. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1602231/DC1
movie S1. 3D rendering of the measured prophase nucleus.
movie S2. 3D rendering of the measured prophase nucleus from another orientation.
table S1. Volume statistics of all the broken chromosomes in the nucleus.
table S2. Reassignment of all the intact chromosomes as a comparison with the assignment in the main text.
fig. S1. SBFSEM slices of the measured (smaller) chromosomes.
fig. S2. Second linear fitting of the chromatid volumes against their base pair numbers.
REFERENCES AND NOTES
- 1.Kornberg R. D., Chromatin structure: A repeating unit of histones and DNA. Science 184, 868–871 (1974). [DOI] [PubMed] [Google Scholar]
- 2.Griffith J. D., Chromatin structure: Deduced from a minichromosome. Science 187, 1202–1203 (1975). [DOI] [PubMed] [Google Scholar]
- 3.E. S. Tobias, J. M. Connor, M. Ferguson-Smith, Essential Medical Genetics (Wiley-Blackwell, ed. 3, 2011), chap. 5. [Google Scholar]
- 4.Horn P. J., Peterson C. L., Chromatin higher order folding–wrapping up transcription. Science 297, 1824–1827 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Scheffer M. P., Eltsov M., Frangakis A. S., Evidence for short-range helical order in the 30-nm chromatin fibers of erythrocyte nuclei. Proc. Natl. Acad. Sci. U.S.A. 108, 16992–16997 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bian Q., Belmont A. S., Revisiting higher-order and large-scale chromatin organization. Curr. Opin. Cell Biol. 24, 359–366 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishino Y., Eltsov M., Joti Y., Ito K., Takata H., Takahashi Y., Hihara S., Frangakis A. S., Imamoto N., Ishikawa T., Maeshima K., Human mitotic chromosomes consist predominantly of irregularly folded nucleosome fibres without a 30-nm chromatin structure. EMBO J. 31, 1644–1653 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song F., Chen P., Sun D., Wang M., Dong L., Liang D., Xu R.-M., Zhu P., Li G., Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science 344, 376–380 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Marsden M. P. F., Laemmli U. K., Metaphase chromosome structure: Evidence for a radial loop model. Cell 17, 849–858 (1979). [DOI] [PubMed] [Google Scholar]
- 10.Earnshaw W. C., Laemmli U. K., Architecture of metaphase chromosomes and chromosome scaffolds. J. Cell Biol. 96, 84–93 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belmont A. S., Mitotic chromosome structure and condensation. Curr. Opin. Cell Biol. 18, 632–638 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Yusuf M., Chen B., Hashimoto T., Estandarte A. K., Thompson G., Robinson I., Staining and embedding of human chromosomes for 3-D serial block-face scanning electron microscopy. Biotechniques 57, 302–307 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Denk W., Horstmann H., Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLOS Biol. 2, e329 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starborg T., Kalson N. S., Lu Y., Mironov A., Cootes T. F., Holmes D. F., Kadler K. E., Using transmission electron microscopy and 3View to determine collagen fibril size and three-dimensional organization. Nat. Protoc. 8, 1433–1448 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arenkiel B. R., Ehlers M. D., Molecular genetics and imaging technologies for circuit-based neuroanatomy. Nature 461, 900–907 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen B., Guizar-Sicairos M., Xiong G., Shemilt L., Diaz A., Nutter J., Burdet N., Huo S., Mancuso J., Monteith A., Vergeer F., Burgess A., Robinson I., Three-dimensional structure analysis and percolation properties of a barrier marine coating. Sci. Rep. 3, 1177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boveri T., Die blastomerenkerne von Ascaris megalocephala und die theorie der chromosomenindividualität. Arch. Zellforsch. 3, 181–268 (1909). [Google Scholar]
- 18.Walter J., Schermelleh L., Cremer M., Tashiro S., Cremer T., Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J. Cell Biol. 160, 685–697 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cvačková Z., Mašata M., Staněk D., Fidlerová H., Raška I., Chromatin position in human HepG2 cells: Although being non-random, significantly changed in daughter cells. J. Struct. Biol. 165, 107–117 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cremer T., Cremer M., Chromosome territories. Cold Spring Harb. Perspect. Biol. 2, a003889 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Speicher M. R., Gwyn Ballard S., Ward D. C., Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat. Genet. 12, 368–375 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Langlois R. G., Yu L.-C., Gray J. W., Carrano A. V., Quantitative karyotyping of human chromosomes by dual beam flow cytometry. Proc. Natl. Acad. Sci. U.S.A. 79, 7876–7880 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gray J. W., Lucas J., Peters D., Pinkel D., Trask B., van den Engh G., Van Dilla M., Flow karyotyping and sorting of human chromosomes. Cold Spring Harb. Symp. Quant. Biol. 51, 141–149 (1986). [DOI] [PubMed] [Google Scholar]
- 24.G. M. Cooper, in The Cell: A Molecular Approach (Sinauer Associates, ed. 2, 2000). [Google Scholar]
- 25.Güttinger S., Laurell E., Kutay U., Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat. Rev. Mol. Cell Biol. 10, 178–191 (2009). [DOI] [PubMed] [Google Scholar]
- 26.G. Karp, in Cell and Molecular Biology: Concepts and Experiments (John Wiley & Sons Inc., ed. 5, 2008), chap. 14. [Google Scholar]
- 27.Wanner G., Formanek H., Imaging of DNA in human and plant chromosomes by high-resolution scanning electron microscopy. Chromosome Res. 3, 368–374 (1995). [DOI] [PubMed] [Google Scholar]
- 28.Archive Ensembl Release 68, All human genome (length) data used in the paper are from this database (2012); http://Jul2012.archive.ensembl.org/Homo_sapiens/Location/Chromosome?r=1 [accessed 10 September 2016].
- 29.L. G. Shaffer, J. McGowan-Jordan, M. Schmid, ISCN 2013: An International System for Human Cytogenetic Nomenclature (2013) (S. Karger AG, 2013). [Google Scholar]
- 30.C. E. Shannon, Communication in the presence of noise. Proc. IRE 37, 10–21 (1949); reprinted as classic paper in Proc. IEEE 86, 447–457 (1998).
- 31.Arnott S., Chandrasekaran R., Birdsall D. L., Leslie A. G. W., Ratliff R. L., Left-handed DNA helices. Nature 283, 743–745 (1980). [DOI] [PubMed] [Google Scholar]
- 32.Kassis A. I., Sastry K. S. R., Adelstein S. J., Kinetics of uptake, retention, and radiotoxicity of 125IUdR in mammalian cells: Implications of localized energy deposition by auger processes. Radiat. Res. 109, 78–89 (1987). [PubMed] [Google Scholar]
- 33.Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J., Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997). [DOI] [PubMed] [Google Scholar]
- 34.Uchiyama S., Kobayashi S., Takata H., Ishihara T., Hori N., Higashi T., Hayashihara K., Sone T., Higo D., Nirasawa T., Takao T., Matsunaga S., Fukui K., Proteome analysis of human metaphase chromosomes. J. Biol. Chem. 280, 16994–17004 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Cremer T., Cremer C., Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2, 292–301 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Croft J. A., Bridger J. M., Boyle S., Perry P., Teague P., Bickmore W. A., Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145, 1119–1131 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun H. B., Shen J., Yokota H., Size-dependent positioning of human chromosomes in interphase nuclei. Biophys. J. 79, 184–190 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1602231/DC1
movie S1. 3D rendering of the measured prophase nucleus.
movie S2. 3D rendering of the measured prophase nucleus from another orientation.
table S1. Volume statistics of all the broken chromosomes in the nucleus.
table S2. Reassignment of all the intact chromosomes as a comparison with the assignment in the main text.
fig. S1. SBFSEM slices of the measured (smaller) chromosomes.
fig. S2. Second linear fitting of the chromatid volumes against their base pair numbers.