

Abstract
Aberrant DNA methylation patterns are a common theme across all cancer types. Specific DNA demethylation of regulatory sequences can result in upregulation of genes that are critical for tumor development and progression. Integrin α6β4 is highly expressed in pancreatic carcinoma and contributes to cancer progression, in part, through the specific DNA demethylation and upregulation of epidermal growth factor receptor (EGFR) ligands amphiregulin (AREG) and epiregulin (EREG). Whole genome bisulfite sequencing (WGBS) revealed that integrin α6β4 signaling promotes an overall hypomethylated state and site specific DNA demethylation of enhancer elements within the proximal promoters of AREG and EREG. Additionally, we find that the base excision repair (BER) pathway is required to maintain expression of AREG and EREG, as blocking DNA repair molecules, TET1 GADD45A, TDG, or PARP-1 decreased gene expression. Likewise, we provide the novel finding that integrin α6β4 confers an enhanced ability on cells to repair DNA lesions and survive insult. Therefore, while many known signaling functions mediated by integrin α6β4 that promote invasive properties have been established, this study demonstrates that integrin α6β4 can dramatically impact the epigenome of cancer cells, direct global DNA methylation levels toward a hypomethylated state, and impact DNA repair and subsequent cell survival.
Introduction
Integrins contribute to essential components of tumor progression such as survival, proliferation, and cell motility1. Specifically, integrin α6β4 is a known driver of tumor cell invasion2, which in turn promotes metastasis3. In cancer cells, integrin α6β4 signaling is activated upon binding to laminin extracellular matrix proteins and in cooperation with growth factor receptors such as EGFR, RON, and c-MET4–6. Activation of integrin α6β4 results in stimulation of downstream signaling pathways including PI3K, MAPK, Src family kinases, Rho family small GTPases, and the Nuclear Factor of Activated T-cells (NFAT)7–9 that contribute to invasion, angiogenesis, anoikis-resistance, cell survival, and proliferation10. Integrin α6β4 enhances these properties in part through transcriptional upregulation of pro-tumorigenic genes including S100A4 in breast cancer11, 12 and the EGFR ligands AREG and EREG in pancreatic carcinomas13.
The importance of AREG and EREG in tumor progression, therapeutic resistance, and as a potential prognostic and predictive biomarker has been well established in multiple cancer types14, 15. Cleavage of pro-AREG and pro-EREG by the MMPs results in protein release and autocrine signaling to activate EGFR13. AREG and EREG are unique in their ability to cause EGFR recycling back to the plasma membrane for reactivation16, 17. EGFR signaling by AREG and EREG is enhanced in pancreatic carcinomas and contributes to the aggressive nature of the disease18, 19. We have shown that AREG and EREG are required for HGF-mediated migration and invasion in response to signaling from integrin α6β4, further demonstrating their importance to invasive properties of cancer cells13. We and others find that AREG13 and EREG13, 20 gene expression is controlled by DNA methylation. However, the mechanisms guiding the demethylation of these promoters have not been elucidated.
Transcriptionally silenced genes have a repressive epigenetic state that compacts chromatin. Repressive epigenetic marks include non-acetylated histones, lysine methylation at H3K27 and H3K4 and cytosine methylation at CpGs21. Active DNA demethylation is tightly regulated and requires a series of enzymatic reactions that proceed through the BER pathway. This mechanism of epigenetic alteration is likely responsible for upregulation of pro-tumorigenic genes, as it has been identified for dynamic, context dependent modification of DNA22, 23.
The ten-eleven translocation methylcytosine dioxygenase (TET1) is the first crucial step in DNA demethylation as this protein recognizes specific 5-mCs to be targeted for removal by DNA repair and conversion from 5-mC to 5-hydroxymethyl cytosine (5-hmC)23. 5-hmC can be further oxidized by TET proteins to 5-carboxycytosine (5-caC) and 5-formylcytosine (5-fmC); however, these derivatives are found less often in the genome, and their complete function is still being characterized24.
5-mC products are identified by growth arrest and DNA damage inducible alpha (GADD45A). GADD45A is responsible for recruitment of other repair factors to CpG sites for removal of methyl groups, and has been implicated as a necessary step in DNA demethylation by providing a link between epigenetics and DNA repair25, 26. GADD45A recruits Activation Induced Cytidine Deaminase (AID) and Apolipoprotein B mRNA Editing Enzyme, Catalytic polypeptide-like (APOBEC) proteins26, which deaminates 5-hmC to 5-hmU, generating a G-U DNA mismatch. This mismatch is removed by thymine DNA glycosylase (TDG) or methyl-binding protein 4 (MBD4). This cleavage activates the normal functions of the BER pathway including cleavage of the DNA backbone by AP-endonuclease and repair back to a non-methylated cytosine by XRCC-1, PARP-1, DNA ligase, and DNA polymerase27.
Here, we sought to determine in mechanistic detail how integrin α6β4 stimulates DNA de methylation of AREG and EREG by systematically examining the NER and BER pathways and define the impact of integrin α6β4 on genome-wide methylation patterns.
Results
Integrin α6β4 promotes laminin deposition in pancreatic cancer cells
Integrin α6β4 signaling has been shown to be ligand-independent in several model systems where the integrin does not require exogenous ligand to mediate its effects7. In our previous studies, we have found that exogenous laminin is not required to see changes in gene expression associated with enhanced invasion and migration13. Interestingly, the Jones group has demonstrated that integrin α6β4 promotes secretion and deposition of laminin-5, a major laminin isoform implicated in integrin α6β4 signaling, in the extracellular matrix, which in turn promotes motility of keratinocytes28. To test if endogenous secretion of laminin-5 is responsible for ligating integrin α6β4 in the Panc1 model, we plated Panc1-2G6 (low α6β4) and Panc1-3D7 (high α6β4) cells were plated onto collagen-coated coverslips for 4 hours and stained for the laminin γ2 subunit, which is unique to laminin-5. We find that in integrin α6β4 high expressing Panc1-3D7 cells, there is enhanced deposition of laminin, which strongly colocalizes with the integrin β4 subunit (Fig. 1A), which is in line with observations from keratinocyte studies. In contrast, cells with low integrin α6β4 have low detectable levels of laminin (Fig. 1D), which is not deposited extracellularly (Fig. 1B), implicating a deficient integrin α6β4 signaling network. Likewise, our previous studies demonstrate the enhanced migratory abilities of integrin α6β4 high versus low expressing pancreatic cancer cell lines when plated on laminin29. We chose to use these stable subpopulations derived from the Panc1 cell line as our model system for studying the impact of integrin α6β4 on the transcriptome as we have clearly demonstrated both variable levels of the integrin α6β4 and its cognate ligand, laminin-5.
Figure 1.

Integrin α6β4 binds to and promotes secretion of laminin-5 in Panc1 cells. (A–C) Panc1 clones 3D7 (A; high α6β4) and 2G6 (B; low α6β4) were plated on collagen I coated coverslips and allowed to adhere under normal culture conditions for 4 hours. Cells were fixed and immunostained for integrin β4 subunit (red), laminin-5 (γ2 subunit; green), or f-actin (blue) as described in the Methods section. Cells were imaged by TIRF microscopy using the same exposure times and settings. Pearson’s coefficient for colocalization between integrin β4 and laminin-5 were 0.8 for Panc1-3D7 (A) and 0.011 for Panc1-2G6 (B). These values are representative for the 30 cells analyzed for each cell line. (C) Represents a rare β4 expressing cell in the Panc-2G6 cell population. (D) Western blot analysis of whole cell extracts from MDA-MB-231 (positive control), Panc1-2G6 and Panc1-3D7 cells for integrin β4, laminin-5 and actin (loading control).
Epigenetic events regulate expression of AREG and EREG
Integrin α6β4 signaling stimulates progression of multiple types of cancer in part by altering the transcriptome. Notably, expression of AREG and EREG positively correlates with expression of and signaling through integrin α6β4 (Fig. 2A), supporting our previous work13. To determine if AREG and EREG expression is regulated by DNA methylation, Panc1-2G6 cells were treated with the DNA methyltransferase inhibitor 5-aza-CdR at indicated concentrations, harvested at indicated time points, and RNA analyzed by QPCR. We found that both AREG and EREG mRNA expression increased in a time and dose dependent manner (Fig. 2B) demonstrating the susceptibility of AREG and EREG to DNA methylation. Furthermore, integrin α6β4 was required for induction of AREG and EREG mediated by 5-aza-CdR, as knocking down the integrin β4 in Panc1-3D7 cells hindered epigenetic induction of AREG and EREG expression (Fig. 2C). Considering that epigenetic changes are reversible, AsPC1 and Suit2 cells, high expressers of integrin α6β4, AREG, and EREG, were treated with the methyl donor S-adenosylmethionine (SAM) and assessed for AREG and EREG expression by QPCR. These data revealed a 50% decrease in expression of AREG and EREG as seen in Fig. 2D. Taken together these data indicate that signaling from integrin α6β4 and DNA demethylation are required to drive AREG and EREG expression.
Figure 2.
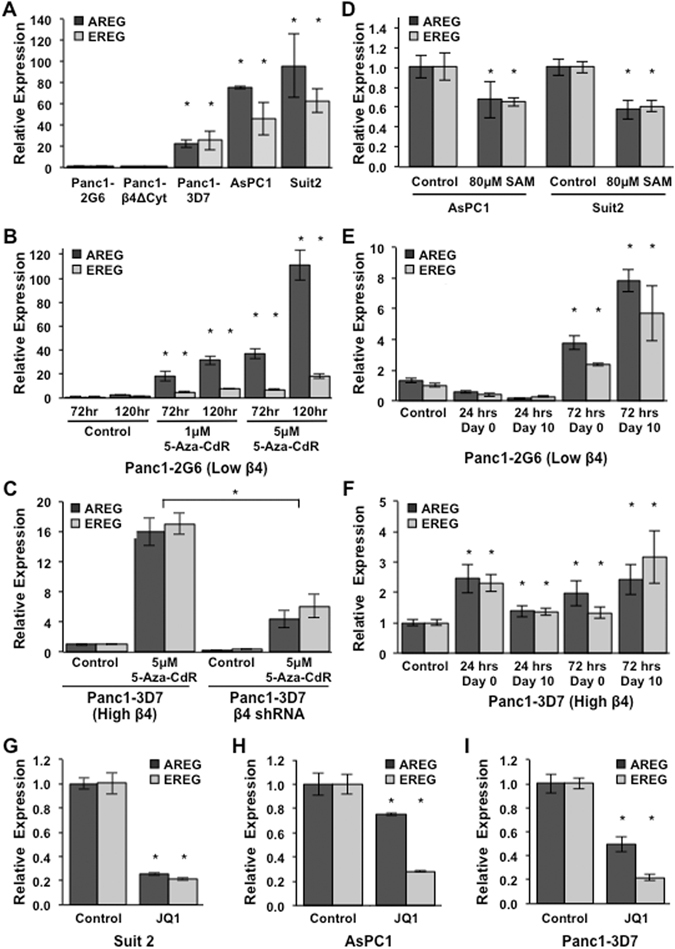
AREG and EREG expression is mediated by DNA demethylation in response to signaling from the integrin α6β4. (A) Expression of AREG and EREG was compared in Panc1-2G6 (low α6β4) and cells expressing a dominant negative α6β4 (Panc1-β4ΔCyt), Panc1-3D7, Suit2, and AsPC1 (high α6β4; in order of increasing expression) cell lines. (B) Panc1-2G6 cells (low α6β4) were treated with vehicle only (control) or with 1 μM or 5 μM 5-aza-2′-deoxycitine (5-aza-CdR) in fresh medium daily for 3 or 5 days. (C) Panc1-3D7 stably expressing an shRNA targeting the β4 subunit or a non-targeting (NT) shRNA control vector were treated with 2 μM 5-aza-CdR for 3 days and then assessed for AREG and EREG expression. (D) AsPC1 and Suit2 (high α6β4) were treated with vehicle only (control) or 80 μM S-adenosylmethione (SAM) in fresh medium daily for 5 days (E,F). Panc1-2G6 (E) and Panc1-3D7 (F) cells were treated with 2 μM 5-aza-CdR for 24 or 72 hours, 5-aza-CdR was removed and cells were either collected immediately or maintained in culture for 10 days. (G–I) Cells with high integrin α6β4 were treated with vehicle only (control) or 0.5 μM JQ1 overnight and harvested for analysis by QPCR. For all experiments RT-PCR was used to convert RNA to cDNA and QPCR was used to assess AREG and EREG expression. Data depicted here are representative of at least three different experiments and represent the mean +/− standard deviation. Statistical significance was calculated using a one-tailed t-test in which * denotes P < 0.05 as compared to controls, unless otherwise indicated.
True epigenetic alterations are stable changes maintained across many generations. Since 5-Aza-CdR can modify the epigenetic landscape30, we assessed the impact of short term 5-aza-CdR treatment on AREG and EREG expression by treating cells with the indicated concentrations of 5-Aza-CdR for 24 or 72 hours. 5-Aza-CdR was removed and cells were either harvested immediately or maintained in culture for 10 days. As shown in Fig. 1E, expression of AREG and EREG was not only induced 20–40 fold and maintained in Panc1-2G6 cells following 5-aza-CdR treatment but continued to increase when kept in culture 10 days post 5-aza-CdR removal. Treatment of Panc1-3D7 cells only slightly increased transcription of AREG and EREG (Fig. 2F), suggesting these stable epigenomic modifications have already taken place. These data confirm that the integrin α6β4 contributes to the stable upregulation of pro-tumorigenic molecules AREG and EREG through epigenetic alterations.
Alterations in DNA methylation strongly impact the activity of enhancers, which activate specific transcriptional profiles through recruitment of transcription factors that interact with the mediator complex31. To determine if enhancer activity is required for AREG and EREG expression in pancreatic cancer cells, we treated cells with JQ1, a BET bromo-domain inhibitor that is specific for BRD432. BRD4 interacts with the elongating factor P-TEFB in Pol II complexes to enhance transcription for both protein-coding and enhancer-derived noncoding RNAs33. We found that AREG and EREG expression markedly decreased with JQ1 treatment, thus indicating their transcriptional dependence on enhancer function (Fig. 2G–I).
Integrin α6β4 impacts genome wide DNA methylation patterns
To define DNA demethylation changes that drive expression of AREG and EREG, sodium bisulfite conversion and whole genome sequencing was performed on genomic DNA from pancreatic cancer cells with either high (Panc1-3D7) or low (Panc1-2G6) integrin α6β4 expression. Sequencing reads were aligned to the reference genome, GRCH37, mapped to the AREG and EREG genes, and visualized using the UCSC genome browser. We found that cells with high integrin α6β4 (Fig. 3A and B; bottom panels) have reduced DNA methylation within intronic regions of both EREG (Fig. 3A) and AREG (Fig. 3B), confirming that the integrin α6β4 drives site-specific DNA demethylation, and defining the critical CpG sites of AREG and EREG that become altered downstream of integrin α6β4.
Figure 3.
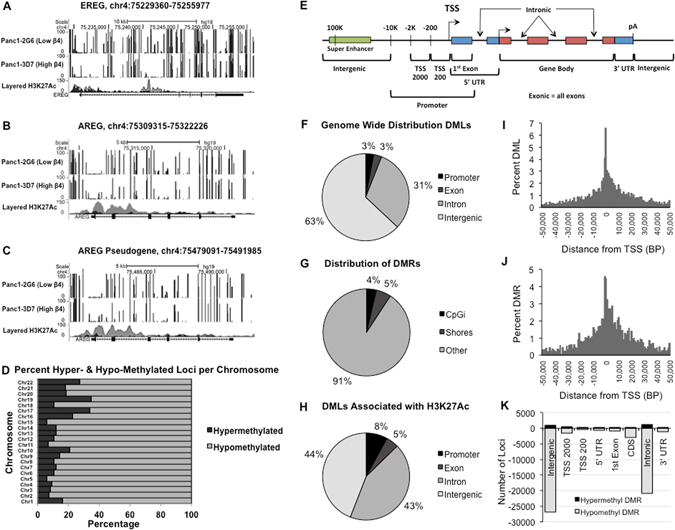
The integrin α6β4 drives both gene specific and global DNA hypomethylation. (A–C) Genomic DNA from Panc1-2G6 (β4 low; upper panels) and Panc1-3D7 (high β4; lower panels) was processed for high-resolution methyl-seq by the NextGen Sequencing Core at the Norris Comprehensive Cancer Center. Samples were analyzed bioinformatically and percent methylation shown for EREG (A), AREG (B), and AREG pseudogene (C). (D) Percent hypomethylation and hypermethylation per chromosome when comparing Panc1-3D7 vs. Panc1-2G6. (E) Defined regions of interest assessed for changes in DNA methylation (F) Location of DMLs across the genome. (G) Percent of methylation changes located in CpG islands and shores. (H) Location of DMLs associated with H3K27Ac. (H) Distance from TSSs for DMLs (I) Distances from TSSs for DMRs (J) Number of both hypomethylated and hypermethylated regions corresponding to genomic features.
Importantly, we also found alterations in DNA methylation in an AREG pseudogene, which lies directly downstream of AREG (Fig. 3C). When examining these two regions, both the sequence structure and regulatory similarity were noted as they are 99% homologous when blasted against the reference genome. Since Bismark only reports unique matches, the multi-mapping scenario of AREG and its pseudogene made it difficult to investigate the methylation alternations in these two regions. However, the analysis was possible by masking AREG pseudogene and mapping AREG, and vice-versa for AREG pseudogene. We attempted to investigate this further by using bisulfite conversion with methylation specific PCR to confirm altered CpGs within this region. However, the sequence similarity between these two regions and difficulty designing unique primers for bisulfite converted DNA proved that this analysis was technically unfeasible.
Regions that had the greatest difference in DNA methylation in both AREG and EREG as a result of integrin α6β4 signaling corresponded to areas enriched in H3K27Ac marks (Fig. 3A,B), as annotated by the ENCODE project, that are reported to mark active enhancer elements34. Additionally, a super-enhancer associated with AREG and EREG expression lies between AREG and the AREG pseudogene35. We found no significant differences in super-enhancer DNA methylation (data not shown), indicating that it is unlikely that DNA methylation of this element is the major driver for enhanced AREG and EREG gene expression. Taken together, these data, along with our observation that BRD4 is required for AREG and EREG expression, indicate that DNA demethylation of enhancer elements localized within the proximal promoters of AREG and EREG drive expression in response to integrin α6β4 signaling.
Next, we examined the genome wide effects of integrin α6β4 on DNA methylation using our WGBS data. A total of 236,371 differentially methylated loci (DML; 207,168 hypomethylated and 29,203 hypermethylated) were identified comparing Panc1-3D7 vs. Panc1-2G6. Figure 3D illustrates the percentage of hypermethylated and hypomethylated events per chromosome as a percent of the number of DMLs. Of the DMLs identified, 87.6% were hypomethylated and 12.4% were hypermethylated, thus indicating that the integrin α6β4 shifts chromatin to a more hypomethylated state. Further analysis of these data revealed that only 3.1% of these loci were located in promoter regions, 2.1% in exonic regions, 31.1% in intronic regions and 63.1% were in intergenic regions (Fig. 3F). 13,889 differentially methylated regions (DMRs) were identified, of which only about 4% were located in CpG islands, and 5% in CpG shores (Fig. 3G). We found that 40,609 DMLs associated with H3K27Ac marks were hypomethylated as opposed to 13,679 DMLs hypermethylated. These events correspond to 4993 genes that have alterations in methylation within enhancer elements. As seen in Fig. 3H, the majority of these altered DML are localized to intronic and intergenic regions (defined in Fig. 3E) of which the majority are hypomethylated (Fig. 3K). This observation is typical of enhancer elements, as many enhancers are part of non-coding regions of the genome36. Additionally, we found that DMLs and DMRs occur predominantly within the first ten thousand base pairs on either side of the TSS with slightly more occurring after the TSS, as expected (Fig. 2I,J).
AREG and EREG expression is not regulated by NER
Since our data suggest that AREG and EREG DNA demethylation is an active process, we tested the hypothesis that DNA repair is required to maintain their expression downstream of integrin α6β4 signaling. The NER pathway, including the Xeroderma pigmentosum complementation group proteins XPA, XPG, and XPF, has been implicated in active DNA demethylation by DNA repair37, 38. Accordingly, we targeted molecules critical for and specific to the NER pathway and examined their impact on AREG and EREG expression. When knockdown of XPA (Fig. 4A) was achieved, transcription of AREG and EREG in Panc-3D7 (high α6β4; Fig. 4C) remained unaffected. AREG and EREG transcription in Panc-2G6 (low α6β4; Fig. 4B) showed a statistically significant increase when XPA was knocked down, which implies negative regulation. However, due to very low basal expression of AREG and EREG in these cells (cT value >35) it is unlikely to be biologically significant. Using specific shRNAs we knocked down ERCC4 (XPF; Fig. 4D) and ERCC5 (XPG; Fig. 4G) and demonstrated that effective knockdown of NER genes had relatively little or no effect on AREG and EREG expression (Fig. 4E–F,I–J). Taken together these data indicate that NER is not required to maintain AREG or EREG expression.
Figure 4.
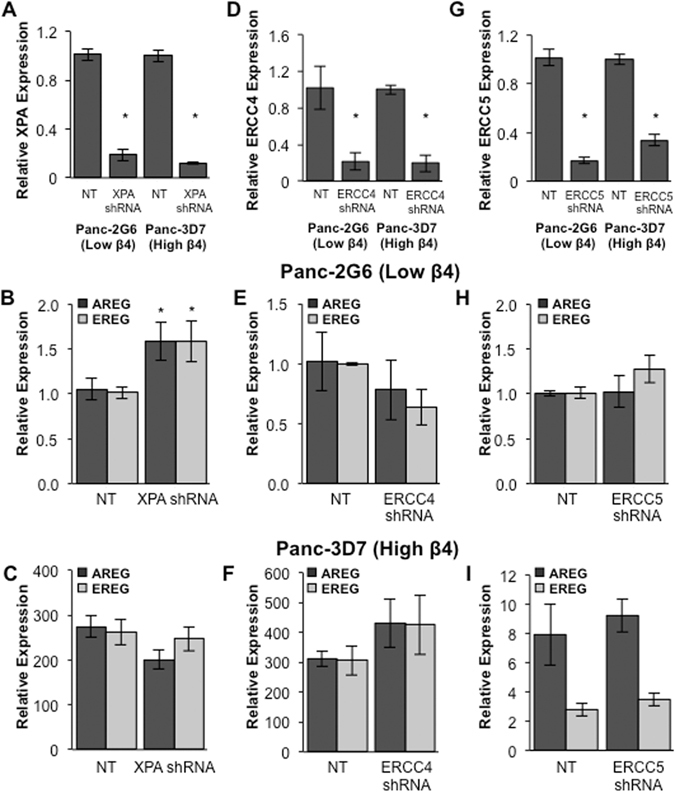
NER is not required for expression of AREG and EREG. Using lentiviral transfection stable knockdown of XPA (A), ERCC4 (XPF) (D), and ERCC5 (XPG) (G) was achieved in Panc1-2G6 (low α6β4) and Panc1-3D7 (high α6β4) cells as confirmed by QPCR. AREG and EREG expression was examined following knockdown in cells with both low α6β4 (B,E,H) and high α6β4 (C,F,I) expression. Data depicted are representative of at least three different experiments and represent the mean +/− standard deviation. Statistical significance was calculated using a one-tailed t-test in which * denotes P < 0.05 as compared to controls.
Alterations in BER impact AREG and EREG expression
Gemcitabine is a chemotherapeutic with multiple proposed mechanisms of action, including depletion of deoxynucleotide triphosphates that are necessary for DNA synthesis and completion of DNA repair39. Interestingly, gemcitabine has been shown to specifically inhibit GADD45A mediated gene activation via DNA demethylation and DNA repair40. To investigate the role of DNA repair in expression of AREG and EREG, cells were treated with 10 μM gemcitabine for 72 hours. As demonstrated in Fig. 5A, AREG and EREG expression dramatically decreased in cells with high integrin α6β4 in response to treatment, thus indicating that DNA repair is required to maintain expression. As summarized in Fig. 5B, GADD45A mediated active DNA demethylation is achieved through BER. Therefore, we next investigated the role of TET1, GADD45A, TDG, and PARP1 in the regulation of AREG and EREG as key regulators of DNA repair-mediated DNA demethylation.
Figure 5.
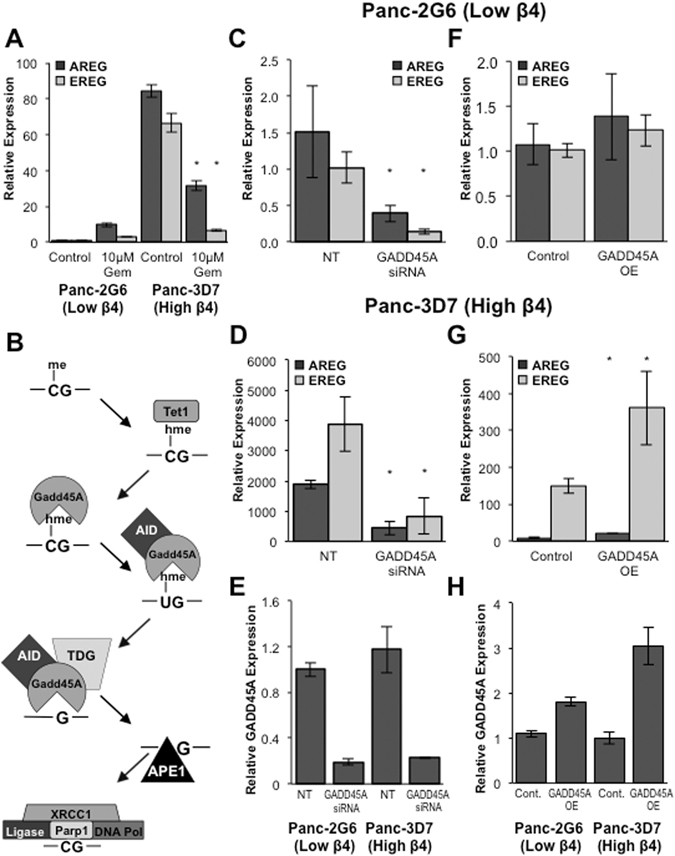
GADD45A is both required for and the rate-limiting step in activation of AREG and EREG expression. (A) Cells were treated with 10 μM Gemcitabine for 72 hours and expression of AREG and EREG measured by QPCR. (B) Summary of current literature for how GADD45A mediated DNA demethylation is achieved. Transient knockdown of GADD45A was achieved using electroporation and specific siRNA (E). Adenovirus was used to overexpress GADD45A in Panc1-2G6 and Panc1-3D7 cells (H). Changes in AREG and EREG expression were measured by QPCR in Panc1-2G6 (C,F) and Panc1-3D7 (D,G). Data depicted here are representative of at least three different experiments and represent the mean +/− standard deviation. Statistical significance was calculated using a one-tailed t-test in which * denotes P < 0.05 as compared to controls.
GADD45A is responsible for identifying residues for DNA demethylation by DNA repair25, 41. We modulated GADD45A in pancreatic cancer cells using either siRNA to knockdown or adenoviral infection to overexpress GADD45A and examined the effects on AREG and EREG expression. As depicted in Fig. 4, knockdown of GADD45A (Fig. 5E) resulted in decreased expression of AREG and EREG regardless of integrin α6β4 expression (Fig. 5C,D). Similarly, overexpression of GADD45A (Fig. 5H) resulted in a further increase in AREG and EREG expression, only in Panc1-3D7 cells (Fig. 5F vs G). These data indicate that GADD45A is a required for and is potentially a rate-limiting step in gene activation of AREG and EREG downstream of integrin α6β4 signaling.
TET proteins are solely responsible for oxidation of 5-mC to 5-hmC, 5-fC and 5-caC in mammalian DNA42, 43, which provide substrates for further processing to a cytosine by the DNA glycosylases and BER44, 45, with 5-hmC being the most common43. To test the role of the TET proteins, we depleted TET1 using specific shRNAs in Panc1-3D7 cells (Fig. 6A). As demonstrated in Fig. 6B, AREG and EREG expression is robustly decreased following a 70% reduction in TET1.
Figure 6.
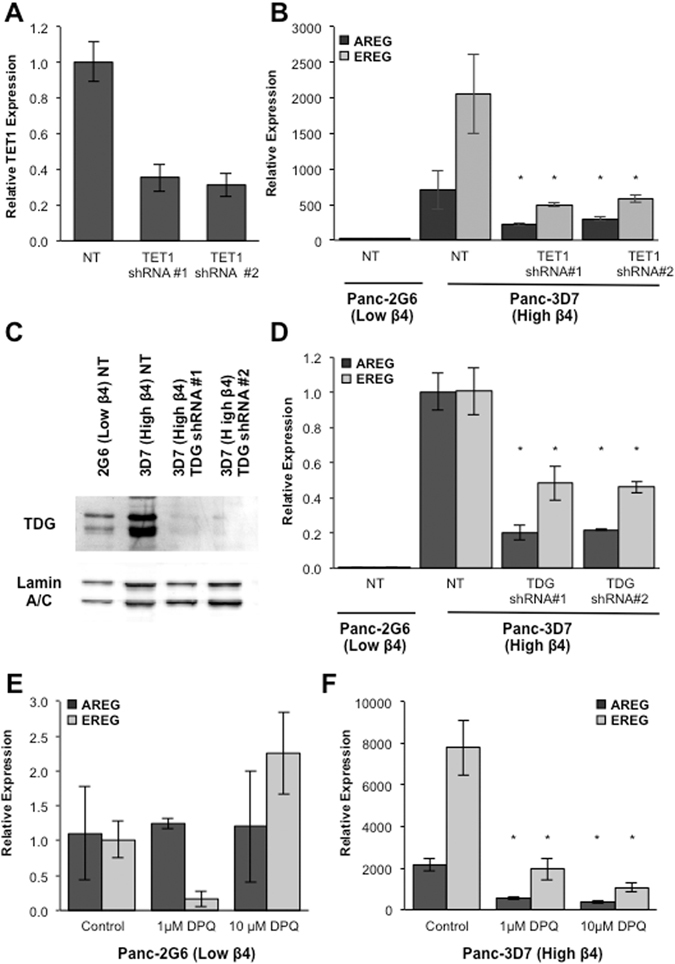
BER is necessary for induction of AREG and EREG expression downstream of integrin α6β4 signaling. (A,B) RNA was isolated from Panc1-3D7 cells stably expressing non-targeting or shRNA specific for TET1. QPCR analysis was used to confirm TET1 knockdown (A) and expression of AREG and EREG (B). (C) Nuclei were isolated from Panc1-2G6, Panc1-3D7, and Panc1-3D7 cells expressing specific lentiviral shRNA for TDG. Western blot analysis was performed on nuclear fractions for TDG and Lamin A/C used as a loading control. (D) Cells were collected and AREG and EREG expression measured by QPCR. (E,F) Cells were treated with either 1 μM or 10 μM 3,4-Dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2 H)-isoquinolinone (DPQ) for 72 hours. Expression of AREG and EREG was measured by QPCR in Panc-2G6 (low α6β4; E) and Panc-3D7 (high α6β4; F) cell lines. Data depicted are representative of at least three different experiments and represent the mean +/− standard deviation. Statistical significance was calculated using a one-tailed t-test in which *denotes P < 0.05 as compared to controls.
TDG has been found in complex with AID and GADD45A in the context of active DNA demethylation and evidence exists that glycosylase activity is necessary for this process45. As shown in Fig. 6C, there was substantially lower nuclear TDG protein expression in Panc1-2G6 compared to Panc1-3D7. As seen in Fig. 6D, this stable knockdown of TDG resulted in marked downregulation of AREG and EREG in Panc1-3D7 cells, indicating that TDG is necessary to maintain expression of AREG and EREG downstream of integrin α6β4, potentially through preferential localization of TDG into the nucleus.
PARP-1 is required for BER and is implicated in genome-wide and locus specific active DNA demethylation in part through epigenetic regulation of TET146. Using a PARP-1 inhibitor, DPQ, we observed a dramatic decrease in AREG and EREG expression in Panc1-3D7 cells (Fig. 6F). However, in Panc1-2G6 cells, expression of AREG and EREG was relatively unaffected by PARP-1 inhibition (Fig. 6E), indicating that PARP-1 is mediator of AREG and EREG induction regulated by the integrin α6β4.
Integrin α6β4 mediates cell survival and repair upon DNA damage
We rationalized that if the integrin α6β4 is using the BER pathway to activate specific genes, the integrin may also enhance DNA repair in response to DNA damage. Therefore, we induced oxidative damage, which is repaired by the BER pathway, by exposing cells to 500 μM H2O2 over seven days and measuring cell viability by MTT assay. We observed a modest decrease in cell number in Panc1-3D7 cells; however, this H2O2 treatment nearly abolished Panc1-2G6 cells, indicating a decreased ability to survive insult by oxidative stress (Fig. 7A). To measure DNA repair more directly, we examined NER dependent DNA repair by exposing cells to 30 J/m2 UV light and measuring resolution of 6-4 photoproducts over time. As illustrated in Fig. 7B, Panc1-3D7 cells resolved UV induced lesions more rapidly than Panc1-2G6 cells, with a difference in half-life of about 1 hour. Taken together, these data indicate that the integrin α6β4 can both utilize DNA repair, and enhance the ability of cells to respond to, repair, and survive DNA damage.
Figure 7.
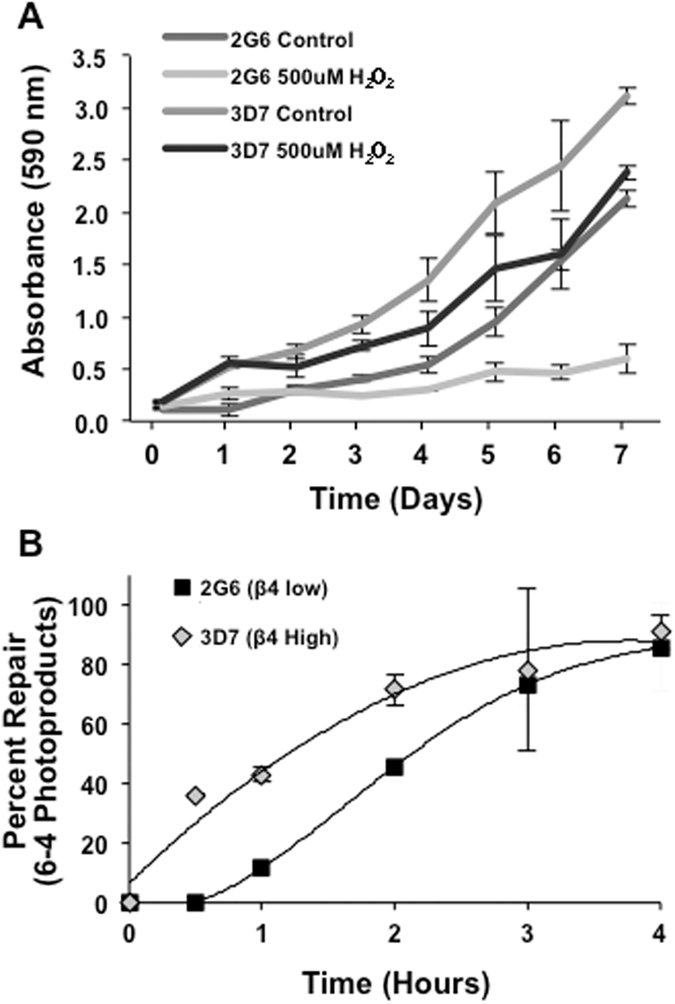
Integrin α6β4 promotes DNA repair and cell survival in response to DNA damage. (A) Cells were treated with 500 μM H2O2 in fresh medium daily for 7 days. Each day cell proliferation was measured by MTT colorimetric assay. (B) Cells were exposed to 30 J/m2 UV light and DNA isolated at indicated time points. Slot blot assay was performed using antibody for 6-4 photoproducts and percent repair compared to damage achieved immediately after exposure (0 hr).
Discussion
While our knowledge of cancer epigenetics has developed rapidly, how dynamic epigenetic regulation is influenced by the tumor microenvironment to foster a metastasis phenotype has yet to be revealed. We find that integrin α6β4 is a critical mediator of DNA demethylation of two pro-invasive molecules, AREG and EREG. These specific changes in DNA demethylation of AREG and EREG occurred at enhancer elements within their proximal promoters that drive their expression downstream of integrin α6β4. Similarly, our data support integrin α6β4 as a modulator of genome-wide DNA methylation patterns, as overexpression of integrin α6β4 resulted in dramatic hypomethylation of the genome, with a significant percentage of these CpGs located in putative enhancer sites. Lastly, our study revealed that integrin α6β4 not only utilizes the BER DNA repair but also facilitates enhanced repair of DNA lesions, as cells with high integrin α6β4 survived better in response to oxidative stress, and directly repaired 6-4 photoproducts more rapidly. Our unique findings provide evidence that places integrin α6β4 as a critical mediator of cancer epigenetics, and thus offer new mechanisms for the integrin’s role in cancer progression.
Upregulation of invasion promoting molecules and subsequent activation of their downstream signaling targets are critical for the progression of cancer. Here, we demonstrate that AREG and EREG, which are established contributors of tumor progression14, 15, are upregulated downstream of signaling from integrin α6β4 and this upregulation is dependent on active DNA demethylation. This observation builds on our previous data showing that integrin α6β4 stimulates specific DNA demethylation of the S100A4 promoter, ultimately contributing to invasive capabilities of breast cancer cells11. Interestingly, work in squamous cell carcinoma and MDA-MB-231 breast cancer cells demonstrates that ECM content, cell-cell interactions, and 3D environment impact the methylation state of the E-cadherin promoter and this dynamic epigenetic plasticity helps drive EMT47, 48. These observations collectively solidify the role of the tumor microenvironment in regulating specific sites of DNA methylation, thus contributing to invasive growth of cancer cells.
Our analysis of genome-wide DNA methylation patterns revealed that integrin α6β4 dramatically reshapes the epigenetic landscape, shifting global DNA methylation patterns to a more hypomethylated state. Furthermore, this study shows that changes in specific CpG methylation within the AREG and EREG genes occurred in intronic regions that are not defined by the presence of CpG islands. These sites of altered DNA demethylation within AREG and EREG regulatory region correspond to known sites of H3K27Ac. Coupled with the requirement of BRD4 activity for AREG and EREG expression implicates the necessity for enhancer elements to drive gene expression. Our previous work on S100A4 yielded similar results as specific changes that control gene expression reside in an enhancer element located in a CpG rich region rather than a CpG island11. Similar to our gene specific data, most hypomethylation events induced by integrin α6β4 are not localized to CpG islands or promoter regions, but are instead found in intronic and intergenic elements. In addition, 23% of these regions corresponded to potential sites of H3K27ac, which is indicative of enhancer location34. These changes in DNA methylation are not surprising as hypomethylation of enhancer elements is tightly linked to overexpression of cancer promoting genes and gene profiles, as opposed to promoter methylation49, 50. Therefore, these data suggest that this shift in methylation patterns mediated by integrin α6β4 is indeed a mechanism driving gene expression and progression to a more malignant phenotype in pancreatic cancer cells. While other evidence exists to suggest that the tumor microenvironment can influence epigenetics47, 51, 52, this study is the first to identify a specific mediator of the microenvironment, the integrin α6β4, as a regulator of this process.
Mounting evidence places the BER pathway as the most common, and context dependent mediator for active DNA demethylation45, 53. Our data support this concept, as we have demonstrated that modulation of multiple components of the BER pathway, including GADD45A, TET1, TDG, and PARP-1, impact transcriptional upregulation of AREG and EREG. Additionally, our confirmation that AREG and EREG enhancers become demethylated downstream of integrin α6β4, supports active DNA demethylation by DNA repair as the mechanism for transcriptional upregulation by the integrin α6β4. More specially, GADD45A acts as an important step in the activation of AREG and EREG and in accordance with the literature, is the coordinating molecule for specific DNA demethylation by BER54. We also show that recruitment of TDG to the nucleus is amplified in cells with high integrin α6β4 expression, suggesting that the integrin coordinates steps in this pathway, potentially through nuclear recruitment or specific targeting of repair factors. These data implicate the integrin α6β4 as a critical amplifier of DNA repair mediated DNA demethylation, identifying a novel mode of transcriptional upregulation in response to this integrin. Finally, we find that not only can the integrin α6β4 utilize BER to promote transcriptional upregulation also enhances the ability of pancreatic cancer cells to respond to and survive in the presence of DNA damage mediated by damaging agents whose damage is repaired by both the BER and NER pathways. This observation supports previous studies demonstrating that tissue architecture mediated by integrin α6β4 promotes resolution of double strand breaks55. Taken together these studies demonstrate that the integrin α6β4 contributes to a multitude of DNA repair pathways, and is a key component for connecting the extracellular environment with enhanced DNA repair.
In conclusion, this study examines a specific sensor of the tumor microenvironment, the integrin α6β4, and provides an exciting new role for this molecule in promoting tumor progression. Our data offer a novel mechanism for the upregulation of tumor promoting genes, alterations in the epigenome, and utilization of DNA repair, and places the integrin α6β4 as a major player in cancer epigenetics. These findings have far reaching impacts on our understanding of pancreatic carcinoma and further analysis of the integrin α6β4’s role in these processes will yield a more comprehensive understanding for how this integrin impacts tumor progression.
Methods
Cell lines and cell culturing
Panc1 cells (ATCC) were grown in Dulbecco’s modified Eagle’s medium (high glucose). Panc1 clones 2G6 (low integrin α6β4) and 3D7 (high integrin α6β4) were characterized and cultured as described previously56. Suit2 (Dr. Takeshi Iwamura, Miyazaki Medical College, Japan) and AsPC1 cells (America Type Culture Collection, ATCC) were maintained in RPMI 1640. Media was supplemented with 10% Fetal Bovine Serum (Sigma-Aldrich, St. Louis, MO), 1% penicillin, 1% streptomycin, and 1% L-glutamine (GIBCO by Life Technologies, Grand Island, NY).
Immunocytochemistry (ICC)
Glass coverslips were coated with 10 μg/ml collagen I (BD Biosciences) at 4 °C overnight, then rinsed three times with PBS. Cells were plated on coverslips in normal culture medium and allowed to adhere for 4 hours before fixation. Cells were fixed, permeabilized, and immunostained as described previously57. Briefly, cells were fixed for 15 minutes with 4% paraformaldehyde containing 10 mM PIPES, pH 6.8, 2 mM EGTA, 2 mM MgCl2, 7% sucrose and 100 mM KCl for 15 min at room temperature, and permeabilized with 0.25% Triton X-100. Cells were blocked for 1 hour with 3% BSA + 1% goat serum in PBS. The following primary antibodies were used at indicated concentrations and incubated at 4 °C overnight: rat anti-CD104 (439-9B, BD Pharmingen, 1:100) mouse anti-Laminin-5 (γ2 chain, clone D4B5, Millipore, 1:500 dilution) in 3% BSA in PBST overnight at 4 °C. Cy3-conjugated goat anti-rat and Cy2-conjugated goat anti-mouse (Jackson Immuno Research, 1:500) and Alexa Fluor 647 phalloidin were incubated with 3% + 1% goat serum BSA for 1 hour at room temperature in dark. Coverslips were mounted on glass slides using 50% glycerol solution and sealed with clear polish. Images were acquired by total internal reflection (TIRF) microscopy using a Nikon Eclipse Ti. Images were processed for colocalization analysis and Pearson’s correlation coefficient by NIS Elements AR 3.2 software.
Drug Treatment
5-Aza-2′deoxycytidine (5-aza-CdR; Sigma-Aldrich) and S-adenosylmethionine (SAM; NEB, Ipswich, MA) was added to cells in fresh medium daily at indicated concentrations for 3 or 5 days or equal volume DMSO or 0.005 M H2SO4 plus 10% ETOH respectively. JQ1 (250–500 nM; Bradner Lab; Dana-Farber Cancer Institute) or DMSO was added to cells for 16 hours. Gemcitabine (Sigma-Aldrich) or 3,4-Dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2 H)-isoquinolinone (DPQ; Sigma-Aldrich) treatment or equal volume vehicle control was added once for 72 hours.
For H2O2 treatment (Sigma-Aldrich), cells were plated in a 96-well plate at 2000 cells/well. Medium was changed each day to normal growth 500 μM H2O2 containing medium. Cell density was measured using 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; Fisher Scientific) according to manufacturer’s protocol.
RNA Extraction and Real-Time Quantitative PCR (QPCR)
Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA) per manufacturer’s protocol. cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and target expression was assessed using available probes, reagents, and the StepOnePlus Real-Time PCR System from Applied Biosystems, as performed previously13. Target expression (2−ΔΔCT) was normalized to endogenous reference (18S or β-actin) and reported relative to control samples. Each QPCR experiment was performed a minimum of three times and consistent trends across biologically replicated experiments were observed. The representative experiments shown in most figures are from experiments that generally best reflected the average QPCR data of these three experiments. We chose representative data over averaging the individual experiments together since significant variability often existed between experiments. This variability occurred because of the amplification of small differences present in the mRNA when values of one of the conditions are exceptionally low (as we see in the Panc-2G6 cells with AREG and EREG expression). The exceptions are data from Figs 4B,H and 5F, which showed less inter-experimental variability and were averaged in order to demonstrate significance of the findings.
Whole Genome Bisulfite Sequencing
Whole genomic DNA was isolated from cell lines using the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich). DNA was processed for high-resolution methyl-seq by the NextGen Sequencing Core at the Norris Comprehensive Cancer Center. Whole genome sequencing was done on an Illumina NextSeq and each library sequenced with paired-end runs for 150-bp read length analysis.
Read Alignment and Differential Methylation Analysis
DNA reads were aligned against GRCH37 using Bismark58 software version 0.14.3, permitting at most one mismatch, considering both sequence and bisulfite conversion mismatches. Methylation calls for each CpG were extracted using Bismark methylation extractor tool. Read alignment revealed that many reads could be mapped to both AREG and the AREG pseudogene due to high degree of homology. To allow mapping of AREG, the AREG pseudogene was masked during analysis, and vice-versa. Differential methylation analysis comparing Panc1-3D7 and Panc1-2G6 was performed using Bioconductor DSS software version 2.10.059. Differentially methylated loci (DML) were determined by >0.99 posterior probability of the difference in mean methylation levels being >0.3. Differentially methylated regions (DMR) were also detected by joining DMLs with p-value less than 0.01. DMRs have a minimum length >50 bps, minimum number of DML >3 and >50% of CpG sites with p-value < 0.01. DMRs with distance less than 100 bps were merged. DMLs and DMRs were annotated using methylKit60 version 0.9.5, where we defined the promoters as +/− 1000 bp from TSS and CpG shores +/− 2000 bp flanking each side of the CpG island.
Gene knockdown by RNAi
For siRNA treatment cells (3 × 106) were electroporated without or with 200 nM non-targeting or specific siRNA (Dharmacon, Inc.) as described previously12.
For shRNA, lentivirus was produced by combining MISSION constructs for packaging (psPAX2), envelope (pDM2G) and targeting shRNA or a non-targeting vector (pLKO.1), at a 4:2:1 ratio (Sigma Aldrich, St. Louis, MO). Polyethylenimine (PEI; Polysciences) was combined with DNA at a 3:1 ratio, and added to 70% confluent HEK 293LTV cells. Conditioned media was collected 24 and 48 hrs post transfection by centrifugation, and viral supernatant added to cells with 8 μg/ml hexadimethrine bromide (polybrene, Sigma-Aldrich). Gene expression was measured by QPCR 24 hrs following puromycin selection (2 μg/ml).
Western Blotting Analysis
Cells were washed 2x with cold PBS and collected with 400 μl cold Buffer A (10 mM Hepes pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF). Nuclei were lysed using 10% NP-40 and nuclear pellet resuspended in cold Buffer C (20 mM Hepes pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF). Nuclear extracts were collected by centrifugation, separated using 10% SDS-PAGE, transferred and immunoblotted for TDG (Genetex, GT622), and Lamin A/C (EMD Millipore). Alternatively, whole cell lysates were collected using RIPA buffer, separated using 10% or 12.5% SDS-PAGE and then immunoblotted for integrin β4 (BD Transduction Labs, #611232), laminin γ2 chain for laminin-5 (Millipore, clone D4B5) or β-actin (Sigma-Aldrich). Uncropped western blot images can be found in the supplemental information.
DNA Repair Analysis
Immuno-slot-blot analysis was performed as described previously61. Briefly, cells (70% confluent) were exposed to 30 J/m2 UV and harvested immediately or indicated times post-treatment. Cells were lysed with 10 mM Tris pH 8.0, 1 mM EDTA, 0.05% SDS, 100 μg/ml fresh proteinase K) and DNA isolated. DNA was bound to a nitrocellulose membrane using a slot blot apparatus and probed using antibody for 6-4 photoproducts (6-4 PP; Cosmobio). Data are reported as percent repair compared to the amount of initial damage (0 hr time point).
Electronic supplementary material
Acknowledgements
We would like to especially thank Dr. Min Chen for helpful advice and support. We appreciate Dr. Isabel Mellon and Nathaniel Holcomb (University of Kentucky) for providing protocols and technical assistance with the slot blotting technique for the NER assay. We thank Dr. Jay Bradner’s group (Dana-Farber Cancer Center) for providing us with JQ1. This research was supported by the National Institutes of Health Grants T32 CA165990 (BLC), R21 CA178753 (LQ, CW and KLO) and P30 CA177558 (JL) as well as the Biostatistics and Bioinformatics Shared Resource Facility of the University of Kentucky Markey Cancer Center (P30 CA177558).
Author Contributions
B.L.C. and K.L.O. conceived and designed the study and wrote the manuscript. B.L.C. performed the experiments except Fig. 1, which was performed by L.Q., J.L. and C.W. performed the analysis from the whole genome bisulfite sequencing data and approved the manuscript text.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-06351-4
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stewart RL, O’Connor KL. Clinical significance of the integrin α6β4 in human malignancies. Lab Invest. 2015;95:976–986. doi: 10.1038/labinvest.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo W, et al. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell. 2006;126:489–502. doi: 10.1016/j.cell.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 4.Yoon SO, Shin S, Mercurio AM. Ras stimulation of E2F activity and a consequent E2F regulation of integrin alpha6beta4 promote the invasion of breast carcinoma cells. Cancer Res. 2006;66:6288–6295. doi: 10.1158/0008-5472.CAN-06-0826. [DOI] [PubMed] [Google Scholar]
- 5.Yu PT, et al. The RON-receptor regulates pancreatic cancer cell migration through phosphorylation-dependent breakdown of the hemidesmosome. Int. J. Cancer. 2012;131:1744–1754. doi: 10.1002/ijc.27447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertotti A, Comoglio PM, Trusolino L. Beta4 integrin is a transforming molecule that unleashes Met tyrosine kinase tumorigenesis. Cancer Res. 2005;65:10674–10679. doi: 10.1158/0008-5472.CAN-05-2827. [DOI] [PubMed] [Google Scholar]
- 7.Lipscomb EA, Mercurio AM. Mobilization and activation of a signaling competent alpha6beta4 integrin underlies its contribution to carcinoma progression. Cancer Metastasis Reviews. 2005;24:413–423. doi: 10.1007/s10555-005-5133-4. [DOI] [PubMed] [Google Scholar]
- 8.O’Connor KL, Nguyen BK, Mercurio AM. RhoA function in lamellae formation and migration is regulated by the alpha6beta4 integrin and cAMP metabolism. J Cell Biol. 2000;148:253–258. doi: 10.1083/jcb.148.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jauliac S, et al. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nature cell biology. 2002;4:540–544. doi: 10.1038/ncb816. [DOI] [PubMed] [Google Scholar]
- 10.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 11.Chen M, Sinha M, Luxon BA, Bresnick AR, O’Connor KL. Integrin alpha6beta4 controls the expression of genes associated with cell motility, invasion, and metastasis, including S100A4/metastasin. J Biol Chem. 2009;284:1484–1494. doi: 10.1074/jbc.M803997200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen M, O’Connor KL. Integrin alpha6beta4 promotes expression of autotaxin/ENPP2 autocrine motility factor in breast carcinoma cells. Oncogene. 2005;24:5125–5130. doi: 10.1038/sj.onc.1208729. [DOI] [PubMed] [Google Scholar]
- 13.Carpenter BL, et al. Integrin alpha6beta4 Promotes Autocrine EGFR Signaling to Stimulate Migration and Invasion toward Hepatocyte Growth Factor (HGF) J Biol Chem. 2015 doi: 10.1074/jbc.M115.686873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Busser B, Sancey L, Brambilla E, Coll JL, Hurbin A. The multiple roles of amphiregulin in human cancer. Biochim Biophys Acta. 2011;1816:119–131. doi: 10.1016/j.bbcan.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 15.Riese DJ, 2nd, Cullum RL. Epiregulin: roles in normal physiology and cancer. Semin Cell Dev Biol. 2014;28:49–56. doi: 10.1016/j.semcdb.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roepstorff K, et al. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic. 2009;10:1115–1127. doi: 10.1111/j.1600-0854.2009.00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baldys A, et al. Essential role of c-Cbl in amphiregulin-induced recycling and signaling of the endogenous epidermal growth factor receptor. Biochemistry. 2009;48:1462–1473. doi: 10.1021/bi801771g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ebert M, et al. Induction and expression of amphiregulin in human pancreatic cancer. Cancer Res. 1994;54:3959–3962. [PubMed] [Google Scholar]
- 19.Zhu Z, et al. Epiregulin is Up-regulated in pancreatic cancer and stimulates pancreatic cancer cell growth. Biochem Biophys Res Commun. 2000;273:1019–1024. doi: 10.1006/bbrc.2000.3033. [DOI] [PubMed] [Google Scholar]
- 20.Yun J, et al. Gene silencing of EREG mediated by DNA methylation and histone modification in human gastric cancers. Lab Invest. 2012;92:1033–1044. doi: 10.1038/labinvest.2012.61. [DOI] [PubMed] [Google Scholar]
- 21.McEwen KR, Ferguson-Smith AC. Distinguishing epigenetic marks of developmental and imprinting regulation. Epigenetics Chromatin. 2010;3:2. doi: 10.1186/1756-8935-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thillainadesan G, et al. TGF-beta-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol Cell. 2012;46:636–649. doi: 10.1016/j.molcel.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 23.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song CX, Yi C, He C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat Biotechnol. 2012;30:1107–1116. doi: 10.1038/nbt.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barreto G, et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- 26.Rai K, et al. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dianova II, et al. XRCC1-DNA polymerase beta interaction is required for efficient base excision repair. Nucleic Acids Res. 2004;32:2550–2555. doi: 10.1093/nar/gkh567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sehgal BU, et al. Integrin β4 Regulates Migratory Behavior of Keratinocytes by Determining Laminin-332 Organization. J. Biol. Chem. 2006;281:35487–35498. doi: 10.1074/jbc.M606317200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cruz-Monserrate Z, O’Connor KL. Integrin alpha6beta4 promotes migration, invasion through Tiam1 upregulation and subsequent Rac activation. Neoplasia. 2008;10:408–417. doi: 10.1593/neo.07868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komashko VM, Farnham PJ. 5-azacytidine treatment reorganizes genomic histone modification patterns. Epigenetics. 2010;5:229–240. doi: 10.4161/epi.5.3.11409. [DOI] [PubMed] [Google Scholar]
- 31.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 32.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanno T, et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol. 2014;21:1047–1057. doi: 10.1038/nsmb.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loven J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kron KJ, Bailey SD, Lupien M. Enhancer alterations in cancer: a source for a cell identity crisis. Genome Med. 2014;6:77. doi: 10.1186/s13073-014-0077-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitz KM, et al. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol Cell. 2009;33:344–353. doi: 10.1016/j.molcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 38.Le May N, Fradin D, Iltis I, Bougneres P, Egly JM. XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol Cell. 2012;47:622–632. doi: 10.1016/j.molcel.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 39.Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17(Suppl 5):v7–12. doi: 10.1093/annonc/mdj941. [DOI] [PubMed] [Google Scholar]
- 40.Schafer A, Schomacher L, Barreto G, Doderlein G, Niehrs C. Gemcitabine functions epigenetically by inhibiting repair mediated DNA demethylation. PLoS One. 2010;5:e14060. doi: 10.1371/journal.pone.0014060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Z, et al. Gadd45a promotes DNA demethylation through TDG. Nucleic Acids Res. 2015;43:3986–3997. doi: 10.1093/nar/gkv283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hajkova P, et al. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cortellino S, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ciccarone F, et al. Poly(ADP-ribosyl)ation acts in the DNA demethylation of mouse primordial germ cells also with DNA damage-independent roles. PLoS One. 2012;7:e46927. doi: 10.1371/journal.pone.0046927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DesRochers TM, et al. The 3D tissue microenvironment modulates DNA methylation and E-cadherin expression in squamous cell carcinoma. Epigenetics. 2012;7:34–46. doi: 10.4161/epi.7.1.18546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JG. Methylation patterns of the E-cadherin 5’ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem. 2000;275:2727–2732. doi: 10.1074/jbc.275.4.2727. [DOI] [PubMed] [Google Scholar]
- 49.Aran D, Hellman A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell. 2013;154:11–13. doi: 10.1016/j.cell.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 50.Aran D, Sabato S, Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013;14:R21. doi: 10.1186/gb-2013-14-3-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamm CA, et al. Microenvironment alters epigenetic and gene expression profiles in Swarm rat chondrosarcoma tumors. BMC Cancer. 2010;10:471. doi: 10.1186/1471-2407-10-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Denko N, et al. Epigenetic regulation of gene expression in cervical cancer cells by the tumor microenvironment. Clin Cancer Res. 2000;6:480–487. [PubMed] [Google Scholar]
- 53.Shen L, et al. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153:692–706. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma DK, Guo JU, Ming GL, Song H. DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle. 2009;8:1526–1531. doi: 10.4161/cc.8.10.8500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vidi PA, et al. Interconnected contribution of tissue morphogenesis and the nuclear protein NuMA to the DNA damage response. J Cell Sci. 2012;125:350–361. doi: 10.1242/jcs.089177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Du J, Ma Y, Ma P, Wang S, Fan Z. Demethylation of epiregulin gene by histone demethylase FBXL11 and BCL6 corepressor inhibits osteo/dentinogenic differentiation. Stem Cells. 2013;31:126–136. doi: 10.1002/stem.1255. [DOI] [PubMed] [Google Scholar]
- 57.O’Connor KL, Shaw LM, Mercurio AM. Release of cAMP gating by the alpha6beta4 integrin stimulates lamellae formation and the chemotactic migration of invasive carcinoma cells. J. Cell Biol. 1998;143:1749–1760. doi: 10.1083/jcb.143.6.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu H, et al. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akalin A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mellon I, Hock T, Reid R, Porter PC, States JC. Polymorphisms in the human xeroderma pigmentosum group A gene and their impact on cell survival and nucleotide excision repair. DNA Repair (Amst) 2002;1:531–546. doi: 10.1016/S1568-7864(02)00053-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.