

Abstract
Sulfonylureas are the most commonly-used second line drug class for treating type 2 diabetes mellitus. While the cardiovascular safety of sulfonylureas has been examined in a number of trials and non-randomized studies, little is known of their specific effects on sudden cardiac arrest and related serious arrhythmic outcomes. This knowledge gap is striking, as persons with diabetes mellitus are at increased risk of sudden cardiac arrest. This review explores sulfonylureas’ influence on the risk of serious arrhythmias, with specific foci on ischemic preconditioning, cardiac excitability, and serious hypoglycemia as putative mechanisms. Elucidating the relationship between individual sulfonylureas and serious arrhythmias is critical, especially as the diabetes epidemic intensifies and sudden cardiac arrest incidence increases in persons with diabetes.
Keywords: sudden cardiac death, cardiac arrhythmias, sulfonylurea compounds, diabetes mellitus type 2, epidemiology, KATP channels
Introduction
Sudden cardiac arrest (SCA; see Glossary) is a global public health problem. Of the 17 million worldwide cardiovascular deaths per year, about half are SCAs.[1] Diabetes mellitus (DM), a major risk factor for SCA, is an uncontrolled epidemic affecting >8% of the world’s population.[2] Given this, there is currently very strong interest in minimizing SCA risk overall[3] and in persons with DM.[4] Antidiabetic sulfonylurea drugs have been a longstanding treatment for type 2 DM (T2DM); agents of this pharmacologic class are still the most commonly used second line therapy (i.e., add-on to metformin).[5] Recent meta-analyses[6–8] have reinvigorated the debate of sulfonylurea’s cardiovascular effects and potential associations with all-cause and cardiovascular death.[9] Yet, there has been little specific focus on arrhythmogenicity. Major ongoing trials such as Glycemia Reduction Approaches in Diabetes (GRADE) and Cardiovascular Outcome Trial of Linagliptin Versus Glimepiride in Type 2 Diabetes (CAROLINA) will not provide data on the arrhythmogenic effects of sulfonylureas. The ongoing Thiazolidinediones or Sulfonylureas and Cardiovascular Accidents Intervention (TOSCA.IT) trial is in fact examining SCA, but only as part of a composite secondary outcome and will not elucidate differences in risk among individual sulfonylureas. Therefore, whether sulfonylureas increase, decrease, or do not affect SCA risk is a major knowledge gap in the treatment persons with T2DM. This review synthesizes what is known about sulfonylurea-associated SCA, explores potential underlying mechanisms, sheds light on knowledge gaps, and supports calls for research to fill the void.
Sudden cardiac arrest, an arrhythmic sequela, in the setting of diabetes mellitus
SCA is the sudden cessation of cardiac activity in which the affected individual becomes unresponsive, with no normal breathing or signs of circulation.[10] Coronary heart disease is the most common pathology underlying SCA.[11] Despite advances in coronary heart disease treatment, SCA accounts for about 450,000 annual deaths in the United States (US),[12] nearly 100,000 of which are in persons with DM.[13] The pathophysiology of SCA is complex, requiring an underlying substrate and a transient event that induces electric instability and a ventricular arrhythmia (VA) followed by hemodynamic collapse.[14]
DM confers a 2- to 4-fold increased risk of SCA.[13] This may be due to a combination of atherosclerotic, thrombotic, neural, and other factors.[15] Interrelated putative determinants of SCA risk in persons with DM are shown in Figure 1; the relative importance of each determinant is unknown, although recent opinion has emphasized the roles of coronary heart disease, myocardial dysfunction, and electrical abnormalities,[16] while downplaying cardiac autonomic dysfunction.[17] The incidence of SCA in persons with DM is approximately 3 per 1,000 person-years[16] and is increasing,[18] the cause of which is unknown.[19] Among persons with DM and clinically-recognized heart disease, it is approximately 14 per 1,000 person-years.[16] The ongoing diabetes epidemic, coupled with this increase in SCA incidence, represents a major and growing public health concern.
Figure 1.
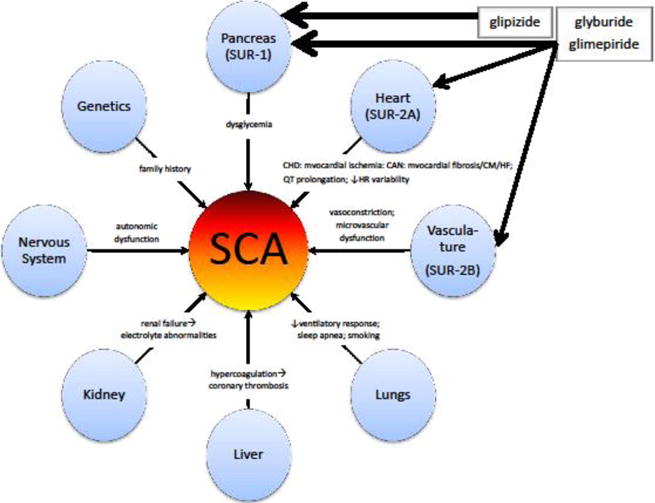
Potential mechanisms of sudden cardiac arrest in persons with diabetes mellitus. This conceptual model was developed from literature findings and in consultation with field experts. The increased risk of sudden cardiac arrest in persons with diabetes is multifactorial. Sulfonylureas may affect this risk primarily via actions at the pancreas, heart, and/or smooth muscle. The presence and width of arrows emanating from the sulfonylurea boxes is broadly indicative of their actions and selectivity. While numerous other drugs may influence the risk of sudden cardiac arrest, this conceptual model simply highlights the drug class discussed herein. CHD = coronary heart disease; CAN = cardiac autonomic neuropathy; CM = cardiomyopathy; HF = heart failure; HR = heart rate; SCA = sudden cardiac arrest; SUR = adenosine triphosphate-binding cassette protein sulfonylurea receptor.
In persons with DM and coronary heart disease, the reduction of mortality from SCA is a major challenge[16] such that cornerstones of therapy in this patient population include β-adrenergic blockers, angiotensin converting enzyme inhibitors, and statins—pharmacologic classes intended to minimize SCA risk.[15] Attention should also be paid to avoiding or discontinuing drugs that may exacerbate mechanisms presented in Figure 1. Obvious examples would include drugs associated with electrocardiographic QT interval prolongation and/or torsade de pointes. An important approach, if available, would be to tailor oral antidiabetic drug therapy to minimize the risk of serious arrhythmia.
Sulfonylureas―a maligned yet commonly-prescribed drug class
Sulfonylureas increase plasma insulin concentrations and are therefore utilized to treat T2DM. Mechanistically, these drugs bind to sulfonylurea receptors on pancreatic β-cells and block the inflow of potassium.[20] As the flow of potassium within the β-cell goes to zero, the cell membrane depolarizes and no longer prevents the diffusion of calcium into the cytosol.[20] The increased inward flow of calcium causes contraction of filaments of actomyosin responsible for the exocytosis of insulin.[20] Sulfonylureas are the most commonly-used add-on to metformin[5] and their use is increasing in the US.[21] Sulfonylureas are at least initially effective, generally well tolerated, and inexpensive; however, they lack durability in their glycosylated hemoglobin (HbA1C)-lowering effects. Further, sulfonylureas compete with an influx of new oral antidiabetic drugs,[5] are deemphasized in some guidelines[22] due to side effects including weight gain and serious hypoglycemia,[23] and have uncertain cardiovascular risks.[24–27] But, given their reliable glucose-lowering effects with low cost and demonstrated ability to reduce microvascular complications,[28] sulfonylureas remain important in the armamentarium of T2DM treatments.[29]
To help inform second line antidiabetic therapy prescribing decisions, it is critical to understand the extrapancreatic effects of sulfonylureas―in particular their impact on arrhythmias. The arrhythmogenicity of drugs has been of major concern to clinicians, patients, caregivers, federal regulators, and drug developers. In fact, drug-induced SCA and VA have been responsible for the market withdrawal of more drugs in recent years than any other adverse drug reaction.[30] Yet, the potential for oral antidiabetic drugs to affect arrhythmogenicity―in a T2DM population already primed for or having cardiovascular disease[31,32]―is not widely considered. Given their common use, biologic basis for putative cardiac effects, and preliminary data on arrhythmia-related endpoints in animals in humans, this review paper will explore the sulfonylureas’ influence on serious arrhythmia risk. Subsequent sections will focus on effects largely or solely mediated by potassium channel antagonism, including ischemic preconditioning (IPC) and shortening of action potential duration, cardiac excitability and QT interval prolongation, and physiologic effects of serious hypoglycemia.
Proarrhythmic and antiarrhythmic actions of sulfonylureas
The primary site of action of sulfonylureas is the adenosine triphosphate-sensitive potassium channels (KATP) located in the pancreatic β-cell membrane; blockade of these channels stimulates insulin secretion.[33] KATP channels are also found at differing densities in cardiac myocytes, smooth muscle myocytes, vascular endothelium, and the brain.[34] While the consequences of extrapancreatic KATP channel antagonism are not fully defined, they may at least partially explain any cardiovascular effects of sulfonylureas.[35]
Potential mechanisms influencing arrhythmogenic risk
Any impact of sulfonylureas on arrhythmogenic risk may be driven by a given agent’s ability to impair or abolish the heart’s innate protective IPC mechanism, effects on cardiac excitability, the likelihood for causing serious hypoglycemia, or other mechanisms. Elucidating the interplay among these mechanisms and their component actions is complicated by a number of factors, including that: a) different mechanistic actions may oppositely influence arrhythmogenicity; b) pathophysiologic changes in DM may modify such actions; c) data supporting some actions are sparse and occasionally conflicting; and d) actions likely differ by individual sulfonylurea rather than being class effects. As described below, mechanisms exist whereby sulfonylureas could both cause and prevent serious arrhythmias.[36,37]
Ischemic preconditioning and cardiac excitability mediated by myocardial KATP blockade
IPC is a self-protective mechanism that allows the heart to minimize a potentially lethal ischemic insult.[38] It is mediated by typically-closed myocardial KATP channels that open in response to metabolic stress, such as ischemia.[39] IPC occurs in two phases, a classical early phase offering peak cardioprotection for an initial 2–3 hour period, and a so-called “second window of protection” beginning about 10 hours later and lasting up to 3 days.[40,41] Some sulfonylureas may attenuate or abolish IPC via antagonism of myocardial KATP channels (Box 1) and therefore lead to larger infarct sizes during acute ischemia (Figure I, Box 1). Further, their blockade of these channels may prevent the shortening of action potential duration, thereby increasing intracellular calcium loading that may precipitate deleterious delayed afterdepolarization-mediated arrhythmias. However, reentrant arrhythmias may be minimized[37] and in turn prevent SCA.[38] It has been hypothesized that the exact mechanisms underlying IPC effects are distinct from those on arrhythmia, such that a given cardioactive sulfonylurea could abolish IPC but retain its impact on arrhythmias.[42] That said, it has been suggested that IPC is already blunted or absent in persons with DM.[43] Given that myocardial KATP channels are closed under physiologic conditions, arrhythmic and/or IPC effects arising from their antagonism may be most relevant in persons with cardiovascular disease[33]―a common comorbidity in T2DM.
Box 1. Sulfonylurea’s blockade of myocardial KATP channels.
Plasma sulfonylurea concentrations in chronic users are sufficient to antagonize myocardial KATP channels,[92] but not all agents do so to the same extent.[40] The effect (or lack thereof) of an individual sulfonylurea on IPC and/or cardiac excitability may be driven by its selectivity for pancreatic vs. myocardial KATP channels. Chlorpropamide, gliclazide, glipizide, and tolbutamide are highly (>100X) selective for pancreatic channels, while glimepiride and glyburide are moderately (approximately 10X) selective.[35] However, the relationship between selectivity and cardiovascular effects is not straightforward. First, a sulfonylurea’s antagonism of myocardial KATP channels is modulated by magnesium adenosine diphosphate.[35] Second, differences may exist in a sulfonylurea’s preference for blocking mitochondrial vs. sarcolemmal myocardial KATP channels,[93] each of which has been debated as mediating IPC effects.[36,94] While the relationship between KATP channels and general cardiovascular disease is beyond the scope of this review, Nichols et al provide a detailed overview for interested readers.[39]
Figure I, Box 1.
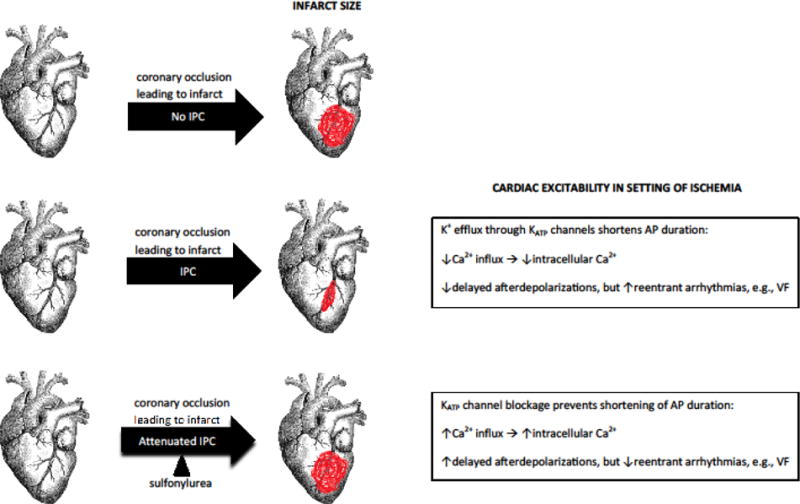
Myocardial KATP-mediated effects on infarct size and cardiac excitability in the setting of sulfonylurea exposure. Ischemic preconditioning, an innate protective mechanism of the heart, minimizes infarct size. This protection may be blunted by some sulfonylureas via blockade of myocardial KATP channels. Channel blockade also affects cardiac excitability. This figure was adapted from Brady PA & Terzic A (J Am Coll Cardiol 1998;31:950–956), with permission from Dr. Brady. The graphic of the heart, downloaded from the openclipart.org media repository, is in the public domain (creative commons license zero 1.0). AP = action potential; IPC = ischemic preconditioning; VF = ventricular fibrillation.
The effects of sulfonylureas on IPC have been examined in numerous animal models (Table 1). The designs and methods of these studies both across and within species are heterogeneous, leading to some challenges in interpretation. That being said, many studies support the hypothesis that glyburide attenuates or abolishes IPC. Interestingly, in a recent study of diabetic rat hearts resistant to IPC, glimepiride was able to elicit cardioprotection.[44] Studies of sulfonylurea effects on IPC in humans are equally difficult to interpret, but also generally demonstrate that glyburide abolishes while glimepiride preserves IPC; very little is known of glipzide’s effects (Table 2). Despite this body of work, it is unknown whether long-term sulfonylurea exposure in persons with T2DM has clinically meaningful effects on IPC or related endpoints.[45]
Table 1.
Effects of sulfonylureas on ischemic preconditioning in animalsa
| Animal model used | First author and publication year | Sulfonylurea | Outcome(s) | Major finding(s) | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|
| Gly | Glim | Glip | Glip | Oth | |||||
| Evidence for abolished IPCb | |||||||||
| dog | Auchampach, J.A. 1992 | x | Infarct size, hemodynamics, myocardial blood flow, blood glucose | Gly abolished IPC | [95] | ||||
| dog | Gross, G.J. 1992 | x | Infarct size, mortality, hemodynamics, regional myocardial blood flow, myeloperoxidase measurements, blood glucose | Gly abolished IPC | [96] | ||||
| dog | Grover, G.J. 1992 | x | Infarct size, hemodynamics, regional myocardial blood flow | Gly abolished IPC | [97] | ||||
| dog | Yao, Z. 1993 | x | Infarct size, mortality, hemodynamics, regional myocardial blood flow | Gly abolished IPC | [98] | ||||
| pig | Schulz, R. 1994 | x | Infarct size, hemodynamics, blood flow, MAP duration, regional myocardial function and metabolism | Gly abolished IPC, but did not impact infarct size | [99] | ||||
| pig | Schulz, R. 1999 | x | Infarct size, hemodynamics | Gly abolished IPC | [100] | ||||
| rabbit | Bernardo, N.L. 1999 | x | Infarct size, mortality, blood gases, hemodynamics, MAP duration | Gly abolished IPC | [101] | ||||
| rabbit | Bernardo, N.L. 1999 | x | Infarct size, mortality, hemodynamics, MAP duration, blood glucose | Gly abolished IPC | [102] | ||||
| rabbit | Elliott, G.T. 1996 | x | Infarct size, hemodynamics | Gly abolished IPC | [103] | ||||
| rabbit | Flynn, D.M. 2005 | x | x | Infarct size, hemodynamics, blood glucose | Gly, but not glip, attenuated IPC | [104] | |||
| rabbit | Hoag, J.B. 1997 | x | Infarct size, mortality, blood gases, hemodynamics | Gly abolished IPC | [105] | ||||
| rabbit | Horimoto, H. 2002 | x | x | Infarct size, pressures, MAP duration, coronary flow | Gly, but not glim, abolished IPC | [106] | |||
| rabbit | Krenz, M. 2001 | x | Infarct size, hemodynamics | Gly abolished IPC | [107] | ||||
| rabbit | Munch-Ellingsen, J. 1996 | x | Infarct size | Gly abolished IPC | [108] | ||||
| rabbit | Nieszner, E. 2002 | x | x | Infarct size, MAP, blood glucose | Gly, but not glim, abolished IPC | [109] | |||
| rabbit | Ohno, Y. 1997 | x | Infarct size, mortality, hemodynamics, | Gly abolished IPC | [110] | ||||
| rabbit | Pain, T. 2000 | x | Infarct size, hemodynamics | Gly abolished IPC | [111] | ||||
| rabbit | Toombs, C.F. 1993 | x | Infarct size, hemodynamics, blood glucose | Gly abolished IPC | [112] | ||||
| rabbit | Walsh, R.S. 1994 | x | Infarct size | Gly abolished IPC | [113] | ||||
| rabbit | Wang, S. 2001 | x | Infarct size, hemodynamics | Gly abolished IPC | [114] | ||||
| rat | D’Souza, S.P. 2003 | x | Infarct size, coronary flow | Gly abolished IPC | [115] | ||||
| rat | Iwai, T. 2000 | x | Cardiac function, coronary flow, metabolite release, myocardial energy metabolites, mitochondrial oxygen consumption | Gly abolished IPC | [116] | ||||
| rat | Joyeux, M. 1998 | x | Infarct size, hemodynamics | Gly abolished IPC | [117] | ||||
| rat | Maddock, H.L. 2004 | x | x | Infarct size, mitochondrial membrane potential | Gly, but not glic, abolished IPC | [118] | |||
| rat | Mocanu, M.M. 2001 | x | x | Infarct size, mitochondrial membrane potential | Gly, but not glim, abolished IPC | [119] | |||
| rat | Qian, Y.Z. 1996 | x | Infarct size, hemodynamics | Gly abolished IPC | [120] | ||||
| rat | Schultz, J.E. 1996 | x | Infarct size, hemodynamics | Gly abolished IPC | [121] | ||||
| rat | Tavackoli, S. 2004 | x | Infarct size | Gly abolished IPC | [122] | ||||
| rat | Wang, G.Y. 2001 | x | Infarct size, hemodynamics | Gly abolished IPC | [123] | ||||
| rat | Wu, G.T. 2007 | x | x | x | Infarct size, hemodynamics | Gly, but not glim, abolished IPC; glic attenuated IPC | [124] | ||
| rat | Ye, Y. 2008 | x | x | Infarct size | Gly, but not glim, abolished IPC | [125] | |||
| Evidence for preserved IPCb | |||||||||
| rabbit | Sato, T. 2006 | x | x | x | mitoKATP channel activity, membrane currents | Glim, glic, and tolbutamide had no effect on mitoKATP channel; these agents did not interfere with IPC in clinical setting | [126] | ||
| rabbit | Takano, H. 2000 | x | Infarct size, hemodynamics, blood glucose, regional myocardial function | Gly did not abolish late IPC | [127] | ||||
| rabbit | Thornton, J.D. 1993 | x | Infarct size, hemodynamics, blood glucose | Gly did not abolish IPC, but still increased infarct size | [128] | ||||
| rat | Fralix, T.A. 1993 | x | Time to ischemic contracture, ionic alterations, recovery of function | Gly did not abolish IPC | [129] | ||||
| rat | Grover, G.J. 1993 | x | Global ischemia, time to contracture | Gly did not abolish IPC | [130] | ||||
| rat | Hausenloy, D.J. 2013 | x | Infarct size, blood glucose, glycosylated hemoglobin | Glim elicited IPC in an IPC- resistant heart | [44] | ||||
| rat | Lu, H. 1993 | x | Infarct size, hemodynamics | Gly did not attenuate IPC | [131] | ||||
Abbreviations: IPC = ischemic preconditioning; glic = gliclazide; glim = glimepiride; glip = glipizide; gly = glyburide (glibenclamide); mitoKATP = mitochondrial adenosine triphosphate-sensitive potassium channel; MAP = monophasic action potential; oth = other antidiabetic sulfonylurea
Primarily for glyburide, as the most commonly studied sulfonylurea
Table 2.
Effects of sulfonylureas on ischemic preconditioning in humansa
| First author and publication year | Sulfonylurea | Study population | Outcome(s) | Major finding(s) | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|
| Gly | Glim | Glip | Glic | Oth | |||||
| Evidence for abolished IPCb | |||||||||
| Bilinska, M. 2007 | x | x | Persons with chronic stable angina, N = 64 (N = 47 with T2DM) | ST segment depression, time to onset of exercise test positivity, exercise duration, rate blood -pressure product | Gly abolished IPC, Glic partially preserved IPC | [132] | |||
| Cleveland, J.C. 1997 | x | x | Right atrial appendages from persons with stable CHD undergoing coronary artery surgery, N = 48 trabeculae from N = 16 persons (5 persons exposed to gly, 1 exposed to glip) | Recovery of contractile function/developed force | SUs (considered together) abolished IPC | [133] | |||
| Ferreira, B.M. 2005 | x | Persons with chronic stable angina and a coronary artery obstruction, N = 40 (N = 20 with DM) | ST segment depression, exercise duration, rate- blood pressure product, HR, BP | Gly abolished IPC | [134] | ||||
| Klepzig, H. 1999 | x | x | Persons with stable CHD undergoing PTCA, N = 45 | ST segment shift, time to onset of angina pain, HR, arterial BP | Gly abolished IPC, Glim preserved IPC | [135] | |||
| Lee, T.M. 2002 | x | Persons with chronic stable angina and a coronary artery obstruction undergoing angioplasty, N = 56 | ST segment shift, cardiac pain severity, myocardial lactate | Gly abolished IPC | [136] | ||||
| Lee, T.M. 2003 | x | x | Persons with chronic stable angina and a coronary artery obstruction, N = 43 (N = 23 with DM) | ST segment shift, rate-blood pressure product, cardiac pain, blood glucose, myocardial lactate | Gly, but not glim, abolished IPC | [137] | |||
| Loubani, M. 2005 | x | x | Right atrial appendages from persons undergoing CABG or aortic valve replacement, N = 160 specimens from N = 31 persons | Tissue injury, tissue viability | Gly, but not glic, abolished IPC | [138] | |||
| Ovunc, K. 2000 | x | Persons with T2DM and chronic stable angina with a coronary artery obstruction, N = 18 | ST segment depression, blood glucose, time to pain, rate-blood pressure product, HR, BP | Gly abolished IPC | [139] | ||||
| Speechly-Dick, M.E. 1995 | x | Right atrial appendages from persons with chronic stable angina undergoing cardiac bypass, N = 47 specimens from N = 47 persons | Developed force, time to onset of contracture, peak contracture | Gly abolished IPC | [140] | ||||
| Tomai, F. 1994 | x | Persons with chronic stable angina and a coronary artery obstruction undergoing angioplasty, N = 20 | ST segment shift, cardiac pain severity, blood glucose | Gly abolished IPC | [141] | ||||
| Tomai, F. 1999 | x | Persons with chronic stable angina and CHD, N = 26 | ST segment depression, exercise duration, blood glucose, HR, BP | Gly abolished IPC | [142] | ||||
| Evidence for preserved IPCb | |||||||||
| Bogaty, P. 1998 | x | Persons with chronic stable angina and significant CHD, N = 24; persons with chronic stable angina and T2DM, N = 10 | ST segment depression, left ventricular wall motion score, rate-blood pressure product, time to angina | Gly preserved IPC | [143] | ||||
| Correa, S.D. 1997 | x | Persons with CHD and a coronary artery obstruction, N = 10 in exercise protocol, N = 12 in pacing protocol | ST depression, exercise time, rate-blood pressure product, angina, blood glucose, HR | Gly preserved IPC | [144] | ||||
Abbreviations: BP = blood pressure; CABG = coronary artery bypass graft; CHD = coronary heart disease; DM = diabetes mellitus; HR = heart rate; PTCA = percutaneous transluminal coronary angioplasty; SU = sulfonylurea; T2DM = type 2 diabetes mellitus
Primarily for glyburide, as the most commonly studied sulfonylurea
The effects of sulfonylureas on cardiac excitability have been extensively studied in animals (Table 3). Much like with studies of IPC, designs and methods vary widely. Findings conflict as to whether or not individual agents consistently affect measures of excitability and therefore serious arrhythmia risk. Many fewer studies have been conducted in humans (Table 4). In the late 1980s, Pogátsa et al reported an antiarrhythmic effect of glyburide in digoxin-treated persons with T2DM;[46] nearly 15 years later, Aroson et al reported similar findings in the setting of acute heart failure decompensation.[47] Lomuscio et al reported that glyburide’s antiarrhythmic actions may be limited to acute ischemic periods.[48] The latter two studies implicated KATP channel blockade as the potential mechanism underlying this observation. Data in a non-ischemic setting and for other sulfonylureas are severely lacking.
Table 3.
Effects of sulfonylureas on myocardial KATP-mediated cardiac excitability in animalsa
| Animal model used | First author and publication year | Antidiabetic SU studied | Outcome(s) | Major finding(s) | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|
| Glip | Gly | Glim | Glic | Oth | |||||
| Evidence for antiarrhythmic effectsb | |||||||||
| dog | Billman, G.E. 1993 | x | Susceptibility to VF in post-MI setting | Gly significantly reduced the incidence of VF induced by the combination of exercise and myocardial ischemia | [145] | ||||
| dog | Billman, G.E. 1998 | x | Susceptibility to VF in post-MI setting | Gly prevented ischemically-induced VF without altering the hemodynamic response to coronary occlusion | [146] | ||||
| guinea pig | Gwilt, M. 1992 | x | x | K+ efflux and VF incidence in setting of myocardial ischemia | Gly and glip suppressed VF without reducing ischemic K+ loss | [147] | |||
| guinea pig | Tosaki, A. 1994 | x | Incidence of VF and VT | Gly dose-dependently reduced the incidence of reperfusion-induced VF and VT | [148] | ||||
| multiple | Pogatsa, G. 1988 | x | x | Incidence of ventricular ectopic beats, duration of temporary VF in rabbit and ratc | Gly reduced, yet carbutamide and tolbutamide enhanced, strophanthidin-evoked ventricular ectopic beats and VF | [46] | |||
| multiple | Wilde, A.A. 1990 | x | Action potential parameters in guinea pig myocytes, extracellular accumulation of K+ and H+ in isolated ischemic hearts of guinea pig, rat, and rabbit | Gly prevented hypoxia induced shortening of the - action potential and therefore may have antiarrhythmic properties | [149] | ||||
| multiple | Zhang, H.L. 1991 | x | x | Mortality, incidence and duration of VT and VF, and action potential duration in rat and guinea pig | Gly and tolbutamide reduced ouabain-induced arrhythmias in a dose-dependent fashion | [150] | |||
| rabbit | Barrett, T.D. 1998 | x | Monophasic action potential duration and VF incidence in setting of ischemia | Gly significantly reduced the incidence of VF, although the effect was not dose related | [151] | ||||
| rabbit | Bellemin-Baurreau, J. 1994 | x | Incidence of VF, VT, and ventricular premature beats | Gly reduced programmed electrical stimulation-induced VF | [152] | ||||
| rabbit | Black, S.C. 1993 | x | Incidence of VF | Gly prevented 4β-phorbol,12,13-dibutyrate-induced VF | [153] | ||||
| rabbit | Chi, L. 1993 | x | Incidence of VF | Gly significantly reduced the incidence of VF associated with pinacidil | [154] | ||||
| rabbit | Dhein, S. 2000 | x | Epicardial activation-recovery interval and VF in isolated hearts, in the course of regional ischemia and reperfusion | Gly reduced shortening of activation-recovery intervals in the ischemic area and exerts an antiarrhythmic effect | [155] | ||||
| rabbit | Le Grand, B. 1992 | x | Action potential duration, suppression of sustained tachycardia | Tolbutamide suppressed reproducible tachycardia induced in the presence of nicorandil | [156] | ||||
| rat | Baczko, I. 1997 | x | Incidence of VF, VT, and other arrhythmias; arrhythmia score, in setting of ischemia | Gly significantly decreased the incidence of sustained VF and markedly increased survival during reperfusion | [157] | ||||
| rat | Ballagi-Pordany, G. 1990 | x | x | x | x | Incidence of ventricular ectopic beats, duration of transitional VF in setting of ischemia | Gly and glip decreased the number of ventricular ectopic beats and duration of transitional VF in a dose-dependent manner; Glic, tolbutamide, and carbutamide increased these parameters | [158] | |
| rat | Bril, A. 1992 | x | Incidence and duration of VT and VF | Gly reduced the duration of VF during reperfusion | [159] | ||||
| rat | Csonka, C. 2003 | x | Cardiac function and spontaneous defibrillation | Gly exerted a defibrillatory action and improved post-VF cardiac function | [160] | ||||
| rat | D’Alonzo, A.J. 1994 | x | VF, VT, and arrhythmia scores in setting of ischemia | Gly markedly reduced ischemic VT and VF | [161] | ||||
| rat | El-Reyani, N.E. 1999 | x | x | Incidence of arrhythmias, including reversible VF, irreversible VF, VT, and other arrhythmias | Gly and glim significantly decreased the incidence of irreversible VF and increase survival rate | [162] | |||
| rat | Hatcher, A.S. 2011 | x | x | VF incidence, time to VF, QT interval | Gly and glim delayed the onset of VF triggered by CCCP | [163] | |||
| rat | Lepran, I. 1996 | x | Incidence of VF and VT, sudden cardiac death | Gly significantly decreased incidence of life-threatening arrhythmias and increased survival | [164] | ||||
| rat | Rees, S.A. 1995 | x | Incidence of VF and sustained VF | Gly reduced the incidence of sustained VF | [165] | ||||
| rat | Tosaki, A. 1993 | x | Incidence of VF and VT | Gly reduced the incidence of reperfusion-induced VF and VT | [166] | ||||
| rat | Vajda, S. 2007 | x | Survival and incidence of arrhythmias during myocardial ischemia | Gly decreased the incidence of irreversible VF and improved survival during acute MI | [167] | ||||
| rat | Wirth, K.J. 1999 | x | Incidence and duration of VF in setting of ischemia | Gly reduced VF duration, but not its incidence | [168] | ||||
| rat | Wolleben, C.D. 1989 | x | x | VF incidence, time to VF, rate of VF | Gly and tolbutamide prevented ischemia-induced fibrillation | [169] | |||
| Evidence for proarrhythmogenic effectsb | |||||||||
| dog | D’Alonzo, A.J. 1994 | x | Ectopy, action potential duration | Gly exacerbated cesium chloride-induced arrhythmias | [170] | ||||
| dog | D’Alonzo, A.J. 1995 | x | VF thresholds | Gly caused a nonsignificant reduction in VF threshold, demonstrating a propensity toward proarrhythmic activity | [171] | ||||
| guinea pig | Cole, W.C. 1991 | x | Action potential duration, arrhythmias, mechanical function in setting of ischemia | Gly elicited extrasystoles and VT | [172] | ||||
| guinea pig | Testai, L. 2010 | x | x | Maximal change in QTc from basal value in anesthetized animals | Gly and tolbutamide caused a potentiating trend on the effects of thioridazine on QTc prolongation | [173] | |||
| rat | Bernauer, W. 1997 | x | Incidence and severity of arrhythmias | Gly intensified ischemic arrhythmias; in hearts without coronary occlusion, gly had proarrhythmic effects | [174] | ||||
| rat | Iskit, A.B. 2007 | x | Mortality, VT incidence and duration, VF incidence and duration | Gly prevented the antiarrhythmic effects of endotoxin | [175] | ||||
| rat | Takahashi, N. 2003 | x | Recovery of cardiac performance, CK release, incidence of VA | Gly increased incidence of reperfusion induced VT - in diabetic rats | [176] | ||||
| sheep | del Valle, H.F. 2001 | x | Epicardial monophasic action potential; incidence of VF, VT, and other arrhythmias in setting of ischemia | Gly intensified the appearance of reperfusion-induced arrhythmias and had proarrhythmic action during ischemia | [177] | ||||
| Evidence for equivocal effectsd | |||||||||
| dog | Chi, L. 1989 | x | Mortality, incidence of VF | Gly failed to prevent acute VF | none | ||||
| dog | Vegh, A. 1993 | x | Incidence of ventricular premature beats, VT, and VF | Gly caused more ventricular premature beats and VT, but did not modify VF during ischemia or reperfusion | [178] | ||||
| dog | Yao, Z. 1993 | x | Incidence of VF | Gly had no effect of VF incidence | [98] | ||||
| dog | Zhu, B.M. 2003 | x | Programmed electrical stimulation-induced arrhythmia scores in setting of MI | Gly had no effect on arrhythmias | [179] | ||||
| guinea pig | Pasnani, J.S. 1992 | x | Action potential duration, incidence and duration of arrhythmias | Gly abolished reperfusion re-entry arrhythmias, but potentiated oscillatory afterpotentials and triggered arrhythmias with characteristics of oscillatory afterpotentials | [180] | ||||
| pig | Harbinson, M.T. 2000 | x | Frequency patterns of VF, defibrillation threshold, QTc interval | Gly caused small reduction in early VF frequency which was not sustained nor produced other important effects; gly did not cause a significant change in defibrillating current | [181] | ||||
| rabbit | Das, B. 2001 | x | Incidence and duration of VF, VT, and other arrhythmias, arrhythmia severity | Tolbutamide offered no protection against arrhythmias | [182] | ||||
| rabbit | Takano, H. 2000 | x | Incidence of VF | Gly had no effect of VF incidence | [127] | ||||
| rat | Ferdinandy, P. 1995 | x | Incidence of VF | Gly did not change the incidence of VF | [183] | ||||
| rat | Lu, H. 1993 | x | Incidence, onset, and duration of VF | Gly had no beneficial or adverse effects on reperfusion-induced arrhythmias in non-preconditioned animals | [131] | ||||
Abbreviations: CCCP = carbonyl cyanide m-chlorophenyl hydrazone; CK = creatine kinase; IPC = ischemic preconditioning; glic = gliclazide; glim = glimepiride; glip = glipizide; gly = glyburide (glibenclamide); MI = myocardial infarction; mitoKATP = mitochondrial adenosine triphosphate-sensitive potassium channel; oth = other antidiabetic sulfonylurea; QTc = corrected QT interval; SU = sulfonylurea; VA = ventricular arrhythmia; VF = ventricular fibrillation; VT = ventricular tachycardia
Primarily for glyburide, as the most commonly studied sulfonylurea
Also examined humans
Table 4.
Effects of sulfonylureas on myocardial KATP-mediated cardiac excitability in humansa
| First author and publication year | Sulfonylurea | Study population | Outcome(s) | Major finding(s) | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|
| Gly | Glim | Glip | Glic | Oth | |||||
| Evidence for antiarrhythmic effectsb | |||||||||
| Aronson, D. 2003 | x | x | Persons admitted for decompensated HF with (N = 100) and without DM (N = 107); among the former N = 10 treated with gly, N = 15 treated with glip, N = 66 treated with other antidiabetic drug or untreated | PVCs, ventricular pairs, repetitive ventricular beats, VT | SUs (considered together) reduced ventricular pairs, repetitive ventricular beats, and VT | [47] | |||
| Cacciapuoti, F. c 1991 | x | Men with T2DM and CHD with evidence of PVCs and/or NSVT induced by ischemia, N = 19 (N = 10 treated with gly) | PVCs, NSVT, blood glucose | Gly reduced PVCs and NSVT | [77] | ||||
| Danchin, N. 2005d | Unspecified | Persons with an MI with (N = 487) and without (N = 1,833) DM; among the former, N = 215 treated with a SU | In-hospital mortality, in-hospital complications (including VF) | Incidence of VF was lower in SU users than non-users | [80] | ||||
| Davis, T.M. 1998d | x | x | Persons with an MI with (N = 745) and without (N = 4,970) DM; among the former, N = 110 treated with gly, N = 111 treated with glic, and N = 524 treated with other antidiabetic drug or diet | Short-term survival, VT, VF, atrial fibrillation, complete heart block, pulmonary edema | Gly users were the least likely to experience VF, with an incidence similar to persons without DM | [76] | |||
| Farid, T.A. 2011 | x | Explanted cardiomyopathic Langendorff-perfused hearts, N = 15 (N = 9 treated with gly, N = 6 control) | Abrupt VF termination, effective refractory period, spatial dispersion of refractoriness | Spontaneous defibrillation occurred in larger proportion of gly-treated hearts vs. control hearts | [184] | ||||
| Halkin, A. 2001d | Unspecified | Persons with T2DM having an MI and being treated with a thrombolytic, N = 245 (N = 121 treated with SU and N = 124 treated with other antidiabetic drug or diet) | Mortality, in-hospital complications (including VT/VF) | SU users were the least likely to experience VT/VF, with an incidence similar to persons treated with diet alone | [79] | ||||
| Jollis, J.G. 1999d | x | Unk | x | Unk | x | Persons with an MI with (N = 64,171) and without (N = 143,248) DM; among the former, N = 17,031 treated with gly, N = 6,983 treated with glip, N = 2,632 treated with chlorpropamide, N = 666 treated with tolazamide, N = 540 treated with tolbutamide, N = 978 treated with other SUs | Mortality, in-hospital complications (including) cardiac arrest and VT | SU users had lower odds or cardiac arrest compared to persons with DM not treated with either insulin or a SU | [78] |
| Lomuscio, A. 1994 | x | x | Persons admitted to the CCU with first MI with (N = 278) and without (N = 830) T2DM; among the former, N = 106 treated with gly, N = 178 treated with other antidiabetic drug or diet | VF, sustained VT, mortality | Gly reduced VF and composite endpoint of VF/sustained VT | [48] | |||
| Pogatsa, G. 1988 | x | x | Digoxin-exposed persons with HF with (N = 278) and without T2DM (N = 278); among the former, N = 80 treated with gly, N = 71 treated with tolbutamide, and N = 61 treated with carbutamide | Premature ventricular ectopic beats | There were no occurrences of ventricular ectopic beats in gly users; this endpoint was more common in carbutamide users | [46] | |||
| Turner, R. (UKPDS) 1998c | x | x | x | Persons with newly-diagnosed T2DM randomly assigned to intensive glucose control (SU or insulin) or conventional control | Any diabetes-related endpoint (including sudden death), diabetes-related death (including sudden death), all-cause mortality, and 21 prespecified single endpoints (including sudden death) | Relative risk for sudden death was lower in intensive vs. conventional group; sudden death incidence lower in gly users and chlorpropamide users than in conventional group | [28] | ||
| Evidence for proarrhythmogenic effectsb | |||||||||
| Soler, N.G. 1974 | x | x | Persons with DM admitted to the CCU with suspected MI, N = 184 (N = 90 treated with an oral hypoglycemic agent, of whom N = 37 with chlorpropamide, N = 15 with tolbutamide, and N = 5 with gly) | Mortality, VF | SU users were the most likely to experience VF, with an incidence greater than persons with DM being treated with insulin or diet alone | [185] | |||
| Evidence for equivocal effects | |||||||||
| Landstedt-Hallin, L. 1999 | x | Persons with T2DM during hypoglycemia, N = 13, and euglycemia, N = 8 | Significant arrhythmias, QT measurements, plasma adrenaline | No significant arrhythmias were seen during hypoglycemia, with no difference between presence and absence of gly | [186] | ||||
| Garratt, K.N. 1999 | Unspecified | Persons with DM undergoing direct coronary angioplasty for treatment of acute MI, N = 185 (N = 67 treated with SU and N = 118 treated with other antidiabetic drug or diet) | In-hospital mortality, VT/VF, MI, severe angina | Incidence of VT/VF was similar in SU users vs. non-users | [187] | ||||
Abbreviations: CCU = coronary care unit; CHD = coronary heart disease; DM = diabetes mellitus; HF = heart failure; MI = myocardial infarction; NSVT = nonsustained ventricular tachycardia; PVC = premature ventricular complex; SU = sulfonylurea; T2DM = type 2 diabetes mellitus; Unk = unknown; VF = ventricular fibrillation; VT = ventricular tachycardia; UKPDS = United Kingdom Prospective Diabetes Study Group
Primarily for glyburide, as the most commonly studied sulfonylurea
Randomized trial discussed subsequently in the manuscript
Non-experimental epidemiologic study discussed subsequently in the manuscript
Cardiac excitability mediated by blockade of other K+ channels and Cl− channels
In addition to KATP, individual sulfonylureas may also inhibit the delayed rectifier potassium channel (IKr) encoded by the human ether-a-go-go-related gene. In in vitro and ex vivo animal cell models, Rosati et al demonstrated that glyburide prominently blocked IKr and prolonged cardiac repolarization; glimepiride was approximately 10x less effective in blocking this current.[49] In human myocytes, Schaffer et al demonstrated that glyburide blocked both the ultrarapid IKr and transient outward potassium current.[50] Yet, given that this inhibition occurred at concentrations greater than glyburide plasma levels, it has been suggested that such effects are unlikely to play a major role in persons with T2DM.[50]
Despite this, there is clinical evidence to support the conclusion that glyburide prolongs the QT interval.[51] Ikeda et al demonstrated that the QT interval was significantly longer in glyburide-treated T2DM subjects than those treated with diet alone;[52] Najeed et al and Curione et al reported similar findings.[53,54] Najeed et al further demonstrated that glyburide increased QT interval dispersion when compared to metformin.[53] Recently, Ninkovic et al found that sulfonylurea exposure was an independent risk factor for QT interval prolongation in persons with T2DM.[55] In contrast, Tentolouris et al found that neither QT interval duration nor dispersion was significantly longer in sulfonylurea vs. insulin users.[56] Yet, the latter two studies comingled users of different sulfonylureas into a single exposure group. Little else is known of the QT effects of sulfonylureas other than glyburide.
Finally, individual sulfonylureas may inhibit myocardial chloride currents such as the cyclic adenosine monophosphate-activated chloride conductance (ICl, cAMP) current through the cystic fibrosis transmembrane conductance regulator (CFTR).[57,58] Sheppard et al demonstrated that glyburide and tolbutamide inhibited CFTR chloride currents in fibroblasts;[59] findings for tolbutamide were confirmed by Venglarik et al.[60] Tominaga et al demonstrated that glyburide inhibited ICl, cAMP in guinea pig myocytes.[61] Antagonism of such channels may be proarrhythmogenic (via abolished protection against focal triggered arrhythmias) or antiarrhythmic (via prevented reentrant arrhythmias).[62–65]
Serious hypoglycemia
Among oral antidiabetic drugs, sulfonylureas carry the greatest risk of hypoglycemia[66]—an event that causes a cascade of physiological responses. At the cellular level, hypoglycemia has effects on myocardial tissue similar to that of proarrhythmic medications,[67] via action potential prolongation (the substrate for QT interval prolongation) and increased myoplasmic calcium concentrations.[68] These pathways interact in a synergistic way to increase the risk of serious arrhythmias. Recent basic science research has tied calcium overload to events that lead to apoptosis and necrosis—the mechanisms of myocardial cell death. Further, hypoglycemia may induce the processes that lead to apoptosis.[69] Given these effects, it is not surprising that hypoglycemia is considered a proarrhythmic event[67] that is uniformly associated with ventricular tachycardia, torsade de pointes, and SCA.[67,70] Yet, because hypoglycemia is common and SCA is rare, abnormal repolarization alone cannot explain how hypoglycemia may lead to sudden death.[71]
The continuum of evidence: from spontaneous reports to clinical trials
Sulfonylureas and spontaneous reports of SCA-related clinical outcomes
Case reports of SCA-related outcomes (i.e., sudden death, sudden arrhythmic death, sudden cardiac death, cardiac arrest, VA, and torsade de pointes) attributed to sulfonylureas began appearing in the literature in the 1950s. An early example described sudden arrhythmic death in a carbutamide user.[72] Episodes of VA attributed to tolbutamide and tolazamide were published years later,[73,74] potentially spurred by unexpected randomized trial findings of increased cardiac mortality in tolbutamide users.[75] Since the mid-1980s, additional published cases are scant. Yet, cases reported to the US Food and Drug Administration abound (Figure 2). Despite the role of such spontaneous reports in signal detection and hypothesis generation, these clinical observations provide a very low level of causal evidence.
Figure 2.
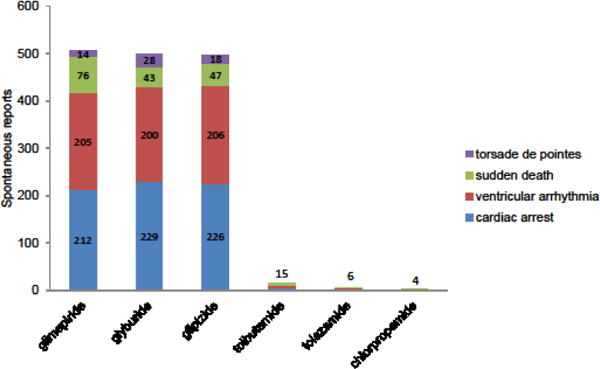
Spontaneous reports of sudden cardiac arrest-related outcomes attributed to a sulfonylurea. These data were collected via DrugInformer queries of United States Food and Drug Administration Adverse Event Reporting System (01/01/2004 through 10/18/2016). Spontaneous reports were dominated by cardiac arrest and ventricular arrhythmia events. These clinical observations, while potentially useful for hypothesis generation, provide a very low level of causal evidence.
Sulfonylureas and SCA-related clinical outcomes in non-experimental epidemiologic studies
A few epidemiologic studies have examined the association between sulfonylureas and SCA-related outcomes. The first example known to us was conducted by Davis et al using 1985–1993 data from the Western Australian Monitoring of Trends and Determinants of Cardiovascular Disease (MONICA) register.[76] This retrospective cohort study was designed to examine 28-day survival, dysrhythmias, heart block, and pulmonary edema in non-elder adults after acute myocardial infarction. Among 745 persons with DM, 110 (14.8%) and 111 (14.9%) were being treated with glyburide and gliclazide, respectively. In a multivariable logistic regression model that adjusted for a small set of covariates, gliclazide had non-statistically significantly elevated effect estimates for ventricular tachycardia (odds ratio [OR] = 1.2, 95% confidence interval [CI] 0.6–2.3) and ventricular fibrillation (OR = 1.9, 0.8–4.4) vs. glyburide. Davis et al concluded that glyburide users were less likely to experience ventricular fibrillation, with an incidence similar to that in persons without DM. This study has a number of limitations, including: a) reliance on self-reported diabetes; b) inadequate sample size for assessing these dysrhythmic outcomes; c) unclear methods of outcome ascertainment; d) lack of consideration of sudden death and cardiac arrest as outcomes; and e) lack of consideration of major risk factors as potential confounders. Despite this, these results are broadly consistent with prospective studies by Cacciapuoti et al[77] and Lomuscio et al.[48]
Subsequently, Jollis et al reported on in-hospital complications and mortality rates for Medicare beneficiaries with DM after acute myocardial infarction, using 1994–1995 data from the Cooperative Cardiovascular Project.[78] Among 64,171 persons with DM, 25,035 (39.0%) were receiving a sulfonylurea—the majority (68.0%) glyburide. In a logistic regression model adjusted for a small set of covariates, on-admission sulfonylurea exposure was associated with a statistically significant reduction in in-hospital cardiac arrest (OR = 0.93, 95% CI not reported) vs. persons with DM not treated with either a sulfonylurea or insulin; analyses were not stratified by individual sulfonylurea agent. Jollis et al did not specifically comment on this protective finding, but rather broadly concluded that sulfonylureas were not associated with clinically-meaningful differences in short-term outcomes. This study has a number of limitations, including: a) unclear methods of outcome ascertainment; b) lack of consideration of VA as an outcome; c) lack of consideration of out-of-hospital events; and d) lack of consideration of major risk factors as potential confounders.
Two later studies simply compared rates of VA between sulfonylureas users and non-users. Halkin et al conducted a secondary analysis of Argatroban in Acute Myocardial Infarction 2 (ARGAMI-2) trial data to elucidate the impact of prior antidiabetic treatment on 7-day, 30-day, and 1-year mortality, as well as rates of in-hospital complications.[79] Among 245 persons with T2DM, 121 (49.4%) were receiving a sulfonylurea prior to hospitalization. There was no difference in the proportion of persons with an in-hospital ventricular tachycardia / ventricular fibrillation between sulfonylurea users and persons treated with diet alone (8.3% vs. 8.9%, p = 0.21 across all treatment arms). Danchin et al examined the impact of sulfonylureas on in-hospital death, using data from a French acute myocardial infarction registry.[80] Among 487 persons with DM, 215 (44.1%) were receiving a sulfonylurea prior to hospitalization. There was a borderline statistically significant difference in the proportion of persons with an in-hospital ventricular fibrillation between sulfonylurea users and sulfonylurea nonusers (2.3% vs. 5.9%, p = 0.05). Given there was no statistical modeling of these SCA-related outcomes, interpreting these findings is difficult.
Sulfonylureas and SCA-related clinical outcomes in trials
Trials examining the risks of SCA-related outcomes among sulfonylureas are scant.[81] In the early 1990s, Cacciapuoti et al conducted a randomized crossover trial of 19 men with T2DM, coronary heart disease, and VAs evidenced on electrocardiogram.[77] Subjects underwent a baseline Holter evaluation then were allocated to an initial 2-week treatment with metformin (N = 9) or glyburide (N = 10). During periods of ischemia, there was a significant reduction in the logarithm of isolated ventricular premature complexes per minute at baseline vs. after glyburide treatment (1.30 vs. 0.21, p < 0.001); this was not evident with metformin (1.30 vs. 1.35, no p-value reported). During periods of ischemia, there was also a significant reduction in the logarithm of number of episodes of nonsustained ventricular tachycardia per minute at baseline vs. after glyburide treatment (0.24 vs. 0.003, p < 0.001); this was not evident with metformin (0.24 vs. 0.25, p-value not reported). Neither of these endpoints was significantly reduced during non-ischemic periods. There was no difference in fasting plasma glucose from baseline vs. after glyburide or metformin treatment. Therefore, Cacciapuoti et al concluded that glyburide had an antiarrhythmic effect that was independent of reductions in glucose. This trial has a number of limitations, including: a) small sample size; b) lack of consideration of sudden death or cardiac arrest outcomes; c) lack of consideration of longer-term therapy; and d) lack of examination of other sulfonylureas.
At the end of the 1990s, results from the United Kingdom Prospective Diabetes Study (UKPDS) were published.[28] This large multicenter randomized trial was designed to establish whether intensive glucose control in new-onset T2DM reduced the risk of macrovascular and microvascular complications. Primary outcomes included any diabetes-related endpoint, diabetes-related death, and all-cause mortality. Secondary outcomes included 21 individual endpoints comprising the former composites; sudden death was such an endpoint. Subjects were allocated to an intensive treatment (with a sulfonylurea [N = 1,573] or insulin [N = 1,156]) or conventional treatment with diet alone (N = 1,138); median follow-up was about 10 years. The relative risk (RR) for sudden death among persons randomized to the intensive (vs. conventional) arm was 0.54 (0.24–1.21). Intensive therapy-specific RRs were 0.57 (0.16–1.97), 0.67 (0.21–2.16), and 0.58 (0.19–1.70), for chlorpropamide, glyburide, and insulin (each vs. the conventional arm), respectively. Notably, there was no statistically significant difference in sudden death risk among the allocated intensive arm therapies (p = 0.92). While UKPDS’ primary outcome findings were very influential, the trial was not powered to examine sudden death as a stand-alone outcome. Unsurprisingly, the CIs for the sudden death effect estimates were very wide and findings therefore inconclusive. Unfortunately, the 10-year post-trial monitoring of UKPDS—published in late 2008—reported only on the original primary composite outcomes.[82]
A number of other trials have examined the relationship between intensity of glucose control and cardiovascular outcomes. Yet, sulfonylurea exposure in these studies was not a component of randomization. A seminal example is the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial.[83] This study’s primary outcome included a composite of nonfatal myocardial infarction, nonfatal stroke, and death from cardiovascular causes; a component of the latter was fatal arrhythmia. Subjects were allocated to an intensive treatment targeting HbA1C < 6.0% (N = 5,128) or conventional treatment targeting 7.0–7.9% (N = 5,123). Since therapeutic regimens were individualized and not randomized, both trial arms had a large proportion (>65%) of subjects treated with glimepiride—the only trial-approved sulfonylurea. Therefore, it is impossible to disentangle the role of sulfonylureas by examining the proportion of subjects in each arm with a fatal arrhythmia (0.1% vs. 0.2%, respectively). Similar limitations in design preclude the interpretation of sulfonylurea and SCA-related outcome findings from DIGAMI 2,[84] VADT,[85] RECORD,[86] ORIGIN,[87] and SAVOR-TIMI 53.[88] This is not surprising, though, as these trials were not intended to answer this specific clinical question.
While the Action in Diabetes and Vascular Disease Pretarax and Diamicron Modified Release Controlled Evaluation (ADVANCE) trial did randomly allocate an intensive treatment group to modified-release gliclazide (N = 5,571), the conventional treatment group (N = 5,569) was permitted to remain on any other formulation of a sulfonylurea. In fact, >63% and >57% of conventional therapy arm subjects began and ended the trial on a sulfonylurea other than modified-release gliclazide. Further, study findings only reported on sudden death incidence within a composite major coronary event outcome.
The TOSCA.IT trial has been underway since 2008,[89] with results expected by the end of 2017. This randomized trial is exploring the cardiovascular effects of adding pioglitazone or a sulfonylurea to metformin in T2DM subjects. The primary outcome is a composite of all-cause mortality, nonfatal myocardial infarction, nonfatal stroke, and unplanned coronary revascularization. The primary secondary outcome is a composite ischemic endpoint of sudden death, fatal and nonfatal myocardial infarction, fatal and nonfatal stroke, major leg amputation, and endovascular or surgical interventions on the coronary, leg or carotid arteries. Subjects (N = 3,040) have been allocated to either pioglitazone or a sulfonylurea (glyburide, gliclazide, or glimepiride, used accordingly to local practice). Most notably, treatment with a given sulfonylurea is not randomized. Further, sudden death is not being examined as a stand-alone outcome in a preplanned secondary analysis.
Other major, ongoing trials will unlikely provide definitive evidence. The GRADE pragmatic trial[90] including sulfonylurea users will not elucidate arrhythmogenic risk, as it was not designed as a cardiovascular outcome study; it is only examining, as a secondary outcome, a major adverse cardiovascular event (MACE) composite outcome, angina, and heart failure hospitalization. The CAROLINA trial[91] including glimepiride users is also examining MACE, but not SCA.
More data are needed
The studies discussed above are insufficient to answer the important clinical question at hand—what are the effects of different sulfonylureas on the clinical outcome of serious arrhythmia? A sufficiently-powered trial designed to answer this question would be a massive (and prohibitively expensive) undertaking, making it unlikely. Yet, T2DM trials under development that plan to examine agent-specific differences in cardiovascular safety endpoints should strongly consider SCA/VA as a stand-alone outcome. Ongoing trials examining SCA/VA as part of a composite endpoint should report findings for individual components of the composite. While these results would lack precision, they may help contextualize findings from existing and sure-to-be-forthcoming epidemiologic studies. It is clear that we will have to rely on non-experimental data. Therefore, we need rigorously-designed observational comparative safety study findings to serve as our evidence base. These studies should use sophisticated methods to minimize confounding by indication and mitigate bias. To help assess causality, these studies should examine dose- and duration-response effects. This will go a long way to close the existing knowledge gap.
Concluding remarks and future perspectives
SCA represents a major and growing public health concern, particularly in persons with DM. As the prevalence of DM skyrockets, providers will face an ever-more urgent responsibility to minimize SCA risk among persons with DM—focusing on risk factor reduction and tailoring of prescription therapies. An open clinical question is whether choice of oral antidiabetic drug therapy can minimize the risk of serious arrhythmic events.
Sulfonylureas remain the most widely used add-on to metformin in persons with T2DM. This class remains relevant because of extensive experience with its use, its low cost, and since UKPDS demonstrated its ability to reduce microvascular complications of DM. Yet, trial findings have also heralded concerns of cardiovascular safety—a debate that has raged for over 40 years. Few such trials (and observational studies) have focused specifically on the risk of SCA-related outcomes in sulfonylurea users. This is a major knowledge gap. Clinicians need sufficient evidence on which to base prescribing decisions in T2DM, especially in light of individualized treatment regimens balancing the tetrad of safety, efficacy, cost, and tolerability. While GRADE, CAROLINA, and TOSCA.IT will go a long way to inform dual therapy drug selection in T2DM, these ongoing trials will not close this knowledge gap. Not only is it likely to remain uncertain what role sulfonylureas should have as a class, it is likely to remain entirely unclear whether some modern sulfonylureas confer better or worse outcomes than others. Future work should elucidate the effects of individual sulfonylureas on SCA-related outcomes (see Outstanding Questions).
OUTSTANDING QUESTIONS.
-
-
To what degree do individual putative mechanisms (e.g., electrocardiographic QT interval prolongation, cardiac autonomic neuropathy, dysglycemia, vasoconstriction, hypercoagulation, renal failure) contribute to sudden cardiac arrest in persons with diabetes mellitus?
-
-
What is the selectivity of individual sulfonylureas for blocking mitochondrial vs. sarcolemmal myocardial KATP channels? Which blockade mediates cardiac excitability and ischemic preconditioning effects?
-
-
What are the effects of individual sulfonylureas on IKr and electrocardiographic QT interval prolongation? Are these adequate predictors of sudden cardiac arrest in persons with diabetes mellitus?
-
-
What is the comparative safety of individual sulfonylureas with regard to the risk of sudden cardiac arrest and related serious arrhythmic outcomes?
-
-
Does glyburide decrease the risk of sudden cardiac arrest and related serious arrhythmic outcomes in persons with type 2 diabetes mellitus? Would glyburide’s greater risk of serious hypoglycemia (vs. other sulfonylureas and other oral antidiabetics) outweigh this potential benefit?
-
-
Should clinicians preferentially consider glyburide as the first add-on option to metformin in type 2 diabetes patient requiring dual therapy? How would the risk-benefit change in an older adult? In an individual with comorbid coronary heart disease? In an individual with a history of rhythm disturbances?
TRENDS.
-
-
An increase in the incidence of sudden cardiac arrest among persons with diabetes mellitus represents a major public health concern.
-
-
Given this, it may be prudent to tailor prescribing to minimize sudden cardiac arrest risk; a potential approach could include the optimization of oral antidiabetic drug therapy.
-
-
Sulfonylureas remain the most commonly-used second line class for treating type 2 diabetes.
-
-
The impact of an individual sulfonylurea on sudden cardiac arrest risk may be driven by its ability to influence cardiac excitability, ischemic preconditioning, and/or serious hypoglycemia (among other mechanisms).
-
-
A few small experimental and observational studies in humans suggest that glyburide may reduce ventricular arrhythmias of varying clinical consequence; this putative effect requires rigorous examination.
Acknowledgments
This work was supported in part by the following grants from the US Department of Health and Human Services’ National Institutes of Health: R01 AG025152 (Dr. Sean Hennessy); and R01 DK102694 (Dr. Sean Hennessy).
The authors would like to thank Dr. Christina Aquilante (University of Colorado; Aurora, Colorado, US), Dr. Peter Brady (Mayo Clinic; Rochester, Minnesota, US), and Dr. Olga Vaccaro (University of Naples Federico II; Naples, IT) for their input.
GLOSSARY
- Adenosine triphosphate-sensitive potassium channel (KATP)
inward-rectifying potassium ion channel that sits at the crossroads of cell metabolism and membrane excitability
- found in ventricular myocytes
pancreatic β-cells, muscle, and the brain
- Delayed rectifier potassium channel (IKr)
major time-dependent outward current in ventricular myocytes that mediates repolarization of the cardiac action potential and is critical in determining action potential duration. IKr and the human ether-a-go-go-related gene (see below) are inextricably linked (and terms occasionally used interchangeably), as the human ether-a-go-go-related gene protein forms the voltage-gated channel responsible for this current
- Human ether-a-go-go-related gene
gene that encodes the pore-forming subunit of the rapid component of the delayed rectifier potassium channel expressed in the heart, various brain regions, smooth muscle cells, endocrine cells, and a wide range of tumor cell lines. Channel inhibition may lead to electrocardiographic QT interval prolongation, a putative precursor to torsade de pointes arrhythmias. Drug-induced QT interval prolongation commonly operates via this mechanism. Not all drugs that inhibit human ether-a-go-go-related gene channels are torsadogenic and inhibition of human ether-a-go-go-related gene channels is not the only mechanism by which drugs may be torsadogenic
- Ischemic preconditioning (IPC)
protection conferred to ischemic myocardium by brief periods of sub-lethal ischemia separated by periods of short bursts of reperfusion delivered before the ischemic insult
- QT interval
electrocardiographic measure of ventricular repolarization
- Serious hypoglycemia
a low blood glucose level that requires assistance from another person to treat
- Sudden cardiac arrest (SCA)
sudden cessation of cardiac activity with hemodynamic collapse, typically due to sustained ventricular tachycardia / ventricular fibrillation
- Sulfonylurea
a sulfonamide derivative hypoglycemic compound used in the treatment of type 2 diabetes mellitus. The 2017 Standards of Medical Care in Diabetes recommend an agent in this class as one high-efficacy option to augment metformin when dual therapy is required. Some agents in this class have been marketed since the 1950s and therefore are often considered a known entity—a potentially important consideration as safety concerns arise with novel oral antidiabetic drugs
- Torsade de pointes
ventricular tachycardia characterized by fluctuation of the QRS complexes around the electrocardiographic baseline and is typically caused by a long QT interval
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of potential conflicts of interest: Dr. Leonard receives salary support from the Harvard Pilgrim Health Care Institute for sulfonylurea-related work conducted on behalf of the United States Food and Drug Administration (US FDA). He is also supported by the US National Institutes of Health (NIH) for sulfonylurea-related work. Dr. Hennessy receives salary support from the Harvard Pilgrim Health Care Institute for sulfonylurea-related work conducted on behalf of the US FDA. He is also supported by the US NIH for sulfonylurea-related work. Further, he has consulted for Eli Lilly, GlaxoSmithKline, and Sanofi. The following authors declare no potential conflicts: Dr. Han; Dr. Siscovick; Dr. Flory; and Dr. Deo.
References
- 1.Mehra R. J Electrocardiol. 2007;40:S118–22. doi: 10.1016/j.jelectrocard.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. 2016 Available at: http://www.who.int/mediacentre/factsheets/fs312/en/ Last updated: 11/2016 Last accessed: 04/18/2017.
- 3.Al-Khatib SM, et al. J Am Coll Cardiol. 2017;69:712–744. doi: 10.1016/j.jacc.2016.09.933. [DOI] [PubMed] [Google Scholar]
- 4.Ryden L, et al. Eur Heart J. 2007;28:88–136. doi: 10.1093/eurheartj/ehl260. [DOI] [PubMed] [Google Scholar]
- 5.Lipska KJ, et al. Diabetes Care. 2017;40:468–475. doi: 10.2337/dc16-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varvaki Rados D, et al. PLoS Med. 2016;13:e100. doi: 10.1371/journal.pmed.1001992. 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pladevall M, et al. BMC Cardiovasc Disord. 2016;16:14-016–0187-5. doi: 10.1186/s12872-016-0187-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simpson SH, et al. Lancet Diabetes Endocrinol. 2015;3:43–51. doi: 10.1016/S2213-8587(14)70213-X. [DOI] [PubMed] [Google Scholar]
- 9.Shimoda M, Kaku K. J Diabetes Investig. 2016;7:674–676. doi: 10.1111/jdi.12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopshire JC, Zipes DP. Circulation. 2006;114:1134–1136. doi: 10.1161/CIRCULATIONAHA.106.647933. [DOI] [PubMed] [Google Scholar]
- 11.Hayashi M, Shimizu W, Albert CM. Circ Res. 2015;116:1887–1906. doi: 10.1161/CIRCRESAHA.116.304521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomaselli GF. Circ Res. 2015;116:1883–1886. doi: 10.1161/CIRCRESAHA.115.306515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergner DW, Goldberger JJ. Cardiol J. 2010;17:117–129. [PubMed] [Google Scholar]
- 14.Deo R, Albert CM. Circulation. 2012;125:620–637. doi: 10.1161/CIRCULATIONAHA.111.023838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Atat FA, et al. Curr Diab Rep. 2004;4:187–193. doi: 10.1007/s11892-004-0022-8. [DOI] [PubMed] [Google Scholar]
- 16.Siscovick DS, et al. Rev Endocr Metab Disord. 2010;11:53–59. doi: 10.1007/s11154-010-9133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suarez GA, et al. J Neurol Neurosurg Psychiatry. 2005;76:240–245. doi: 10.1136/jnnp.2004.039339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eranti A, et al. BMC Cardiovasc Disord. 2016;16:51-016–0231-5. doi: 10.1186/s12872-016-0231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Menyar AA. J Cardiovasc Med (Hagerstown) 2006;7:580–585. doi: 10.2459/01.JCM.0000237904.95882.c8. [DOI] [PubMed] [Google Scholar]
- 20.Sola D, et al. Arch Med Sci. 2015;11:840–848. doi: 10.5114/aoms.2015.53304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kantor ED, et al. JAMA. 2015;314:1818–1831. doi: 10.1001/jama.2015.13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garber AJ, et al. Endocr Pract. 2017;23:207–238. doi: 10.4158/EP161682.CS. [DOI] [PubMed] [Google Scholar]
- 23.Leonard CE, et al. Clin Pharmacol Ther. 2016;99:538–547. doi: 10.1002/cpt.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bell DS. CMAJ. 2006;174:185–186. doi: 10.1503/cmaj.051237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phung OJ, et al. Diabet Med. 2013;30:1160–1171. doi: 10.1111/dme.12232. [DOI] [PubMed] [Google Scholar]
- 26.Monami M, Genovese S, Mannucci E. Diabetes Obes Metab. 2013;15:938–953. doi: 10.1111/dom.12116. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, et al. Diabetes Care. 2014;37:3106–3113. doi: 10.2337/dc14-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.UK Prospective Diabetes Study Group. Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- 29.Anonymous. Diabetes Care. 2017;40:S4–S5. doi: 10.2337/dc17-S003. [DOI] [PubMed] [Google Scholar]
- 30.Shah RR. Br J Clin Pharmacol. 2002;54:188–202. doi: 10.1046/j.1365-2125.2002.01627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Centers for Disease Control and Prevention. 2013 Available at: https://www.cdc.gov/DIABETES/statistics/cvd/fig1.htm Last updated: 11/06/2012 Last accessed: 04/18/2017.
- 32.Kanavos P, van den Aardweg S, Schurer W. 2012:1–113. [Google Scholar]
- 33.Ashcroft FM, Gribble FM. J Diabetes Complications. 2000;14:192–196. doi: 10.1016/s1056-8727(00)00081-7. [DOI] [PubMed] [Google Scholar]
- 34.Nichols CG, Singh GK, Grange DK. Circ Res. 2013;112:1059–1072. doi: 10.1161/CIRCRESAHA.112.300514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zunkler BJ. Pharmacol Ther. 2006;112:12–37. doi: 10.1016/j.pharmthera.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 36.Baczko I, et al. Curr Med Chem. 2011;18:3640–3661. doi: 10.2174/092986711796642472. [DOI] [PubMed] [Google Scholar]
- 37.Brady PA, Terzic A. J Am Coll Cardiol. 1998;31:950–956. doi: 10.1016/s0735-1097(98)00038-2. [DOI] [PubMed] [Google Scholar]
- 38.Engler RL, Yellon DM. Circulation. 1996;94:2297–2301. doi: 10.1161/01.cir.94.9.2297. [DOI] [PubMed] [Google Scholar]
- 39.Nichols CG, Lederer WJ. Am J Physiol. 1991;261:H1675–86. doi: 10.1152/ajpheart.1991.261.6.H1675. [DOI] [PubMed] [Google Scholar]
- 40.Rahmi Garcia RM, Rezende PC, Hueb W. World J Diabetes. 2014;5:258–266. doi: 10.4239/wjd.v5.i3.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Negroni JA, Lascano EC, del Valle HF. Cardiovasc Hematol Agents Med Chem. 2007;5:43–53. doi: 10.2174/187152507779315868. [DOI] [PubMed] [Google Scholar]
- 42.Riveline JP, et al. Diabetes Metab. 2003;29:207–222. doi: 10.1016/s1262-3636(07)70030-7. [DOI] [PubMed] [Google Scholar]
- 43.Valensi P, Slama G. Br J Diabetes Vasc Dis. 2006;6:159–165. [Google Scholar]
- 44.Hausenloy DJ, et al. J Cardiovasc Pharmacol Ther. 2013;18:263–269. doi: 10.1177/1074248412468945. [DOI] [PubMed] [Google Scholar]
- 45.Ye Y, et al. Basic Res Cardiol. 2011;106:925–952. doi: 10.1007/s00395-011-0216-6. [DOI] [PubMed] [Google Scholar]
- 46.Pogatsa G, et al. Acta Physiol Hung. 1988;71:243–250. [PubMed] [Google Scholar]
- 47.Aronson D, Mittleman MA, Burger AJ. Pacing Clin Electrophysiol. 2003;26:1254–1261. doi: 10.1046/j.1460-9592.2003.t01-1-00177.x. [DOI] [PubMed] [Google Scholar]
- 48.Lomuscio A, et al. Coron Artery Dis. 1994;5:767–771. [PubMed] [Google Scholar]
- 49.Rosati B, et al. FEBS Lett. 1998;440:125–130. doi: 10.1016/s0014-5793(98)01444-6. [DOI] [PubMed] [Google Scholar]
- 50.Schaffer P, et al. Br J Pharmacol. 1999;128:1175–1180. doi: 10.1038/sj.bjp.0702904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Ponti F, et al. Br J Clin Pharmacol. 2002;54:171–177. doi: 10.1046/j.1365-2125.2002.01617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ikeda T. Diabete Metab. 1994;20:565–567. [PubMed] [Google Scholar]
- 53.Najeed SA, et al. Am J Cardiol. 2002;90:1103–1106. doi: 10.1016/s0002-9149(02)02776-5. [DOI] [PubMed] [Google Scholar]
- 54.Curione M, et al. Acta Diabetol. 2014;51:31–33. doi: 10.1007/s00592-012-0438-6. [DOI] [PubMed] [Google Scholar]
- 55.Ninkovic VM, et al. Acta Diabetol. 2016 doi: 10.1007/s00592-016-0864-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tentolouris N, et al. Exp Clin Endocrinol Diabetes. 2005;113:298–301. doi: 10.1055/s-2005-865641. [DOI] [PubMed] [Google Scholar]
- 57.Schotborgh CE, Wilde AA. Cardiovasc Res. 1997;34:73–80. doi: 10.1016/s0008-6363(97)00036-9. [DOI] [PubMed] [Google Scholar]
- 58.Maertens C, et al. Br J Pharmacol. 2000;129:791–801. doi: 10.1038/sj.bjp.0703102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheppard DN, Welsh MJ. J Gen Physiol. 1992;100:573–591. doi: 10.1085/jgp.100.4.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Venglarik CJ, et al. Biophys J. 1996;70:2696–2703. doi: 10.1016/S0006-3495(96)79839-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tominaga M, et al. Circ Res. 1995;77:417–423. doi: 10.1161/01.res.77.2.417. [DOI] [PubMed] [Google Scholar]
- 62.Duan DD. Compr Physiol. 2013;3:667–692. doi: 10.1002/cphy.c110014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duan D. J Physiol. 2009;587:2163–2177. doi: 10.1113/jphysiol.2008.165860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hiraoka M, et al. Cardiovasc Res. 1998;40:23–33. doi: 10.1016/s0008-6363(98)00173-4. [DOI] [PubMed] [Google Scholar]
- 65.Yamazaki J, Hume JR. Circ Res. 1997;81:101–109. doi: 10.1161/01.res.81.1.101. [DOI] [PubMed] [Google Scholar]
- 66.Bolen S, et al. 2007 [Google Scholar]
- 67.Nordin C. Acta Diabetol. 2014;51:5–14. doi: 10.1007/s00592-013-0528-0. [DOI] [PubMed] [Google Scholar]
- 68.Nordin C. Diabetologia. 2010;53:1552–1561. doi: 10.1007/s00125-010-1752-6. [DOI] [PubMed] [Google Scholar]
- 69.Arad M, Seidman CE, Seidman JG. Circ Res. 2007;100:474–488. doi: 10.1161/01.RES.0000258446.23525.37. [DOI] [PubMed] [Google Scholar]
- 70.Snell-Bergeon JK, Wadwa RP. Diabetes Technol Ther. 2012;14(Suppl 1):S51–8. doi: 10.1089/dia.2012.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frier BM, Schernthaner G, Heller SR. Diabetes Care. 2011;34(Suppl 2):S132–7. doi: 10.2337/dc11-s220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Federman DD, Field JB. Diabetes. 1957;6:67–70. doi: 10.2337/diab.6.1.67. discussion, 85–90. [DOI] [PubMed] [Google Scholar]
- 73.Leonard R, Robinson RS. JAMA. 1978;239:190. [PubMed] [Google Scholar]
- 74.Poffenbarger PL, Scott J. JAMA. 1980;244:811–812. [PubMed] [Google Scholar]
- 75.Schwartz TB, Meinert CL. Perspect Biol Med. 2004;47:564–574. doi: 10.1353/pbm.2004.0071. [DOI] [PubMed] [Google Scholar]
- 76.Davis TM, et al. Diabetes Care. 1998;21:637–640. doi: 10.2337/diacare.21.4.637. [DOI] [PubMed] [Google Scholar]
- 77.Cacciapuoti F, et al. Am J Cardiol. 1991;67:843–847. doi: 10.1016/0002-9149(91)90617-t. [DOI] [PubMed] [Google Scholar]
- 78.Jollis JG, et al. Am Heart J. 1999;138:S376–80. doi: 10.1016/s0002-8703(99)70038-4. [DOI] [PubMed] [Google Scholar]
- 79.Halkin A, et al. J Thromb Thrombolysis. 2001;12:177–184. doi: 10.1023/a:1012979622945. [DOI] [PubMed] [Google Scholar]
- 80.Danchin N, et al. Diabetes Metab Res Rev. 2005;21:143–149. doi: 10.1002/dmrr.498. [DOI] [PubMed] [Google Scholar]
- 81.Holman RR, Sourij H, Califf RM. Lancet. 2014;383:2008–2017. doi: 10.1016/S0140-6736(14)60794-7. [DOI] [PubMed] [Google Scholar]
- 82.Holman RR, et al. N Engl J Med. 2008;359:1577–1589. doi: 10.1056/NEJMoa0806470. [DOI] [PubMed] [Google Scholar]
- 83.Action to Control Cardiovascular Risk in Diabetes Study Group et al. N Engl J Med. 2008;358:2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Malmberg K, et al. Eur Heart J. 2005;26:650–661. doi: 10.1093/eurheartj/ehi199. [DOI] [PubMed] [Google Scholar]
- 85.Duckworth W, et al. N Engl J Med. 2009;360:129–139. doi: 10.1056/NEJMoa0808431. [DOI] [PubMed] [Google Scholar]
- 86.Home PD, et al. Lancet. 2009;373:2125–2135. doi: 10.1016/S0140-6736(09)60953-3. [DOI] [PubMed] [Google Scholar]
- 87.ORIGIN Trial Investigators et al. N Engl J Med. 2012;367:319–328. doi: 10.1056/NEJMoa1203858. [DOI] [PubMed] [Google Scholar]
- 88.Scirica BM, et al. N Engl J Med. 2013;369:1317–1326. doi: 10.1056/NEJMoa1307684. [DOI] [PubMed] [Google Scholar]
- 89.Vaccaro O, et al. Nutr Metab Cardiovasc Dis. 2012;22:997–1006. doi: 10.1016/j.numecd.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 90.Nathan DM, et al. Diabetes Care. 2013;36:2254–2261. doi: 10.2337/dc13-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marx N, et al. Diab Vasc Dis Res. 2015;12:164–174. doi: 10.1177/1479164115570301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Smits P, Thien T. Diabetologia. 1995;38:116–121. doi: 10.1007/BF02369361. [DOI] [PubMed] [Google Scholar]
- 93.Briscoe VJ, Griffith ML, Davis SN. Expert Opin Drug Metab Toxicol. 2010;6:225–235. doi: 10.1517/17425250903512955. [DOI] [PubMed] [Google Scholar]
- 94.Sato T, et al. Circulation. 2000;101:2418–2423. doi: 10.1161/01.cir.101.20.2418. [DOI] [PubMed] [Google Scholar]
- 95.Auchampach JA, Grover GJ, Gross GJ. Cardiovasc Res. 1992;26:1054–1062. doi: 10.1093/cvr/26.11.1054. [DOI] [PubMed] [Google Scholar]
- 96.Gross GJ, Auchampach JA. Circ Res. 1992;70:223–233. doi: 10.1161/01.res.70.2.223. [DOI] [PubMed] [Google Scholar]
- 97.Grover GJ, Sleph PG, Dzwonczyk S. Circulation. 1992;86:1310–1316. doi: 10.1161/01.cir.86.4.1310. [DOI] [PubMed] [Google Scholar]
- 98.Yao Z, Gross GJ. Am J Physiol. 1993;264:H2221–5. doi: 10.1152/ajpheart.1993.264.6.H2221. [DOI] [PubMed] [Google Scholar]
- 99.Schulz R, Rose J, Heusch G. Am J Physiol. 1994;267:H1341–52. doi: 10.1152/ajpheart.1994.267.4.H1341. [DOI] [PubMed] [Google Scholar]
- 100.Schulz R, et al. Circulation. 1999;99:305–311. doi: 10.1161/01.cir.99.2.305. [DOI] [PubMed] [Google Scholar]
- 101.Bernardo L, et al. Am J Physiol. 1999;276:H1323–30. doi: 10.1152/ajpheart.1999.276.4.H1323. [DOI] [PubMed] [Google Scholar]
- 102.Bernardo L, et al. Am J Physiol. 1999;277:H128–35. doi: 10.1152/ajpheart.1999.277.1.H128. [DOI] [PubMed] [Google Scholar]
- 103.Elliott GT, et al. Cardiovasc Res. 1996;32:1071–1080. doi: 10.1016/s0008-6363(96)00154-x. [DOI] [PubMed] [Google Scholar]
- 104.Flynn DM, et al. Cardiovasc Drugs Ther. 2005;19:337–346. doi: 10.1007/s10557-005-4970-2. [DOI] [PubMed] [Google Scholar]
- 105.Hoag JB, et al. Am J Physiol. 1997;273:H2458–64. doi: 10.1152/ajpheart.1997.273.5.H2458. [DOI] [PubMed] [Google Scholar]
- 106.Horimoto H, et al. J Surg Res. 2002;105:181–188. doi: 10.1006/jsre.2002.6379. [DOI] [PubMed] [Google Scholar]
- 107.Krenz M, et al. J Mol Cell Cardiol. 2001;33:2015–2022. doi: 10.1006/jmcc.2001.1465. [DOI] [PubMed] [Google Scholar]
- 108.Munch-Ellingsen J, Bugge E, Ytrehus K. Basic Res Cardiol. 1996;91:382–388. doi: 10.1007/BF00788718. [DOI] [PubMed] [Google Scholar]
- 109.Nieszner E, et al. Exp Clin Endocrinol Diabetes. 2002;110:212–218. doi: 10.1055/s-2002-33069. [DOI] [PubMed] [Google Scholar]
- 110.Ohno Y, et al. Int J Cardiol. 1997;62:181–190. doi: 10.1016/s0167-5273(97)00270-2. [DOI] [PubMed] [Google Scholar]
- 111.Pain T, et al. Circ Res. 2000;87:460–466. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- 112.Toombs CF, Moore TL, Shebuski RJ. Cardiovasc Res. 1993;27:617–622. doi: 10.1093/cvr/27.4.617. [DOI] [PubMed] [Google Scholar]
- 113.Walsh RS, et al. Cardiovasc Res. 1994;28:1337–1341. doi: 10.1093/cvr/28.9.1337. [DOI] [PubMed] [Google Scholar]
- 114.Wang S, Cone J, Liu Y. Am J Physiol Heart Circ Physiol. 2001;280:H246–55. doi: 10.1152/ajpheart.2001.280.1.H246. [DOI] [PubMed] [Google Scholar]
- 115.D’Souza SP, et al. Am J Physiol Heart Circ Physiol. 2003;284:H1592–600. doi: 10.1152/ajpheart.00902.2002. [DOI] [PubMed] [Google Scholar]
- 116.Iwai T, et al. Br J Pharmacol. 2000;129:1219–1227. doi: 10.1038/sj.bjp.0703148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Joyeux M, Godin-Ribuot D, Ribuot C. Br J Pharmacol. 1998;123:1085–1088. doi: 10.1038/sj.bjp.0701710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maddock HL, Siedlecka SM, Yellon DM. Cardiovasc Drugs Ther. 2004;18:113–119. doi: 10.1023/B:CARD.0000029028.75316.5e. [DOI] [PubMed] [Google Scholar]
- 119.Mocanu MM, et al. Circulation. 2001;103:3111–3116. doi: 10.1161/01.cir.103.25.3111. [DOI] [PubMed] [Google Scholar]
- 120.Qian YZ, et al. Am J Physiol. 1996;271:H23–8. doi: 10.1152/ajpheart.1996.271.1.H23. [DOI] [PubMed] [Google Scholar]
- 121.Schultz JE, Hsu AK, Gross GJ. Circ Res. 1996;78:1100–1104. doi: 10.1161/01.res.78.6.1100. [DOI] [PubMed] [Google Scholar]
- 122.Tavackoli S, et al. Coron Artery Dis. 2004;15:53–58. doi: 10.1097/00019501-200402000-00008. [DOI] [PubMed] [Google Scholar]
- 123.Wang GY, et al. Am J Physiol Heart Circ Physiol. 2001;280:H384–91. doi: 10.1152/ajpheart.2001.280.1.H384. [DOI] [PubMed] [Google Scholar]
- 124.Wu GT, et al. Chin Med Sci J. 2007;22:162–168. [PubMed] [Google Scholar]
- 125.Ye Y, et al. Cardiovasc Drugs Ther. 2008;22:429–436. doi: 10.1007/s10557-008-6138-3. [DOI] [PubMed] [Google Scholar]
- 126.Sato T, et al. Diabetes Metab Res Rev. 2006;22:341–347. doi: 10.1002/dmrr.621. [DOI] [PubMed] [Google Scholar]
- 127.Takano H, Tang XL, Bolli R. Am J Physiol Heart Circ Physiol. 2000;279:H2350–9. doi: 10.1152/ajpheart.2000.279.5.H2350. [DOI] [PubMed] [Google Scholar]
- 128.Thornton JD, et al. Circ Res. 1993;72:44–49. doi: 10.1161/01.res.72.1.44. [DOI] [PubMed] [Google Scholar]
- 129.Fralix TA, et al. Cardiovasc Res. 1993;27:630–637. doi: 10.1093/cvr/27.4.630. [DOI] [PubMed] [Google Scholar]
- 130.Grover GJ, et al. J Pharmacol Exp Ther. 1993;265:559–564. [PubMed] [Google Scholar]
- 131.Lu H, Remeysen P, De Clerck F. Coron Artery Dis. 1993;4:649–657. doi: 10.1097/00019501-199307000-00010. [DOI] [PubMed] [Google Scholar]
- 132.Bilinska M, et al. Coron Artery Dis. 2007;18:455–462. doi: 10.1097/MCA.0b013e3282a30676. [DOI] [PubMed] [Google Scholar]
- 133.Cleveland JC, Jr, et al. Circulation. 1997;96:29–32. doi: 10.1161/01.cir.96.1.29. [DOI] [PubMed] [Google Scholar]
- 134.Ferreira BM, et al. Ann Noninvasive Electrocardiol. 2005;10:356–362. doi: 10.1111/j.1542-474X.2005.00650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Klepzig H, et al. Eur Heart J. 1999;20:439–446. doi: 10.1053/euhj.1998.1242. [DOI] [PubMed] [Google Scholar]
- 136.Lee TM, et al. Circulation. 2002;105:334–340. doi: 10.1161/hc0302.102572. [DOI] [PubMed] [Google Scholar]
- 137.Lee TM, Chou TF. J Clin Endocrinol Metab. 2003;88:531–537. doi: 10.1210/jc.2002-020904. [DOI] [PubMed] [Google Scholar]
- 138.Loubani M, et al. Eur J Pharmacol. 2005;515:142–149. doi: 10.1016/j.ejphar.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 139.Ovunc K. Clin Cardiol. 2000;23:535–539. doi: 10.1002/clc.4960230713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Speechly-Dick ME, Grover GJ, Yellon DM. Circ Res. 1995;77:1030–1035. doi: 10.1161/01.res.77.5.1030. [DOI] [PubMed] [Google Scholar]
- 141.Tomai F, et al. Circulation. 1994;90:700–705. doi: 10.1161/01.cir.90.2.700. [DOI] [PubMed] [Google Scholar]
- 142.Tomai F, et al. Eur Heart J. 1999;20:196–202. doi: 10.1053/euhj.1998.1311. [DOI] [PubMed] [Google Scholar]
- 143.Bogaty P, et al. J Am Coll Cardiol. 1998;32:1665–1671. doi: 10.1016/s0735-1097(98)00431-8. [DOI] [PubMed] [Google Scholar]
- 144.Correa SD, Schaefer S. Am J Cardiol. 1997;79:75–78. doi: 10.1016/s0002-9149(96)00681-9. [DOI] [PubMed] [Google Scholar]
- 145.Billman GE, et al. J Cardiovasc Pharmacol. 1993;21:197–204. doi: 10.1097/00005344-199302000-00003. [DOI] [PubMed] [Google Scholar]
- 146.Billman GE, Englert HC, Scholkens BA. J Pharmacol Exp Ther. 1998;286:1465–1473. [PubMed] [Google Scholar]
- 147.Gwilt M, et al. Eur J Pharmacol. 1992;220:231–236. doi: 10.1016/0014-2999(92)90752-p. [DOI] [PubMed] [Google Scholar]
- 148.Tosaki A, Hellegouarch A. J Am Coll Cardiol. 1994;23:487–496. doi: 10.1016/0735-1097(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 149.Wilde AA, et al. Circ Res. 1990;67:835–843. doi: 10.1161/01.res.67.4.835. [DOI] [PubMed] [Google Scholar]
- 150.Zhang HL, et al. Zhongguo Yao Li Xue Bao. 1991;12:398–402. [PubMed] [Google Scholar]
- 151.Barrett TD, Walker MJ. J Mol Cell Cardiol. 1998;30:999–1008. doi: 10.1006/jmcc.1998.0664. [DOI] [PubMed] [Google Scholar]
- 152.Bellemin-Baurreau J, et al. Eur J Pharmacol. 1994;256:115–124. doi: 10.1016/0014-2999(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 153.Black SC, et al. J Mol Cell Cardiol. 1993;25:1427–1438. doi: 10.1006/jmcc.1993.1159. [DOI] [PubMed] [Google Scholar]
- 154.Chi L, et al. J Cardiovasc Pharmacol. 1993;21:179–190. doi: 10.1097/00005344-199302000-00001. [DOI] [PubMed] [Google Scholar]
- 155.Dhein S, Pejman P, Krusemann K. Eur J Pharmacol. 2000;398:273–284. doi: 10.1016/s0014-2999(00)00322-8. [DOI] [PubMed] [Google Scholar]
- 156.Le Grand B, et al. Eur J Pharmacol. 1992;229:91–96. doi: 10.1016/0014-2999(92)90290-k. [DOI] [PubMed] [Google Scholar]
- 157.Baczko I, Lepran I, Papp JG. Eur J Pharmacol. 1997;324:77–83. doi: 10.1016/s0014-2999(97)00064-2. [DOI] [PubMed] [Google Scholar]
- 158.Ballagi-Pordany G, et al. Diabetes Res Clin Pract. 1990;8:109–114. doi: 10.1016/0168-8227(90)90020-t. [DOI] [PubMed] [Google Scholar]
- 159.Bril A, Laville MP, Gout B. Cardiovasc Res. 1992;26:1069–1076. doi: 10.1093/cvr/26.11.1069. [DOI] [PubMed] [Google Scholar]
- 160.Csonka C, et al. J Cardiovasc Pharmacol. 2003;41:916–922. doi: 10.1097/00005344-200306000-00013. [DOI] [PubMed] [Google Scholar]
- 161.D’Alonzo AJ, et al. Cardiovasc Res. 1994;28:881–887. doi: 10.1093/cvr/28.6.881. [DOI] [PubMed] [Google Scholar]
- 162.El-Reyani NE, et al. Eur J Pharmacol. 1999;365:187–192. doi: 10.1016/s0014-2999(98)00866-8. [DOI] [PubMed] [Google Scholar]
- 163.Hatcher AS, Alderson JM, Clements-Jewery H. J Cardiovasc Pharmacol. 2011;57:439–446. doi: 10.1097/FJC.0b013e31820d5342. [DOI] [PubMed] [Google Scholar]
- 164.Lepran I, et al. J Pharmacol Exp Ther. 1996;277:1215–1220. [PubMed] [Google Scholar]
- 165.Rees SA, Curtis MJ. J Cardiovasc Pharmacol. 1995;26:280–288. doi: 10.1097/00005344-199508000-00014. [DOI] [PubMed] [Google Scholar]
- 166.Tosaki A, et al. J Pharmacol Exp Ther. 1993;267:1355–1362. [PubMed] [Google Scholar]
- 167.Vajda S, Baczko I, Lepran I. Eur J Pharmacol. 2007;577:115–123. doi: 10.1016/j.ejphar.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 168.Wirth KJ, et al. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:295–300. doi: 10.1007/s002109900084. [DOI] [PubMed] [Google Scholar]
- 169.Wolleben CD, Sanguinetti MC, Siegl PK. J Mol Cell Cardiol. 1989;21:783–788. doi: 10.1016/0022-2828(89)90717-7. [DOI] [PubMed] [Google Scholar]
- 170.D’Alonzo AJ, et al. J Cardiovasc Pharmacol. 1994;23:446–452. [PubMed] [Google Scholar]
- 171.D’Alonzo AJ, et al. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:222–228. doi: 10.1007/BF00176778. [DOI] [PubMed] [Google Scholar]
- 172.Cole WC, McPherson CD, Sontag D. Circ Res. 1991;69:571–581. doi: 10.1161/01.res.69.3.571. [DOI] [PubMed] [Google Scholar]
- 173.Testai L, et al. J Pharm Pharmacol. 2010;62:924–930. doi: 10.1211/jpp.62.06.0014. [DOI] [PubMed] [Google Scholar]
- 174.Bernauer W. Eur J Pharmacol. 1997;326:147–156. doi: 10.1016/s0014-2999(97)85409-x. [DOI] [PubMed] [Google Scholar]
- 175.Iskit AB, et al. Vascul Pharmacol. 2007;46:129–136. doi: 10.1016/j.vph.2006.08.415. [DOI] [PubMed] [Google Scholar]
- 176.Takahashi N, et al. Exp Biol Med (Maywood) 2003;228:1234–1238. doi: 10.1177/153537020322801021. [DOI] [PubMed] [Google Scholar]
- 177.del Valle HF, et al. Cardiovasc Res. 2001;50:474–485. doi: 10.1016/s0008-6363(01)00209-7. [DOI] [PubMed] [Google Scholar]
- 178.Vegh A, et al. Cardiovasc Res. 1993;27:638–643. doi: 10.1093/cvr/27.4.638. [DOI] [PubMed] [Google Scholar]
- 179.Zhu BM, et al. J Pharmacol Sci. 2003;93:106–113. doi: 10.1254/jphs.93.106. [DOI] [PubMed] [Google Scholar]
- 180.Pasnani JS, Ferrier GR. J Pharmacol Exp Ther. 1992;262:1076–1084. [PubMed] [Google Scholar]
- 181.Harbinson MT, Allen JD, Adgey AA. Int J Cardiol. 2000;76:187–197. doi: 10.1016/s0167-5273(00)00378-8. [DOI] [PubMed] [Google Scholar]
- 182.Das B, Sarkar C, Karanth KS. Pharmacology. 2001;63:134–141. doi: 10.1159/000056124. [DOI] [PubMed] [Google Scholar]
- 183.Ferdinandy P, et al. Cardiovasc Res. 1995;30:781–787. [PubMed] [Google Scholar]
- 184.Farid TA, et al. Circ Res. 2011;109:1309–1318. doi: 10.1161/CIRCRESAHA.110.232918. [DOI] [PubMed] [Google Scholar]
- 185.Soler NG, et al. Lancet. 1974;1:475–477. doi: 10.1016/s0140-6736(74)92785-8. [DOI] [PubMed] [Google Scholar]
- 186.Landstedt-Hallin L, et al. J Intern Med. 1999;246:299–307. doi: 10.1046/j.1365-2796.1999.00528.x. [DOI] [PubMed] [Google Scholar]
- 187.Garratt KN, et al. J Am Coll Cardiol. 1999;33:119–124. doi: 10.1016/s0735-1097(98)00557-9. [DOI] [PubMed] [Google Scholar]