

Abstract
Regulation of expression of HTLV-1 gene products from integrated proviruses plays an important role in HTLV-1-associated disease pathogenesis. Previous studies have shown that T cell receptor (TCR)- and phorbol ester (PMA) stimulation of chronically infected CD4 T cells increases the expression of integrated HTLV-1 proviruses in latently infected cells, however the mechanism remains unknown. Analysis of HTLV-1 RNA and protein species following PMA treatment of the latently HTLV-1-infected, FS and SP T cell lines demonstrated rapid induction of tax/rex mRNA. This rapid increase in tax/rex mRNA was associated with markedly enhanced tax/rex mRNA stability while the stability of unspliced or singly spliced HTLV-1 RNAs did not increase. Tax/rex mRNA in the HTLV-1 constitutively expressing cell lines exhibited high basal stability even without PMA treatment. Our data support a model whereby T cell activation leads to increased HTLV-1 gene expression at least in part through increased tax/rex mRNA stability.
Keywords: HTLV-1, latency, RNA stability, retroviruses, arginosuccinate lyase
Graphical abstract
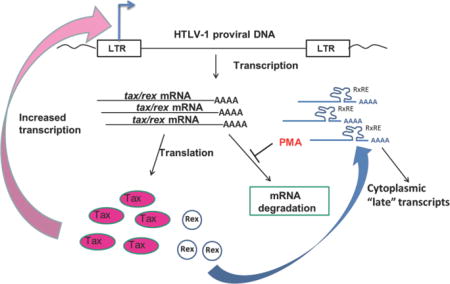
1. Introduction
The human T cell leukemia virus type 1 (HTLV-1) was the first discovered human retrovirus and is the causative agent of adult T cell leukemia/lymphoma (ATL) and HTLV-associated myelopathy/tropical spastic paraparesis (HAM/TSP) (Gallo, 2005; Yoshida, 2005). HTLV-1 infects an estimated 5–10 million people worldwide (Gessain and Cassar, 2012; Goncalves et al., 2010). Approximately 2%–6% of HTLV-1-infected individuals develop ATL, a devastating malignancy of CD4 T cells, generally after a prolonged period (20–60 years) of clinical latency [reviewed in (Iwanaga et al., 2012; Yasunaga and Matsuoka, 2007)]. HAM/TSP has been estimated to occur in 5%–10% of infected patients with recent studies suggesting neurological symptoms can be seen in > 30% of patients over an eight year follow-up (Tanajura et al., 2015). HAM/TSP is associated with expansions of both infected CD4 T cells and activated CD8 T cells [reviewed in (Araya et al., 2011; Nagai and Osame, 2003)]. The pathogenesis of both ATL and HAM/TSP remains unclear, particularly with respect to the factors that determine whether infected individuals develop disease, the determinants of the disease phenotype, and the events that occur during the clinically latent period.
HTLV-1 infects primarily CD4 T cells. As with other retroviruses, the HTLV-1 genome integrates into host DNA. Following integration, infected cells can undergo a productive infection with generation of progeny virus, or can enter a state of low or no virion production (i.e. viral latency) [reviewed in (Carpentier et al., 2015)]. The existence and nature of HTLV-1 latency has been somewhat controversial. The continued induction of high levels of anti-HTLV-1 cytotoxic T lymphocyte (CTL) response, particularly directed against the HTLV-1 Tax transactivator protein as well as the HBZ protein, is evidence that there is significant production of viral antigens even during clinically quiescent periods. The strong CTL response has been argued to be responsible for the lack of evidence of viral gene expression in blood (Asquith et al., 2000). On the other hand, at any given time, the number of cells bearing the HTLV-1 genome exceeds the number of cells in which viral RNA species and viral proteins can be detected (Gessain et al., 1991; Hollsberg, 1999). Even a brief period of ex vivo culture is sufficient to detect markedly increased HTLV-1 expression in cells (Hanon et al., 2000). By analogy with HIV and other retroviruses, these data suggest that a significant number of HTLV-1 infected cells are latently infected in vivo. Furthermore, considerable DNA sequencing data show that the major route of HTLV-1 expansion in vivo is proliferation of preexisting infected T cells, rather than the de novo infection of new T cells. This also suggests that active production of infectious virions is not the major contributor to HTLV-1 pathogenesis (Gillet et al., 2011; Wattel et al., 1995). Finally, a number of groups have isolated and characterized CD4 T cell lines derived from HTLV-1 infected patients that express either no or only very low levels of HTLV-1 RNA and proteins constitutively (Lin et al., 1998; Richardson et al., 1997), suggesting that such latently infected cells also exist in vivo. Development of disease however appears to require Tax expression (Nomura et al., 2004; Peloponese et al., 2007; Yamagishi and Watanabe, 2012), at least in the early stages. Thus, activation of HTLV-1 expression from latently-infected cells and escape from the CTL response would likely be essential steps in HTLV-1 disease pathogenesis. A key question is how does HTLV-1 expression become activated in latent/low level-expressing, infected T cells?
The regulation of HTLV-1 gene expression is complex and involves both cellular and virally-encoded regulators. In particular, the viral Tax gene product, required for HTLV-1 transformation of CD4 T cells [reviewed in (Matsuoka and Jeang, 2011)], is a transcriptional activator of the HTLV-1 long terminal repeat (LTR), as well as of thousands of cellular genes (Ng et al., 2001). Tax has been extensively studied and transactivates the LTR by binding to cyclic AMP response element binding protein (CREB/ATF) family members [reviewed in (Kashanchi and Brady, 2005)], recruiting transcriptional coactivators including CBP and p300, and clearing occluding nucleosomes from the HTLV LTR (Lemasson et al., 2006; Nyborg et al., 2010). HTLV-1 gene expression is also controlled at the RNA level through the effects of the HTLV-1 Rex protein, which promotes nuclear export of unspliced and singly spliced RNAs (Baydoun et al., 2008; Nakano and Watanabe, 2012), and through the effects of p30 which inhibits nuclear export of tax/rex mRNAs (Anupam et al., 2013; Younis et al., 2006). The sum of these effects is early expression of the Tax and Rex proteins due to early export of tax/rex mRNAs, with later expression of structural and enzymatic proteins (Baydoun et al., 2008; Cavallari et al., 2013; Hidaka et al., 1988; Li et al., 2009). Important roles for the HTLV-1 anti-sense encoded gene, HBZ, have also been proposed (Barbeau and Mesnard, 2011; Matsuoka and Jeang, 2011; Mesnard et al., 2006). HBZ inhibits Tax-induced activation of the 5′LTR through inhibiting interactions with CREB proteins and CBP (Clerc et al., 2008; Gaudray et al., 2002; Lemasson et al., 2007), antagonizes the senescence-inducing properties of Tax (Zhi et al., 2011), which potentiates viral persistence (Arnold et al., 2006) and cellular proliferation (Arnold et al., 2008; Satou et al., 2006). Thus, the interplay of Tax, Rex, and HBZ is essential for transformation and disease progression (Matsuoka and Yasunaga, 2013), and plays a key role in regulation of HTLV-1 expression in some models of viral latency (Philip et al., 2014).
Several groups have studied the activation of expression of integrated HTLV-1 proviruses in latently infected CD4 T cells. These cell lines exhibit only low levels of HTLV-1 protein expression basally, but show marked activation of HTLV-1 protein expression upon treatment with various mimetics of different aspects of T cell activation. These include stimulation through the T cell receptor (TCR) by anti-CD3 antibody (Lin et al., 2005; Swaims et al., 2010), anti-CD2 antibody (Guyot et al., 1997), lectin [phytohemagglutinin-P (PHA), a surrogate for TCR cross-linking], phorbol ester [phorbol 12-myristate 13-acetate (PMA) which activates PKC pathways downstream of TCR activation (Lin et al., 1998)], and anti-CD28 antibody plus prostaglandins (Dumais et al., 2003). Cellular stresses (including arsenic, oxidative stress, heavy metal) (Andrews et al., 1995) and direct chromatin remodeling agents such as histone deacetylase inhibitors (Lemasson et al., 2004; Lin et al., 1998; Villanueva et al., 2006) also activate the HTLV-1 LTR.
In our current study, we observed a rapid but transient induction of tax/rex mRNA and protein following activation of infected T cells. We further examined the mechanism of HTLV-1 RNA induction and identified a marked enhancement of tax/rex mRNA stability following PMA treatment of chronically-infected, low-level expressing CD4 T cells, but not in infected cells with high basal levels of HTLV-1 expression. Thus, we propose that PMA activation may enhance the stability of tax/rex mRNA in latent/low-level expressing HTLV-1 infected cells, which in turn results in increased transcription from the HTLV-1 LTR, contributing to the exit of infected cells from latency and the induction of events ultimately contributing to HTLV-1 pathogenesis.
2. Materials & Methods
2.1 Plasmids
Construction of pLTR-Luc (luciferase) was described previously (Lin et al., 1998). The human ASL promoter (−310 to +59) was amplified by polymerase chain reaction performed on genomic DNA purified from Jurkat T cells using Taq DNA polymerase (Qiagen,) and the following primer pair: cacctcgagcgggcctgatgtcatagcctctacc (forward) and gtcaagcttctccgcctggccgcacg (reverse). The amplified product was ligated into the XhoI and HindIII sites of pGL3-Basic (Promega) to create pASL-Luc. The NcoI/BamHI fragment encoding the Tax gene, 3′LTR and SV40 polyadenylation site from pHTLV-tat1 (Nerenberg et al., 1987) was cloned in pLTR-Luc replacing the Luc gene, to create pLTR-Tax. The Tax gene and 3′UTR were deleted from pLTR-Tax by NcoI/BglII digestion, followed by treatment with Klenow enzyme and religation to create pLTR-deltaTax (pLTR-DTax). Restriction enzymes and DNA modifying enzymes were obtained from New England Biolabs.
2.2 Cell Lines and Media
Except as noted below, cell lines were grown in RPMI 1640 (Life Technologies) supplemented with 10% fetal bovine serum (Gemini Bio-Products), 1% Penicillin/Streptomycin (Life Technologies), 1% L-Glutamine (Life Technologies), and 10 mM HEPES (Life Technologies). JPX-9 cells (Nagata et al., 1989) were supplemented with 0.6 mg/ml G418 (Life Technologies). When indicated, JPX-9 and Jurkat cells [JE6-1 clone, (Weiss et al., 1984)] were treated with 5 μg/ml PHA (Sigma-Aldrich) and/or 120 μM zinc chloride (Sigma-Aldrich) for 24 hours. FS and SP cells (Rowe et al., 1995) were grown in media containing 20% FBS and 20 U/ml IL-2. Transfections were performed using GenePORTER (Gene Therapy Systems) according to the manufacturer’s protocol. Unless otherwise indicated, each reaction contained 400ng luciferase reporter plasmid and 100 ng Tax (or deltaTax) plasmid. Transfected cells were treated with PHA (5 μg/ml; Sigma-Aldrich) or PMA (50 ng/ml; Sigma-Aldrich) four hours after transfection. Cells were harvested 24 hours after transfection, washed in DPBS, resuspended in lysis buffer and assayed for luciferase activity using the manufacturer’s protocol (Promega). Protein concentration was determined by the BCA method (Pierce). Luciferase activity was normalized to the amount of recovered protein for each sample. FS, SP, MT2, MT4 and HUT102 cells were treated with PMA (50 ng/ml) or vorinostat (5 μM; Sigma-Aldrich) for the indicated times, spun, washed and resuspended in fresh media. Cells were harvested at the indicated time points.
For studies of the effects of downstream pathways of protein kinase C (PKC) signaling on HTLV-1 activation, FS cells were pre-treated with the NF-κB inhibitor, Bay 11-7082 (5μM, Cayman Chemical) (Pierce et al., 1997), the NFAT inhibitor, Cyclosporin A (0.5μM, Cayman Chemical) (McCaffrey et al., 1993), or the AP-1 inhibitor, SR 11302 (1μM, Cayman Chemical) (Fanjul et al., 1994) for 1 hr, followed by a 15 minute PMA treatment. Cells were then washed, resuspended with fresh media and cultured for 4 hours. RNA were prepared as described.
2.3 RT-qPCR
Total RNA was harvested (RNeasy Mini Kit; Qiagen), reverse transcribed (SuperScript; Invitrogen/Life Technologies) and subjected to semi-quantitative PCR using primers to ASL (gtggatgttcaaggcagcaaagc and gctcattggctgtgtggatgtcc), IL2Rα (gctctgccactcggaacacaacg and gcagacgctctcagcaggacc) and Tax (cacctgtccagagcatcaga and gggaacattggtgaggaagg). For quantitative PCR reactions, SYBR Green qPCR SuperMix (Invitrogen) was used with mRNA specific primers as listed in Supplemental file 2: Supplementary Table S1. Reactions were performed using an MX4000 quantitative PCR system (Agilent Technologies).
2.4 Western Blot
Cells were harvested, washed twice in 1× DPBS and extracted with RIPA Lysis Buffer (50mM Tris pH 7.6, 150mM NaCl, 5mM EDTA, 1% NP-40, 0.5% deoxycholate, 0.1% SDS) with freshly added PMSF (0.1mM) and protease inhibitor cocktail (P8340; Sigma-Aldrich,). Protein extracts (15μg) were subjected to SDS-PAGE and transferred to PVDF (Hybond P; GE Healthcare). Blots were probed with antibodies to Actin at 1:5000 dilution (A2066; Sigma-Aldrich), Tax at 1:1000 (Tab 172; NIH-AIDS research and reference reagent program, Bethesda, Maryland, NIH, (Jeang et al., 1987; Jeang et al., 1990), Env (PRH-21) at 10 μg/ml (gift of S. Foung, (Hadlock et al., 1997)), or Rex at 1:4000 (gift of P. Green, (Ye et al., 2003)). To detect evidence of senescence and apoptosis by western blot assays, antibodies against p21 (CDKN1A) at1:500 dilution (cat # 556431; BD Pharmingen), p27 (CDKN1B) at 1: 1000 dilution (#2552; Cell Signaling technology), PARP 0.6μg/ml (R &D system, MAB600), Caspase 3 at 1:1000 dilution (#9662; Cell Signaling technology), and Cleaved Caspase 3 (ASP175) at 1:1000 dilution (#9661; Cell Signaling technology) were used. Chemiluminescent detection (GE Healthcare) of signal was captured and analyzed with the BioRad Gel Doc using Quantity One software (Bio-Rad Laboratories).
2.5 RNA decay
Cells were treated with stimuli as indicated, followed by PBS wash and resuspension in fresh media containing Actinomycin D (10 μg/ml; Sigma-Aldrich) at time zero. Total RNA was collected at different time points and measured by RT-qPCR as described. Data are shown as percentage mRNA remaining compared to time zero (set as 100%). The data were analyzed by nonlinear regression fit (one phase exponential decay), and mRNA half-lives (t1/2) were determined from first-order decay constants (k) utilizing PRISM (GraphPad Software, Inc) as previously described (Ysla et al., 2008). Three independent experiments were performed and data are presented as mean ± standard deviation. Comparisons between half-lives were performed with the unpaired two-tailed t test with three independent experiments.
2.6 Nuclear run on assay
The nuclear run-on assay was based on the incorporation of biotin-16-uridine-5′-triphosphate (biotin-16-UTP) in nascent transcripts as described (Patrone et al., 2000), with minor modifications. In brief, 20×106 cells per sample were centrifuged at 216×g for 10 minutes, washed once with cold PBS, resuspended in 10 ml Cell Lysis Buffer (20 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 20 mM NaCl, 100 mM sucrose, 0.25% NP-40) and incubated on ice for 5 min. Nuclei were collected by centrifugation at 216×g, and resuspended in 100 μl Nuclei Storage Buffer (50.0 mM Tris-HCl, pH 8.0, 5.0 mM MgCl2, 0.1 mM EDTA, 45% Glycerol), snap frozen in liquid N2 and stored at −80°C. For UTP incorporation reactions, 100 μl of nuclei were incubated with 100 μl 2× NRO Reaction Buffer (300 mM KCl, 10 mM MgCl2, 2 mM of cold rATP, rCTP, rGTP and 1 mM Biotin-16-UTP (Roche Life Science), 100 units RNaseOUT™ (Life Technologies) at 30°C for 30 min; then cold rUTP was added to a final concentration of 1 mM, incubation continued for another 15 min. The reaction was stopped by adding 6 μl of 250 mM CaCl2 and 100 units RNase-free DNase I (Roche), and incubating for 10 minutes at 37°. Total RNA was extracted as described and biotinylated RNA was purified using streptavidin-conjugated magnetic beads (Dynal KilobaseBINDER; Life Technologies). Isolated biotinylated RNA was used for RT-qPCR as described.
3. Results
3.1 Identification of arginosuccinate lyase as a housekeeping gene unresponsive to T cell activation or HTLV-1 Tax
In order to characterize the events leading to activation of integrated HTLV-1, we analyzed the induction of HTLV-1 mRNA species in chronically-infected CD4 T cells with low level HTLV-1 expression, following treatment with T cell activation stimuli. To normalize the RNA expression data, it was critical to identify a cellular “housekeeping” gene whose expression was Tax- and PMA-insensitive, as our past experiments had suggested that these stimuli have effects in T cells on the levels of commonly used housekeeping genes, such as actin and GAPDH (data not shown). During a study of changes in HTLV-1 gene expression (Ng et al., 2001), it was noted that some cellular genes showed relatively invariant expression in the presence of Tax. We hypothesized that one or more of those genes could therefore serve as a useful housekeeping control. Out of a set of 229 genes whose expression varied by less than 20% following the induction of Tax from the JPX-9 cell line [a Jurkat CD4 T cell line with an inducible Tax gene under the control of the metallothionein promoter (Nagata et al., 1989)] [(Ng et al., 2001) and data not shown], we identified the arginosuccinate lyase (ASL) gene (Abramson et al., 1991; O’Brien et al., 1986) as a candidate gene whose expression was unaffected by Tax. Total RNA was harvested from the Jurkat cell subclone, JE6-1 and JPX-9 cells,, stimulated with zinc chloride and/or PHA and analyzed by semi-quantitative, reverse transcription PCR with primers for ASL, Tax or the interleukin 2-receptor α (IL-2Rα), a gene known to be sensitive to PHA and Tax stimulation (Siekevitz et al., 1987) (Supplementary file 1: Supplementary Fig. S1A). Levels of ASL mRNA did not change upon addition of PHA (in Jurkat cells) or zinc-induced expression of Tax (in JPX-9 cells, a Jurkat cell line with an inducible Tax gene under the control of the metallothionein promoter. We further characterized the ASL promoter (Abramson et al., 1991) by cloning it 5′ to the luciferase gene. This vector was co-transfected into CD4 JE6-1 Jurkat cells with either an LTR-driven Tax (pLTR-Tax) or control LTR plasmid from which Tax sequences had been deleted (pLTR-deltaTax). After treatment with PHA or PMA, cells were harvested and protein lysates assayed for luciferase activity (Supplementary file 1: Supplementary Fig. S1B). The ASL promoter was insensitive to co-transfection with the Tax expression plasmid and to treatment with PHA or PMA, whereas pLTR-Luc exhibited induction by Tax, PHA and PMA. These experiments support the use of ASL as a Tax, PHA and PMA-insensitive control gene to examine induction of HTLV-1 mRNA species in T cells.
3.2 Treatment of chronically infected T cells leads to a rapid induction of Tax and Rex mRNA and protein
In order to study the kinetics of induction of HTLV-1 RNAs in chronically infected, low level-expressing cells following T cell activation stimuli, we used two chronically infected T cell lines shown to have low, but inducible levels of HTLV-1 gene expression. FS cells are CD4 T cells which contain one full-length integrated HTLV-1 provirus, as well as several deleted proviruses [(Dezzutti et al., 1993) and H.C. Lin, unpublished observations], and which we have previously shown to increase Tax protein levels in response to various T cell activation stimuli (Lin et al., 1998; Lin et al., 2005). SP cells are dual CD4, CD8 T cells that contain a single integrated HTLV-1 provirus (Rowe et al., 1995). Using reverse transcription -quantitative PCR (RT-qPCR), we analyzed the relative levels of various HTLV-1 mRNA species at multiple time points following one hour of PMA induction. Our previous studies had shown that the effects of PMA on HTLV-1 expression were similar to those induced by crosslinking of the T cell receptor using anti-CD3 antibody (Lin et al., 2005), and LTR activation induced by PMA was downstream of TCR signaling. Thus, for studies in these cell lines, we used PMA treatment as a surrogate for TCR activation of the HTLV-1 LTR. The primers used for these studies are shown in Supplementary file 1: Supplementary Fig. S2 and Supplementary file 2: Supplementary Table S1. Cycle threshold (Ct) values for each mRNA species were normalized to those of ASL and the calculated difference was normalized to the data from the zero hour time point.
We first analyzed the kinetics of induction of HTLV-1 mRNA species in PMA-treated FS cells (Fig. 1A and Supplementary file 1: Supplementary Fig. S3). FS cells showed a rapid induction of the tax/rex message, by almost 7-fold even at 1 hour after the removal of PMA and resuspension in fresh media, the earliest time point examined, peaking between 4 to 8 hours with an increase of approximately 50-fold, and then markedly decreasing by 24 hours. The increase in tax/rex mRNA preceded the more delayed induction of unspliced (full-length RNA detected by gag/pol primers) and singly-spliced env messages. Although p30 mRNA is also multiply spliced, interestingly, its induction more closely resembled that of the unspliced and singly spliced full-length and env mRNAs, peaking 8 hours after induction, with increases of ~12–17-fold in FS cells. Only very low levels of HBZ mRNA (spliced form) were detected basally and exhibited a very small (< 2fold) increase upon PMA treatment. The consistency of these results was validated in three separate experiments (Supplementary file 1: Supplementary Fig. S3, showing results for each HTLV-1 gene separately). We also analyzed the induction of HTLV-1 RNAs following PMA induction of SP cells (Fig. 1B), which showed an overall similar pattern of HTLV-1 RNA expression as FS cells. Doubly spliced tax/rex message was strongly induced by 4 hours, and rapidly declined by 8 hours. Tax/rex mRNA also increased prior to that of the singly spliced env or of unspliced full-length mRNA. In SP cells, the induction of p30 mRNA was more pronounced ~40-fold), however, like FS cells, HBZ RNA levels were extremely low and essentially unchanged by PMA.
Figure 1. Kinetics of HTLV-1 gene expression in latently infected cell lines following PMA treatment.
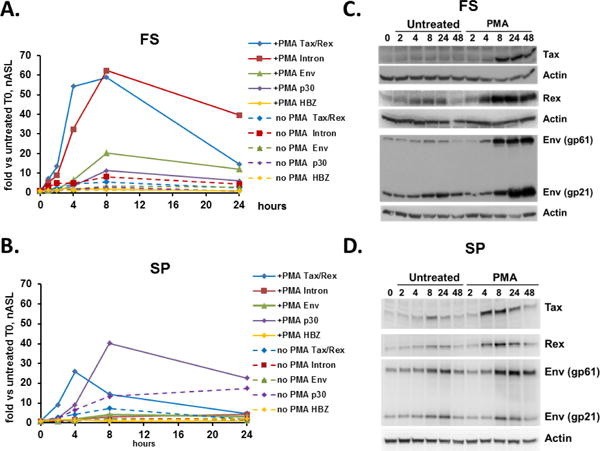
(A & B) Time course of individual HTLV-1 RNA transcripts for PMA-treated (solid lines) or untreated (dashed lines) FS cells (A) and SP cells (B), measured by quantitative reverse transcription-PCR (RT-qPCR) using splice site-specific primers (Supplementary file 2: Supplementary Table S1). Blue diamonds, tax/rex mRNA; Red squares, gag/pol (unspliced) mRNA; green triangles, env mRNA; purple diamonds, p30 mRNA; and yellow diamonds, HBZ mRNA (spliced form). Data are normalized to Ct values for ASL mRNA, and divided by the value obtained for the zero time point. Cells were treated with PMA for one hour, washed and resuspended in fresh media, and harvested for RNA and protein isolation at indicated time points. Stimulation of latently infected cell lines induces a rapid, but transient, increase in tax/rex mRNA that precedes other HTLV-1 mRNA species. (C & D) Western blots for protein extracts of PMA-treated and untreated FS cells (C) and SP cells (D). Overall, induction of HTLV-1 protein levels resembles that of mRNA induction.
Based on the critical roles of the HTLV-1 Tax and Rex proteins in the HTLV-1 life cycle, and the role of Rex as a temporal regulator that separates an early regulatory phase of the viral life cycle from the later production of viral structural proteins and enzymes (Cavallari et al., 2013; Hidaka et al., 1988), we performed western blots on protein extracts following PMA induction of FS and SP cells (Fig. 1C, 1D, and Supplementary file 1: Supplementary Fig. S4, showing quantitation of SP cell results). Overall, the levels of expression of different HTLV-1 proteins correlated with those of the corresponding mRNAs determined by RT-qPCR analysis. Increases in Rex protein were readily apparent by 4 hours, preceding increased levels of Env. SP cells showed an early increase in Tax, which was less prominent in FS cells. Tax protein levels fell from their peak over 24–48 hours in both FS and SP cells, although the decrease was more pronounced in SP cells. Taken together, these data support a model in which PKC activation rapidly induces tax/rex mRNA and protein expression in HTLV-1 chronically infected cells, followed by increases in other viral mRNA and protein species. Furthermore, Tax induction is rapidly attenuated.
3.3 Induction of HTLV-1 RNA expression in chronically infected T cells is associated with increased transcription and stabilization of tax/rex mRNA
The induction of increased levels of HTLV-1 mRNAs could result from several factors, including new RNA transcription and increased RNA stability. Therefore, we analyzed the contributions of these two processes to the increased levels of HTLV-1 RNAs in FS cells. In order to assess the contributions of new HTLV-1 RNA transcription to the induction of HTLV-1 gene expression, we measured newly transcribed mRNA using the nuclear run-on assay (Fig. 2). As shown previously in Fig. 1, PMA treatment led to a rapid increase in tax/rex mRNA (Fig. 2A). In order to study new transcription, FS cells were treated with PMA for 15 minutes or 5 hours, and nuclei were isolated. RNA produced by isolated nuclei was metabolically labeled with biotin-UTP and purified. The labeled, newly synthesized RNA was bound to streptavidin beads and RT-qPCR was used to quantitate levels of biotin-labeled RNA. As shown in Figure 2B, PMA treatment led to an approximately 2-fold increase in transcription of full length HTLV-1 RNA after 15 minutes, increasing to over 12-fold after 5 hours. Thus, at early time points, PMA has a modest effect on new HTLV-1 transcription that is amplified to much higher levels over time. As a control, we also investigated the effects of treatment with the histone deacetylase (HDAC) inhibitor, vorinostat, based on previous observations that HDAC inhibitors induced HTLV-1 transcription (Lin et al., 1998; Lu et al., 2004; Villanueva et al., 2006). Vorinostat treatment led to a slow increase in tax/rex mRNA over a 24 hour period (Fig. 2C). Vorinostat also led to increased HTLV-1 transcription (Fig. 2D), although to a lower level with slower kinetics than PMA (reaching 2-fold induction by 5 hours and 6-fold induction by 9 hours).
Figure 2. PMA and vorinostat increase tax/rex mRNA and induce new HTLV-1 transcription.
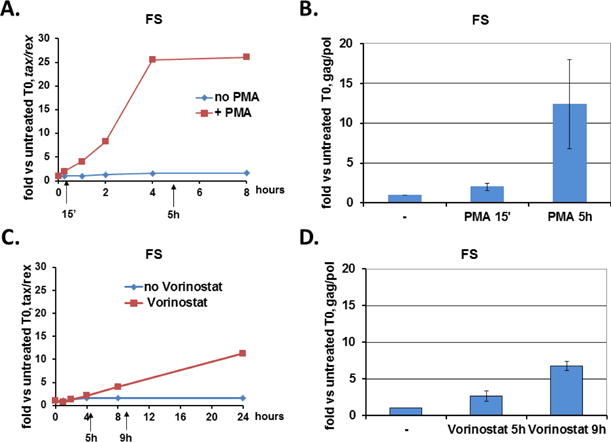
(A & C) RT-qPCR analysis of tax/rex mRNA levels from untreated (blue) or stimuli treated (red) FS cells. FS cells were incubated with PMA (A) or vorinostat (C) over the times indicated, prior to harvest for RNA analysis. Data are normalized to Ct values for ASL and divided by the value obtained for the zero time point. Arrows indicate time points of harvest for nuclear run-on assays. (B & D) Nuclear run-on assay for measuring newly transcribed mRNA. Nuclei were isolated at the indicated times [15 minute and 5 hours of PMA treatment (B), 5 hours and 9 hours of vorinostat treatment (D)], and incubated with biotinylated uridine, and biotinylated transcripts were isolated. Transcripts corresponding to full length HTLV-1 and ASL were detected by RT-qPCR, and the data normalized to ASL. Results represent the fold change over the value obtained for time zero. Average fold change with standard deviation (error bars) from triplicate inductions are shown.
T cell activation signaling and phorbol ester signaling have been shown to increase the stability of certain cellular mRNAs in T cells, thereby increasing their steady state levels (Chen et al., 1998; Ford et al., 1999; Lindstein et al., 1989; Powell et al., 1998; Zhu et al., 2010). Furthermore, the sharp decrease in tax/rex mRNA levels following the initial induction that we observed in Fig. 1 was striking, suggesting that degradation of tax/rex mRNA may play an important role in regulating tax/rex mRNA levels in these cells. Therefore, we were interested in whether PMA might have an effect on HTLV-1 mRNA stability (Fig. 3A). FS cells were treated with PMA for 15 minutes, washed, resuspended in fresh media and Actinomycin D (Act D) was added (time zero) to inhibit new transcription. RNAs were collected at different time points, and analyzed by RT-qPCR for the levels of gag/pol, tax/rex and env sequences. The percentage of RNA remaining at each time point compared to time zero was calculated for three independent experiments, and the average and standard deviations for each time point are shown in Fig. 3A. Compared to the untreated control, PMA treatment significantly increased the estimated half-life (t1/2) of tax/rex mRNA from 4.1±1.3 hr to 20.3 ± 4.2 hr (P = .02) concomitant with the major increase in total tax/rex mRNA (Fig. 1, 2A). Enhanced stability was only observed for tax/rex mRNA. The half-life of the singly-spliced env HTLV-1 messages remained essentially unchanged by PMA treatment when compared with the untreated control. Interestingly, PMA treatment appeared to have a small effect in destabilizing unspliced genomic and gag/pol RNAs, but this effect did not reach statistical significance (control t1/2: 3.2± 0.5; PMA-treated t1/2:1.9±0.1; P=.07). As expected, the HDAC inhibitor, vorinostat had no effect on tax mRNA stability (Fig. 3B), consistent with its known effects on transcription through chromatin modulation.
Figure 3. PMA increases tax/rex mRNA stability in FS cells.
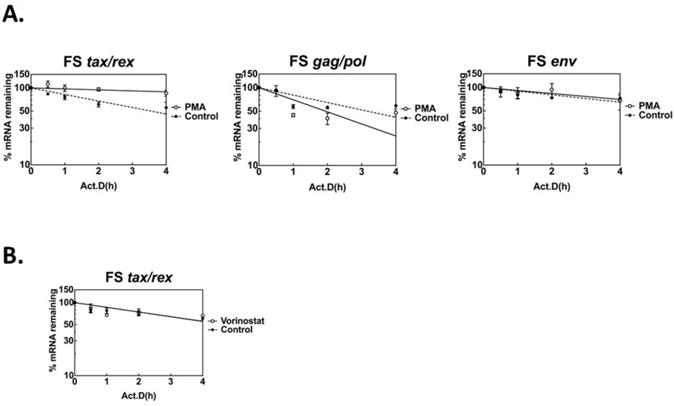
(A) RNA decay assay for RNA stability of different HTLV-1 transcripts upon PMA treatment. FS cells were treated with PMA for 15 min, washed, and followed by Act D treatment for 4 hours. RNAs were collected at the time indicated, and measured by RT-qPCR with specific primers for tax/rex, env, and gag/pol (full-length genome). Data are normalized to ASL, and presented as percentage mRNA remaining compared with time zero. Data shown are average of triplicates with standard deviation (error bars). PMA increases tax/rex mRNA stability, but does not change the stability of env or gag/pol mRNAs. (B) RNA decay assay for RNA stability of tax/rex mRNA upon vorinostat treatment. FS cells were treated with vorinostat for 4 hours, washed, and followed by Act D treatment for 4 hours. RNAs were collected at the times indicated, and analyzed as described in (A). Vorinostat does not change the stability of HTLV-1 tax/rex mRNA.
To determine if enhanced tax/rex mRNA stability upon PMA treatment is also observed in other HTLV-1 infected T cells, we compared tax/rex RNA stability between FS cells, SP cells (a second inducible cell line), and the HTLV-1 constitutively expressing, HUT102 cells. Similar to FS cells (Fig. 4A), PMA treatment also increased the stability of the tax/rex mRNA in SP cells (Fig. 4B), increasing the estimated t1/2 from approximately 4.5±1.3 hours to >24 hours (P=.0001). We also examined tax/rex mRNA stability in HUT102 cells (Fig. 4C), an HTLV-1-infected CD4 T cell line which constitutively expresses high levels of Tax protein (Bunn and Foss, 1996). PMA treatment had little effect on steady state levels of tax/rex mRNA in HUT102 cells (less than 1.2-fold induction, data not shown). The untreated tax/rex mRNA half-life in HUT102 cells was considerably longer (estimated t1/2 = 6 hrs) than that seen in the inducible FS and SP cells, and was unchanged by PMA treatment. To confirm the generality of these results for HTLV-1-constitutively expressing cell lines, we also examined the basal and PMA-treated t1/2 of tax/rex mRNA in two additional constitutive HTLV-1 producing cell lines; MT2 and MT4 cells (Miyoshi et al., 1981). As shown in Supplementary File 1, Supplementary Fig. S5, both of these cell lines had basal tax/rex mRNA half-lives longer than that observed for FS or SP cells (MT4=16.4±4.4 hrs.; MT2=8.4±2.9 hrs.). PMA treatment of these HTLV-1-expressing cell lines resulted in insignificant changes in tax/rex mRNA half-lives (MT4=18.2±3.3 hrs., P=.75; MT2=8.6±3.6 hrs., P=.96). Thus, MT2, MT4 and HUT102 cells constitutively expressing high levels of HTLV-1 RNA had a longer basal mRNA half-life that was not significantly affected by PMA treatment. In contrast, PMA-mediated induction of tax/rex mRNA levels in low-level expressing/latently infected cells. was associated with a marked increase in RNA stability.
Figure 4. PMA increases tax/rex RNA stability in inducible HTLV-1 T Cell Lines.
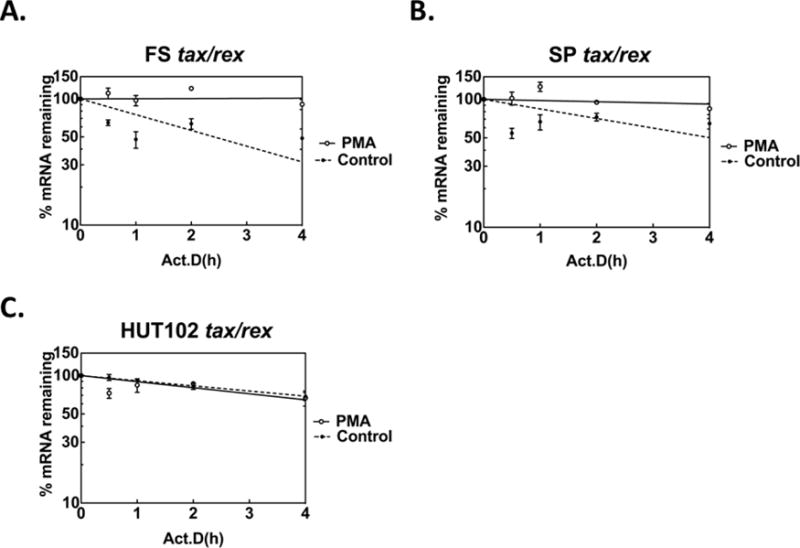
HTLV-1 infected FS (A), SP (B) and HUT102 (C) cells were treated with PMA for 30 min, washed, then treated with Act D, and RNA decay assays were performed. Tax/rex mRNAs were measured with splice site-specific primers (closed circle: untreated, open circle: PMA treated). Similar to results observed in FS cells, PMA also increased the tax/rex RNA stability in SP cells. In contrast, in HUT102 cells, which constitutively express high levels of Tax protein, PMA fails to change the RNA degradation rate.
The relatively lower levels of tax/rex mRNA observed after the initial early peak following PMA treatment of FS cells prompted us to examine the effects of PMA on HTLV-1 transcription and tax/rex mRNA stability at a later time point following treatment. (Fig. 5). For these studies, FS cells were treated with PMA for 15 minutes, washed, resuspended and then harvested at 16 hours after treatment for analysis of tax/rex RNA levels, new gag/pol transcription (by nuclear run-on assay), and tax/rex mRNA stability. In this experiment, at the 16 hr. time point, tax/rex total mRNA levels were elevated approximately 25 fold compared to untreated cells (Fig. 5A). New transcription of full-length (gag/pol-containing) RNA was also elevated compared to untreated cells (approximately 14 fold, Fig. 5B) consistent with the effects of the Tax positive feedback loop transactivating expression of other HTLV-1 RNAs. In contrast, tax/rex mRNA stability in PMA-treated cells was decreased compared with early times after treatment (Fig. 5C), and was no longer statistically different from that of untreated control cells (PMA treated=7.6±3.1 hrs., untreated=5.0±0.9 hrs.; P=0.47). Thus, although the PMA-induced increase in tax/rex mRNA half-life was transient, there was a sustained increase in HTLV-1 transcription, likely due to the continued effects of the Tax protein on the HTLV-1 LTR.
Figure 5. Late effects of PMA on tax/rex mRNA stability and new HTLV-1 transcription.
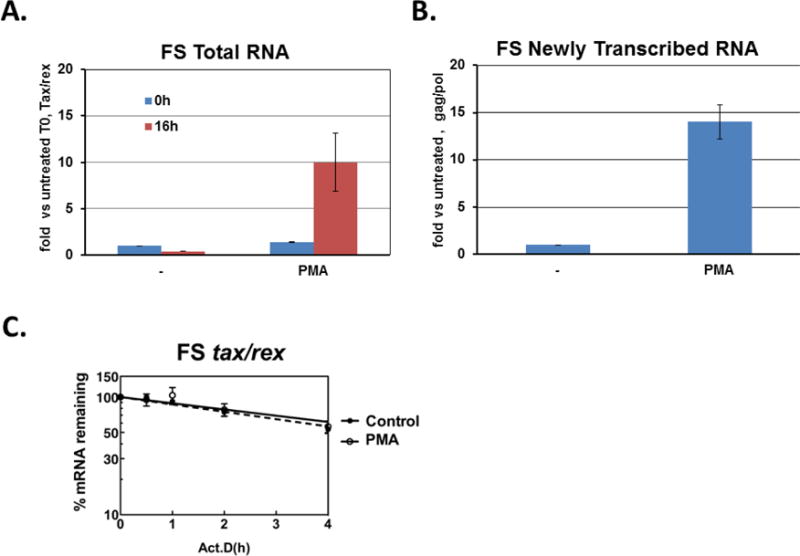
FS cells were treated with PMA for 15 min, washed, and re-cultured with fresh media. At 16 hours after resuspension, cells were either harvested for total RNA (A), Nuclear run on assay (B) or treated with Act D for RNA decay assay (C). (A) RT-qPCR analysis of tax/rex mRNA levels. Data are normalized to Ct values for ASL and divided by the value obtained for the zero time point. Average fold change with standard deviation (error bars) from triplicate inductions are shown. (B) Nuclear run-on assay for measuring newly transcribed mRNA. Nuclei were isolated, and incubated with biotinylated uridine and biotinylated transcripts were isolated. Transcripts corresponding to full length HTLV-1 and ASL were detected by RT-qPCR, and the data normalized to ASL. Results represent the fold change over the value obtained for control treatment. Average fold change with standard deviation (error bars) from triplicate inductions are shown. (C) RNA decay assay for RNA stability. RNAs were collected at the times after Act D treatment, as indicated, and measured by RT-qPCR with specific primers for tax/rex mRNA. Data are normalized to ASL, and presented as percentage mRNA remaining compared with time zero (closed circle: untreated, open circle: PMA treated). Data shown are average of triplicates with standard deviations (error bars).
To further understand the basis of these changes in HTLV-1 RNA and protein levels, it was important to know if PMA treatment induced changes in cell proliferation and viability. A 15 minute PMA treatment resulted in a significant decrease in the number of viable FS cells at 24 hours as compared with untreated cells (PMA treated: 1 × 105 cells/ml; untreated: 5 × 105 cells/ml). This reduced cell number is in part due to the induction of a senescent phenotype in PMA-treated cells population (Supplementary File 1, Supplementary Fig. S6). Western blots showed that PMA treatment, with the resultant increased expression of Tax, induced increased expression of p21 (CDKN1A) and p27 (CDKN1B), classic markers of senescence, previously shown to be induced by HTLV-1 Tax (Zhang et al., 2009; Zhi et al., 2011). FS cells showed a low, constitutive level of PARP cleavage, unchanged by PMA treatment, and no evidence of caspase 3 cleavage, basally or following PMA treatment. Thus, PMA treatment and the resultant Tax protein expression resulted in expression of senescent markers, but no evidence of apoptosis was observed.
As PKC activation leads to activation of a number of key downstream signaling pathways in T cells, including NF-κB, NFAT, and AP-1 [reviewed in (Isakov and Altman, 2002)], we were interested in whether inhibition of these would, in turn, block PMA-induced HTLV-1 gene expression. Pretreatment of FS cells with small molecule inhibitors of the NF-κB, NFAT, and AP-1 pathways did not reduce the PMA-induced increase in tax/rex mRNA (Supplementary File S1, Supplementary Fig. S7). Thus, these critical cellular signaling pathways are not likely to play a role in the PMA-induced, stabilization of tax/rex mRNA.
4. Discussion
Our studies have focused on understanding the mechanisms through which signaling in CD4 T cells can induce expression of the HTLV-1 Tax protein in latently infected cells. We have previously shown that signals induced by TCR activation can induce HTLV-1 gene expression (Lin et al., 1998; Lin et al., 2005) providing a possible mechanism for induction of HTLV-1 gene expression in chronically infected cells in vivo. In the current studies, we have observed that PMA, a T cell signaling mimetic acting through PKC activation, can increase the stability of the HTLV-1 tax/rex mRNA. In the context of a latently infected cell, we hypothesize that TCR activation will induce PKC activation, which leads to specific stabilization of the viral tax/rex mRNA (Fig. 6). Increased tax/rex mRNA would then mediate increased production of these two viral “early” proteins. Increased Tax would lead to a positive feedback loop through effects on the HTLV-1 LTR, resulting in markedly enhanced de novo transcription of HTLV-1 mRNAs (as seen in our nuclear run-on experiments). Furthermore, increased Tax could also induce the large numbers of cellular genes that are targets of Tax-activated transcription, such as NF-κB targets and many others with multiple potential effects on infected cell biology. Increases in Rex would result in the transition from “early” to “late” HTLV-1 gene expression, and production of viral structural and enzymatic proteins (Cavallari et al., 2013; Hidaka et al., 1988; Li et al., 2009), as well as potentially of more Tax and Rex themselves (Bai et al., 2012). Induction of HTLV-1 gene expression could in turn lead to diverse biological outcomes for the infected cell as described above, which could either facilitate immune clearance of infected cells or enhance the ability of the cell to survive and proliferate.
Figure 6. Model of PMA induction of HTLV-1 gene expression in inducible HTLV-1 T cell lines.
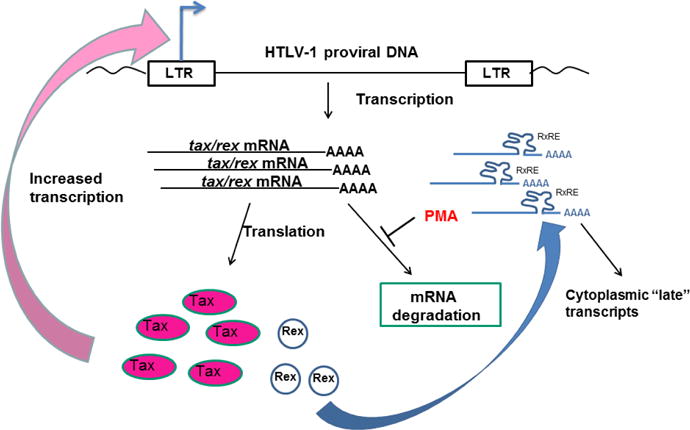
In this model, PMA treatment (as a surrogate for T cell activation signals) results in increased tax/rex mRNA stability with resultant increases in translation of Tax and Rex proteins. Increased Tax in turn feeds back to the LTR increasing de novo HTLV-1 transcription, while Rex increases the nuclear-cytoplasmic transport of unspliced and singly spliced HTLV-1 RNAs.
Based on analogy with other retroviruses, such as HIV, we initially assumed that the induction of tax/tex mRNA by T cell activation-related stimuli was due to transcriptional activation. Surprisingly, our nuclear run-on experiments demonstrated only a relatively modest degree of de novo transcription induced by phorbol ester treatment compared to a robust increase in RNA levels. A striking feature of the HTLV-1 RNA induction studies was the dramatic decrease in tax/rex mRNA levels over time following the initial stimulation. Together, these results suggested that T cell activation-related stimuli, such as PKC activation, might be exerting their predominant effect through regulation of HTLV-1 RNA stability. In fact, a central observation of our studies was a marked increase in the half-life of the HTLV-1 tax/rex mRNA, which was not seen for envelope or full-length RNAs.
At this time, we do not yet understand the mechanisms responsible for increased tax/rex RNA stability or for the specificity of this effect. It will be important to determine the RNA nucleotide sequences that are critical for increased tax/rex mRNA stability. With the exception of discontinuous sequence juxtapositions at splice sites, all of the nucleotide sequences in the tax/rex mRNA are present in full-length and env mRNAs. Thus, either the specific unique splice junction sequences or unique secondary structure elements in the tax/rex mRNAs are likely responsible for the specific stabilization of this message. An RNA element recently shown to be required for Rex dependent mRNA stability is not likely to play a role in the stabilization of the tax/rex mRNA, as it maps to an intronic region not present in this message (Cavallari et al., 2016).
Relatively little is know about the roles of RNA stability in the general regulation of viral latency and reactivation. RNA stability was reported to be important for activation of latent HIV (Micheva-Viteva et al., 2011), however cis-acting RNA stability-regulating sequences were identified in gag-pol, but not in tat mRNA (Schwartz et al., 1992). More recently, viral RNA stability as a key mediator of latency and reactivation was also described for the Kaposi’s associated herpesvirus (KSHV). The KSHV noncoding polyadenylated nuclear RNA (PAN) accumulates to very high levels during the activation of the lytic cycle out of latency, due to both transcriptional induction and increased PAN RNA stability (Conrad, 2016).
The molecular mechanisms responsible for both the relative instability of tax/rex RNA in unstimulated SP or FS cells, and its markedly increased stability following stimulation of these cells, as well in unstimulated, constitutively expressing cells (HUT102, MT2 and MT4) are also not known. Increased RNA stability is an important mechanism for the control of gene expression in the immune system and during the process of normal T cell activation (Chen et al., 1998; Khabar, 2007; Lindstein et al., 1989; Miller et al., 2009; Turner and Hodson, 2012; Wu and Brewer, 2012). It is likely that HTLV-1 has adapted these normal T cell mechanisms for control of its gene expression, and constitutive mRNA stability in HTLV-1-expressing cells could reflect a more activated T cell phenotype as compared with FS and SP cells. Likely candidate mechanisms would include cellular pathways regulating RNA stability through AU-rich, GU-rich, CA-rich or CU-rich sequences in the 3′ untranslated RNA sequences, all of which have been implicated in activation-associated stabilization of cytokine- and T cell activation marker RNAs. In particular, AU-binding proteins play an important role in stabilizing RNAs following T cell activation (Raghavan et al., 2004; Raghavan et al., 2001), and PKC activation has been associated with phosphorylation of AUF1 isoforms and mRNA stabilization (White et al., 2013). PKC activation, as induced by PMA, is known to induce the stabilization of cellular mRNAs containing AU-rich elements bound by AUF1 and other AU-binding proteins. Phorbol ester treatment with resultant PKC activation is associated with increased mRNA stability of a number of cellular genes. For example, phorbol ester treatment of monocytic cells induced rapid stabilization of ARE-containing cytokine mRNAs through changes in phosphorylation of AUF1, changes in binding of the AUF1 complex to these mRNAs and remodeling of RNA structure (Wilson et al., 2003a; Wilson et al., 2003b). PKC activation has also been associated with phosphorylation and altered function of other proteins regulating mRNA stability, such as HuR, and modulation of PKC-regulated mRNA stability has been suggested as a therapeutic target for a number of malignant and degenerative diseases (Eberhardt et al., 2007).
Another potential mechanism regulating the stability of tax/rex mRNA is nonsense-mediated decay (NMD), a process which poses a serious handicap for viruses such as HTLV-1, with multiple overlapping reading frames and premature termination codons. NMD is inhibited in HTLV-1 constitutively-expressing cell lines, including HUT102, and tax/rex as well as full-length and env transcripts were found to be sensitive to NMD (Mocquet et al., 2012; Nakano et al., 2013). Both HTLV-1 Tax (Mocquet et al., 2012) and Rex (Nakano et al., 2013) have been shown to inhibit NMD, thus, in our system there exists the potential for a positive feedback loop in which expression of small amounts of Tax and Rex proteins may inhibit NMD. The cellular microRNA (miRNA) machinery has also emerged as a critical modulator of the stability of endogenous cellular mRNAs [reviewed in (Jonas and Izaurralde, 2015; Wu and Brewer, 2012)]. Previous reports have suggested that the HTLV-1 tax/rex mRNA maybe the target of cellular miRNAs (Hakim et al., 2008; Ruggero et al., 2010), which could lead to instability of this message. On the other hand, both Tax and Rex have been shown to interfere with the cellular microRNA machinery (Ruggero et al., 2010; Van Duyne et al., 2012), again suggesting potential positive feedback loops that would enhance tax/rex mRNA stability.
HTLV-1 has devoted considerable effort to regulating the stability and processing of its RNA transcripts. The Rex protein has well-known roles in enhancing the nuclear export of unspliced and singly spliced RNAs thus ensuring their stability and availability for translation (reviewed in (Baydoun et al., 2008; Nakano and Watanabe, 2012)). In contrast, the p30 gene (encoded by a multiply spliced RNA from the X region), has both transcriptional and post-transcriptional effects, including retention of the tax/rex mRNA in the nucleus through inhibition of export (Anupam et al., 2013; Younis et al., 2006). This latter activity has been proposed to have an important role in maintaining viral latency (Baydoun et al., 2008). In our experiments the levels of p30 RNA were very low at baseline, and then markedly increased following activation of latently infected cells (Fig 1). Thus, p30 is not likely to be a determinant of the maintenance of FS and SP cell latency.
The marked reduction of the HTLV-1 tax/rex mRNA after the initial dramatic and transient increase following PMA treatment is particularly interesting. While some of this effect may be accounted for by more general cellular effects of PMA (including the induction of senescence, and likely a significant degree of non-apoptotic cell death (Supplementary File 1, Supplementary Fig. S6), these more general effects are unlikely to explain the loss of tax/rex mRNA. Other HTLV-1 RNAs, such as intronic gag/pol, p30, and env, did not show this dramatic loss, and furthermore, RNA levels were normalized to the constitutive cellular gene ASL, again suggesting some specificity in the decrease of tax/rex mRNA. The functional effects of Rex in inhibiting splicing through rapid nuclear export of Rex response element (RRE)-containing RNAs (Nakano and Watanabe, 2012) may be a strong contributor to the loss of tax/rex mRNA. In this model, as increasing amounts of Rex are available to bind to the HTLV-1 RNA RRE, the complete splicing of HTLV-1 RNA required to generate tax/rex mRNA would be reduced leading to the observed loss in abundance of this message following its initial induction.
Although HBZ has been suggested to be an important regulator of HTLV-1 latency (Clerc et al., 2008; Gaudray et al., 2002; Lemasson et al., 2007; Philip et al., 2014), only very low levels of HBZ RNA were detected basally and exhibited a very small (< 2-fold) increase upon PMA treatment, suggesting that HBZ expression is not a major contributor to either maintenance of HTLV-1 latency or induction of expression in these chronically-infected cells.
What role might immune activation-induced HTLV-1 expression play in HTLV-1 biology and pathogenesis? HTLV-1 latency in vivo represents a way for virally-infected cells to escape potent anti-HTLV-1 CTL responses and persist in the infected individual. However, expression of viral gene products does play a key role in the pathogenesis of HTLV-1-induced diseases. Thus, mechanisms that lead to activation of viral gene expression are critically important. These studies coupled with our previous work (Lin et al., 1998; Lin et al., 2005) suggest a model whereby TCR-mediated activation (potentially either through cognate antigen or superantigen) of infected CD4 T cells leads to induced tax/rex mRNA stability with generation of increased Tax and Rex proteins. We have hypothesized that specific TCR activation of infected cells may promote viral gene expression and clonal expansion of these subsets of infected cells (Lin et al., 1998; Lin et al., 2005). Bangham and colleagues have presented a considerable evidence that the site and direction of HTLV-1 integration is important in determining HTLV-1 biology (Cook et al., 2014; Gillet et al., 2011; Melamed et al., 2013; Niederer et al., 2014). Importantly, this does not conflict with our model, which focuses on post-transcriptional regulation of HTLV-1 RNA stability. T cell activation would result in stabilization of tax/rex mRNAs expressed due to any of multiple mechanisms.
Although the functional importance of latent HTLV-1 infection remains unclear, the induction of HTLV-1 gene products, particularly Tax, from latently (or low level-expressing) HTLV-1 infected cells may alter the biology of these cells leading to alternative outcomes ranging from apoptosis, senescence, or death due to cytotoxic T cells, to cellular proliferation and transformation. In other retroviral infections, such as HIV infection, activation of viral gene expression from the pool of latently infected cells is an important contributor to the long-term biology of infection and the inability to “cure” patients (Archin et al., 2014; Blankson et al., 2014). Thus, better understandings of the mechanisms that regulate the expression of HTLV-1 genes in chronically- or latently- infected cells may provide important clues to both pathogenesis and to therapy.
Supplementary Material
Supplementary file 1: Figure S1: The arginosuccinate lyase (ASL) gene promoter is insensitive to Tax or PHA/PMA activation. (A) Semi-quantitative PCR amplifications of cDNA derived from JPX-9 and JE6-1 Jurkat cell lines treated with zinc chloride and/or PHA. Aliquots of the PCR reaction, using primers specific for tax/rex mRNA, ASL or IL-2Rα, were removed at the indicated cycle number and electrophoresed on an agarose gel. Zinc-mediated induction of Tax (JPX-9) or PHA treatment (JPX-9 and JE6-1) fail to increase ASL RNA levels. (B) Transient co-transfection of JE6-1 cells with either pLTR-Luc or pASL-Luc, and pLTR-Tax or the respective control, pLTR -DeltaTax (denoted as DTax). The results are displayed as luciferase activity normalized to total protein. Error bars indicate standard deviation of triplicate reactions. The ASL promoter is essentially unresponsive to Tax, PHA, or PMA.
Supplementary file 1: Figure S2: Structure of HTLV-1 genome and transcripts. Schematic representation of HTLV-1 genome and transcripts. Positions of primer sets for RT-qPCR are indicated as red arrows.
Supplementary file 1: Figure S3: Time courses of HTLV-1 mRNA expression with PMA treatment. Three independent treatments of FS cells with PMA were performed, as described in Fig. 1. Cells were treated with PMA for one hour, washed and re-suspended in fresh media. RNAs were collected at time points indicated and levels of individual HTLV-1 transcripts were measured as described. Time courses for untreated parallel samples were also performed for repeats 2 and 3.
Supplementary file 1: Figure S4: Western blots for protein extracts of untreated and PMA treated SP cells. The western blots of HTLV-1 proteins in SP cells (left panel, from Figure 1) were subjected to quantitative analysis (right panels), normalized to actin and divided by the value obtained for the zero time point.
Supplementary file 1: Figure S5: PMA does not change tax/rex RNA stability in MT4 and MT2 HTLV-1 T cell lines. HTLV-1 infected MT4 (A) and MT2 (B) cells were treated with PMA for 30 min, washed, and then treated with Act D, and RNA decay assays were performed as described above. Tax/rex mRNAs were measured with splice site-specific primers. Data were normalized to ASL, and presented as percentage mRNA remaining compared with time zero (closed circle: untreated, open circle: PMA treated). Data shown are average of triplicates with standard deviations (error bars). Similar to the results for HUT102 cells (Fig. 4), PMA does not change tax/rex mRNA stability in MT2 and MT4 cells.
Supplementary file 1: Figure S6: Western blot analysis of PMA-treated FS cells for cell senescence and apoptosis markers. FS cells were treated with PMA for 15′, washed, and cultured with fresh media for 24 hours. Protein lysates were prepared and western blot analyses were performed. (A) Western blots for expression of p21 (CDKN1A) and p27 (CDKN1B) proteins, markers of senescence. (B) Western blots for expression of apoptosis markers; cleaved PARP and cleaved caspase-3. Treatment of Jurkat cells with anti-CD95 (2mg/ml for 4 hours) induced cleaved PARP and caspase-3, and was used as a positive control for apoptosis. PMA treatment resulted in expression of senescent markers, p21 and p27 (A), but did not induce apoptosis (B).
Supplementary file1: Figure S7: Effects of inhibitors of the NFAT, NF-κB or AP-1 pathways on levels of PMA-induced tax/rex mRNA. FS cells were pre-treated with Bay 11-7082 (NF-κB pathway inhibitor), Cyclosporin A (NFAT pathway inhibitor) or SR 11302 (AP1 pathway inhibitor) for 1 hr, followed by 15 minutes of PMA treatment. Cells were then washed, resuspended with fresh media and cultured for 4 hours. Tax/rex mRNA was detected by RT –PCR. Data are normalized to Ct values for ASL mRNA, and divided by the value obtained for the untreated control. Average fold change with standard deviation (error bars) from triplicate treatments are shown. Inhibitors of these pathways did not reduce the levels of PMA-induced tax/rex mRNA
Highlights.
Induction of HTLV-1 expression from latently-infected CD4 T cells by phorbol ester stimulation leads to a rapid, transient increase in tax/rex mRNA levels.
Increased tax/rex mRNA in latently infected T cells is due to increased tax/rex mRNA stability following T cell stimulation.
Increased tax/rex mRNA leads to increased Tax and Rex protein expression with resulting feedback induction of the RNAs encoding HTLV-1 structural and enzymatic proteins.
These results demonstrate a new mechanism for activation of latent HTLV-1 from infected T cells, potentially contributing to the pathogenesis of HTLV-1-associated diseases.
Acknowledgments
We thank Dr. Patrick Ng (formerly at NIH) for sharing data from Tax-induced gene microarray experiments, Dr. Benoit Barbeau (Université of Québec at Montreal) for sharing HBZ primer sequences and helpful discussions, Dr. Steven Foung (Stanford University) for anti-HTLV-1 envelope antibody, and Dr. Patrick Green (Ohio State University) for anti-HTLV-1 Rex antibody. We are very grateful to Dr. Joseph Dougherty (Robert Wood Johnson Medical School) and Dr. Steven Jacobson (NINCDS, NIH) for helpful discussions
Funding information
This research was supported by grants to ABR from the National Cancer Institute National Institutes of Health, RO1 CA94148-05, to GB from the National Cancer Institute CA052443, and by research support of the Child Health Institute of New Jersey provided by the Robert Wood Johnson Foundation, grant # 67038. The funders had no role in study design, data collection and interpretation, or decisions regarding submission for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they have no conflict of interest.
References
- Abramson RD, Barbosa P, Kalumuck K, O’Brien WE. Characterization of the human argininosuccinate lyase gene and analysis of exon skipping. Genomics. 1991;10:126–132. doi: 10.1016/0888-7543(91)90492-w. [DOI] [PubMed] [Google Scholar]
- Andrews JM, Oglesbee MJ, Trevino AV, Guyot DJ, Newbound GC, Lairmore MD. Enhanced human T-cell lymphotropic virus type I expression following induction of the cellular stress response. Virology. 1995;208:816–820. doi: 10.1006/viro.1995.1218. [DOI] [PubMed] [Google Scholar]
- Anupam R, Doueiri R, Green PL. The need to accessorize: molecular roles of HTLV-1 p30 and HTLV-2 p28 accessory proteins in the viral life cycle. Frontiers in microbiology. 2013;4:275. doi: 10.3389/fmicb.2013.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya N, Sato T, Yagishita N, Ando H, Utsunomiya A, Jacobson S, Yamano Y. Human T-lymphotropic virus type 1 (HTLV-1) and regulatory T cells in HTLV-1-associated neuroinflammatory disease. Viruses. 2011;3:1532–1548. doi: 10.3390/v3091532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin NM, Sung JM, Garrido C, Soriano-Sarabia N, Margolis DM. Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nat Rev Microbiol. 2014;12:750–764. doi: 10.1038/nrmicro3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold J, Yamamoto B, Li M, Phipps AJ, Younis I, Lairmore MD, Green PL. Enhancement of infectivity and persistence in vivo by HBZ, a natural antisense coded protein of HTLV-1. Blood. 2006;107:3976–3982. doi: 10.1182/blood-2005-11-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold J, Zimmerman B, Li M, Lairmore MD, Green PL. Human T-cell leukemia virus type-1 antisense-encoded gene, Hbz, promotes T-lymphocyte proliferation. Blood. 2008;112:3788–3797. doi: 10.1182/blood-2008-04-154286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asquith B, Hanon E, Taylor GP, Bangham CR. Is human T-cell lymphotropic virus type I really silent? Philos Trans R Soc Lond B Biol Sci. 2000;355:1013–1019. doi: 10.1098/rstb.2000.0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XT, Sinha-Datta U, Ko NL, Bellon M, Nicot C. Nuclear export and expression of human T-cell leukemia virus type 1 tax/rex mRNA are RxRE/Rex dependent. J Virol. 2012;86:4559–4565. doi: 10.1128/JVI.06361-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeau B, Mesnard JM. Making sense out of antisense transcription in human T-cell lymphotropic viruses (HTLVs) Viruses. 2011;3:456–468. doi: 10.3390/v3050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baydoun HH, Bellon M, Nicot C. HTLV-1 Yin and Yang: Rex and p30 master regulators of viral mRNA trafficking. AIDS Rev. 2008;10:195–204. [PMC free article] [PubMed] [Google Scholar]
- Blankson JN, Siliciano JD, Siliciano RF. Finding a cure for human immunodeficiency virus-1 infection. Infect Dis Clin North Am. 2014;28:633–650. doi: 10.1016/j.idc.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunn PA, Jr, Foss FM. T-cell lymphoma cell lines (HUT102 and HUT78) established at the National Cancer Institute: history and importance to understanding the biology, clinical features, and therapy of cutaneous T-cell lymphomas (CTCL) and adult T-cell leukemia-lymphomas (ATLL) J Cell Biochem Suppl. 1996;24:12–23. doi: 10.1002/jcb.240630503. [DOI] [PubMed] [Google Scholar]
- Carpentier A, Barez PY, Hamaidia M, Gazon H, de Brogniez A, Perike S, Gillet N, Willems L. Modes of Human T Cell Leukemia Virus Type 1 Transmission, Replication and Persistence. Viruses. 2015;7:3603–3624. doi: 10.3390/v7072793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallari I, Rende F, Bender C, Romanelli MG, D’Agostino DM, Ciminale V. Fine tuning of the temporal expression of HTLV-1 and HTLV-2. Frontiers in microbiology. 2013;4:235. doi: 10.3389/fmicb.2013.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallari I, Rende F, Bona MK, Sztuba-Solinska J, Silic-Benussi M, Tognon M, LeGrice SF, Franchini G, D’Agostino DM, Ciminale V. Expression of Alternatively Spliced Human T-Cell Leukemia Virus Type 1 mRNAs Is Influenced by Mitosis and by a Novel cis-Acting Regulatory Sequence. J Virol. 2016;90:1486–1498. doi: 10.1128/JVI.02298-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Del Gatto-Konczak F, Wu Z, Karin M. Stabilization of interleukin-2 mRNA by the c-Jun NH2-terminal kinase pathway. Science. 1998;280:1945–1949. doi: 10.1126/science.280.5371.1945. [DOI] [PubMed] [Google Scholar]
- Clerc I, Polakowski N, Andre-Arpin C, Cook P, Barbeau B, Mesnard JM, Lemasson I. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of tax-dependent viral transcription by HBZ. J Biol Chem. 2008;283:23903–23913. doi: 10.1074/jbc.M803116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad NK. New insights into the expression and functions of the Kaposi’s sarcoma-associated herpesvirus long noncoding PAN RNA. Virus Res. 2016;212:53–63. doi: 10.1016/j.virusres.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook LB, Melamed A, Niederer H, Valganon M, Laydon D, Foroni L, Taylor GP, Matsuoka M, Bangham CR. The role of HTLV-1 clonality, proviral structure, and genomic integration site in adult T-cell leukemia/lymphoma. Blood. 2014;123:3925–3931. doi: 10.1182/blood-2014-02-553602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dezzutti CS, Rudolph DL, Roberts CR, Lal RB. Characterization of human T-lymphotropic virus type I- and II-infected T-cell lines: antigenic, phenotypic, and genotypic analysis. Virus Res. 1993;29:59–70. doi: 10.1016/0168-1702(93)90125-7. [DOI] [PubMed] [Google Scholar]
- Dumais N, Pare ME, Mercier S, Bounou S, Marriot SJ, Barbeau B, Tremblay MJ. T-cell receptor/CD28 engagement when combined with prostaglandin E2 treatment leads to potent activation of human T-cell leukemia virus type 1. J Virol. 2003;77:11170–11179. doi: 10.1128/JVI.77.20.11170-11179.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt W, Doller A, Akool el S, Pfeilschifter J. Modulation of mRNA stability as a novel therapeutic approach. Pharmacol Ther. 2007;114:56–73. doi: 10.1016/j.pharmthera.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Fanjul A, Dawson MI, Hobbs PD, Jong L, Cameron JF, Harlev E, Graupner G, Lu XP, Pfahl M. A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature. 1994;372:107–111. doi: 10.1038/372107a0. [DOI] [PubMed] [Google Scholar]
- Ford GS, Barnhart B, Shone S, Covey LR. Regulation of CD154 (CD40 ligand) mRNA stability during T cell activation. J Immunol. 1999;162:4037–4044. [PubMed] [Google Scholar]
- Gallo RC. History of the discoveries of the first human retroviruses: HTLV-1 and HTLV-2. Oncogene. 2005;24:5926–5930. doi: 10.1038/sj.onc.1208980. [DOI] [PubMed] [Google Scholar]
- Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol. 2002;76:12813–12822. doi: 10.1128/JVI.76.24.12813-12822.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessain A, Cassar O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Frontiers in microbiology. 2012;3:388. doi: 10.3389/fmicb.2012.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessain A, Louie A, Gout O, Gallo RC, Franchini G. Human T-cell leukemia-lymphoma virus type I (HTLV-I) expression in fresh peripheral blood mononuclear cells from patients with tropical spastic paraparesis/HTLV-I-associated myelopathy. J Virol. 1991;65:1628–1633. doi: 10.1128/jvi.65.3.1628-1633.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet NA, Malani N, Melamed A, Gormley N, Carter R, Bentley D, Berry C, Bushman FD, Taylor GP, Bangham CR. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood. 2011;117:3113–3122. doi: 10.1182/blood-2010-10-312926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves DU, Proietti FA, Ribas JG, Araujo MG, Pinheiro SR, Guedes AC, Carneiro-Proietti AB. Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin Microbiol Rev. 2010;23:577–589. doi: 10.1128/CMR.00063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyot DJ, Newbound GC, Lairmore MD. Signaling via the CD2 receptor enhances HTLV-1 replication in T lymphocytes. Virology. 1997;234:123–129. doi: 10.1006/viro.1997.8636. [DOI] [PubMed] [Google Scholar]
- Hadlock KG, Rowe J, Perkins S, Bradshaw P, Song GY, Cheng C, Yang J, Gascon R, Halmos J, Rehman SM, McGrath MS, Foung SK. Neutralizing human monoclonal antibodies to conformational epitopes of human T-cell lymphotropic virus type 1 and 2 gp46. J Virol. 1997;71:5828–5840. doi: 10.1128/jvi.71.8.5828-5840.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim ST, Alsayari M, McLean DC, Saleem S, Addanki KC, Aggarwal M, Mahalingam K, Bagasra O. A large number of the human microRNAs target lentiviruses, retroviruses, and endogenous retroviruses. Biochem Biophys Res Commun. 2008;369:357–362. doi: 10.1016/j.bbrc.2008.02.025. [DOI] [PubMed] [Google Scholar]
- Hanon E, Hall S, Taylor GP, Saito M, Davis R, Tanaka Y, Usuku K, Osame M, Weber JN, Bangham CR. Abundant tax protein expression in CD4+ T cells infected with human T-cell lymphotropic virus type I (HTLV-I) is prevented by cytotoxic T lymphocytes. Blood. 2000;95:1386–1392. [PubMed] [Google Scholar]
- Hidaka M, Inoue J, Yoshida M, Seiki M. Post-transcriptional regulator (rex) of HTLV-1 initiates expression of viral structural proteins but suppresses expression of regulatory proteins. EMBO J. 1988;7:519–523. doi: 10.1002/j.1460-2075.1988.tb02840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollsberg P. Mechanisms of T-cell activation by human T-cell lymphotropic virus type I. Microbiology and molecular biology reviews: MMBR. 1999;63:308–333. doi: 10.1128/mmbr.63.2.308-333.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakov N, Altman A. Protein kinase C(theta) in T cell activation. Annu Rev Immunol. 2002;20:761–794. doi: 10.1146/annurev.immunol.20.100301.064807. [DOI] [PubMed] [Google Scholar]
- Iwanaga M, Watanabe T, Yamaguchi K. Adult T-cell leukemia: a review of epidemiological evidence. Frontiers in microbiology. 2012;3:322. doi: 10.3389/fmicb.2012.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeang KT, Giam CZ, Nerenberg M, Khoury G. Abundant synthesis of functional human T-cell leukemia virus type I p40x protein in eucaryotic cells by using a baculovirus expression vector. J Virol. 1987;61:708–713. doi: 10.1128/jvi.61.3.708-713.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeang KT, Widen SG, Semmes OJT, Wilson SH. HTLV-I trans-activator protein, tax, is a trans-repressor of the human beta-polymerase gene. Science. 1990;247:1082–1084. doi: 10.1126/science.2309119. [DOI] [PubMed] [Google Scholar]
- Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- Kashanchi F, Brady JN. Transcriptional and post-transcriptional gene regulation of HTLV-1. Oncogene. 2005;24:5938–5951. doi: 10.1038/sj.onc.1208973. [DOI] [PubMed] [Google Scholar]
- Khabar KS. Rapid transit in the immune cells: the role of mRNA turnover regulation. Journal of leukocyte biology. 2007;81:1335–1344. doi: 10.1189/jlb.0207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasson I, Lewis MR, Polakowski N, Hivin P, Cavanagh MH, Thebault S, Barbeau B, Nyborg JK, Mesnard JM. Human T-cell leukemia virus type 1 (HTLV-1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV-1 transcription. J Virol. 2007;81:1543–1553. doi: 10.1128/JVI.00480-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasson I, Polakowski NJ, Laybourn PJ, Nyborg JK. Transcription regulatory complexes bind the human T-cell leukemia virus 5′ and 3′ long terminal repeats to control gene expression. Molecular and cellular biology. 2004;24:6117–6126. doi: 10.1128/MCB.24.14.6117-6126.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasson I, Polakowski NJ, Laybourn PJ, Nyborg JK. Tax-dependent displacement of nucleosomes during transcriptional activation of human T-cell leukemia virus type 1. J Biol Chem. 2006;281:13075–13082. doi: 10.1074/jbc.M512193200. [DOI] [PubMed] [Google Scholar]
- Li M, Kesic M, Yin H, Yu L, Green PL. Kinetic analysis of human T-cell leukemia virus type 1 gene expression in cell culture and infected animals. J Virol. 2009;83:3788–3797. doi: 10.1128/JVI.02315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Dezzutti CS, Lal RB, Rabson AB. Activation of human T-cell leukemia virus type 1 tax gene expression in chronically infected T cells. J Virol. 1998;72:6264–6270. doi: 10.1128/jvi.72.7.6264-6270.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Hickey M, Hsu L, Medina D, Rabson AB. Activation of human T cell leukemia virus type 1 LTR promoter and cellular promoter elements by T cell receptor signaling and HTLV-1 Tax expression. Virology. 2005;339:1–11. doi: 10.1016/j.virol.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Lindstein T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- Lu H, Pise-Masison CA, Linton R, Park HU, Schiltz RL, Sartorelli V, Brady JN. Tax relieves transcriptional repression by promoting histone deacetylase 1 release from the human T-cell leukemia virus type 1 long terminal repeat. J Virol. 2004;78:6735–6743. doi: 10.1128/JVI.78.13.6735-6743.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M, Jeang KT. Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: viral infectivity, Tax, HBZ and therapy. Oncogene. 2011;30:1379–1389. doi: 10.1038/onc.2010.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M, Yasunaga J. Human T-cell leukemia virus type 1: replication, proliferation and propagation by Tax and HTLV-1 bZIP factor. Current opinion in virology. 2013;3:684–691. doi: 10.1016/j.coviro.2013.08.010. [DOI] [PubMed] [Google Scholar]
- McCaffrey PG, Perrino BA, Soderling TR, Rao A. NF-ATp, a T lymphocyte DNA-binding protein that is a target for calcineurin and immunosuppressive drugs. J Biol Chem. 1993;268:3747–3752. [PubMed] [Google Scholar]
- Melamed A, Laydon DJ, Gillet NA, Tanaka Y, Taylor GP, Bangham CR. Genome-wide determinants of proviral targeting, clonal abundance and expression in natural HTLV-1 infection. PLoS Pathog. 2013;9:e1003271. doi: 10.1371/journal.ppat.1003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesnard JM, Barbeau B, Devaux C. HBZ, a new important player in the mystery of adult T-cell leukemia. Blood. 2006;108:3979–3982. doi: 10.1182/blood-2006-03-007732. [DOI] [PubMed] [Google Scholar]
- Micheva-Viteva S, Kobayashi Y, Edelstein LC, Pacchia AL, Lee HL, Graci JD, Breslin J, Phelan BD, Miller LK, Colacino JM, Gu Z, Ron Y, Peltz SW, Dougherty JP. High-throughput screening uncovers a compound that activates latent HIV-1 and acts cooperatively with a histone deacetylase (HDAC) inhibitor. J Biol Chem. 2011;286:21083–21091. doi: 10.1074/jbc.M110.195537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J, Baker C, Cook K, Graf B, Sanchez-Lockhart M, Sharp K, Wang X, Yang B, Yoshida T. Two pathways of costimulation through CD28. Immunologic research. 2009;45:159–172. doi: 10.1007/s12026-009-8097-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi I, Kubonishi I, Yoshimoto S, Akagi T, Ohtsuki Y, Shiraishi Y, Nagata K, Hinuma Y. Type C virus particles in a cord T-cell line derived by co-cultivating normal human cord leukocytes and human leukaemic T cells. Nature. 1981;294:770–771. doi: 10.1038/294770a0. [DOI] [PubMed] [Google Scholar]
- Mocquet V, Neusiedler J, Rende F, Cluet D, Robin JP, Terme JM, Duc Dodon M, Wittmann J, Morris C, Le Hir H, Ciminale V, Jalinot P. The human T-lymphotropic virus type 1 tax protein inhibits nonsense-mediated mRNA decay by interacting with INT6/EIF3E and UPF1. J Virol. 2012;86:7530–7543. doi: 10.1128/JVI.07021-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M, Osame M. Human T-cell lymphotropic virus type I and neurological diseases. J Neurovirol. 2003;9:228–235. doi: 10.1080/13550280390194028. [DOI] [PubMed] [Google Scholar]
- Nagata K, Ohtani K, Nakamura M, Sugamura K. Activation of endogenous c-fos proto-oncogene expression by human T-cell leukemia virus type I-encoded p40tax protein in the human T-cell line, Jurkat. J Virol. 1989;63:3220–3226. doi: 10.1128/jvi.63.8.3220-3226.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Ando T, Yamagishi M, Yokoyama K, Ishida T, Ohsugi T, Tanaka Y, Brighty DW, Watanabe T. Viral interference with host mRNA surveillance, the nonsense-mediated mRNA decay (NMD) pathway, through a new function of HTLV-1 Rex: implications for retroviral replication. Microbes Infect. 2013;15:491–505. doi: 10.1016/j.micinf.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Nakano K, Watanabe T. HTLV-1 Rex: the courier of viral messages making use of the host vehicle. Frontiers in microbiology. 2012;3:330. doi: 10.3389/fmicb.2012.00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerenberg M, Hinrichs SH, Reynolds RK, Khoury G, Jay G. The tat gene of human T-lymphotropic virus type 1 induces mesenchymal tumors in transgenic mice. Science. 1987;237:1324–1329. doi: 10.1126/science.2888190. [DOI] [PubMed] [Google Scholar]
- Ng PW, Iha H, Iwanaga Y, Bittner M, Chen Y, Jiang Y, Gooden G, Trent JM, Meltzer P, Jeang KT, Zeichner SL. Genome-wide expression changes induced by HTLV-1 Tax: evidence for MLK-3 mixed lineage kinase involvement in Tax-mediated NF-kappaB activation. Oncogene. 2001;20:4484–4496. doi: 10.1038/sj.onc.1204513. [DOI] [PubMed] [Google Scholar]
- Niederer HA, Laydon DJ, Melamed A, Elemans M, Asquith B, Matsuoka M, Bangham CR. HTLV-1 proviral integration sites differ between asymptomatic carriers and patients with HAM/TSP. Virol J. 2014;11:172. doi: 10.1186/1743-422X-11-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Ohashi T, Nishikawa K, Nishitsuji H, Kurihara K, Hasegawa A, Furuta RA, Fujisawa J, Tanaka Y, Hanabuchi S, Harashima N, Masuda T, Kannagi M. Repression of tax expression is associated both with resistance of human T-cell leukemia virus type 1-infected T cells to killing by tax-specific cytotoxic T lymphocytes and with impaired tumorigenicity in a rat model. J Virol. 2004;78:3827–3836. doi: 10.1128/JVI.78.8.3827-3836.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyborg JK, Egan D, Sharma N. The HTLV-1 Tax protein: revealing mechanisms of transcriptional activation through histone acetylation and nucleosome disassembly. Biochim Biophys Acta. 2010;1799:266–274. doi: 10.1016/j.bbagrm.2009.09.002. [DOI] [PubMed] [Google Scholar]
- O’Brien WE, McInnes R, Kalumuck K, Adcock M. Cloning and sequence analysis of cDNA for human argininosuccinate lyase. Proc Natl Acad Sci U S A. 1986;83:7211–7215. doi: 10.1073/pnas.83.19.7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrone G, Puppo F, Cusano R, Scaranari M, Ceccherini I, Puliti A, Ravazzolo R. Nuclear run-on assay using biotin labeling, magnetic bead capture and analysis by fluorescence-based RT-PCR. Biotechniques. 2000;29:1012–1014. 1016–1017. doi: 10.2144/00295st02. [DOI] [PubMed] [Google Scholar]
- Peloponese JM, Jr, Kinjo T, Jeang KT. Human T-cell leukemia virus type 1 Tax and cellular transformation. Int J Hematol. 2007;86:101–106. doi: 10.1532/IJH97.07087. [DOI] [PubMed] [Google Scholar]
- Philip S, Zahoor MA, Zhi H, Ho YK, Giam CZ. Regulation of human T-lymphotropic virus type I latency and reactivation by HBZ and Rex. PLoS Pathog. 2014;10:e1004040. doi: 10.1371/journal.ppat.1004040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- Powell JD, Ragheb JA, Kitagawa-Sakakida S, Schwartz RH. Molecular regulation of interleukin-2 expression by CD28 co-stimulation and anergy. Immunol Rev. 1998;165:287–300. doi: 10.1111/j.1600-065x.1998.tb01246.x. [DOI] [PubMed] [Google Scholar]
- Raghavan A, Dhalla M, Bakheet T, Ogilvie RL, Vlasova IA, Khabar KS, Williams BR, Bohjanen PR. Patterns of coordinate down-regulation of ARE-containing transcripts following immune cell activation. Genomics. 2004;84:1002–1013. doi: 10.1016/j.ygeno.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Raghavan A, Robison RL, McNabb J, Miller CR, Williams DA, Bohjanen PR. HuA and tristetraprolin are induced following T cell activation and display distinct but overlapping RNA binding specificities. J Biol Chem. 2001;276:47958–47965. doi: 10.1074/jbc.M109511200. [DOI] [PubMed] [Google Scholar]
- Richardson JH, Hollsberg P, Windhagen A, Child LA, Hafler DA, Lever AM. Variable immortalizing potential and frequent virus latency in blood-derived T-cell clones infected with human T-cell leukemia virus type I. Blood. 1997;89:3303–3314. [PubMed] [Google Scholar]
- Rowe T, Dezzutti C, Guenthner PC, Lam L, Hodge T, Lairmore MD, Lal RB, Folks TM. Characterization of a HTLV-I-infected cell line derived from a patient with adult T-cell leukemia with stable co-expression of CD4 and CD8. Leuk Res. 1995;19:621–628. doi: 10.1016/0145-2126(95)00030-r. [DOI] [PubMed] [Google Scholar]
- Ruggero K, Corradin A, Zanovello P, Amadori A, Bronte V, Ciminale V, D’Agostino DM. Role of microRNAs in HTLV-1 infection and transformation. Mol Aspects Med. 2010;31:367–382. doi: 10.1016/j.mam.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Satou Y, Yasunaga J, Yoshida M, Matsuoka M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci U S A. 2006;103:720–725. doi: 10.1073/pnas.0507631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Felber BK, Pavlakis GN. Distinct RNA sequences in the gag region of human immunodeficiency virus type 1 decrease RNA stability and inhibit expression in the absence of Rev protein. J Virol. 1992;66:150–159. doi: 10.1128/jvi.66.1.150-159.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siekevitz M, Feinberg MB, Holbrook N, Wong-Staal F, Greene WC. Activation of interleukin 2 and interleukin 2 receptor (Tac) promoter expression by the trans-activator (tat) gene product of human T-cell leukemia virus, type I. Proc Natl Acad Sci U S A. 1987;84:5389–5393. doi: 10.1073/pnas.84.15.5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaims AY, Khani F, Zhang Y, Roberts AI, Devadas S, Shi Y, Rabson AB. Immune activation induces immortalization of HTLV-1 LTR-Tax transgenic CD4+ T cells. Blood. 2010;116:2994–3003. doi: 10.1182/blood-2009-07-231050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanajura D, Castro N, Oliveira P, Neto A, Muniz A, Carvalho NB, Orge G, Santos S, Glesby MJ, Carvalho EM. Neurological Manifestations in Human T-Cell Lymphotropic Virus Type 1 (HTLV-1)-Infected Individuals Without HTLV-1-Associated Myelopathy/Tropical Spastic Paraparesis: A Longitudinal Cohort Study. Clin Infect Dis. 2015;61:49–56. doi: 10.1093/cid/civ229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner M, Hodson D. Regulation of lymphocyte development and function by RNA-binding proteins. Current opinion in immunology. 2012;24:160–165. doi: 10.1016/j.coi.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Van Duyne R, Guendel I, Klase Z, Narayanan A, Coley W, Jaworski E, Roman J, Popratiloff A, Mahieux R, Kehn-Hall K, Kashanchi F. Localization and sub-cellular shuttling of HTLV-1 tax with the miRNA machinery. PLoS One. 2012;7:e40662. doi: 10.1371/journal.pone.0040662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva R, Iglesias AH, Camelo S, Sanin LC, Gray SG, Dangond F. Histone deacetylase 3 represses HTLV-1 tax transcription. Oncol Rep. 2006;16:581–585. [PubMed] [Google Scholar]
- Wattel E, Vartanian JP, Pannetier C, Wain-Hobson S. Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J Virol. 1995;69:2863–2868. doi: 10.1128/jvi.69.5.2863-2868.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss A, Wiskocil RL, Stobo JD. The role of T3 surface molecules in the activation of human T cells: a two-stimulus requirement for IL 2 production reflects events occurring at a pre-translational level. J Immunol. 1984;133:123–128. [PubMed] [Google Scholar]
- White EJ, Brewer G, Wilson GM. Post-transcriptional control of gene expression by AUF1: mechanisms, physiological targets, and regulation. Biochim Biophys Acta. 2013;1829:680–688. doi: 10.1016/j.bbagrm.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GM, Lu J, Sutphen K, Suarez Y, Sinha S, Brewer B, Villanueva-Feliciano EC, Ysla RM, Charles S, Brewer G. Phosphorylation of p40AUF1 regulates binding to A + U-rich mRNA-destabilizing elements and protein-induced changes in ribonucleoprotein structure. J Biol Chem. 2003a;278:33039–33048. doi: 10.1074/jbc.M305775200. [DOI] [PubMed] [Google Scholar]
- Wilson GM, Lu J, Sutphen K, Sun Y, Huynh Y, Brewer G. Regulation of A + U-rich element-directed mRNA turnover involving reversible phosphorylation of AUF1. J Biol Chem. 2003b;278:33029–33038. doi: 10.1074/jbc.M305772200. [DOI] [PubMed] [Google Scholar]
- Wu X, Brewer G. The regulation of mRNA stability in mammalian cells: 2.0. Gene. 2012;500:10–21. doi: 10.1016/j.gene.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi M, Watanabe T. Molecular hallmarks of adult T cell leukemia. Frontiers in microbiology. 2012;3:334. doi: 10.3389/fmicb.2012.00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga J, Matsuoka M. Human T-cell leukemia virus type I induces adult T-cell leukemia: from clinical aspects to molecular mechanisms. Cancer control: journal of the Moffitt Cancer Center. 2007;14:133–140. doi: 10.1177/107327480701400206. [DOI] [PubMed] [Google Scholar]
- Ye J, Silverman L, Lairmore MD, Green PL. HTLV-1 Rex is required for viral spread and persistence in vivo but is dispensable for cellular immortalization in vitro. Blood. 2003;102:3963–3969. doi: 10.1182/blood-2003-05-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M. Discovery of HTLV-1, the first human retrovirus, its unique regulatory mechanisms, and insights into pathogenesis. Oncogene. 2005;24:5931–5937. doi: 10.1038/sj.onc.1208981. [DOI] [PubMed] [Google Scholar]
- Younis I, Boris-Lawrie K, Green PL. Human T-cell leukemia virus open reading frame II encodes a posttranscriptional repressor that is recruited at the level of transcription. J Virol. 2006;80:181–191. doi: 10.1128/JVI.80.1.181-191.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ysla RM, Wilson GM, Brewer G. Chapter 3. Assays of adenylate uridylate-rich element-mediated mRNA decay in cells. Methods Enzymol. 2008;449:47–71. doi: 10.1016/S0076-6879(08)02403-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhi H, Liu M, Kuo YL, Giam CZ. Induction of p21(CIP1/WAF1) expression by human T-lymphotropic virus type 1 Tax requires transcriptional activation and mRNA stabilization. Retrovirology. 2009;6:35. doi: 10.1186/1742-4690-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi H, Yang L, Kuo YL, Ho YK, Shih HM, Giam CZ. NF-kappaB hyper-activation by HTLV-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein HBZ. PLoS Pathog. 2011;7:e1002025. doi: 10.1371/journal.ppat.1002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Jiang W, Cao L, Yu W, Pei Y, Yang X, Wan B, Liu JO, Yi Q, Yu L. IL-2 mRNA stabilization upon PMA stimulation is dependent on NF90-Ser647 phosphorylation by protein kinase CbetaI. J Immunol. 2010;185:5140–5149. doi: 10.4049/jimmunol.1000849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file 1: Figure S1: The arginosuccinate lyase (ASL) gene promoter is insensitive to Tax or PHA/PMA activation. (A) Semi-quantitative PCR amplifications of cDNA derived from JPX-9 and JE6-1 Jurkat cell lines treated with zinc chloride and/or PHA. Aliquots of the PCR reaction, using primers specific for tax/rex mRNA, ASL or IL-2Rα, were removed at the indicated cycle number and electrophoresed on an agarose gel. Zinc-mediated induction of Tax (JPX-9) or PHA treatment (JPX-9 and JE6-1) fail to increase ASL RNA levels. (B) Transient co-transfection of JE6-1 cells with either pLTR-Luc or pASL-Luc, and pLTR-Tax or the respective control, pLTR -DeltaTax (denoted as DTax). The results are displayed as luciferase activity normalized to total protein. Error bars indicate standard deviation of triplicate reactions. The ASL promoter is essentially unresponsive to Tax, PHA, or PMA.
Supplementary file 1: Figure S2: Structure of HTLV-1 genome and transcripts. Schematic representation of HTLV-1 genome and transcripts. Positions of primer sets for RT-qPCR are indicated as red arrows.
Supplementary file 1: Figure S3: Time courses of HTLV-1 mRNA expression with PMA treatment. Three independent treatments of FS cells with PMA were performed, as described in Fig. 1. Cells were treated with PMA for one hour, washed and re-suspended in fresh media. RNAs were collected at time points indicated and levels of individual HTLV-1 transcripts were measured as described. Time courses for untreated parallel samples were also performed for repeats 2 and 3.
Supplementary file 1: Figure S4: Western blots for protein extracts of untreated and PMA treated SP cells. The western blots of HTLV-1 proteins in SP cells (left panel, from Figure 1) were subjected to quantitative analysis (right panels), normalized to actin and divided by the value obtained for the zero time point.
Supplementary file 1: Figure S5: PMA does not change tax/rex RNA stability in MT4 and MT2 HTLV-1 T cell lines. HTLV-1 infected MT4 (A) and MT2 (B) cells were treated with PMA for 30 min, washed, and then treated with Act D, and RNA decay assays were performed as described above. Tax/rex mRNAs were measured with splice site-specific primers. Data were normalized to ASL, and presented as percentage mRNA remaining compared with time zero (closed circle: untreated, open circle: PMA treated). Data shown are average of triplicates with standard deviations (error bars). Similar to the results for HUT102 cells (Fig. 4), PMA does not change tax/rex mRNA stability in MT2 and MT4 cells.
Supplementary file 1: Figure S6: Western blot analysis of PMA-treated FS cells for cell senescence and apoptosis markers. FS cells were treated with PMA for 15′, washed, and cultured with fresh media for 24 hours. Protein lysates were prepared and western blot analyses were performed. (A) Western blots for expression of p21 (CDKN1A) and p27 (CDKN1B) proteins, markers of senescence. (B) Western blots for expression of apoptosis markers; cleaved PARP and cleaved caspase-3. Treatment of Jurkat cells with anti-CD95 (2mg/ml for 4 hours) induced cleaved PARP and caspase-3, and was used as a positive control for apoptosis. PMA treatment resulted in expression of senescent markers, p21 and p27 (A), but did not induce apoptosis (B).
Supplementary file1: Figure S7: Effects of inhibitors of the NFAT, NF-κB or AP-1 pathways on levels of PMA-induced tax/rex mRNA. FS cells were pre-treated with Bay 11-7082 (NF-κB pathway inhibitor), Cyclosporin A (NFAT pathway inhibitor) or SR 11302 (AP1 pathway inhibitor) for 1 hr, followed by 15 minutes of PMA treatment. Cells were then washed, resuspended with fresh media and cultured for 4 hours. Tax/rex mRNA was detected by RT –PCR. Data are normalized to Ct values for ASL mRNA, and divided by the value obtained for the untreated control. Average fold change with standard deviation (error bars) from triplicate treatments are shown. Inhibitors of these pathways did not reduce the levels of PMA-induced tax/rex mRNA