

Abstract
Background
Marfan syndrome (MFS) is caused by mutations in the gene encoding fibrillin‐1 (FBN1); however, the mechanisms through which fibrillin‐1 deficiency causes MFS‐associated aortopathy are uncertain. Recently, attention was focused on the hypothesis that MFS‐associated aortopathy is caused by increased transforming growth factor‐β (TGF‐β) signaling in aortic medial smooth muscle cells (SMC). However, there are many reasons to doubt that TGF‐β signaling drives MFS‐associated aortopathy. We used a mouse model to test whether SMC TGF‐β signaling is perturbed by a fibrillin‐1 variant that causes MFS and whether blockade of SMC TGF‐β signaling prevents MFS‐associated aortopathy.
Methods and Results
MFS mice (Fbn1 C1039G/+ genotype) were genetically modified to allow postnatal SMC‐specific deletion of the type II TGF‐β receptor (TBRII; essential for physiologic TGF‐β signaling). In young MFS mice with and without superimposed deletion of SMC‐TBRII, we measured aortic dimensions, histopathology, activation of aortic SMC TGF‐β signaling pathways, and changes in aortic SMC gene expression. Young Fbn1 C1039G/+ mice had ascending aortic dilation and significant disruption of aortic medial architecture. Both aortic dilation and disrupted medial architecture were exacerbated by superimposed deletion of TBRII. TGF‐β signaling was unaltered in aortic SMC of young MFS mice; however, SMC‐specific deletion of TBRII in Fbn1 C1039G/+ mice significantly decreased activation of SMC TGF‐β signaling pathways.
Conclusions
In young Fbn1 C1039G/+ mice, aortopathy develops in the absence of detectable alterations in SMC TGF‐β signaling. Loss of physiologic SMC TGF‐β signaling exacerbates MFS‐associated aortopathy. Our data support a protective role for SMC TGF‐β signaling during early development of MFS‐associated aortopathy.
Keywords: fibrillin‐1, gene expression, genetically altered mice, Marfan syndrome, signaling pathways, transforming growth factor‐β pathway aneurysm
Subject Categories: Animal Models of Human Disease, Growth Factors/Cytokines, Aneurysm, Vascular Disease, Genetically Altered and Transgenic Models
Introduction
Marfan syndrome (MFS; OMIM 154700) is an autosomal dominant connective tissue disorder that is caused by mutations in the fibrillin‐1 gene (FBN1).1 Cardiovascular manifestations of MFS include mitral valve prolapse and aortic aneurysmal dilation, dissection, and rupture.2 It is well accepted that abnormal or deficient fibrillin‐1 causes MFS3; however, the mechanisms through which fibrillin‐1 deficiency causes aortopathy have been elusive. Because fibrillin‐1 is a major protein component of microfibrils,4 because microfibrils are associated with assembly of elastic fibers,5 and because aortas of individuals with MFS have fragmented elastic laminae,2 the aortopathy of MFS was initially attributed to defective embryonic elastogenesis leading to impaired aortic structural integrity.3, 6, 7 However, mice with defective or deficient fibrillin‐1 develop MFS‐like aortopathy despite normal aortic elastogenesis.8, 9, 10 This observation engendered the hypothesis that MFS‐associated aortopathy results from deficiencies in pathways through which fibrillin‐1 normally maintains postnatal aortic homeostasis including: maintaining connections between aortic smooth muscle cells (SMC) and adjacent elastic laminae; maintaining the differentiated SMC phenotype; regulating cell growth; suppressing synthesis of proteases; and protecting aortic elastin from proteolysis.11, 12, 13
Over 10 years ago, a novel hypothesis was proposed to explain how fibrillin‐1 deficiency causes aortopathy. Based on studies showing that antibody‐mediated neutralization of TGF‐β prevented pulmonary and atrioventricular valvular pathology in Fbn1‐deficient mice,14, 15 Habashi et al treated mice heterozygous for a mutant Fbn1 allele (Fbn1 C1039G/+ genotype) with the same TGF‐β neutralizing antibody.16 Treatment with this antibody significantly improved aortic root pathology, leading the authors to hypothesize that MFS‐related aortopathy is caused by excess TGF‐β signaling in the aortic media and that this excess TGF‐β signaling is a consequence of low levels of normal fibrillin‐1 protein both in Fbn1 C1039G/+ mice and in humans with MFS. This hypothesis was supported by detection of elevated levels of phosphorylated Smad2 (pSmad2, a molecule that can mediate TGF‐β signaling) in aortic SMC in the Fbn1 C1039G/+ mice and by the homology between a repeated 8‐cysteine‐containing motif in fibrillin‐1 and a similar motif in latent TGF‐β binding proteins.6 According to this hypothesis, fibrillin‐1 normally interacts with latent TGF‐β binding proteins (LTBPs) to limit TGF‐β activation. When levels of normal fibrillin‐1 are decreased, TGF‐β activation is excessive and drives MFS‐associated aortopathy.17
This is an attractive hypothesis; however, there are several reasons to question it. These include the following: (1) phosphorylation of Smad2 is neither a sensitive nor a specific indicator of increased TGF‐β signaling18, 19; (2) when investigators looked for increased active TGF‐β in aneurysmal tissue from MFS patients, they did not find it20; (3) the rabbit anti‐TGF‐β antibody used in the initial studies appears to have limited activity in mice,21 and infusion of a more potent murine anti‐TGF‐β antibody exacerbates aortopathy in young MFS mice22; and (4) introduction of Smad4 haploinsufficiency or loss of Tgfb2 (which would both decrease Smad2/3‐mediated TGF‐β signaling that is reportedly upregulated and pathogenic in MFS mice) worsens rather than improves aneurysmal disease in MFS mice.23, 24
Here we report experiments in MFS mice (Fbn1 C1039G/+ genotype) that directly assess the hypothesis that MFS‐related aortopathy is caused by increased aortic SMC TGF‐β signaling. We first deleted the type II TGF‐β receptor in aortic SMC of postnatal Fbn1 C1039G/+ mice and determined whether the associated loss of physiologic SMC TGF‐β signaling improved their aortopathy. We then measured TGF‐β signaling pathway activity in aortic SMC of young Fbn1 C1039G/+ mice to assess whether TGF‐β signaling was increased during the early stages of aortopathy development. Results of our experiments do not support a role for increased SMC TGF‐β signaling in the initiation of MFS‐associated aortopathy.
Methods
Mice and Genotyping
Mice with a floxed Tgfbr2 allele (exon 4) were from Drs Per Levéen and Stefan Karlsson,25 Acta2‐CreERT2 transgenic mice were from Drs Daniel Metzger and Pierre Chambon,26 and mice with Fbn1 C1039G/+ were from Dr Harry Dietz.10 Mice with these alleles were provided to us by these investigators after extensive backcrossing into the C57BL/6 background. After their arrival, the mice were mated exclusively with other mice of the C57BL/6 background. Based on data provided to us by the above‐named investigators, as well as our own records, we estimate that the experimental mice reported on here are products of at least 10 generations of backcrossing (>99.9% C57BL/6 background). Mice were maintained in specific‐pathogen‐free housing and fed normal chow. All experimental mice were of the Tgfbr2 flox/flox genotype. Comparisons were made among 4 groups of mice: (1) Fbn1 C1039G/+ Acta2‐CreERT2 +/0 mice treated with tamoxifen (MFS mice with loss of the type II TGF‐β receptor [TBRII] in SMC, termed “MFS‐TBRII−/−”); (2) Fbn1 C1039G/+ Acta2‐CreERT2 +/0 mice treated with vehicle [MFS mice controlled for effects of Cre expression in SMC, termed “MFS(V)”]; (3) Fbn1 C1039G/+ Acta2‐CreERT2 0/0 mice treated with tamoxifen [MFS mice controlled for administration of tamoxifen, termed “MFS(T)”]; and (4) Fbn1 +/+ Acta2‐CreERT2 0/0 mice treated with tamoxifen (littermates of the MFS mice; essentially tamoxifen‐treated wild‐type C57BL/6 mice, termed “WT”). Genotyping was performed on tail‐tip DNA using primers reported previously for the Tgfbr2 flox, Tgfbr2 null , Acta2‐CreERT2, and Fbn1 C1039G/+ alleles.10, 27, 28 All animal protocols were approved by the University of Washington Office of Animal Welfare.
Tamoxifen Injections
Forty milligrams of tamoxifen free base (Sigma‐Aldrich Corp, St. Louis, MO; #T5648) was dissolved in 0.5 mL of 100% ethanol, and 9.5 mL autoclaved olive oil was added. The mixture was vortexed and then sonicated until completely dissolved, and aliquots stored at −20°C. Two hundred fifty microliters of this solution (1 mg tamoxifen) or of a control solution (autoclaved olive oil/ethanol, 20:1 v/v) was injected intraperitoneally into experimental mice each day for 5 days.
Harvest, Processing, and Analysis of Ascending Aortas
Mice were deeply anesthetized with ketamine and xylazine and exsanguinated by saline perfusion. Hearts and aortas were fixed in situ by perfusion at physiologic pressure with 10% formalin, dissected free, and removed. The tissues were then incubated in 10% formalin overnight, followed by storage in 70% ethanol at 4°C. Perivascular fat surrounding heart and aorta was then removed with the aid of a dissecting microscope. The ascending aorta was photographed with a reference ruler using a Leica S6D microscope and digital camera (model DFC295; Leica Microsystems, Buffalo Grove, IL). Image J software Version 1.48 (NIH, Bethesda, MD) was used to measure the external diameter of the ascending aorta at the takeoff of the innominate artery (perpendicular to the axis of the ascending aorta).
We used a random‐number generator to select a subset of aortas in each group for sectioning and staining. These aortas were selected without consideration as to whether gross pathology was present or absent. A segment of the ascending aorta was obtained by transverse sectioning of the aorta above the aortic root and at the takeoff of the innominate artery. This segment was embedded in OCT compound (4583, Sakura Finetek USA, Torrance, CA) with the cranial end positioned at the OCT block edge. A total of 60 serial 8‐μm‐thick sections were cut, covering 480 μm along the ascending aorta. For each stain, 5 sections per ascending aorta at 96‐μm steps were stained for each mouse. Measurements from each of the 5 sections were used to calculate a single mean value for that mouse.
Sections of ascending aorta were stained with hematoxylin and eosin (H&E) and Prussian blue. Stained sections were photographed using a Leica DM4000B microscope and digital camera (model DFC295; Leica Microsystems, Buffalo Grove, IL). Planimetry (measurement of internal elastic lamina [IEL] and external elastic lamina [EEL] length, calculation of medial thickness and area) was performed on H&E‐stained sections, using ImagePro Plus (Media Cybernetics, Rockville, MD). Mean medial thickness was calculated by assuming circular geometry of the IEL and EEL and calculating the difference between the radii of these circles. Medial area was calculated by subtracting the area within the IEL from the area within the EEL.
Aortic medial elastin damage was quantified by counting elastin breaks. H&E‐stained sections of ascending aorta were illuminated with fluorescein filters, causing autofluorescence of the elastic laminae. Images were acquired with a Leica DM4000B microscope and a digital camera (model DFC295; Leica Microsystems, Buffalo Grove, IL). An observer blinded to genotype counted the number of elastin breaks per section. A break was defined as the presence of 2 free ends of what seemed otherwise to be a continuous elastin fiber. Mean numbers of breaks per section were calculated from 5 step sections per mouse and were used as a value for that mouse.
Architecture of the ascending aortic wall was independently graded by 3 observers blinded to genotype and treatment according to an “aortic wall architecture score” (AWAS) ranging from 1 to 5: 1=normal elastic lamina morphology; 2=increased space between adjacent elastic laminae, noticeable elastin breaks; 3=penetrating aortic ulcer(s), large gaps between adjacent elastic laminae, but most of circumference includes a normal number of elastic laminae (6‐7 layers); 4=penetrating aortic ulcer(s), large gaps between adjacent elastic laminae, some areas with loss of elastic laminae, and medial thinning; 5=highly dysmorphic aorta, severe reduction of elastic laminae with medial thinning and diffuse elastic lamina fragmentation. Observers were trained on a set of images and were then asked to assign scores to sets of experimental samples. The score for each aorta was calculated as the mean of the 3 observers' scores. We developed and applied the AWAS based on our impression that an increase in medial elastic breaks was an early event in medial tissue destruction (ie, easily detected in young mice), whereas, in older mice, medial thinning and substantial loss of elastic laminae rendered counting of elastic breaks less informative. Accordingly, counting elastin breaks is a more sensitive method for evaluating medial histopathology in young mice, and AWAS is a more sensitive means of quantifying medial degeneration in older mice.
Prussian Blue Staining
To detect hemosiderin, sections from the ascending aorta were stained with Prussian blue. Briefly, working solution was prepared by mixing equal parts of 10% hydrochloric acid and 10% potassium ferrocyanide solution (P3289; Sigma‐Aldrich, St. Louis, MO) just before use. Sections were dried at room temperature for 1 hour and then incubated in working solution for 20 minutes followed by 3 washes with distilled water. Sections were then counterstained with nuclear fast red (R5463200‐500A; Ricca Chemical, Arlington, TX) for 5 minutes and washed 3 times with distilled water, followed by dehydration through 70% and 95% ethanol solutions for 2 minutes each. Sections were then air‐dried and coverslipped.
Extraction of Protein and RNA From Full‐Length Aortic Media and Protein From Ascending Aorta
Mice were deeply anesthetized then saline perfused and exsanguinated via cardiac puncture. To isolate the full‐length aortic media, the aorta was excised from the root to the bifurcation, placed in cold PBS on a clear dissection Petri dish (Living Systems Instrumentation, St. Albans, VT; #DD‐90‐S), cut open longitudinally, and pinned with the luminal side up. Endothelium was removed by abrasion with a cotton swab, and a scalpel was used to cut through the medial layer, stopping short of the adventitia. The medial layer was then peeled off of the adventitia with a fine‐point forceps, snap‐frozen in liquid nitrogen, and stored at −80°C. This protocol yields samples of aortic tissue that—compared to the adventitia—is highly enriched in SMC‐specific mRNA.29 For protein extraction, the medial layer was ground in liquid nitrogen using mortar and pestle and resuspended in 100 μL of cOmplete™ Lysis‐M with protease inhibitors (Roche, #4719956001). The suspension was then vortexed twice, incubated on ice for 30 minutes, centrifuged at 14 000g for 15 minutes at 4°C, and supernatant was collected for use in Western blotting. For RNA extraction, the medial layer was ground as described above and resuspended in 1 mL of Trizol (Invitrogen, Carlsbad, CA). The Trizol‐tissue suspension was passed through a 20‐gauge needle 10 times, and 200 μL of chloroform was added and mixed vigorously. The mixture was incubated at room temperature for 5 minutes, then centrifuged at 14 000g for 15 minutes at 4°C. Supernatant was collected, mixed with an equal volume of 70% ethanol, then loaded onto an RNeasy column (Qiagen, RNeasy Mini Kit #74104, Hilden, Germany). The column was washed and RNA eluted according to the manufacturer's protocol.
For some of the mice, we extracted protein from the ascending aorta only. These mice were saline perfused as described above, and the ascending aorta was excised from the aortic root to the innominate artery takeoff. This segment was trimmed of fat, and protein was extracted from the full‐thickness aorta, as described above.
Western Blot Detection of TBRII Protein and TGF‐β Signaling Molecules in Aortic Media
For each sample, 20 μg of protein was separated by SDS/PAGE and transferred to PVDF membranes.30 The membranes were blocked for 1 hour at room temperature with 5% powdered skim milk in Tris‐buffered saline pH 7.4 with 0.05% Tween (TBST) and incubated with antibodies for TGFBR2 (#SC400; 1:1000; Santa Cruz Biotechnology, Dallas, TX), phospho‐SMAD2 (S465‐467, #3101S; 1:1000; Cell Signaling, Danvers, MA), SMAD2 (#3122S; 1:1000; Cell Signaling), phospho‐ERK1/2 (T202‐Y204, #9101S; 1:1000; Cell Signaling), ERK1/2 (#9102S; 1:1000; Cell Signaling), phospho‐p38 (T180‐Y182, #4511S; 1:2000; Cell Signaling), p38 (#9212S; 1:2000; Cell Signaling), or β‐actin (#A5316; 1:5000; Sigma, St. Louis, MO). Antibodies were diluted in TBST with 5% milk and incubated overnight at 4°C. Blots were then washed 3 times (5 minutes each) in TBST and incubated with HRP‐conjugated secondary antibody (goat antirabbit or antimouse; Biorad, Hercules, CA; #170‐6515 or #170‐6516; 1:3000‐1:5000 in TBST with 5% milk) for 1 hour at room temperature. Bound antibodies were detected using enhanced chemiluminescence (ECL) (Thermo, Rockford, IL). Band density was analyzed by densitometry, using the Image J software Version 1.48 (NIH, Bethesda, MD).
Measurement of mRNA Levels in Aortic Media
Levels of mRNA in the aortic medial layer (2 weeks after the first tamoxifen injection) were measured with the Verso 1‐step RT‐qPCR Kit with SYBR Green and low ROX (Life Technologies, Carlsbad, CA; #AB‐4106), using the ΔΔCt method,31 with normalization to 18S RNA. We used qRT‐PCR and SYBR Green to measure aortic medial mRNA levels for genes integral to TGF‐β signaling (Tgfb1, Tgfb2, Tgfb3, Tgfbr1, and Tgfbr3), SMC marker genes (Myh11, Tagln, and Smtn), genes encoding matrix components or otherwise involved in extracellular matrix metabolism (Eln, Fbn1, Lox, Loxl1, Mmp2, Mmp9, and Mmp12), and canonical TGF‐β‐responsive genes (Ctgf and Serpine1) (Table S1).
Statistics
For experiments including only 2 groups, we used t tests after confirming that the groups were normally distributed with equal variances. For experiments that included 3 groups, we tested only 2 a priori null hypotheses (both of these hypotheses involved a comparison of only 2 of the 3 groups): (1) that MFS‐TBRII+/+ mice did not differ from WT mice; and (2) that MFS‐TBRII+/+ mice did not differ from MFS‐TBRII−/− mice. For 3‐group experiments with continuous data, we used t tests when the 2 groups being compared were normally distributed with equal variances and the Mann‐Whitney rank‐sum test when these conditions were not met. We then used the Bonferroni correction to adjust the P values to control for performing multiple (ie, 2) comparisons on the same data set. For 3‐group experiments with categorical data, we used the Fisher exact test to compare 2 groups, with P values again adjusted with the Bonferroni correction. Group data are shown as mean±SEM. All tests were carried out with the Graph Pad Prism program (GraphPad Software, Inc, La Jolla, CA).
Results
Deletion of TBRII Protein in Aortic Media of MFS Mice Worsens Aortopathy
To assess efficiency of deletion of Tgfbr2 in tamoxifen‐treated Fbn1 C1039G/+ Acta2‐CreERT2 +/0 Tgfbr2 flox/flox mice, we measured type II TGF‐β receptor (TBRII) protein in aortic media of these mice (henceforth termed “MFS‐TBRII−/−” mice), tamoxifen‐treated Fbn1 +/+ Acta2‐CreERT2 0/0 Tgfbr2 flox/flox mice (henceforth termed “WT” mice), and tamoxifen‐treated Fbn1 C1039G/+ Acta2‐CreERT2 0/0 Tgfbr2 flox/flox mice [henceforth termed “MFS(T)” mice]. Western blots of aortic medial protein obtained 2 weeks after the first tamoxifen injections showed significantly reduced TBRII protein in MFS‐TBRII−/− mice compared to MFS(T) mice (76% reduction; P<0.001; Figure 1A and 1B). TBRII protein was increased in aortic media of MFS(T) compared to WT mice (43% increase; P<0.05).
Figure 1.

Knockdown of TBRII in aortic medial SMC exacerbates aortic dilation in MFS mice. A and B, Protein was extracted from aortic media of Acta2‐Cre0/0 Fbn1 +/+ mice (WT), Acta2‐Cre0/0 Fbn1 C1039G/+ mice [MFS(T)], or Acta2‐Cre+/0 Fbn1 C1039G/+ mice (MFS‐TBRII −/−). All mice were Tgfbr2 flox/flox, were injected with tamoxifen beginning at 6 weeks of age, and were euthanized 2 weeks later. A, Western blots were probed with antibodies to TBRII or β‐actin. Each lane is from a single mouse. B, Densitometry of Western blots, as shown in A. The TBRII signal was normalized to β‐actin in the same samples. Data are from 4 mice per group; 2 of each group are shown in A. C through G, Ten weeks after treatment with tamoxifen or vehicle (with treatment begun at 6 weeks of age), aortas were harvested from Acta2‐Cre0/0 Fbn1 +/+ mice treated with tamoxifen (WT), Acta2‐Cre0/0 Fbn1 C1039G/+ mice treated with tamoxifen [MFS (T)], Acta2‐Cre+/0 Fbn1 C1039G/+ mice treated with vehicle [MFS (V)], or Acta2‐Cre+/0 Fbn1 C1039G/+ mice treated with tamoxifen (MFS‐TBRII −/−). After confirmation that there was no difference between the MFS(T) and MFS(V) groups, these groups were combined into a single MFS‐TBRII +/+ group for statistical analyses. C through F, Representative images of ascending aorta and arch; ruler is in millimeters. G, External diameter of the ascending aorta measured at the takeoff of the innominate artery, as indicated by the red line in C; n=9 to 12 per group. B and G, Data are mean±SEM; t tests with Bonferroni correction; *P<0.05; ***P<0.001. AU indicates arbitrary units; KD, kilodaltons; MFS, Marfan syndrome; SMC, smooth muscle cells; (T), tamoxifen; TBRII, type II TGF‐β receptor; (V), vehicle; WT, wild‐type.
We next assessed the impact of the MFS‐causing Fbn1 C1039G variant—and of superimposed loss of SMC TBRII—on ascending aortic diameter, measured 10 weeks after the first tamoxifen injection (ie, at 16 weeks of age). This experiment included an additional control group: Fbn1 C1039G/+ Acta2‐CreERT2 +/0 Tgfbr2 flox/flox mice treated with vehicle [henceforth termed “MFS(V)” mice; when MFS(T) and MFS(V) groups are combined, they are termed “MFS‐TBRII+/+” mice]. Ascending aortas of MFS‐TBRII+/+ mice were significantly dilated (27% increase vs WT; P<0.001; Figure 1C through 1G). MFS‐TBRII−/− mice had an additional significant increase in aortic dilation (37% versus MFS‐TBRII+/+ mice; P<0.001). Analysis of histologic sections of the ascending aorta confirmed and extended these results. Compared to WT mice, aortas of MFS‐TBRII+/+ mice had significantly larger circumferences of both IEL and EEL (27% and 25% increases, respectively; Figure 2A through 2C, 2E, and 2F; P<0.01) and a significantly larger medial area (37%; P<0.05; Figure 2G), consistent with full‐thickness aortic dilation and increased medial mass. MFS‐TBRII−/− mice had further significant increases in IEL and EEL circumferences (57% and 48%, respectively vs MFS‐TBRII+/+ mice; P<0.001 for both; Figure 2D through 2F). However, MFS‐TBRII−/− mice had less medial area than MFS‐TBRII+/+ mice (46%; P<0.01; Figure 2G). Therefore, loss of SMC TBRII in MFS mice exacerbates aortic dilation and decreases medial mass.
Figure 2.

Knockdown of TBRII in aortic SMC exacerbates aortopathy in MFS mice. A through D, Representative sections of ascending aortas of 16‐week‐old mice, 10 weeks after beginning treatment with tamoxifen or vehicle. Sections are from perfusion‐fixed mice: (A) Acta2‐Cre0/0 Fbn1 +/+ mice treated with tamoxifen (WT), (B) Acta2‐Cre0/0 Fbn1 C1039G/+ mice treated with tamoxifen [MFS (T)]; (C) Acta2‐Cre+/0 Fbn1 C1039G/+ mice treated with vehicle [MFS (V)]; or (D) Acta2‐Cre+/0 Fbn1 C1039G/+ mice treated with tamoxifen (MFS‐TBRII −/−). All mice were Tgfbr2 flox/flox. E through G, Measurements made on sections of ascending aorta of groups in A through D. E, Length of internal elastic lamina (IEL). F, length of external elastic lamina (EEL), G, medial area. After confirming no difference between the MFS(T) and MFS(V) groups, these groups were combined into a single MFS‐TBRII +/+ group for statistical analyses. A through D, Scale bar: 100 μm. E through G, Data are mean±SEM; n=7 to 8 per group; t tests with Bonferroni correction; *P<0.05; **P<0.01; ***P<0.001. EEL indicates external elastic lamina; IEL, internal elastic lamina; MFS, Marfan syndrome; SMC, smooth muscle cells; (T), tamoxifen; TBRII, type II TGF‐β receptor; (V), vehicle; WT, wild‐type.
We examined the histologic sections for the presence of penetrating aortic ulcers (PAU; defined as a break in the intima and media that exposes the inner media to the lumen), intramural hematomas (IMH; defined as free red blood cells in the media), and evidence of past aortic wall hemorrhage (Prussian blue stain). Ascending aortas of WT mice had none of these features (Table and Figure S1A, S1C, and S1E). Aortas of MFS‐TBRII+/+ mice had occasional PAU and Prussian blue stain, but the increases versus WT mice did not achieve significance (Table). All 3 features were common in aortas of MFS‐TBRII−/− mice (Figure S1B, S1D, and S1F), and 2 of 3 were significantly increased compared to the MFS‐TBRII+/+ mice (Table; P<0.01 for PAU; P<0.001 for Prussian blue stain; P=0.06 for IMH).
Table 1.
Microscopic Aortic Pathology After Smooth Muscle Cell Loss of TBRII
| 10 Weeks After Tamoxifen | WT | MFS(T) | MFS(V) | MFS‐TBRII−/− |
|---|---|---|---|---|
| Penetrating ulcer | 0/7 | 3/8 | 2/7 | 8/8* |
| Intramural hematoma | 0/7 | 0/8 | 0/7 | 3/8ߥ |
| Prussian blue | 0/7 | 1/8 | 2/7 | 8/8† |
| 2 Weeks After Tamoxifen | WT | MFS(T) | MFS‐TBRII−/− |
|---|---|---|---|
| Penetrating ulcer | 0/8 | 0/7 | 5/7§ |
| Intramural hematoma | 0/8 | 0/7 | 5/7§ |
| Prussian blue | 0/8 | 0/7 | 4/7|| |
All pathologies were evaluated in the ascending aorta. All P values are from a Fisher exact test with the Bonferroni correction. MFS indicates Marfan syndrome; (T), tamoxifen; TBRII, type II transforming growth factor β receptor; (V), vehicle; WT, wild‐type.
*P<0.01; † P<0.001 for comparison between MFS TBRII−/− group and pooled MFS(T) and MFS(V) groups.
‡ P=0.06 for comparison of MFS TBRII−/− group and pooled MFS(T) and MFS(V) groups.
§ P<0.05 for comparison of MFS TBRII−/− group and MFS(T) group.
|| P=0.1 for comparison of MFS TBRII−/− group and MFS(T) group.
Compared to WT mice, MFS‐TBRII+/+ mice also had significantly more medial elastin breaks (4‐fold; P<0.01; Figure 3A and Figure S2). There was no further increase in elastin breaks in aortas of MFS‐TBRII−/− mice, likely due to substantial medial thinning and the accompanying loss of some of the elastic laminae in these aortas (Figure 2G). We also quantified aortic histopathology with an aortic wall architecture score (AWAS; see Methods). MFS‐TBRII+/+ mice had a 2‐fold higher AWAS than WT mice (P<0.05; Figure 3B and Figure S3). The mean AWAS for MFS‐TBRII−/− mice was increased by an additional 2‐fold (P<0.001; Figure 3B). Therefore, as assessed by several measures, the aortopathy of murine MFS is exacerbated—not prevented—by superimposed loss of SMC TBRII.
Figure 3.

Elastin fragmentation and medial degeneration in aortas of MFS mice with and without knockdown of SMC TBRII. Ten weeks after treatment of 6‐week‐old mice with tamoxifen or vehicle, aortas were harvested from: Acta2‐Cre0/0 Fbn1 +/+ mice treated with tamoxifen (WT); Acta2‐Cre0/0 Fbn1 C1039G/+ mice treated with tamoxifen [MFS (T)]; Acta2‐Cre+/0 Fbn1 C1039G/+ mice treated with vehicle [MFS (V)]; or Acta2‐Cre+/0 Fbn1 C1039G/+ mice treated with tamoxifen (MFS‐TBRII −/−). All mice were Tgfbr2 flox/flox. After confirmation that there was no difference between the MFS(T) and MFS(V) groups, these groups were combined into a single MFS‐TBRII +/+ group for statistical analyses. A, Aortic medial elastin damage was quantified by counting elastin breaks on sections of ascending aortas. B, Aortic medial degeneration was graded with an aortic wall architecture score. A and B, Data are mean±SEM; n=7 to 8 per group; t tests with Bonferroni correction; *P<0.05; **P<0.01; ***P<0.001. MFS indicates Marfan syndrome; SMC, smooth muscle cells; (T), tamoxifen; TBRII, type II TGF‐β receptor; (V), vehicle; WT, wild‐type.
SMC TGF‐β Signaling Pathways Are Affected by Loss of TBRII, Not by the Pathogenic Fbn1 C1039G Variant
Others have reported evidence of increased SMC TGF‐β signaling in aortas of humans and mice with MFS.16, 23, 32 However, human aortic tissue is typically available only after its excision from patients with advanced disease. Similarly, MFS mouse aortas in which evidence of elevated TGF‐β signaling was reported were from older individuals (16‐52 weeks) with advanced aortic disease.16, 23 We therefore used younger mice (8 weeks old) to determine whether development of early aortopathy in Fbn1 C1039G/+ mice is accompanied by alterations in SMC TGF‐β signaling. We began by examining the activities of canonical and noncanonical TGF‐β signaling in aortic media of 8‐week‐old MFS(T) and WT mice. This time point is 2 weeks after the first tamoxifen injection and 8 weeks before development of the aortic pathology described above. In these 8‐week‐old MFS(T) aortas (from MFS‐TBRII+/+ mice), activation of SMC SMAD2, ERK1/2, and P38 signaling (measured by ratios of pSMAD2, pERK1/2, and pP38 to their unphosphorylated counterparts) were not significantly different from those in WT mice (Figure 4). Moreover, both pSMAD2 and pP38 levels tended to be lower—not higher—in MFS(T) versus WT aortas (30% to 40% lower; Figure 4B and 4C). In contrast, aortic SMC of MFS‐TBRII−/− mice had decreased pSMAD2 and pP38 levels [70% to 80% vs MFS(T) mice; P<0.01 and P=0.06, respectively]. Deletion of SMC TBRII in MFS mice did not affect SMC pERK1/2 levels. These results, combined with the anatomic findings presented above, do not support the hypothesis that aortopathy in MFS mice is caused by increased TGF‐β signaling in medial SMC. Rather, they suggest that loss of physiological SMC TGF‐β signaling (in MFS‐TBRII−/− aortas) worsens MFS‐associated aortopathy. Accordingly, normal levels of SMC TGF‐β signaling appear to protect against MFS‐associated aortopathy.
Figure 4.

TGF‐β signaling pathways in aortic media are affected by knockdown of SMC TBRII, but not by the Fbn1 C1039G/+ genotype. Two weeks after treatment with tamoxifen, protein was extracted from aortic media of 8‐week‐old Acta2‐Cre0/0 Fbn1 +/+ mice (WT), Acta2‐Cre0/0 Fbn1 C1039G/+ mice (MFS(T)), or Acta2‐Cre+/0 Fbn1 C1039G/+ mice (MFS‐TBRII −/−). All mice were Tgfbr2 flox/flox. A, Western blots were probed with antibodies to phospho‐SMAD2, SMAD2, phospho‐P38, P38, phospho‐ERK1/2, ERK1/2, or β‐actin. Each lane is from a single mouse. B through D, Densitometry of Western blots, as shown in A. Levels of phosphorylated signaling proteins were normalized to the corresponding unphosphorylated proteins in the same samples. Data are from 4 mice per group; 2 of each group are shown in A. B through D, Data are mean±SEM; t tests with Bonferroni correction; **P<0.01. KD indicates kilodaltons; MFS, Marfan syndrome; SMC, smooth muscle cells; (T), tamoxifen; TBRII, type II TGF‐β receptor; WT, wild‐type.
We considered the possibility that our analyses of SMC TGF‐β signaling—performed on extracts of whole aortic media—might not detect alterations of TGF‐β signaling pathways that were confined to the proximal aorta. Therefore, we extracted protein from ascending aortas of 8‐week‐old WT and MFS mice (without tamoxifen or vehicle treatment of either group) and measured phosphorylation of SMAD2, P38, and ERK1/2. Similar to results obtained with entire aortas, levels of pSMAD2, pERK1/2, and pP38 in ascending aortas of MFS mice did not differ from levels in WT mice (Figure S4, P>0.3 for all 3).
MFS Mice Develop Aortopathy in the Absence of Altered TGF‐β Signaling
We next tested the hypothesis that—despite the apparent absence of altered TGF‐β signaling in the aortic media of 8‐week‐old MFS mice (Figure 4)—aortopathy has already begun to develop. We measured the diameters of ascending aortas of 8‐week‐old WT, MFS(T), and MFS‐TBRII−/− mice (ie, 2 weeks after the first tamoxifen injection). Ascending aortas of MFS(T) mice were already significantly dilated (15% larger than WT mice; P<0.01; Figure 5A, 5B, and 5D). Examination of histologic sections revealed that both the IEL and EEL circumferences of MFS ascending aortas were increased compared to WT aortas (17% and 22% increases; P<0.05 and P=0.07, respectively; Figure 6A, 6B, 6D, and 6E), and there were 3 times more elastin breaks in MFS(T) ascending aorta than in WT ascending aorta (P<0.001; Figure 6F). Despite this increase in elastin breaks, the AWAS was not elevated in the 8‐week‐old MFS mice (Figure 6G).
Figure 5.

MFS mice develop aortic dilation at an early age. Two weeks after treatment with tamoxifen, aortas were harvested from 8‐week‐old Acta2‐Cre0/0 Fbn1 +/+ mice (WT), Acta2‐Cre0/0 Fbn1 C1039G/+ mice [MFS(T)], or Acta2‐Cre+/0 Fbn1 C1039G/+ mice (MFS‐TBRII −/−). All mice were Tgfbr2 flox/flox. A through C, Representative images of ascending aorta and arch; ruler is in millimeters. D, External diameter of the ascending aorta measured at the takeoff of the innominate artery, as indicated by red line in A; data are mean±SEM; n=7 to 9 per group; t test with Bonferroni correction; **P<0.01. MFS indicates Marfan syndrome; (T), tamoxifen; TBRII, type II TGF‐β receptor; WT, wild‐type.
Figure 6.
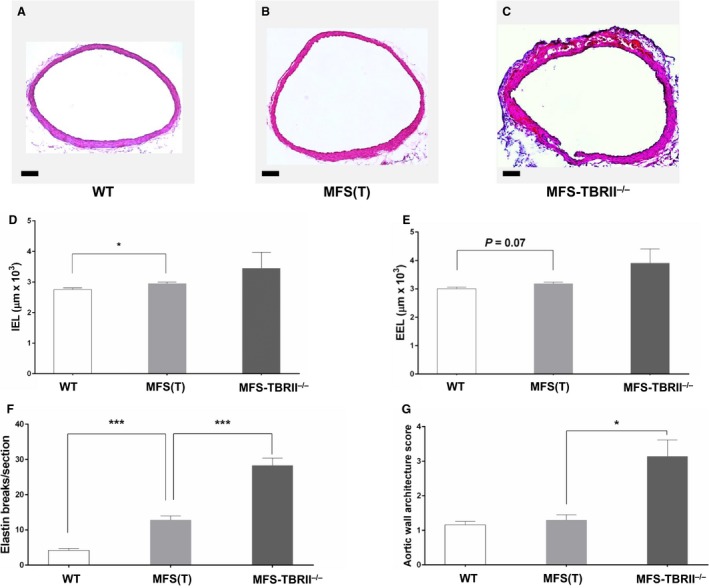
Aortopathy in young MFS mice is worsened by loss of SMC TBRII. A through C, Representative sections of ascending aortas of 8‐week‐old mice, 2 weeks after treatment with tamoxifen. Sections are from (A) an Acta2‐Cre0/0 Fbn1 +/+ mouse (WT), (B) an Acta2‐Cre0/0 Fbn1 C1039G/+ mouse [MFS(T)]; or (C) an Acta2‐Cre+/0 Fbn1 C1039G/+ mouse (MFS‐TBRII −/−). All mice were Tgfbr2 flox/flox. A through C, Scale bar: 100 μm. D and E, Measurements made on sections of ascending aorta of groups in A through C. D, Length of internal elastic lamina (IEL); (E) length of external elastic lamina (EEL); (F) medial elastin damage was quantified by counting elastin breaks on sections of ascending aorta of groups in A through C. G, Aortic medial degeneration was graded with an aortic wall architecture score. D through G, Data are mean±SEM; n=7 to 8 per group; t tests with Bonferroni correction; *P<0.05; ***P<0.001. MFS indicates Marfan syndrome; SMC, smooth muscle cells; (T), tamoxifen; TBRII, type II TGF‐β receptor; WT, wild‐type.
At this early time point, deletion of TBRII in SMC in MFS mice (ie, in MFS‐TBRII−/− mice) had only modest, statistically insignificant effects on ascending aortic external diameter, but in all cases deletion of TBRII tended to exacerbate MFS‐associated aortic dilation (Figures 5C, 5D, and 6C through 6E). Deletion of TBRII increased elastin breaks in aortas of MFS mice by 2‐ to 3‐fold (P<0.001; Figure 6F and Figure S3) significantly increased the prevalence of PAU and IMH (Table) and significantly worsened the AWAS (P<0.02; Figure 6G).
SMC Gene Expression in Young Mice Is Not Affected by the Fbn1 C1039G Variant
Others reported increased expression of TGF‐β‐regulated genes (including SERPINE1 and CTGF) in humans with MFS and in a mouse model of MFS and cited these data as evidence that TGF‐β signaling contributes to MFS pathogenesis.15, 23 We therefore measured aortic medial mRNA levels of Serpine1 and Ctgf as well as genes in 3 other categories potentially affected by altered TGF‐β: TGF‐β ligands and receptors, markers of SMC differentiation (Myh11, Smtn, and Tagln), and mediators of extracellular matrix integrity and metabolism (Mmp2, Mmp9, Mmp12, Eln, Fbn1, Lox, and Loxl1). Compared to WT mice, MFS(T) mice had relatively small increases in expression of virtually all of these genes (≤2‐fold), but none of these differences approached statistical significance (P>0.1 for all; Figure 7). In contrast, MFS‐TBRII−/− mice had altered expression of several of these genes (Tgfb2, Tgfb3, Tgfbr3, Tagln, Mmp12, and Eln) that were large (50% decrease for Eln; 3‐ to 6‐fold increases for all others) and statistically significant [P<0.04 for all, compared to MFS(T) mice; Figure 7]. These gene expression changes were similar to those we reported in older non‐MFS mice in which Tgfbr2 was deleted in SMC.29 For 6 of the 17 mRNAs we measured, modest trends toward increased expression in MFS mice were reversed by loss of Tgfbr2. However, for 9 of the genes, loss of Tgfbr2 further increased expression (2 were essentially unchanged). Therefore, most of the gene expression changes are in the same direction for both MFS mice and MFS‐TBRII−/− mice. The concordance of these changes does not support a model in which the Fbn1 C1039G/+ variant results in increased aortic SMC TGF‐β that is reversed by loss of SMC TGF‐β signaling in MFS‐TBRII−/− mice.
Figure 7.

Loss of TBRII alters aortic medial gene expression in MFS mice. mRNA was purified from aortic media of 8‐week‐old mice, 2 weeks after treatment with tamoxifen. Groups included Acta2‐Cre0/0 Fbn1 +/+ mice (WT); Acta2‐Cre0/0 Fbn1 C1039G/+ mice [MFS(T)], or Acta2‐Cre+/0 Fbn1 C1039G/+ mice (MFS‐TBRII −/−). All mice were Tgfbr2 flox/flox. Gene expression was measured by qRT‐PCR, with normalization to 18S rRNA. Data are mean±SEM; n=8 to 12 per group; t tests with Bonferroni correction; † P<0.05; †† P<0.01; ††† P<0.001 for comparison of MFS‐TBRII −/− and MFS(T) mice. AU indicates arbitrary units; MFS, Marfan syndrome; (T), tamoxifen; TBRII, type II TGF‐β receptor; WT, wild‐type.
Discussion
We used genetically modified mice to investigate the relationship between MFS‐associated aortopathy and TGF‐β signaling in aortic SMC. Our major findings are these: (1) the pathogenic variant Fbn1 C1039G has no significant effects on canonical or noncanonical TGF‐β signaling in aortic SMC of young mice; (2) young Fbn1 C1039G/+ mice develop aortopathy in the absence of detectable activation of SMC TGF‐β signaling; (3) SMC‐specific deletion of Tgfbr2 in vivo results in downregulation of some—but not all—signaling pathways associated with TGF‐β stimulation; (4) SMC Tgfbr2 deletion accelerates aortopathy in young Fbn1 C1039G/+ mice. Our results do not support a causal relationship between increased SMC TGF‐β signaling and MFS‐associated aortopathy. Instead, physiologic SMC TGF‐β signaling appears to protect against development of MFS‐associated aortopathy.
A role for aortic SMC TGF‐β signaling in the pathogenesis of MFS‐associated aortopathy was suggested by data showing that fibrillin‐1 interacts with the TGF‐β‐binding proteins LTBP‐1 and LTBP‐4.33, 34, 35 LTBP‐1 and LTBP‐4 are extracellular matrix proteins that bind latent TGF‐β propeptide (otherwise known as latency‐associated peptide, LAP). After separation from the mature TGF‐β peptide by proteolysis, LAP remains noncovalently attached to mature TGF‐β. The complex of LTBP, LAP, and mature TGF‐β is known as the large latent complex, and active tissue TGF‐β is generated by release of mature TGF‐β peptide from this complex. The association of fibrillin‐1 with LTBPs led to the hypothesis that lower amounts of normal fibrillin‐1 (in MFS) could perturb the binding of mature TGF‐β to LTBP/LAP and thereby increase local levels of active TGF‐β.14, 35 According to this hypothesis, increased active TGF‐β in the aortic media leads to aortic dilation, matrix accumulation, dissection, and rupture.16
The Dietz group tested this hypothesis in a series of elegant studies that build a compelling case for involvement of TGF‐β in MFS‐associated aortopathy. Major supportive findings include: detection of elevated pSmad2 in aortic media of mice and humans with MFS;16 amelioration of aortopathy in MFS mice treated with neutralizing antibodies to TGF‐β;16 and elevated levels of plasma TGF‐β in humans with MFS.36 This hypothesis was later expanded to include TGF‐β activation of pathogenic noncanonical (ie, non‐Smad‐mediated) signaling pathways. Specifically, ERK1/2 signaling was elevated in aortas of 12‐month‐old Fbn1 C1039G/+ mice, and pharmacologic inhibition of MEK1/2 (an activator of ERK1/2) between 2 and 4 months of age prevented aortic root dilation.23 Largely due to these studies, a central role for TGF‐β in the pathogenesis of MFS‐associated aortopathy is now so widely accepted that disappointing results of a recent clinical trial of losartan in MFS were attributed to its failure to sufficiently block TGF‐β.37, 38 In addition, the Marfan Foundation website states that clinical manifestations of MFS are due to “an increase in a protein called transforming growth factor beta” (www.marfan.org/about/marfan).
Nevertheless, as outlined earlier, there are many reasons to doubt that elevated TGF‐β signaling in aortic SMC is a primary driver of MFS‐associated aortopathy. For example, it remains uncertain whether vascular wall TGF‐β activation is enhanced in fibrillin‐1‐deficient humans and mice, and a diligent search for increased TGF‐β activity in aortas of humans with MFS produced negative results.20 Thirteen years after deficient fibrillin‐1–LTBP interactions were hypothesized to increase activation of TGF‐β in the large latent complex17, 35 we could find no biochemical data that support this hypothesis,39 whereas some data cast doubt on it.40 Moreover, in contrast to the initial report of efficacy of anti‐TGF‐β antibodies in MFS mice,16 a recent study found that treatment of young (16‐day‐old) MFS mice with a more effective TGF‐β‐neutralizing antibody21 significantly exacerbated their aortopathy.22 In a separate study, infusion of a different pan‐TGF‐β neutralizing antibody in 8‐ to 12‐week‐old wild‐type mice exacerbated thoracic and abdominal aortic disease caused by angiotensin II infusion.41 In addition, increased SMC production of active TGF‐β in mice (expected to phenocopy the hypothetical enhanced TGF‐β activation in blood vessels of MFS mice) is embryonically lethal due to vascular defects,42 whereas mice with germ‐line fibrillin‐1 mutations that cause MFS‐like aortopathy are born in Mendelian ratios with no reports of abnormal vasculature.8, 9, 10 Finally, a recent clinical study of MFS patients correlated decreased plasma TGF‐β levels with increased rates of aneurysmal dilation, leading the authors to discount TGF‐β as an “initiator of aortic dilatation.”43 The present study, which found no evidence of elevated aortic SMC TGF‐β signaling in young Fbn1 C1039G/+ mice, fits in well with this body of work in suggesting that elevated aortic wall TGF‐β activity is not the primary cause of MFS‐associated aortopathy.
Our central conclusion that aortopathy in young Fbn1 C1039G/+ mice develops in the absence of elevated SMC TGF‐β signaling is based on finding significant aortic dilation and medial elastin damage in the absence of alterations in 3 well‐characterized mediators of TGF‐β signaling. It is particularly significant that we found no elevations of aortic SMC pSmad2 and pERK1/2 because these 2 molecules are firmly implicated as effectors of pathogenic TGF‐β signaling in Fbn1 C1039G/+ mice.16, 23, 44 Although not commented on specifically, data from another group also show lack of pSmad2 elevation in aortas of young (5‐week‐old) Fbn1 C1039G/+ mice (Figure 6F in that study; compare the vehicle‐treated Fbn1 +/+ and Fbn1 C1039G/+ mice).45 We do not doubt others' reports that levels of both pSmad2 and pERK1/2 are elevated in older Fbn1 C1039G/+ mice and in young mice with a more severe form of MFS.16, 22, 23, 44, 45 These elevations could be accurate readouts of increased TGF‐β signaling; however, they could also be epiphenomena that are unrelated to disease pathogenesis, or they could reflect activation of compensatory healing pathways. Lack of causality between TGF‐β signaling activity and pSmad2 elevation in aortic SMC is also suggested by a study showing that epigenetic mechanisms (rather than increased TGF‐β activity) can account for high levels of pSmad2 in human thoracic aortic aneurysms of diverse etiology.46
It remains possible that TGF‐β signaling is increased in aortic SMC of young MFS mice but that we were unable to detect this increase by Western blotting of tissue lysates. This concern is based on the limitations of Western blotting, which measures protein expression averaged across large numbers of cells. Small numbers of aortic SMC with highly activated TGF‐β signaling might be undetectable on Western blots but might nevertheless be sufficient to initiate aortic pathology.47 These cells could potentially be detected by immunostaining of aortic sections; however, immunostaining signals are challenging to quantify,48, 49 and it would be even more difficult to normalize these signals to levels of unphosphorylated signaling molecules present in the same cells. In addition, data collection from tissue sections is subject to sampling errors. Instead of attempting immunostaining, we used 2 approaches to increase our ability to detect elevated/pathogenic SMC TGF‐β signaling by Western blotting. First, to decrease the background of our initial Western blots, instead of measuring proteins in whole aortic lysates (including endothelium and adventitia) we analyzed only aortic medial tissue (which is highly enriched in SMC).29 Second, we performed Western blots of ascending aortic tissue (an area of relatively severe pathology in Fbn1 C1039G/+ mice),10 and we documented significant histopathology in the same areas of ascending aorta that we analyzed with Western blots. Although we cannot prove the absence of focal elevations of SMC TGF‐β signaling in aortic media of Fbn1 C1039G/+ mice, we found no evidence that they exist.
We also used a molecular genetic approach to investigate whether elevated SMC TGF‐β signaling is a primary driver of aortopathy in Fbn1 C1039G/+ mice: blockade of physiological TGF‐β signaling in aortic SMC by deleting Tgfbr2 specifically in SMC. If elevated SMC TGF‐β signaling was the primary driver of aortic disease in Fbn1 C1039G/+ mice, TGF‐β signaling blockade in SMC should mitigate their aortic disease. To verify loss of TGF‐β signaling in SMC‐Tgfbr2 −/− mice, we measured levels of SMC pSMAD2, pP38, and pERK1/2. Surprisingly, deletion of Tgfbr2 resulted in loss of pSMAD2 and pP38 but preservation of pERK1/2. These results support reliance on pSMAD2 and pP38 as reporters of TGF‐β signaling in this model and suggest that pERK1/2 is generated independently of physiological TGF‐β signaling. Contrary to expectations based on models of TGF‐β‐induced aortopathy, deletion of Tgfbr2 and loss of downstream TGF‐β signals unequivocally worsened aortopathy.
While this study was under way, another group reported similar experiments, in which they deleted Tgfbr2 in Fbn1 C1039G/+ mice using a different Cre driver and Tgbfr2 flox allele.45 In Fbn1 C1039G/+ mice, they did not measure aortic SMC TBRII protein (to confirm its loss) and measured only pSmad2 as a read‐out of impaired SMC TGF‐β signaling (it was decreased); however, they also observed accelerated aortic pathology in Fbn1 C1039G/+ mice with Cre‐mediated deletion of Tgfbr2. In the present study we measured 3 potential mediators of SMC TGF‐β signaling and found that they either decreased (2) or did not change (1) after Tgfbr2 deletion; none was elevated. Moreover, mRNA measurements in aortic media of MFS‐TBRII−/− mice did not reveal a signature of increased TGF‐β signaling after deletion of Tgfbr2 (Serpine1 and Ctgf mRNA are decreased compared to MFS‐TBRII+/+ mice). Therefore, our data do not support a hypothesis that worsened aortopathy in MFS‐TBRII−/− mice is driven by paradoxically enhanced TGF‐β signaling. This is important because paradoxically enhanced TGF‐β signaling is cited to explain why haploinsufficiency for Smad4 or Tgfb2 (which should both decrease TGF‐β signaling) worsens aortopathy in Fbn1 C1039G/+ mice.23, 24 An alternative explanation, supported by both our study and its precedent,45 is that physiologic SMC TGF‐β signaling is protective in young Fbn1 C1039G/+ mice.
If increased SMC TGF‐β signaling does not cause MFS‐associated aortopathy, then what does? It is our hope that this study—and others that have questioned a causal role for TGF‐β signaling in MFS22, 41, 50—will stimulate reexamination of potential etiologies of MFS‐associated aortopathy. This reexamination should include investigation of postnatal homeostatic roles for fibrillin‐1 in relation to both aortic SMC and elastin fibers11, 12, 13 as well as a possible role for fibrillin‐1 in supporting normal mechanotransduction.51 The TGF‐β hypothesis may yet prove correct, but there are many reasons to question it. The time is ripe for novel hypotheses to explain the irrefutable connection between low levels of normal fibrillin‐1 and thoracic aortic aneurysmal disease in MFS.3
Sources of Funding
This work was supported by a Grant in Aid from the American Heart Association, R01HL116612, and the John L. Locke, Jr Charitable Trust. Dr Angelov was supported by T32HL007828.
Disclosures
None.
Supporting information
Table S1. Primers Used for qRT‐PCR
Figure S1. Pathologic sequelae of knockdown of TBRII in aortic SMC of MFS mice. Representative images of sections of ascending aortas of 16‐week‐old mice 10 weeks after treatment with tamoxifen. A, C, and E, Sections from aortas of Acta2‐Cre0/0 Fbn1 +/+ mice (WT). B, D, and F, Sections from aortas of Acta2‐Cre+/0 Fbn1 C1039G/+ mice (MFS‐TBRII−/−). All mice were Tgfbr2 flox/flox. Sections were chosen to illustrate specific pathologic features, not to provide a formal comparison among the 3 groups in this experiment. For a formal comparison of all 3 groups of experimental mice (WT, MFS‐TBRII+/+, and MFS‐TBRII−/−), see Table. B, Arrow: penetrating aortic ulcer. D, Arrowheads: aortic intramural hematomas. F, arrowheads: Prussian blue stain. Dashed line indicates the boundary between media and adventitia. A through F, scale bars: 100 μm. A through D, Hematoxylin and eosin stain. E and F, Prussian blue stain with eosin counterstain. MFS indicates Marfan syndrome; TBRII, type II TGF‐β receptor.
Figure S2. Abnormal elastic laminae in 16‐week‐old MFS and MFS‐TBRII−/− mice. Representative images of sections of ascending aortas 10 weeks after treatment with tamoxifen (A, B, and D) or vehicle (C). Hematoxylin and eosin‐stained sections were illuminated with fluorescein wavelengths. Sections are from (A) an Acta2‐Cre0/0 Fbn1 +/+ mouse (WT), (B) an Acta2‐Cre0/0 Fbn1 C1039G/+ mouse [MFS (T)], (C) an Acta2‐Cre+/0 Fbn1 C1039G/+ mouse [MFS (V)], or (D) an Acta2‐Cre+/0 Fbn1 C1039G/+ mouse (MFS‐TBRII−/−). All mice were Tgfbr2 flox/flox. Arrows: elastin breaks. A through D, scale bars: 100 μm. MFS indicates Marfan syndrome; (T), tamoxifen; TBRII, type II TGF‐β receptor; (V), vehicle.
Figure S3. Sixteen‐week‐old MFS mice develop aortic medial degeneration that is worsened by loss of SMC TBRII. A through D, Images of representative sections of ascending aortas of 16‐week‐old mice 10 weeks after treatment with tamoxifen or vehicle. Hematoxylin and eosin–stained sections were illuminated with fluorescein wavelengths. Sections are from (A) an Acta2‐Cre0/0 Fbn1 +/+ mouse (WT), (B) an Acta2‐Cre0/0 Fbn1 C1039G/+ mouse [MFS (T)], (C) an Acta2‐Cre+/0 Fbn1 C1039G/+ mouse that received vehicle [MFS (V)], and (D) an Acta2‐Cre+/0 Fbn1 C1039G/+ mouse (MFS‐TBRII−/−). All mice were Tgfbr2 flox/flox. Boxed areas in A through D are enlarged in E through I with corresponding colored frames. F and G, Arrows: moderate elastin damage. H and I, arrowheads: severe media damage leaving 0 to 1 elastic laminae intact. Scale bars: 100 μm. MFS indicates Marfan syndrome; (T), tamoxifen; TBRII, type II TGF‐β receptor; (V), vehicle.
Figure S4. Eight‐week‐old MFS mice do not have alterations in 3 TGF‐β signaling pathways in the ascending aortic media. Protein was extracted from aortic media of 8‐week‐old Acta2‐Cre0/0 Fbn1 +/+ mice (WT) or Acta2‐Cre0/0 Fbn1 C1039G/+ mice (MFS). All mice were Tgfbr2 flox/flox and were not injected with either tamoxifen or vehicle. A, Western blots were probed with antibodies to phospho‐SMAD2, SMAD2, phospho‐P38, P38, phospho‐ERK1/2, ERK1/2, or β‐actin. Each lane is from a single mouse. B, Densitometry of Western blots shown in A. Levels of phosphorylated signaling proteins were normalized to the corresponding unphosphorylated proteins in the same samples. Data are from 3 mice per group; all are shown in A. Data are mean±SEM; P>0.3 for all 3; unpaired t tests. AU indicates arbitrary units; MFS, Marfan syndrome.
Acknowledgments
We thank Minghui Shi for technical assistance, Dr Nathan Airhart for helpful discussions, and Julia Feyk for administrative assistance.
(J Am Heart Assoc. 2017;6:e004968. DOI: 10.1161/JAHA.116.004968.)
References
- 1. Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, Stetten G, Meyers DA, Francomano CA. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. [DOI] [PubMed] [Google Scholar]
- 2. Pyeritz RE. The Marfan syndrome. Annu Rev Med. 2000;51:481–510. [DOI] [PubMed] [Google Scholar]
- 3. McKusick VA. The defect in Marfan syndrome. Nature. 1991;352:279–281. [DOI] [PubMed] [Google Scholar]
- 4. Sakai LY, Keene DR, Engvall E. Fibrillin, a new 350‐kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103:2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Milewicz DM, Pyeritz RE, Crawford ES, Byers PH. Marfan syndrome: defective synthesis, secretion, and extracellular matrix formation of fibrillin by cultured dermal fibroblasts. J Clin Invest. 1992;89:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maslen CL, Corson GM, Maddox BK, Glanville RW, Sakai LY. Partial sequence of a candidate gene for the Marfan syndrome. Nature. 1991;352:334–337. [DOI] [PubMed] [Google Scholar]
- 7. Dietz HC. Molecular etiology, pathogenesis and diagnosis of the Marfan syndrome. Prog Pediatr Cardiol. 1996;5:159–166. [Google Scholar]
- 8. Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R, Reinhardt DP, Sakai LY, Biery NJ, Bunton T, Dietz HC, Ramirez F. Targetting of the gene encoding fibrillin‐1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17:218–222. [DOI] [PubMed] [Google Scholar]
- 9. Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Biery NJ, Dietz HC, Sakai LY, Ramirez F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin‐1. Proc Natl Acad Sci USA. 1999;96:3819–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114:172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bunton TE, Biery NJ, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88:37–43. [DOI] [PubMed] [Google Scholar]
- 12. Marque V, Kieffer P, Gayraud B, Lartaud‐Idjouadiene I, Ramirez F, Atkinson J. Aortic wall mechanics and composition in a transgenic mouse model of Marfan syndrome. Arterioscler Thromb Vasc Biol. 2001;21:1184–1189. [DOI] [PubMed] [Google Scholar]
- 13. Ramirez F, Pereira L, Zhang H, Lee B. The fibrillin‐Marfan syndrome connection. Bioessays. 1993;15:589–594. [DOI] [PubMed] [Google Scholar]
- 14. Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF‐β activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. [DOI] [PubMed] [Google Scholar]
- 15. Ng CM, Cheng A, Myers LA, Martinez‐Murillo F, Jie C, Bedja D, Gabrielson KL, Hausladen JM, Mecham RP, Judge DP, Dietz HC. TGF‐β‐dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaartinen V, Warburton D. Fibrillin controls TGF‐β activation. Nat Genet. 2003;33:331–332. [DOI] [PubMed] [Google Scholar]
- 18. Derynck R, Zhang YE. Smad‐dependent and Smad‐independent pathways in TGF‐β family signalling. Nature. 2003;425:577–584. [DOI] [PubMed] [Google Scholar]
- 19. Rodriguez‐Vita J, Sanchez‐Lopez E, Esteban V, Ruperez M, Egido J, Ruiz‐Ortega M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor‐β‐independent mechanism. Circulation. 2005;111:2509–2517. [DOI] [PubMed] [Google Scholar]
- 20. Gomez D, Al Haj Zen A, Borges LF, Philippe M, Gutierrez PS, Jondeau G, Michel JB, Vranckx R. Syndromic and non‐syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol. 2009;218:131–142. [DOI] [PubMed] [Google Scholar]
- 21. Chen X, Rateri DL, Howatt DA, Balakrishnan A, Moorleghen JJ, Cassis LA, Daugherty A. TGF‐β neutralization enhances AngII‐induced aortic rupture and aneurysm in both thoracic and abdominal regions. PLoS One. 2016;11:e0153811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, Nelson CA, Cheng SH, Wentworth BM, Ramirez F. Dimorphic effects of transforming growth factor‐β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35:911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, Loeys BL, Thomas CJ, Patnaik S, Marugan JJ, Judge DP, Dietz HC. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert‐Bober J, Kempers MJ, Fishman EK, Chen Y, Myers L, Bjeda D, Oswald G, Elias AF, Levy HP, Anderlid BM, Yang MH, Bongers EM, Timmermans J, Braverman AC, Canham N, Mortier GR, Brunner HG, Byers PH, Van Eyk J, Van Laer L, Dietz HC, Loeys BL. Loss‐of‐function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levéen P, Larsson J, Ehinger M, Cilio CM, Sundler M, Sjostrand LJ, Holmdahl R, Karlsson S. Induced disruption of the transforming growth factor beta type II receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood. 2002;100:560–568. [DOI] [PubMed] [Google Scholar]
- 26. Wendling O, Bornert JM, Chambon P, Metzger D. Efficient temporally‐controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis. 2009;47:14–18. [DOI] [PubMed] [Google Scholar]
- 27. Jaffe M, Sesti C, Washington I, Du L, Dronadula N, Chin MT, Stolz DB, Davis EC, Dichek DA. Transforming growth factor β signaling in myogenic cells regulates vascular morphogenesis, differentiation, and matrix synthesis. Arterioscler Thromb Vasc Biol. 2012;32:e1–e11. [DOI] [PubMed] [Google Scholar]
- 28. Frutkin AD, Shi H, Otsuka G, Leveen P, Karlsson S, Dichek DA. A critical developmental role for tgfbr2 in myogenic cell lineages is revealed in mice expressing SM22‐Cre, not SMMHC‐Cre. J Mol Cell Cardiol. 2006;41:724–731. [DOI] [PubMed] [Google Scholar]
- 29. Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN, Yan J, Allen JK, Kang I, Wight TN, Fox K, Smith A, Enstrom R, Dichek DA. Postnatal deletion of the type II transforming growth factor‐β receptor in smooth muscle cells causes severe aortopathy in mice. Arterioscler Thromb Vasc Biol. 2015;35:2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Farris SD, Hu JH, Krishnan R, Emery I, Chu T, Du L, Kremen M, Dichek HL, Gold E, Ramsey SA, Dichek DA. Mechanisms of urokinase plasminogen activator (uPA)‐mediated atherosclerosis: role of the uPA receptor and S100A8/A9 proteins. J Biol Chem. 2011;286:22665–22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. [DOI] [PubMed] [Google Scholar]
- 32. Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. [DOI] [PubMed] [Google Scholar]
- 33. Dallas SL, Miyazono K, Skerry TM, Mundy GR, Bonewald LF. Dual role for the latent transforming growth factor‐beta binding protein in storage of latent TGF‐beta in the extracellular matrix and as a structural matrix protein. J Cell Biol. 1995;131:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dallas SL, Keene DR, Bruder SP, Saharinen J, Sakai LY, Mundy GR, Bonewald LF. Role of the latent transforming growth factor β‐binding protein 1 in fibrillin‐containing microfibrils in bone cells in vitro and in vivo. J Bone Miner Res. 2000;15:68–81. [DOI] [PubMed] [Google Scholar]
- 35. Isogai Z, Ono RN, Ushiro S, Keene DR, Chen Y, Mazzieri R, Charbonneau NL, Reinhardt DP, Rifkin DB, Sakai LY. Latent transforming growth factor β‐binding protein 1 interacts with fibrillin and is a microfibril‐associated protein. J Biol Chem. 2003;278:2750–2757. [DOI] [PubMed] [Google Scholar]
- 36. Matt P, Schoenhoff F, Habashi J, Holm T, Van Erp C, Loch D, Carlson OD, Griswold BF, Fu Q, De Backer J, Loeys B, Huso DL, McDonnell NB, Van Eyk JE, Dietz HC; GenTAC Consortium . Circulating transforming growth factor‐β in Marfan syndrome. Circulation. 2009;120:526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lacro RV, Dietz HC, Mahony L. Atenolol versus Losartan in Marfan's syndrome. N Engl J Med. 2015;372:980–981. [DOI] [PubMed] [Google Scholar]
- 38. Bowen JM, Connolly HM. Of Marfan's syndrome, mice, and medications. N Engl J Med. 2014;371:2127–2128. [DOI] [PubMed] [Google Scholar]
- 39. Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. Latent TGF‐β‐binding proteins. Matrix Biol. 2015;47:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koli K, Wempe F, Sterner‐Kock A, Kantola A, Komor M, Hofmann WK, von Melchner H, Keski‐Oja J. Disruption of LTBP‐4 function reduces TGF‐β activation and enhances BMP‐4 signaling in the lung. J Cell Biol. 2004;167:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Ait‐Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, Huang J, Offenstadt G, Combadiere C, Renia L, Johnson JL, Tharaux PL, Tedgui A, Mallat Z. TGF‐β activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II‐infused mice. J Clin Invest. 2010;120:422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Agah R, Prasad KSS, Linnemann R, Firpo MT, Quertermous T, Dichek DA. Cardiovascular overexpression of transforming growth factor‐β1 causes abnormal yolk sac vasculogenesis and early embryonic death. Circ Res. 2000;86:1024–1030. [DOI] [PubMed] [Google Scholar]
- 43. Franken R, Radonic T, den Hartog AW, Groenink M, Pals G, van Eijk M, Lutter R, Mulder BJ, Zwinderman AH, de Waard V; COMPARE Study Group . The revised role of TGF‐β in aortic aneurysms in Marfan syndrome. Neth Heart J. 2015;23:116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124:755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gomez D, Coyet A, Ollivier V, Jeunemaitre X, Jondeau G, Michel JB, Vranckx R. Epigenetic control of vascular smooth muscle cells in Marfan and non‐Marfan thoracic aortic aneurysms. Cardiovasc Res. 2011;89:446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473:308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Taylor CR, Levenson RM. Quantification of immunohistochemistry—issues concerning methods, utility and semiquantitative assessment II. Histopathology. 2006;49:411–424. [DOI] [PubMed] [Google Scholar]
- 49. Waters JC. Accuracy and precision in quantitative fluorescence microscopy. J Cell Biol. 2009;185:1135–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen X, Lu H, Rateri DL, Cassis LA, Daugherty A. Conundrum of angiotensin II and TGF‐β interactions in aortic aneurysms. Curr Opin Pharmacol. 2013;13:180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res. 2015;116:1448–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers Used for qRT‐PCR
Figure S1. Pathologic sequelae of knockdown of TBRII in aortic SMC of MFS mice. Representative images of sections of ascending aortas of 16‐week‐old mice 10 weeks after treatment with tamoxifen. A, C, and E, Sections from aortas of Acta2‐Cre0/0 Fbn1 +/+ mice (WT). B, D, and F, Sections from aortas of Acta2‐Cre+/0 Fbn1 C1039G/+ mice (MFS‐TBRII−/−). All mice were Tgfbr2 flox/flox. Sections were chosen to illustrate specific pathologic features, not to provide a formal comparison among the 3 groups in this experiment. For a formal comparison of all 3 groups of experimental mice (WT, MFS‐TBRII+/+, and MFS‐TBRII−/−), see Table. B, Arrow: penetrating aortic ulcer. D, Arrowheads: aortic intramural hematomas. F, arrowheads: Prussian blue stain. Dashed line indicates the boundary between media and adventitia. A through F, scale bars: 100 μm. A through D, Hematoxylin and eosin stain. E and F, Prussian blue stain with eosin counterstain. MFS indicates Marfan syndrome; TBRII, type II TGF‐β receptor.
Figure S2. Abnormal elastic laminae in 16‐week‐old MFS and MFS‐TBRII−/− mice. Representative images of sections of ascending aortas 10 weeks after treatment with tamoxifen (A, B, and D) or vehicle (C). Hematoxylin and eosin‐stained sections were illuminated with fluorescein wavelengths. Sections are from (A) an Acta2‐Cre0/0 Fbn1 +/+ mouse (WT), (B) an Acta2‐Cre0/0 Fbn1 C1039G/+ mouse [MFS (T)], (C) an Acta2‐Cre+/0 Fbn1 C1039G/+ mouse [MFS (V)], or (D) an Acta2‐Cre+/0 Fbn1 C1039G/+ mouse (MFS‐TBRII−/−). All mice were Tgfbr2 flox/flox. Arrows: elastin breaks. A through D, scale bars: 100 μm. MFS indicates Marfan syndrome; (T), tamoxifen; TBRII, type II TGF‐β receptor; (V), vehicle.
Figure S3. Sixteen‐week‐old MFS mice develop aortic medial degeneration that is worsened by loss of SMC TBRII. A through D, Images of representative sections of ascending aortas of 16‐week‐old mice 10 weeks after treatment with tamoxifen or vehicle. Hematoxylin and eosin–stained sections were illuminated with fluorescein wavelengths. Sections are from (A) an Acta2‐Cre0/0 Fbn1 +/+ mouse (WT), (B) an Acta2‐Cre0/0 Fbn1 C1039G/+ mouse [MFS (T)], (C) an Acta2‐Cre+/0 Fbn1 C1039G/+ mouse that received vehicle [MFS (V)], and (D) an Acta2‐Cre+/0 Fbn1 C1039G/+ mouse (MFS‐TBRII−/−). All mice were Tgfbr2 flox/flox. Boxed areas in A through D are enlarged in E through I with corresponding colored frames. F and G, Arrows: moderate elastin damage. H and I, arrowheads: severe media damage leaving 0 to 1 elastic laminae intact. Scale bars: 100 μm. MFS indicates Marfan syndrome; (T), tamoxifen; TBRII, type II TGF‐β receptor; (V), vehicle.
Figure S4. Eight‐week‐old MFS mice do not have alterations in 3 TGF‐β signaling pathways in the ascending aortic media. Protein was extracted from aortic media of 8‐week‐old Acta2‐Cre0/0 Fbn1 +/+ mice (WT) or Acta2‐Cre0/0 Fbn1 C1039G/+ mice (MFS). All mice were Tgfbr2 flox/flox and were not injected with either tamoxifen or vehicle. A, Western blots were probed with antibodies to phospho‐SMAD2, SMAD2, phospho‐P38, P38, phospho‐ERK1/2, ERK1/2, or β‐actin. Each lane is from a single mouse. B, Densitometry of Western blots shown in A. Levels of phosphorylated signaling proteins were normalized to the corresponding unphosphorylated proteins in the same samples. Data are from 3 mice per group; all are shown in A. Data are mean±SEM; P>0.3 for all 3; unpaired t tests. AU indicates arbitrary units; MFS, Marfan syndrome.