

Abstract
Background
The structural elements of the vascular wall, namely, extracellular matrix and smooth muscle cells (SMCs), contribute to the overall stiffness of the vessel. In this study, we examined the crosslinking‐dependent and crosslinking‐independent roles of tissue transglutaminase (TG2) in vascular function and stiffness.
Methods and Results
SMCs were isolated from the aortae of TG2−/− and wild‐type (WT) mice. Cell adhesion was examined by using electrical cell–substrate impedance sensing and PicoGreen assay. Cell motility was examined using a Boyden chamber assay. Cell proliferation was examined by electrical cell–substrate impedance sensing and EdU incorporation assays. Cell micromechanics were studied using magnetic torsion cytometry and spontaneous nanobead tracer motions. Aortic mechanics were examined by tensile testing. Vasoreactivity was studied by wire myography. SMCs from TG2−/− mice had delayed adhesion, reduced motility, and accelerated de‐adhesion and proliferation rates compared with those from WT. TG2−/− SMCs were stiffer and displayed fewer cytoskeletal remodeling events than WT. Collagen assembly was delayed in TG2−/− SMCs and recovered with adenoviral transduction of TG2. Aortic rings from TG2−/− mice were less stiff than those from WT; stiffness was partly recovered by incubation with guinea pig liver TG2 independent of crosslinking function. TG2−/− rings showed augmented response to phenylephrine‐mediated vasoconstriction when compared with WT. In human coronary arteries, vascular media and plaque, high abundance of fibronectin expression, and colocalization with TG2 were observed.
Conclusions
TG2 modulates vascular function/tone by altering SMC contractility independent of its crosslinking function and contributes to vascular stiffness by regulating SMC proliferation and matrix remodeling.
Keywords: crosslinking, matrix remodeling, tissue transglutaminase, vascular biology, vascular disease, vascular smooth muscle, vascular stiffness, vessel
Subject Categories: Animal Models of Human Disease, Cell Biology/Structural Biology, Vascular Biology
Introduction
Stiffness of the central vasculature increases with normal aging and accompanies various morbidities such as hypertension and obesity. Loss of compliance in the large vessels results in left ventricular hypertrophy, isolated systolic hypertension, and persistence of pulsatility along the vascular tree, which can ultimately damage more distal microvasculature and compromise end‐organ perfusion and function.1 Indeed, vascular stiffening is highly predictive of major adverse cardiovascular events.2 The disease burden of vascular stiffening continues to grow with the predicted aging of the population as well as the increasing incidence of hypertension and obesity. Therefore, there is significant interest in the discovery and characterization of therapeutic targets.
The development and progression of vascular stiffening is multifactorial and can occur as a result of alterations in the vascular matrix or the function and behavior of vascular smooth muscle cells (SMCs).3, 4, 5, 6 The nuanced bidirectional communication between the SMCs and the matrix further impacts vascular stiffness.7, 8 At the molecular level, loss of nitric oxide (NO) bioavailability and increased redox stress contribute to the upregulation of matrix synthesis and deposition.9, 10, 11 Such changes can also affect the stiffness, tone, and functional contractility of the SMCs themselves, in what is being increasingly recognized as “smooth muscle cell stiffness syndrome.”3, 4, 5, 6 The SMCs not only regulate vascular tone by responding to chemical and biomechanical stimuli, but also maintain and remodel the matrix in which they reside. Alterations to matrix stiffness can further cause changes in SMC behavior, leading to a complex system. Obtaining insights into the nuanced interplay between matrix stiffness and SMC stiffness will assist us in developing novel targeted therapies to manage and treat vascular stiffening.
Tissue transglutaminase (TG2) is an enzyme of the transglutaminase superfamily that is ubiquitously expressed in the vasculature.12, 13, 14 TG2 is secreted through a Golgi‐independent mechanism to the extracellular matrix, where it can be activated to a Ca2+‐bound open conformation to catalyze the formation of isopeptide bonds.15, 16 The secretion of TG2 to the extracellular matrix is redox‐ and NO‐dependent.15, 17, 18 Loss of NO bioavailability (for example, with aging in rats or genetic ablation of endothelial NO synthase in mice) increases TG2 protein secretion and TG‐catalyzed crosslinks in the vascular matrix and coincides with higher pulse wave velocity (PWV), a measure of vascular stiffness.18, 19 In addition to conduit vessels, TG2 has been shown to mediate mesenteric artery remodeling in response to reduced flow and hypertension in a redox‐dependent manner.20, 21, 22
Interestingly, TG2 is a multifunctional protein whose function is dependent on its localization, conformation, and redox state as well as availability of Ca2+ and GTP.12, 13 At the cell surface, TG2 mediates cell–matrix interaction(s) by binding to integrins, fibronectin, and syndecans.23, 24, 25, 26, 27, 28 In various cell types, including fibroblasts, breast cancer cells, and osteoblasts, TG2 mediates cell adhesion and proliferation independent of its crosslinking function.28, 29, 30 Moreover, as a GTPase (G(h)α), TG2 can mediate signaling via the α1B adrenergic receptor, resulting in inositol phosphate 3 release and phospholipase C δ activation, which in turn can lead to increased SMC tone.31, 32, 33, 34 Though these functions of TG2 have been described in the literature, their contribution to vascular function and mechanical properties remain to be fully elucidated. Therefore, in this study, we tested the hypothesis that TG2 can modulate vascular function and stiffness by crosslinking‐dependent and ‐independent mechanisms.
Materials and Methods
Experimental Animals
Male Fischer 344 rats (6 months old), TG2−/− mice on a BL6/129S mixed background, and wild‐type (WT) BL6/129S mixed background mice were used in this study. All animals were maintained in the Johns Hopkins University School of Medicine animal care facility according to Animal Care and Use Committee recommendations. Animals were fed and watered ad libitum and maintained on a 12‐hour light/dark cycle.
Isolation and Culture of Mouse Aortic SMCs
Aortae of mice were dissected and cleaned. SMCs were isolated as previously described.19 Cells were maintained in DMEM supplemented with 10% fetal bovine serum, subcultured at a ratio of 1:2, and used within 3 passages. For recovery‐of‐function experiments, TG2−/− SMCs were transduced with 15 multiplicity of infection of adenovirus encoding the TG2 gene (AdTG2; Vector Biolabs). Adenovirus containing empty vector was used as control at multiplicity of infection of 15.
Cell Adhesion (PicoGreen™ Assay)
Equal numbers of cells (5×105) were seeded into the wells of a 6‐well plate and allowed to adhere for 1 ‐ 3 hours. The media containing nonadhered cells was centrifuged at 105 g for 5 minutes for collection of the cell pellet, which was then lysed in radioimmunoprecipitation assay buffer. The adhered cells were scraped into 1x radioimmunoprecipitation assay buffer containing protease inhibitors (Roche Bioscience). The PicoGreen DNA quantitation assay (Invitrogen) was used to quantify DNA in the bound and unbound fractions according to the vendor's recommendations. Adhesion was defined as the fraction bound, ie, (bound/[bound+unbound]).
Cell De‐Adhesion Assay
Cells were grown to 80% confluence and used to monitor de‐adhesion as described by Sen and Kumar.35 After briefly rinsing the cell monolayer with sterile PBS, we severed the cell–matrix adhesive contacts with 0.05% trypsin. We monitored the cellular retraction that precedes cell detachment by imaging the cell monolayer every 10 s for 2 minutes at ×10 magnification. Total area occupied by cells was then calculated for each image with ImageJ software. The time‐dependent normalized area was quantified as:
where Ainitial=initial spread area; A(t)=spread area at time t, Afinal=final spread area. The normalized area versus time data were then fit to the following Boltzmann sigmoidal curve:
where τ1 and τ2 are time constants of de‐adhesion with lower values representing rapid detachment of cells. τ1 is the time at which 50% retraction in cell spread area has occurred and τ2 describes the steepness (slope) of the curve.
Cell Migration
We determined SMC migration by a Boyden chamber assay (Fluorimetric QCM 8 μm 24‐well Chemotaxis assay; EMD Millipore) using 10% serum as chemo‐attractant according to the vendor's instructions. Before experiments, SMCs were serum starved for 18 hours and then seeded (2×105 in 300 μL) in the top chamber. The bottom chamber contained 500 μL of medium with or without 10% serum (chemoattractant). Samples were incubated for 5 hours in a cell culture incubator, and cells on both sides of the membrane were quantified by CyQUANT fluorimetric dye.
Cell Proliferation
SMC proliferation was determined with the Click‐iT® EdU Cell Proliferation assay (Invitrogen) according to the manufacturer's protocol. SMCs were serum starved for 18 hours and then seeded at 50% confluence on fibronectin‐coated coverslips in SMC medium supplemented with 5% serum. EdU reagent was added 3 hours after seeding, and the cells were cultured for an additional 24 hours. Samples were gently rinsed with 1% bovine serum albumin in sterile PBS 3 times and fixed with the Click‐iT® fixative solution (15 minutes, room temperature). The samples were gently rinsed twice in 1% bovine serum albumin in sterile PBS, and permeabilized with 1x Click‐iT® sponin‐based permeabilization and wash reagent. EdU‐positive cells were labeled with Cy5 dye by using a click reaction. After excess reagent was rinsed, nuclei were labeled with DAPI. The coverslips were mounted, sealed, and imaged by epifluorescence microscopy on a Nikon 80i microscope equipped with a CoolSnap HQ camera. Four images were captured per coverslip at ×4 magnification. EdU‐positive and DAPI‐positive nuclei were quantified (Nikon NIS Elements software).
Electrical Cell–Substrate Impedance Sensing (ECIS)
Cell adhesion and proliferation were also measured by ECIS (Applied Biophysics). We coated 8W10E+ arrays with l‐cysteine according to the manufacturer's recommendation. For cell adhesion, we added 80 000 cells to each well and analyzed the capacitive portion of the impedance at 40 000 Hz.36 For cell proliferation, we added 40 000 cells to each well and measured resistance at 4000 Hz until steady state was achieved in all samples.37 In both sets of experiments, cell‐free wells were used as controls.
Live Cell Micromechanical Methods
Using spontaneous and forced motions of RGD‐coated ferromagnetic microbeads (≈4.5 μm in diameter) anchored to the cytoskeleton through cell surface integrin receptors of the adherent living cell, we detected cytoskeleton remodeling dynamics and the mechanical properties (elastic modulus and loss modulus) of isolated SMCs. These methods, spontaneous nanoscale tracer motions and magnetic twisting cytometry, are described in detail elsewhere.38, 39
Collagen Assembly Assay
We examined the assembly of fluorescein isothiocyanate (FITC)–conjugated collagen I by live SMCs using the assay described by Johnson and Galis.40 Briefly, SMCs isolated from TG2−/− and WT mouse aortae were seeded on cell culture coverslips (Fisher) at 80% confluence and allowed to adhere for 8 hours. The TG2 gene was delivered by adenovirus to a subset of TG2−/− SMCs. Samples were then incubated for 1 day followed by serum starvation for 18 hours. The serum‐free medium was then replaced with DMEM containing 2% serum and 50 μg/mL FITC‐labeled bovine type I collagen. At the indicated times, the coverslips were rinsed 3 times in sterile PBS and fixed in 4% buffered paraformaldehyde. Nuclei were stained with DAPI. Cell‐free coverslips treated in the same manner were used as controls. The coverslips were mounted and imaged at 3 distinct locations with a Nikon Eclipse 80i fluorescence microscope equipped with a CoolSnap HQ2 CCD camera at ×10 magnification. Each experiment was performed in triplicate.
In Vitro Remodeling
The aorta was dissected out, cleaned free of connective tissue, and cut into 1‐ to 2‐mm rings. Rings were maintained in extracellular matrix medium (ScienCell Research Labs) with guinea pig liver TG2 (gpTG2, 1 mU; Sigma) in the presence and absence of dithiothreitol (DTT; 100 μmol/L) and the TG2 inhibitor L682.777 (10 μmol/L; Zedira) for 48 hours in a cell culture incubator. Samples were then gently rinsed twice with PBS and used for either tensile testing or wire myography.
Tensile Testing
The elastic properties of the samples were analyzed by tensile testing as previously described.19, 41 Mouse and rat aortae were harvested, cut into 2‐mm rings and used. A subset of the samples was decellularized as previously described.19 Samples were incubated with the indicated treatments for 16 hours in endothelial cell media (ScienCell Research Labs) in a cell culture incubator. Samples were then mounted onto the pins of an electromechanical puller (DMT560; Danish Myo Technology A/S, Aarhus, Denmark). After calibration and alignment, the pins were slowly moved apart using an electromotor at a rate of 50 μm/s to apply radial stress on the specimen until breakage. Displacement and force were recorded continuously. The sample to be tested was imaged longitudinally and the cross section of a 0.5‐mm segment proximal to the test sample ring was imaged at ×10 magnification along with a graticule. Vessel lumen diameter (Di), wall thickness (t), and sample length were calculated using ImageJ software (National Institutes of Health, Bethesda, MD). Engineering stress (S) was calculated by normalizing force (F) to the initial stress‐free area of the specimen (S=F/2t×l; where t=thickness and l=length of the sample). Engineering strain (λ) was calculated as the ratio of displacement to the initial stress‐free diameter. The stress–strain relationship was represented by the equation S=α exp (βλ), where α and β are constants. α and β were determined by nonlinear regression for each sample and used to generate stress–strain curves by treating the x‐axis as a continuous variable.
Wire Myography
Vasoconstriction in response to phenylephrine treatment was examined by wire myography as previously described.19, 41 Briefly, after careful excision and cleaning from the surrounding soft tissues, the thoracic aorta was cut into 2‐mm rings. The endothelium was removed by mechanical scraping for a subset of the rings. Each ring was placed in Krebs (containing [in mmol/L] 118.3 NaCl, 4.7 KCl, 1.6 CaCl2, 1.2 KH2PO4, 25 NaHCO3, 1.2 MgSO4, and 11.1 dextrose at a pH of 7.4) and then transferred to a myograph chamber (DMT, Denmark), continuously bubbled with 95% O2 and 5% CO2 (37°C). The rings were stretched in 100‐mg increments to a final tension of 600 mg. After passive stretching of the rings, KCl (60 mmol/L) was added to determine the viability of the vascular preparation and to obtain maximal contractility. Concentration–response curves were constructed for phenylephrine (10−9–10−5 mol/L). Next, endothelial‐mediated vasorelaxation was studied using increasing doses of acetylcholine (10−9–10−5 mol/L) in vessels preconsticted with phenylephrine. Finally, endothelial‐independent vasorelaxation mediated by increasing doses of sodium nitroprusside (10−9–10−5 mol/L) was examined in vessels preconstricted with phenylephrine.
Pulse Wave Velocity Measurement
In vivo vascular stiffness was examined by obtaining invasive PWV measurements at mean arterial pressures varying from 55 to 130 mm Hg as previously described.19, 41, 42 We used a high‐fidelity dual‐pressure catheter sensor to measure aortic PWV. Mice were anesthetized with an intraperitoneal injection of 1.2% Avertin (2,2,2‐tribromoethanol, 240 mg/kg) in 0.9% saline. The animal was positioned supine on the heating pad, with water temperature set to 40°C. Anesthesia was maintained by mask ventilation with 1.0% to 1.5% isoflurane (in 100% O2), and the reflex response to hind‐paw pinching was assessed to monitor depth of anesthesia. After midline neck incision from mandible to sternum, a 1.2‐Fr, dual‐pressure sensor catheter (Scisense, London, Ontario, Canada) was introduced into the descending thoracic aorta through the left carotid artery without opening the chest cavity. The distance between 2 sensors is fixed at 1 cm. A 30‐gauge cannulation needle connected to polyethylene tubing was inserted into the left femoral vein for infusion of fluid/drugs. After stabilization of the signal for 10 to 15 minutes, baseline blood pressures were recorded. Mean arterial pressure was raised and lowered to obtain a full physiological range of blood pressure using intravenous infusion of phenylephrine and sodium nitroprusside, respectively. PWV at corresponding mean arterial pressure was calculated using the foot‐to‐foot method, the foot being defined by the peak of the second time derivative of 2 aortic pressures measured simultaneously during each pulse. PWV was plotted against mean arterial pressure to construct phase plots to characterize PWV over a wide range of mean arterial pressures from 50 to 150 mm Hg in the aorta.
Fluorescence Polarization Activity Assay for TG2
The crosslinking of FITC‐cadaverine with N′,N′‐dimethyl casein (DMC; Sigma), as described by Lorand et al,43 was used with minor modifications as a measure of TG2 activity (Figure S1). Reactions were performed in black 96‐well plates as follows: A 10 mg/mL solution of DMC was prepared in a Tris‐HCl buffer (100 mmol/L, pH 8.0) containing 5 mmol/L calcium chloride. FITC‐cadaverine (Invitrogen; 200 nmol/L final concentration) was added to the DMC solution, and 100 μL of this mix was dispensed into each well. To initiate the reaction, we added 100 μL of gpTG2 (4 mU/mL in 100 mmol/L Tris‐HCl, pH 8.0). The plate was incubated for 4 hours at 37°C, and fluorescence polarization was measured on a Flex Station III (Molecular Devices; excitation at 485 nm, emission at 515 nm, G=1). Experiments were performed in the presence and absence of DTT and L682.777.
Human Coronary Tissue Microarray (TMA) Construction
Coronary artery segments were obtained from orthotopic heart transplant specimens that were collected over an 11‐year period. Coronary arteries were selected to capture a range of ages, extent, and etiologies of cardiovascular disease. As previously described, all specimen block information was entered into TMAJ, a Java‐based tool for TMA creation and management.44 We selected a feature (punch) size of 1.5 mm to capture the full‐thickness of the vessel wall (tunica intima and tunica media) and assembled a 99‐spot TMA using a manual tissue arrayer (Beecher Instruments, Silver Spring, MD). The TMA consisted of 4 control and 95 coronary artery segments. Four hearts provided 8 cores (using 2 different coronary segments). The 95 coronary features were composed of coronary arteries from subjects transplanted for dilated cardiomyopathy (39), ischemic cardiomyopathy (26), valve disease (10), hypertrophic cardiomyopathy (7), congenital heart disease (5), myocarditis (5), arrhythmogenic right ventricular cardiomyopathy (1), giant cell myocarditis (1), or sarcoidosis (1). Four control samples (skin, intestine, placenta, and esophagus) were also present for orientation. Four‐micrometer‐thick sections were cut from each block and placed on charged slides stored in a sealed container at −20°C until use for immunofluorescence. The Johns Hopkins School of Medicine IRB granted approval for this study (NA_00036222).
Immunohistochemistry and Immunofluorescent Staining
TG2 (mouse monoclonal T100, 1:250; ThermoFisher) and TG2 crosslinks (Nε‐γ‐glutamyl lysine antibody) were examined by immunohistochemistry in the TMA as previously described.18, 45 TMAJ software was used to quantify the staining. TG2–fibronectin interaction was examined by immunofluorescence staining. After being incubated with TG2 (mouse monoclonal) and fibronectin (rabbit polyclonal) primary antibodies, the samples were stained with FITC‐conjugated mouse secondary and Cy3‐conjugated rabbit secondary antibodies. Nuclei were stained with DAPI. Each sample was imaged at ×20 and ×40 magnification on a Nikon 80i Epifluorescence microscope equipped with a CoolSnap HQ2 camera. A pathologist at the Johns Hopkins University School of Medicine scored fibronectin expression (scale: none=0, high=5) and TG2‐fibronectin colocalization (no=0, rare=0.5, high=1).
Western Blotting
TG2 expression was examined by Western blotting. Briefly, equal amounts of proteins from each sample were resolved by SDS‐PAGE and electrotransferred to a nitrocellulose membrane. After being blocked with 3% nonfat milk in Tris‐buffered saline/Tween‐20 for 1 hour, blots were incubated with primary antibodies targeting TG2 (rabbit polyclonal CUB7402, 1:5000; ThermoFisher) or glyceraldehyde 3‐phosphate dehydrogenase (GAPDH, mouse monoclonal, 1:5000; Novus Biological) for 1 hour at room temperature. Blots were then rinsed and incubated with horseradish peroxidase–conjugated secondary antibodies (1:5000; BioRad) for 1 hour at room temperature. Western blots were developed with West Pico Chemiluminescence substrate (BioRad) and quantified by densitometry with ImageJ software (National Institutes of Health).
Statistical Analysis
Cell micromechanical data are presented as geometric mean±SE of measurement. To satisfy the assumptions of normal distributions associated with ANOVA, micromechanical data were converted to log scale before analysis. All other data are presented as arithmetic mean±SE of measurement. Box and whisker plots are shown as mean±min‐to‐max. Sample size (n) is indicated for each reported value. Statistical evaluation was performed using the nonparametric Wilcoxon rank‐sum test to compare 2 means. When more than 2 means were compared, the Kruskal–Wallis nonparametric test with Dunn's comparison was used. For multiple comparisons, 2‐way ANOVA with Bonferroni analysis was used. Means were considered to be statistically different at P<0.05.
Results
TG2 Regulates Cell Behavior
To investigate the role of TG2 in SMC behavior, we used mouse aortic SMCs isolated from WT and TG2−/− mice. The quality of SMC isolation was determined prior to use. Immunofluorescent staining for α‐smooth muscle actin showed that the isolated cells were >98% SMCs (Figure S2). Western blotting confirmed that TG2 was expressed in WT but not TG2−/− cells (Figure 1A). Next we examined SMC adhesion and proliferation. Lack of TG2 expression markedly delayed SMC adhesion on uncoated tissue culture dishes as determined by the PicoGreen (Figure 1B) and ECIS (Figure 1C) assays. There was a small but statistically significant delay in TG2−/− SMC adhesion on fibronectin and collagen I supports (Figure 1B). SMC proliferation was examined by 2 complementary approaches: the EdU incorporation assay, which measures DNA synthesis, and ECIS, which measures cell counts. Cells lacking TG2 expression had significantly higher proliferation rates (Figure 1D and 1E) than did WT cells that showed robust TG2 expression. Moreover, the resistance in confluent TG2−/− SMCs was strikingly lower than that of WT SMCs, suggesting weaker cell–cell contacts.
Figure 1.
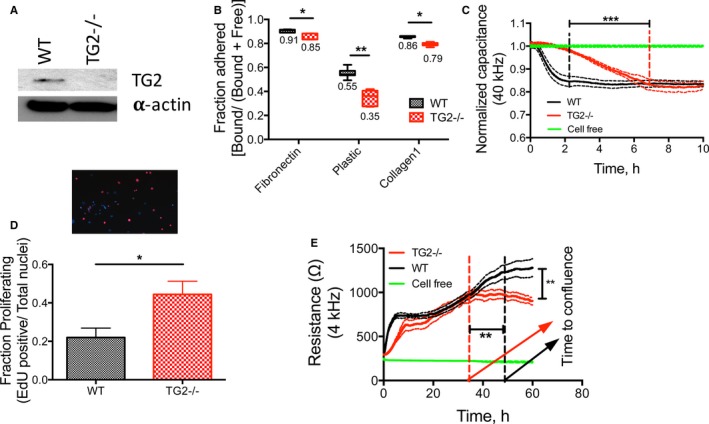
TG2 regulates smooth muscle cell (SMC) function. SMCs were isolated from wild‐type (WT) and TG2−/− mouse aortae. A, Representative Western blot from 8 independent experiments showing that TG2 expression was present in WT SMCs but absent from TG2−/− SMCs; α‐actin was used as a loading control. B, Adhesion of TG2−/− SMCs on plastic, fibronectin, and collagen I supports was lower than that of WT SMCs by PicoGreen assay (*P<0.05, **P<0.01; 2‐way ANOVA with Bonferroni post hoc correction; n=7). C, Adhesion of TG2−/− SMCs was delayed on uncoated supports as examined by electrical cell‐substrate impedance sensing (ECIS). Data are shown as mean (solid line)±SE of measurement (broken lines); ***P<0.001 by Kruskal–Wallis test; n=6 for TG2−/− and WT SMCs, n=4 for cell‐free. D, EdU incorporation assay showed that proliferation of TG2−/− cells was elevated (*P<0.05; by Wilcoxon rank‐sum test; n=6 for WT, n=9 for TG2−/−). E, TG2−/− cells reached confluence earlier than WT cells and had a lower steady‐state resistance as examined by ECIS (**P<0.01; Kruskal–Wallis test; n=6 for TG2−/− and WT SMCs, n=4 for cell‐free condition). TG2 indicates tissue transglutaminase.
TG2 Regulates Cytoskeletal Dynamics and Cell Stiffness
Cytoskeletal dynamics contribute to physical processes, including cell motility/migration. The method of spontaneous nanoscale tracer motions showed that TG2−/− SMCs had significantly fewer cytoskeleton remodeling events than did WT SMCs (Figure 2A). Mean square displacements were significantly lower in TG2−/− cells over a broad time scale from 10 s (Figure 2B) to 300 s (Figure 2C). Consistent with slower cytoskeleton remodeling, TG2−/− cells also showed diminished migration in response to 10% serum in a Boyden chamber assay (Figure 2D). Next, SMC stiffness was examined by magnetic twisting cytometry. SMCs isolated from TG2−/− mouse aorta exhibited greater stiffness (g′) and internal friction (g″) over 5 decades of frequency than did WT SMCs (Figure 2E and 2F). Moreover, TG2−/− cells displayed accelerated de‐adhesion in response to 0.05% trypsin as reflected in a reduced τ1 (time to attain 50% retraction of initial cell spread area) when compared with that of WT cells on uncoated dishes, fibronectin, and collagen I supports (Figure 2G, Figure S3). This finding is in accord with increased stiffness of these cells. Fibronectin support delayed de‐adhesion of WT SMCs but not TG2−/− SMCs (Figure 2G).
Figure 2.
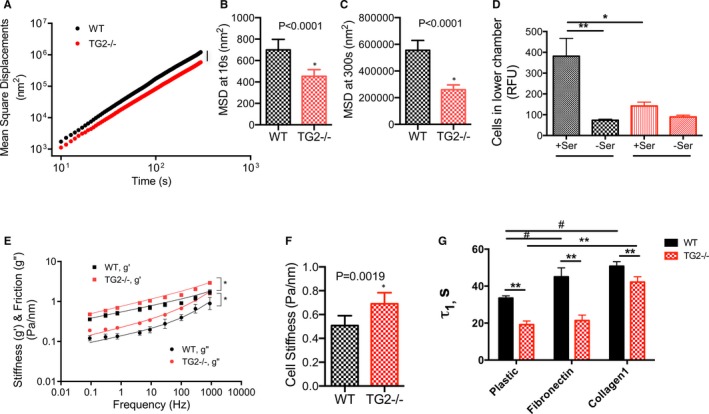
TG2 contributes to smooth muscle cell (SMC) micromechanics. A, TG2−/− SMCs showed strikingly lower cytoskeletal remodeling dynamics when compared with wild‐type (WT) SMCs (n=540 WT cells, 525 TG2−/− cells; 4 independent measurements; *P<0.05). B and C, Mean square displacements were significantly lower in TG2−/− cells at 10 s (B) and 300 s (C). D, TG2−/− SMCs showed diminished motility in response to 10% serum as chemoattractant in a Boyden chamber assay when compared with that of WT SMCs (*P<0.05; by Kruskal–Wallis test; n=9). E, Both storage (g′) and loss (g″) moduli of TG2−/− SMCs were markedly higher than those of WT SMCs over 5 decades of frequency (n=186 WT cells, 227 TG2−/− cells; 4 independent measurements). F, Overall cell stiffness was greater in TG2−/− cells than in WT cells. G, De‐adhesion from uncoated, fibronectin‐coated, and collagen‐coated cell culture dishes was faster in TG2−/− SMCs than in WT SMCs in response to treatment with 0.05% trypsin (**P<0.01 vs WT; # P<0.05 vs plastic WT by 2‐way ANOVA with Bonferroni post‐hoc correction; n=6). RFU indicates relative fluorescence units; TG2, tissue transglutaminase.
TG2 Mediates Early Stages of Collagen Assembly
To further elucidate the contribution of TG2 to extracellular matrix remodeling by SMCs, we examined the assembly of exogenous FITC‐labeled collagen I by TG2−/− and WT SMCs (Figure 3). Collagen assembly was evident in WT cells at the end of 1 day (panel I), and robust structures were visible at the end of 3 days (panel II). In contrast, TG2−/− SMCs exhibited no distinct structures at 1 day (panel III). However, by 3 days, the collagen assembly around TG2−/− cells was indistinguishable from that of WT samples (panel IV). After adenoviral delivery of the TG2 gene to TG2−/− cells, Western blotting showed that TG2 expression was restored (Figure 3B). Restoration of TG2 expression recovered collagen assembly at day 1 similar to that in WT cells (panel V), with robust structures visible at 3 days (panel VI). Cell‐free samples did not show any significant structures (panels VII, VIII), confirming that the collagen assembly observed in panels I to VI is cell dependent.
Figure 3.
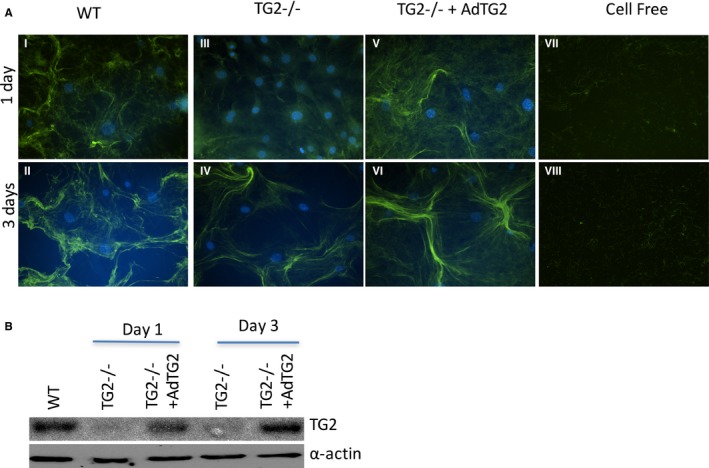
TG2 participates in collagen assembly. A, Collagen assembly was delayed in TG2−/− smooth muscle cells (SMCs; panels III, IV) when compared with that of WT SMCs (I, II). Adenoviral delivery of TG2 to TG2−/− SMCs (panels V, VI) recapitulated the WT time course for collagen assembly. Cell‐free samples did not show any assembled collagen fibers (panels VII, VIII). Images are representative of 6 independent experiments. B, Representative Western blot showing TG2 and α‐actin expression at days 1 and 3. TG2 indicates tissue transglutaminase; WT, wild type.
TG2 Contributes to the Aortic Modulus
Next, we examined the contribution of TG2 at the tissue level in aortic rings ex vivo by tensile testing. Decellularized aortic matrices from WT mice were stiffer than those from TG2−/− mice (Figure 4A). Interestingly, despite the greater stiffness of TG2−/− SMCs, intact aortae from TG2−/− mice remained less stiff than those from WT mice ex vivo (Figure 4B). On the other hand, PWV, a measure of in vivo vascular stiffness, was similar in TG2−/− and WT mice over a range of mean arterial pressures (Figure 4G). In the tensile testing experiments, strain at sample failure were lower in the TG2−/− decellularized segments when compared with TG2−/− intact segments and in WT decellularized segments when compared with WT intact segments (Figure 4C and 4D). Stress at sample failure was higher in TG2−/− decellularized samples when compared with intact TG2−/− rings. Values were similar in intact TG2−/− versus intact WT rings as well as decellularized TG2−/− versus decellularized WT rings. Lumen diameters were similar in the 2 cohorts (Figure 4E). Wall thickness was significantly lower in the decellularized samples than in intact samples in WT aorta (Figure 4F). TG2 was expressed in WT but not TG2−/− mouse aorta (Figure 4H).
Figure 4.
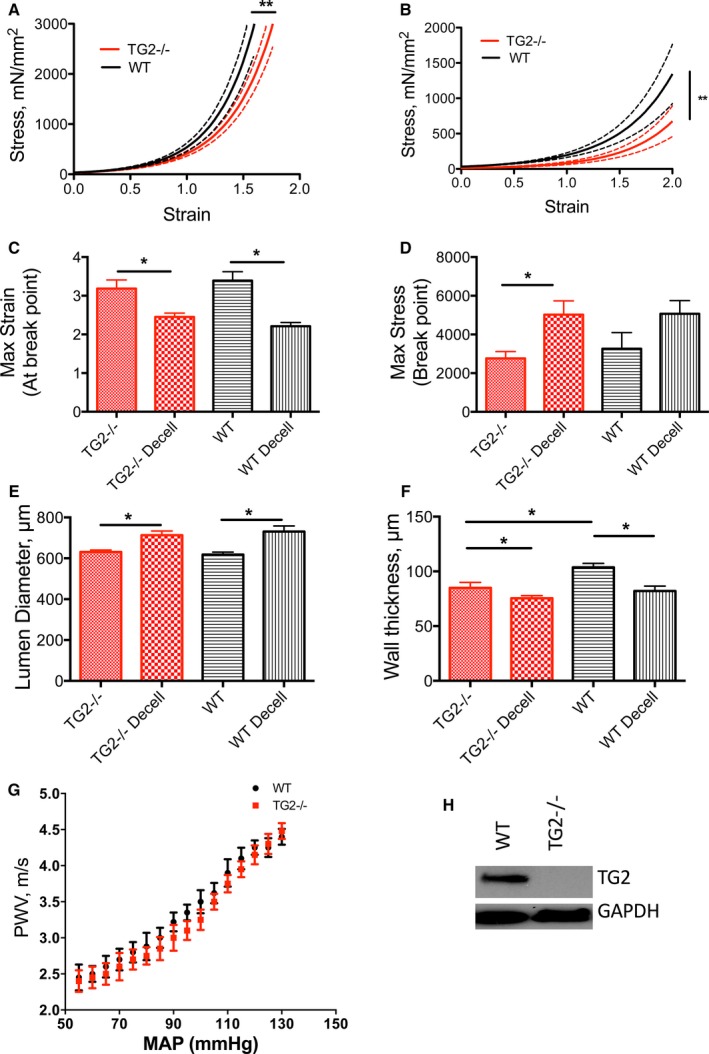
TG2 contributes to aortic modulus. A, Decellularized and (B) intact aortic segments from WT mice were significantly stiffer than those from TG2−/− mice; solid lines represent arithmetic mean and dotted lines represent SE of measurement (*P<0.05). C, Maximum strain at sample rupture was lower in the decellularized segments than in intact segments and similar between genotypes (*P<0.05). D, Maximum stress was higher in the decellularized segments than in intact aorta for the TG2−/− genotype but not the WT genotype. E, Lumen diameters were larger in decellularized segments than in intact segments for WT aorta (*P<0.05). F, Walls were significantly thinner in decellularized samples than in intact samples, in WT aorta (n=12 per group; *P<0.05). G, Pulse wave velocity (PWV)–mean arterial pressure (MAP) correlation was similar in 12‐ to 14‐week‐old WT and TG2−/− mice (n=8). H, Representative Western blot showing that TG2 protein was expressed in aorta from WT but not TG2−/− mice; GAPDH was used as loading control. TG2 indicates tissue transglutaminase; WT, wild type.
TG2 Protein Increases Vascular Stiffness In Vitro Independent of Its Crosslinking Function
We examined whether exogenous, purified gpTG2 protein could restore the mechanical properties of the aorta ex vivo. Guinea pig liver TG2 is commercially available and has 82% sequence identity with mouse TG2 and 81% identity with rat TG2 (Figure S4C). We first verified the activity of gpTG2 by monitoring the incorporation of FITC‐cadaverine with N,N′‐dimethylcasein by fluorescence polarization in the presence or absence of DTT and L682.777, an inhibitor of TG2's crosslinking function. DTT is required to activate the crosslinking function of gpTG2 in vitro, as in its absence, gpTG2 had negligible crosslinking activity. L682.777, the small molecule inhibitor of TG2's crosslinking function, inhibited the crosslinking function of DTT‐activated gpTG2 (Figure 5A). Thus, we can examine crosslinking‐independent functions of TG2 by using gpTG2 in the absence of DTT or in the presence of L682.777. We then verified delivery and activity of gpTG2 in the vascular media using TG2−/− mouse aorta. Confocal microscopy revealed robust FITC‐cadaverine incorporation by gpTG2 in the vascular media around vascular SMCs in the presence of DTT (Figure 5B, top left) and minimal activity in the absence of DTT (Figure 5B, bottom left). L682.777 inhibited gpTG2‐dependent FITC‐cadaverine incorporation in the vascular media (Figure 5B, top right). To investigate potential compensation by other TGases, we examined the basal TGase activity in the aortae isolated from TG2−/− mice. Negligible FITC‐cadaverine incorporation was observed in the absence of gpTG2, suggesting TG2 is the primary TGase in the mouse aorta (Figure 5B, bottom right).
Figure 5.
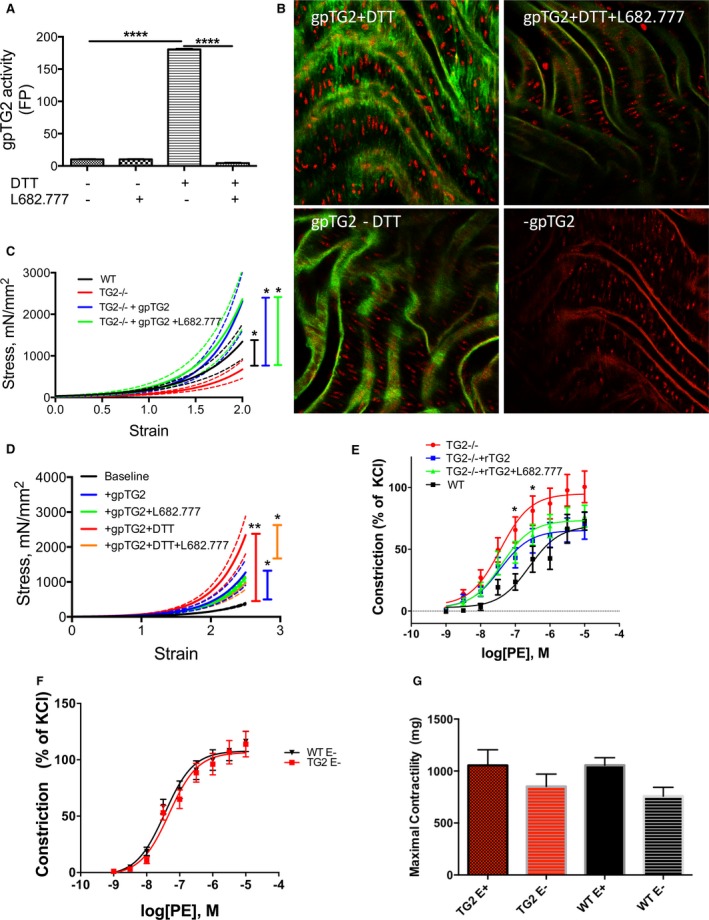
Exogenous TG2 affects vascular stiffness and vasoreactivity independent of crosslinking function. A, Activity of exogenous guinea pig TG2 (gpTG2) was examined by quantifying the crosslinking of FITC‐cadaverine and N,N′ dimethyl casein by fluorescence polarization. Dithiothreitol (DTT) activated and L682.777 inhibited gpTG2 (*P<0.05 by Kruskal–Wallis test; n=12 per group). (B) GpTG2 activity was examined by confocal microscopy in TG2−/− mouse aorta. GpTG2‐dependent incorporation of FITC‐cadaverine occurred in the presence of DTT and was inhibited by L682.777. Negligible FITC‐cadaverine incorporation was observed in samples lacking DTT and those lacking gpTG2 treatment. Images are representative of 8 independent experiments. C, Aortic modulus was examined by tensile testing; solid lines represent arithmetic mean and dotted lines represent SE of measurement. Incubation of TG2−/− mouse aorta with gpTG2 increased aortic stiffness to levels similar that of WT aorta both in the absence (blue) and presence (green) of TG2 inhibitor L682.777 (*P<0.05; n=8 per group). D, Incubation of rat aorta with DTT‐activated gpTG2 significantly increased stiffness (red). GpTG2 in the absence of DTT (blue) also resulted in a significant increase in stiffness when compared with baseline (black), but the magnitude of increase was reduced. L682.777 partially reversed the increase in stiffness in DTT‐activated gpTG2 (orange) and had no effect in the absence of DTT (green) n=8 per group; *P<0.05, **P<0.01. E, Vasoconstriction of aortic rings in response to increasing doses of phenylephrine (PE) was examined by wire myography. The response of rings from TG2−/− mice (red) was significantly left‐shifted compared with those from WT mice (black). Incubation of TG2−/− aortic rings with gpTG2 restored contractility toward that of WT in the absence (blue) and presence (green) of L682.777. n=8 per group; *P<0.05. (F) Endothelium‐denuded rings of TG2−/− and WT mice show similar responses to PE, suggesting an endothelial component to vascular responses. (G) Maximal contractility with KCl (60 mmol/L) is similar in intact TG2−/− and WT vessels and in denuded TG2−/− and WT vessels; denuded vessels had lower contractility than their endothelium‐intact counterparts. FITC indicates fluorescein isothiocyanate; TG2, tissue transglutaminase; WT, wild type.
Next, we incubated aortic rings from TG2−/− mice with recombinant gpTG2 in the presence and absence of L682.777 for 48 hours to examine crosslinking‐dependent and ‐independent effects (Figure 5C). Incubation with gpTG2 resulted in a large increase in aortic stiffness to levels similar to those of WT, even in the presence of the TG2 inhibitor L682.777, suggesting that the crosslinking function of TG2 is not required to elicit increases in vascular stiffness. The same experiments were also performed in rat aortic rings (Figure 5D). In these experiments, DTT‐activated TG2 caused a substantial increase in stiffness compared with baseline stiffness of the rat aorta. TG2 lacking crosslinking function (ie, in the absence of DTT and inhibition in the presence of L682.777) also caused a large increase in stiffness, but to a lower magnitude than observed with catalytically active TG2.
TG2 Modulates the Contractile Response of the Aorta
To study the contribution of TG2 to aortic function, we examined vasoreactivity of aortic rings from WT and TG2−/− mice in response to increasing doses of phenylephrine by wire myography (Figure 5E). Aortic rings from TG2−/− mice had increased contractility and a lower EC50 than those from WT mice (0.37±0.02e‐7 versus 2.54±0.19e‐7; P<0.05). Preincubation of TG2−/− rings with gpTG2 partially restored vessel function toward WT in the presence and absence of L682.777. Responses of endothelial cell–denuded vessels were similar (Figure 5F). Maximal contractility in response to KCl (60 mmol/L) was similar between TG2−/− and WT aorta (Figure 5G). Endothelial‐dependent (acetylcholine‐mediated) and ‐independent (sodium nitroprusside‐mediated) vasorelaxation in rings preconstricted with phenylephrine were similar in both cohorts (Figure S4).
TG2 and Fibronectin Colocalize in Human Coronary Arteries
The relevance of the findings in cell and mouse models to human disease was examined with a 99‐feature TMA. Vascular stiffening is commonly observed in patients with heart failure.46, 47 We chose to sample coronary arteries because it is possible to observe the entire wall of the coronary artery in a feature size of 1.5 mm. The TMA included samples from patients 0.25 to 70 years of age with dilated cardiomyopathy, congenital heart disease, hypertrophic cardiomyopathy, ischemic cardiomyopathy, myocarditis, and valve disease. Control samples included skin, intestine, placenta, and esophagus. Hematoxylin & eosin (H&E) staining was used to determine the quality of the tissues (Figure 6A). A single feature with representative H&E and smooth muscle actin staining is shown in Figures 6B and 6C, respectively. Fibrosis and plaque size did not correlate with age or disease (Figure S5A through S5D). TG2 expression was high in the vascular media and variable in plaque (Figure 6D, Figure S5E). Fibronectin expression (Figure 6E, Figure S5F) and fibronectin‐TG2 colocalization (Figure 6F) were present in the media of coronary arteries. Both fibronectin expression and colocalization with TG2 were greater in plaque than in media. TGase crosslink frequency, which was examined with the n‐ε‐γ‐glutamyl lysine antibody, did not correlate with age, disease, or location in the coronary artery (Figure 6G, Figure S5G). Representative images of TG2 (green) and fibronectin (red) immunofluorescence staining in plaque (Figure 6H) and vascular media (Figure 6I) are shown; nuclei were stained with DAPI (blue).
Figure 6.
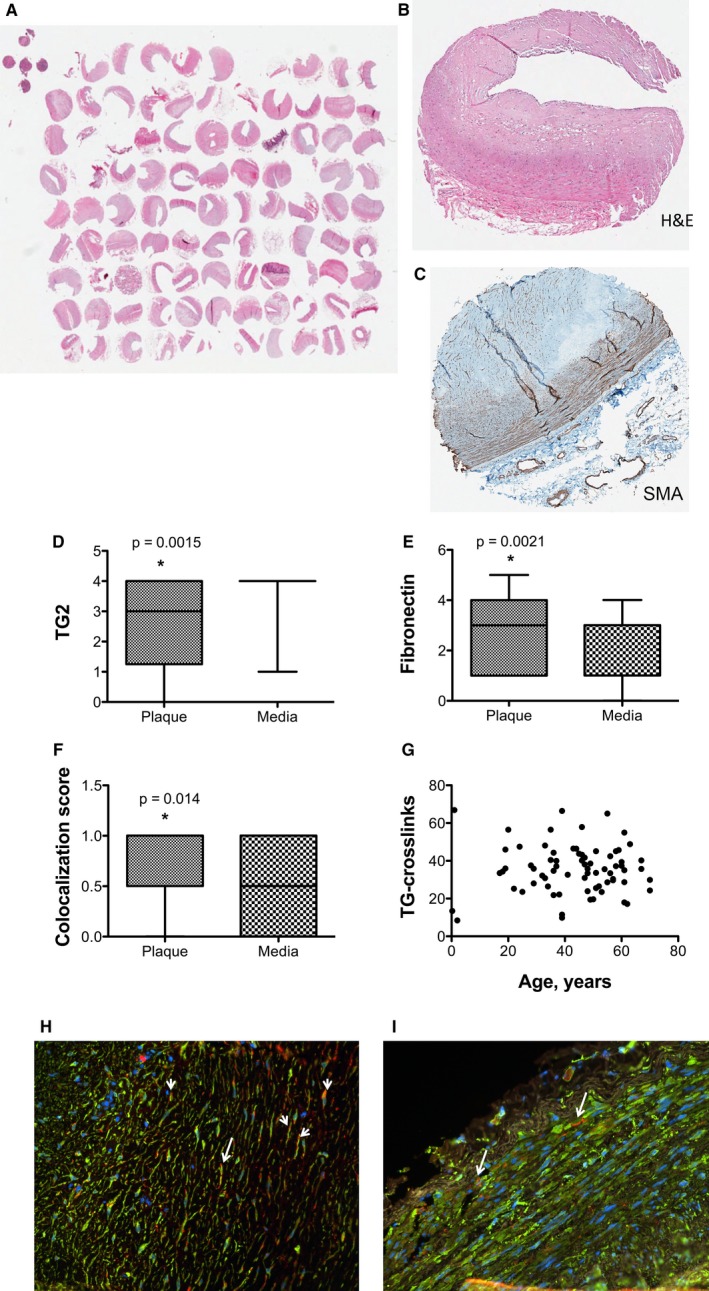
TG2 and fibronectin colocalize in plaque in human coronary arteries. A 99‐feature human coronary artery tissue microarray (TMA) was used to examine the role of TG2 in human disease. A, A photograph of the TMA with hematoxylin & eosin (H&E) staining. B, Sample H&E and (C) smooth muscle actin stains of a single feature. D, TG2 expression was high in the vascular media but varied in plaque from very rare to highly abundant. E, Fibronectin expression was higher in plaque than in media. F, Colocalization of TG2 and fibronectin was more abundant in plaque than in media. G, TG crosslinking was similar in all samples. Representative immunofluorescence images in plaque (H) and vascular media (I; green=TG2, red=fibronectin, blue=nuclei); arrows indicate sample areas of colocalization. *P<0.05. TG2 indicates tissue transglutaminase.
Discussion
The results from this study indicate that (1) TG2 plays a role in the fundamental properties of aortic SMCs including adhesion, motility, proliferation, cell stiffness, and cytoskeletal dynamics; (2) TG2 participates in matrix deposition by SMCs in the early but not later phases; (3) TG2 contributes to vascular function and stiffness through both crosslinking‐dependent and crosslinking‐independent functions; and (4) TG2‐fibronectin colocalization accumulates in humans with coronary artery disease, suggesting a crosslinking‐independent role for TG2 in human disease. TG2 is an enzyme that participates in matrix deposition by catalyzing the formation of isopeptide bonds between the side chains of lysines and glutamines via the classical transglutaminase reaction. The catalytic activity of TG2 is necessary but not sufficient to remodel the matrix, as matrix proteins that are the substrates for TG2 must also be actively synthesized and secreted. In the vasculature, TG2 itself can contribute to matrix synthesis via its crosslinking‐independent functions, for example, by acting through the transforming growth factor‐β pathway.48
In the vascular media, SMCs maintain and remodel the matrix in which they reside. Indeed, SMC hypertrophy and hyperplasia are hallmarks of many vascular pathologies, such as hypertension and atherosclerosis. In this study, genetic ablation of TG2 delayed collagen assembly, suggesting that TG2 participates in early matrix deposition but that other enzymes (such as lysyl oxidases) may compensate in later phases.49 Through its noncovalent interactions with fibronectin, integrins, and syndecans, TG2 can participate in SMC adhesion, motility, and proliferation. Lack of TG2 expression delayed TG2 adhesion and diminished cell migration; these results are consistent with our findings of decreased cytoskeletal remodeling in TG2−/− cells as well as previous work in which aggressive cancer cells were shown to have increased cytoskeleton remodeling.50 Two complementary approaches (DNA synthesis and cell counting) showed that cell proliferation was higher in SMCs lacking TG2 expression. Moreover, cell–cell contacts appeared to be weaker in the TG2−/− cells than in WT cells, as reflected in the significantly lower trans‐well resistance monitored by ECIS (Figure 1E). Previous studies have shown that under basal conditions, TG2 stabilizes the contractile phenotype of SMCs and would thus support lower proliferation rates.51 TG2 protein expression is lost in the SMCs of hypertensive rats (corresponding to proliferative SMC phenotype).52 These findings are supportive of our current results. However, in response to stress or growth factors, cell‐surface TG2 can promote dedifferentiation and proliferation of SMCs. For example, WT SMCs were shown to proliferate more rapidly than TG2−/− SMCs in response to treatment with platelet‐derived growth factor BB.53 Taken together, these studies underscore the complexity of TG2 signaling in normal vascular physiology and pathophysiology.
In vitro, it is possible to assess the crosslinking‐independent effects of TG2 using the small‐molecule inhibitor L682.777, which targets the crosslinking function but not the other functions of TG2. In aortic rings from TG2−/− mice, gpTG2 elicited a substantial increase in stiffness in vitro independent of crosslinking function as evidenced by the lack of any effect of L682.777. In the rat aorta, addition of DTT to activate gpTG2's crosslinking function resulted in a striking increase in stiffness. Rat TG2 may also be activated by DTT in vitro and contribute to this process. In the present study, inhibition with L682.777 did not fully prevent the increase in gpTG2‐mediated stiffness; instead the elastic properties of these samples coincided with those observed in the samples treated with TG2 that lacked crosslinking function. These observations demonstrate a significant contribution of TG2 to vascular modulus through crosslinking independent mechanisms.
Intriguingly, in cell culture, TG2−/− SMCs are stiffer than their WT counterparts and have fewer cytoskeletal remodeling events. The de‐adhesion dynamics of TG2−/− SMCs are also accelerated, a fact that further supports an increase in cell stiffness.35 In vitro, despite the elevated cellular stiffness, the overall modulus of the intact TG2−/− aorta remains lower than that of the WT aorta. In vivo, however, PWV, a measure of vascular stiffness, was similar in adult (12–14‐week‐old) TG2−/− and WT mice over a range of mean arterial pressures. Given these observations, we postulate that increased cell stiffness and SMC tone in the absence of TG2 expression is a compensatory mechanism to counteract the loss in mechanical modulus of the matrix. Moreover, our studies suggest that the contribution of cell stiffness to vascular compliance requires integrated physiological signaling, including mechanical and biochemical cues, underscoring the importance of in vivo compliance measurements.
Ex vivo, in the intact aorta, exogenous TG2 caused an increase in the stiffness of mouse and rat aorta and acutely altered phenylephrine‐mediated vasoconstriction even in the absence of crosslinking function. TG2−/− mouse aorta demonstrated increased sensitivity to phenylephrine‐mediated vasoconstriction that was partly reversed by incubation with gpTG2 independent of crosslinking function. We propose that this could be attributed to the GTPase function of TG2, which is shown to signal via PLCδ and activate the BK channel,31, 54 which in turn would result in SMC hyperpolarization and decrease SMC tone. This effect is in part mediated by endothelial cells as endothelium‐denuded vessels from TG2−/− mice demonstrate phenylephrine responses that are similar to those from WT. Taken together, these findings suggest that TG2 modulates SMC tone/function via crosslinking‐independent mechanisms, which in turn contribute to vascular function and stiffness.
Aortic stiffening and endothelial dysfunction are observed in patients with heart failure. Moreover, endothelial dysfunction and reduced NO bioavailability are triggers for increased TG2 secretion in the vasculature.15, 17, 18, 19, 41 In human coronary arteries obtained from individuals with various cardiomyopathies, TG2 and fibronectin are not only highly expressed in the vascular media, but there is also a strong colocalization of the two. We have previously shown a greater abundance of TG2‐mediated cell adhesion via integrin/fibronectin binding in adult eNOS‐deficient mice when compared with age‐matched WT mice.19 The present study demonstrates that a similar interaction exists in human vasculature. The abundance of N‐ε‐γ‐glutamyl lysine crosslinks was similar in all samples and did not correlate with age, likely because the presence of cardiomyopathy masked the effect of age. We also examined the abundance of proteins and co‐localization in the plaque in these samples. Intriguingly, there is increasing accumulation of fibronectin protein as well as increased colocalization of fibronectin with TG2 in the neo‐intima and plaque when compared with the media, suggesting that this interaction could be important in plaque formation. The relevance of this observation in the development of atherosclerosis remains to be studied. Overall, the presence of the TG2‐fibronectin interaction in vivo in humans with cardiomyopathy is suggestive of a crosslinking‐independent role for TG2 in vascular stiffness. To the best of our knowledge, this is the first study demonstrating TG2‐fibronectin colocalization in human vasculature.
In conclusion, under pathophysiological conditions such as elevated blood pressure, NO dysregulation, and increased oxidative stress, TG2 can contribute to mechanical modulus and contractile function of the aorta through crosslinking‐dependent and ‐independent mechanisms. The specific contribution of possible mechanisms (cell adhesion versus G‐protein signaling) remains to be elucidated.
Sources of Funding
This work was supported by American Heart Association Grants 09BGIA2220181 (to Santhanam) and 13GRNT16420015 (to Halushka), an intramural Stimulating and Advancing ACCM Research (StAAR) grant sponsored by the Department of ACCM (to Santhanam), a Foundation for Anesthesia Education and Research FAER‐MRTG #117853 (to Steppan), National Heart, Lung, and Blood Institute Grants R01‐HL105296 (to Berkowitz) and R01‐HL107361 (to An), and an Australian Research Council Grant DP110101134 (to Avolio).
Disclosures
None.
Supporting information
Figure S1. Fluorescence polarization (FP)‐based assay for transglutaminase (TG) activity. A, The principle of the assay is that as the low‐molecular‐weight fluorescent molecule FITC‐cadaverine is incorporated by transamidation into the high‐molecular‐weight N,N′‐dimethylcasein, the FP signal of FITC‐cadaverine increases. B, His‐tagged recombinant human TG2 was expressed in HEK293 cells and purified with a BD‐Talon purification kit before use. Representative Coomassie blue staining of the purified product is shown in the inset. FP signal increased with increasing TG2 protein in the reaction mixture. C, The IC50 of cystamine was calculated to be 0.025±0.005 mmol/L in the FP assay. This value is in good agreement with the published value of 0.02±0.05 mmol/L (n=12 per group). D, The IC50 of L682.777 was calculated to be 0.86±0.1 μmol/L, which is in good agreement with the value of 0.5 μmol/L provided by the vendor (Zedira; n=12 per group). E, Unlabeled cadaverine, which was used as a competitive inhibitor for FITC‐cadaverine, had an IC50 of 56±0.1 nmol/L (n=12 per group). F, The assay was used to examine activity in the protein lysates/homogenates obtained from human aortic endothelial cells (HAEC), rat liver, and rat heart prepared in 1× radioimmunoprecipitation assay (RIPA) lysis buffer with protease inhibitors. FP signal increased in a protein concentration–dependent manner in all of the samples (n=8 per group). G, The assay tolerated DMSO up to 4% v/v with ≈5% to 7% decrease in activity when compared with 0% DMSO. H, We evaluated the assay by carrying out 24 replicates each of baseline reaction (Rxn); 4% DMSO, 10 μmol/L L682.777, and RIPA buffer negative control. The Z′ values were 0.76 (Rxn vs RIPA), 0.71 (Rxn vs L682.777), 0.71 (DMSO vs RIPA), and 0.62 (DMSO vs L682.777). A Z′ value of 0.5 to 1 represents a robust assay (n=24 per group).
Figure S2. Smooth muscle cells (SMCs) isolated from TG2−/− and wild‐type (WT) mouse aortae (MASMC) were stained with smooth muscle actin to verify the quality of SMC isolation. More than 98% of the cells stained positive for smooth muscle actin. Images are representative of 10 SMC preparations.
Figure S3. De‐adhesion dynamics of TG2−/− and wild‐type (WT) smooth muscle cells (SMCs) were examined by using 0.05% trypsin to sever the cell–matrix interactions. Time course of de‐adhesion on (A) tissue culture plastic; (B) fibronectin; and (C) collagen I‐coated dishes. D, the τ2 time constant of the de‐adhesion curves was similar for TG2−/− and WT SMCs on all supports. Ai=initial spread area; At=spread area at time t; Af=final spread area.
Figure S4. Dose response (D/R) curves for induction of vasorelaxation by acetylcholine (Ach: left) and sodium nitroprusside (SNP; right) in aortic rings from wild‐type (WT) and TG2−/− mice preconstricted with phenylephrine. No difference is apparent between the 2 groups (n=8 per group).
Figure S5. Results from human coronary artery tissue microarray construction (TMA). A, Significant fibrosis was present in samples and %Fibrosis (scored using the Aperio software) did not correlate with age or (B) disease. (C) Plaque was present in all the samples; plaque size was scored by a pathologist at the Johns Hopkins University School of Medicine and did not correlate with age or (D) disease. (E) TG2, (F) Fibronectin, and (G) TG‐crosslinks were highly expressed in the vascular media and did not correlate with disease.
Acknowledgments
The authors would like to thank Karen Fox‐Talbot for her technical assistance with immunostaining of the human coronary artery tissue microarray.
(J Am Heart Assoc. 2017;6:e004161. DOI: 10.1161/JAHA.116.004161.)
References
- 1. Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25:932–943. [DOI] [PubMed] [Google Scholar]
- 2. Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all‐cause mortality with arterial stiffness: a systematic review and meta‐analysis. J Am Coll Cardiol. 2010;55:1318–1327. [DOI] [PubMed] [Google Scholar]
- 3. Lacolley P, Challande P, Osborne‐Pellegrin M, Regnault V. Genetics and pathophysiology of arterial stiffness. Cardiovasc Res. 2009;81:637–648. [DOI] [PubMed] [Google Scholar]
- 4. Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, Resuello RR, Natividad FF, Hunter WC, Genin GM, Elson EL, Vatner DE, Meininger GA, Vatner SF. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res. 2010;107:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sehgel NL, Sun Z, Hong Z, Hunter WC, Hill MA, Vatner DE, Vatner SF, Meininger GA. Augmented vascular smooth muscle cell stiffness and adhesion when hypertension is superimposed on aging. Hypertension. 2015;65:370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sehgel NL, Vatner SF, Meininger GA. “Smooth muscle cell stiffness syndrome”‐revisiting the structural basis of arterial stiffness. Front Physiol. 2015;6:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouissou C, Lacolley P, Dabire H, Safar ME, Gabella G, Duchatelle V, Challande P, Bezie Y. Increased stiffness and cell‐matrix interactions of abdominal aorta in two experimental nonhypertensive models: long‐term chemically sympathectomized and sinoaortic denervated rats. J Hypertens. 2014;32:652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sazonova OV, Isenberg BC, Herrmann J, Lee KL, Purwada A, Valentine AD, Buczek‐Thomas JA, Wong JY, Nugent MA. Extracellular matrix presentation modulates vascular smooth muscle cell mechanotransduction. Matrix Biol. 2015;41:36–43. [DOI] [PubMed] [Google Scholar]
- 9. Gurtner GH, Burke‐Wolin T. Interactions of oxidant stress and vascular reactivity. Am J Physiol. 1991;260:L207–L211. [DOI] [PubMed] [Google Scholar]
- 10. Irani K. Oxidant signaling in vascular cell growth, death, and survival: a review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ Res. 2000;87:179–183. [DOI] [PubMed] [Google Scholar]
- 11. Puhlmann U, Ziemann C, Ruedell G, Vorwerk H, Schaefer D, Langebrake C, Schuermann P, Creutzig U, Reinhardt D. Impact of the cyclooxygenase system on doxorubicin‐induced functional multidrug resistance 1 overexpression and doxorubicin sensitivity in acute myeloid leukemic HL‐60 cells. J Pharmacol Exp Ther. 2005;312:346–354. [DOI] [PubMed] [Google Scholar]
- 12. Eckert RL, Kaartinen MT, Nurminskaya M, Belkin AM, Colak G, Johnson GV, Mehta K. Transglutaminase regulation of cell function. Physiol Rev. 2014;94:383–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nurminskaya MV, Belkin AM. Cellular functions of tissue transglutaminase. Int Rev Cell Mol Biol. 2012;294:1–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. [DOI] [PubMed] [Google Scholar]
- 15. Santhanam L, Berkowitz DE, Belkin AM. Nitric oxide regulates non‐classical secretion of tissue transglutaminase. Commun Integr Biol. 2011;4:584–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zemskov EA, Mikhailenko I, Hsia RC, Zaritskaya L, Belkin AM. Unconventional secretion of tissue transglutaminase involves phospholipid‐dependent delivery into recycling endosomes. PLoS One. 2011;6:e19414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jandu SK, Webb AK, Pak A, Sevinc B, Nyhan D, Belkin AM, Flavahan NA, Berkowitz DE, Santhanam L. Nitric oxide regulates tissue transglutaminase localization and function in the vasculature. Amino Acids. 2013;44:261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Santhanam L, Tuday EC, Webb AK, Dowzicky P, Kim JH, Oh YJ, Sikka G, Kuo M, Halushka MK, Macgregor AM, Dunn J, Gutbrod S, Yin D, Shoukas A, Nyhan D, Flavahan NA, Belkin AM, Berkowitz DE. Decreased S‐nitrosylation of tissue transglutaminase contributes to age‐related increases in vascular stiffness. Circ Res. 2010;107:117–125. [DOI] [PubMed] [Google Scholar]
- 19. Jung SM, Jandu S, Steppan J, Belkin A, An SS, Pak A, Choi EY, Nyhan D, Butlin M, Viegas K, Avolio A, Berkowitz DE, Santhanam L. Increased tissue transglutaminase activity contributes to central vascular stiffness in eNOS knockout mice. Am J Physiol Heart Circ Physiol. 2013;305:H803–H810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bakker EN, Buus CL, Spaan JA, Perree J, Ganga A, Rolf TM, Sorop O, Bramsen LH, Mulvany MJ, Vanbavel E. Small artery remodeling depends on tissue‐type transglutaminase. Circ Res. 2005;96:119–126. [DOI] [PubMed] [Google Scholar]
- 21. Bakker EN, Pistea A, Spaan JA, Rolf T, de Vries CJ, van Rooijen N, Candi E, VanBavel E. Flow‐dependent remodeling of small arteries in mice deficient for tissue‐type transglutaminase: possible compensation by macrophage‐derived factor XIII. Circ Res. 2006;99:86–92. [DOI] [PubMed] [Google Scholar]
- 22. van den Akker J, VanBavel E, van Geel R, Matlung HL, Guvenc Tuna B, Janssen GM, van Veelen PA, Boelens WC, De Mey JG, Bakker EN. The redox state of transglutaminase 2 controls arterial remodeling. PLoS One. 2011;6:e23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Akimov SS, Belkin AM. Cell‐surface transglutaminase promotes fibronectin assembly via interaction with the gelatin‐binding domain of fibronectin: a role in TGFbeta‐dependent matrix deposition. J Cell Sci. 2001;114:2989–3000. [DOI] [PubMed] [Google Scholar]
- 24. Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin‐binding adhesion coreceptor for fibronectin. J Cell Biol. 2000;148:825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hang J, Zemskov EA, Lorand L, Belkin AM. Identification of a novel recognition sequence for fibronectin within the NH2‐terminal beta‐sandwich domain of tissue transglutaminase. J Biol Chem. 2005;280:23675–23683. [DOI] [PubMed] [Google Scholar]
- 26. Telci D, Wang Z, Li X, Verderio EA, Humphries MJ, Baccarini M, Basaga H, Griffin M. Fibronectin‐tissue transglutaminase matrix rescues RGD‐impaired cell adhesion through syndecan‐4 and beta1 integrin co‐signaling. J Biol Chem. 2008;283:20937–20947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Z, Collighan RJ, Gross SR, Danen EH, Orend G, Telci D, Griffin M. RGD‐independent cell adhesion via a tissue transglutaminase‐fibronectin matrix promotes fibronectin fibril deposition and requires syndecan‐4/2 alpha5beta1 integrin co‐signaling. J Biol Chem. 2010;285:40212–40229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Z, Telci D, Griffin M. Importance of syndecan‐4 and syndecan‐2 in osteoblast cell adhesion and survival mediated by a tissue transglutaminase‐fibronectin complex. Exp Cell Res. 2011;317:367–381. [DOI] [PubMed] [Google Scholar]
- 29. Mangala LS, Fok JY, Zorrilla‐Calancha IR, Verma A, Mehta K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene. 2007;26:2459–2470. [DOI] [PubMed] [Google Scholar]
- 30. Stephens P, Grenard P, Aeschlimann P, Langley M, Blain E, Errington R, Kipling D, Thomas D, Aeschlimann D. Crosslinking and G‐protein functions of transglutaminase 2 contribute differentially to fibroblast wound healing responses. J Cell Sci. 2004;117:3389–3403. [DOI] [PubMed] [Google Scholar]
- 31. Baek KJ, Kang S, Damron D, Im M. Phospholipase Cdelta1 is a guanine nucleotide exchanging factor for transglutaminase II (Galpha h) and promotes alpha 1B‐adrenoreceptor‐mediated GTP binding and intracellular calcium release. J Biol Chem. 2001;276:5591–5597. [DOI] [PubMed] [Google Scholar]
- 32. Feng JF, Rhee SG, Im MJ. Evidence that phospholipase delta1 is the effector in the Gh (transglutaminase II)‐mediated signaling. J Biol Chem. 1996;271:16451–16454. [DOI] [PubMed] [Google Scholar]
- 33. Kang SK, Kim DK, Damron DS, Baek KJ, Im MJ. Modulation of intracellular Ca(2+) via alpha(1B)‐adrenoreceptor signaling molecules, G alpha(h) (transglutaminase II) and phospholipase C‐delta 1. Biochem Biophys Res Commun. 2002;293:383–390. [DOI] [PubMed] [Google Scholar]
- 34. Kang SK, Yi KS, Kwon NS, Park KH, Kim UH, Baek KJ, Im MJ. Alpha1B‐adrenoceptor signaling and cell motility: GTPase function of Gh/transglutaminase 2 inhibits cell migration through interaction with cytoplasmic tail of integrin alpha subunits. J Biol Chem. 2004;279:36593–36600. [DOI] [PubMed] [Google Scholar]
- 35. Sen S, Kumar S. Cell‐matrix de‐adhesion dynamics reflect contractile mechanics. Cell Mol Bioeng. 2009;2:218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wegener J, Keese CR, Giaever I. Electric cell‐substrate impedance sensing (ECIS) as a noninvasive means to monitor the kinetics of cell spreading to artificial surfaces. Exp Cell Res. 2000;259:158–166. [DOI] [PubMed] [Google Scholar]
- 37. Zudaire E, Cuesta N, Murty V, Woodson K, Adams L, Gonzalez N, Martinez A, Narayan G, Kirsch I, Franklin W, Hirsch F, Birrer M, Cuttitta F. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 2008;118:640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. An SS, Fabry B, Trepat X, Wang N, Fredberg JJ. Do biophysical properties of the airway smooth muscle in culture predict airway hyperresponsiveness? Am J Respir Cell Mol Biol. 2006;35:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bursac P, Fabry B, Trepat X, Lenormand G, Butler JP, Wang N, Fredberg JJ, An SS. Cytoskeleton dynamics: fluctuations within the network. Biochem Biophys Res Commun. 2007;355:324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson C, Galis ZS. Quantitative assessment of collagen assembly by live cells. J Biomed Mater Res A. 2003;67:775–784. [DOI] [PubMed] [Google Scholar]
- 41. Steppan J, Sikka G, Jandu S, Barodka V, Halushka MK, Flavahan NA, Belkin AM, Nyhan D, Butlin M, Avolio A, Berkowitz DE, Santhanam L. Exercise, vascular stiffness, and tissue transglutaminase. J Am Heart Assoc. 2014;3:e000599 doi: 10.1161/JAHA.113.000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Butlin M, Lindesay G, Viegas KD, Avolio AP. Pressure dependency of aortic pulse wave velocity in vivo is not affected by vasoactive substances that alter aortic wall tension ex vivo. Am J Physiol Heart Circ Physiol. 2015;308:H1221–H1228. [DOI] [PubMed] [Google Scholar]
- 43. Lorand L, Lockridge OM, Campbell LK, Myhrman R, Bruner‐Lorand J. Transamidating enzymes. II. A continuous fluorescent method suited for automating measurements of factor XIII in plasma. Anal Biochem. 1971;44:221–231. [DOI] [PubMed] [Google Scholar]
- 44. Halushka MK, Cornish TC, Lu J, Selvin S, Selvin E. Creation, validation, and quantitative analysis of protein expression in vascular tissue microarrays. Cardiovasc Pathol. 2010;19:136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Weisbrod RM, Shiang T, Al Sayah L, Fry JL, Bajpai S, Reinhart‐King CA, Lob HE, Santhanam L, Mitchell G, Cohen RA, Seta F. Arterial stiffening precedes systolic hypertension in diet‐induced obesity. Hypertension. 2013;62:1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Borlaug BA, Kass DA. Ventricular‐vascular interaction in heart failure. Heart Fail Clin. 2008;4:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boonyasirinant T, Rajiah P, Setser RM, Lieber ML, Lever HM, Desai MY, Flamm SD. Aortic stiffness is increased in hypertrophic cardiomyopathy with myocardial fibrosis: novel insights in vascular function from magnetic resonance imaging. J Am Coll Cardiol. 2009;54:255–262. [DOI] [PubMed] [Google Scholar]
- 48. Telci D, Collighan RJ, Basaga H, Griffin M. Increased TG2 expression can result in induction of transforming growth factor beta1, causing increased synthesis and deposition of matrix proteins, which can be regulated by nitric oxide. J Biol Chem. 2009;284:29547–29558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Simon DD, Niklason LE, Humphrey JD. Tissue transglutaminase, not lysyl oxidase, dominates early calcium‐dependent remodeling of fibroblast‐populated collagen lattices. Cells Tissues Organs. 2014;200:104–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shiraishi T, Verdone JE, Huang J, Kahlert UD, Hernandez JR, Torga G, Zarif JC, Epstein T, Gatenby R, McCartney A, Elisseeff JH, Mooney SM, An SS, Pienta KJ. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget. 2015;6:130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vincan E, Neylon CB, Jacobsen AN, Woodcock EA. Reduction in Gh protein expression is associated with cytodifferentiation of vascular smooth muscle cells. Mol Cell Biochem. 1996;157:107–110. [DOI] [PubMed] [Google Scholar]
- 52. Petersen‐Jones HG, Johnson KB, Hitomi K, Tykocki NR, Thompson JM, Watts SW. Transglutaminase activity is decreased in large arteries from hypertensive rats compared with normotensive controls. Am J Physiol Heart Circ Physiol. 2015;308:H592–H602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nurminskaya M, Beazley KE, Smith EP, Belkin AM. Transglutaminase 2 promotes PDGF‐mediated activation of PDGFR/Akt1 and beta‐catenin signaling in vascular smooth muscle cells and supports neointima formation. J Vasc Res. 2014;51:418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee MY, Chung S, Bang HW, Baek KJ, Uhm D. Modulation of large conductance Ca2+‐activated K+ channel by Galphah (transglutaminase II) in the vascular smooth muscle cell. Pflugers Arch. 1997;433:671–673. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Fluorescence polarization (FP)‐based assay for transglutaminase (TG) activity. A, The principle of the assay is that as the low‐molecular‐weight fluorescent molecule FITC‐cadaverine is incorporated by transamidation into the high‐molecular‐weight N,N′‐dimethylcasein, the FP signal of FITC‐cadaverine increases. B, His‐tagged recombinant human TG2 was expressed in HEK293 cells and purified with a BD‐Talon purification kit before use. Representative Coomassie blue staining of the purified product is shown in the inset. FP signal increased with increasing TG2 protein in the reaction mixture. C, The IC50 of cystamine was calculated to be 0.025±0.005 mmol/L in the FP assay. This value is in good agreement with the published value of 0.02±0.05 mmol/L (n=12 per group). D, The IC50 of L682.777 was calculated to be 0.86±0.1 μmol/L, which is in good agreement with the value of 0.5 μmol/L provided by the vendor (Zedira; n=12 per group). E, Unlabeled cadaverine, which was used as a competitive inhibitor for FITC‐cadaverine, had an IC50 of 56±0.1 nmol/L (n=12 per group). F, The assay was used to examine activity in the protein lysates/homogenates obtained from human aortic endothelial cells (HAEC), rat liver, and rat heart prepared in 1× radioimmunoprecipitation assay (RIPA) lysis buffer with protease inhibitors. FP signal increased in a protein concentration–dependent manner in all of the samples (n=8 per group). G, The assay tolerated DMSO up to 4% v/v with ≈5% to 7% decrease in activity when compared with 0% DMSO. H, We evaluated the assay by carrying out 24 replicates each of baseline reaction (Rxn); 4% DMSO, 10 μmol/L L682.777, and RIPA buffer negative control. The Z′ values were 0.76 (Rxn vs RIPA), 0.71 (Rxn vs L682.777), 0.71 (DMSO vs RIPA), and 0.62 (DMSO vs L682.777). A Z′ value of 0.5 to 1 represents a robust assay (n=24 per group).
Figure S2. Smooth muscle cells (SMCs) isolated from TG2−/− and wild‐type (WT) mouse aortae (MASMC) were stained with smooth muscle actin to verify the quality of SMC isolation. More than 98% of the cells stained positive for smooth muscle actin. Images are representative of 10 SMC preparations.
Figure S3. De‐adhesion dynamics of TG2−/− and wild‐type (WT) smooth muscle cells (SMCs) were examined by using 0.05% trypsin to sever the cell–matrix interactions. Time course of de‐adhesion on (A) tissue culture plastic; (B) fibronectin; and (C) collagen I‐coated dishes. D, the τ2 time constant of the de‐adhesion curves was similar for TG2−/− and WT SMCs on all supports. Ai=initial spread area; At=spread area at time t; Af=final spread area.
Figure S4. Dose response (D/R) curves for induction of vasorelaxation by acetylcholine (Ach: left) and sodium nitroprusside (SNP; right) in aortic rings from wild‐type (WT) and TG2−/− mice preconstricted with phenylephrine. No difference is apparent between the 2 groups (n=8 per group).
Figure S5. Results from human coronary artery tissue microarray construction (TMA). A, Significant fibrosis was present in samples and %Fibrosis (scored using the Aperio software) did not correlate with age or (B) disease. (C) Plaque was present in all the samples; plaque size was scored by a pathologist at the Johns Hopkins University School of Medicine and did not correlate with age or (D) disease. (E) TG2, (F) Fibronectin, and (G) TG‐crosslinks were highly expressed in the vascular media and did not correlate with disease.