

Abstract
Small molecules that bind to RNA potently and specifically are relatively rare. The study of molecules that bind to the HIV-1 transactivation response (TAR) hairpin, a cis-acting HIV genomic element, has long been an important model system for the chemistry of targeting RNA. Here we report the synthesis, biochemical and structural evaluation of a series of molecules that bind to HIV-1 TAR RNA. A promising analog, 15, retained the TAR binding affinity of the initial hit and displaced a Tat-derived peptide with an IC50 of 40 >M. NMR characterization of a soluble analog, 2, revealed a non-canonical binding mode for this class of compounds. Finally, evaluation of 2 and 15 by Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) indicates specificity in binding to TAR within the context of an in vitro-synthesized 365-nt HIV-1 5′-untranslated region (UTR). Thus, these compounds exhibit a novel and specific mode of interaction with TAR, providing important implications for RNA ligand design.
Keywords: HIV-1 transactivation response RNA (TAR), RNA-binding small molecules, thienopyridine compounds, NMR, SHAPE
Graphical Abstract
Introduction
RNA-binding small molecules are an important and highly challenging area of therapeutic research. The discovery of non-coding RNAs and their widespread role in human disease strongly suggests that opportunities to redress disease conditions by targeting RNA should be plentiful.1 Structural and analytical techniques over the last few decades have provided information about RNA structure and function, yet examples of small drug-like molecules that interact with RNA are still rare relative to proteins. The polyanionic nature of RNA, coupled with its highly flexible and dynamic structure and repetitive nature of its surface, present challenges for inhibitor design. Many known RNA-binding small molecules are polycationic, binding tightly (with Kds in the mid nM range) but with limited specificity. Furthermore, they tend to be larger than desirable for typical drugs and do not satisfy the characteristic physicochemical properties of modern successful drug candidates.
A well-characterized system to study small molecule RNA antagonists is transactivation response (TAR) element from HIV type 1 (HIV-1), a 59-nucleotide stem-loop RNA located in the 5′-untranslated region (UTR) of the viral genome. TAR plays a pivotal role in HIV replication via its interaction with Tat, a virus-encoded protein that recruits the host transcriptional machinery to facilitate efficient transcription of the viral DNA.2–5 While the TAR structure is evolutionary conserved, sequence heterogeneity exists among various virus isolates.6 However, in the case of HIV-1 subtypes, base pair co-variations, e.g., A-U conversion into G-C has been shown to maintain TAR structural integrity.7 In addition to being a transcriptional activator, TAR has also been shown to play a role in initiation of reverse transcription.8–10 More recently, several groups have demonstrated that TAR also functions as a miRNA that downregulates cellular apoptotic genes and leads to extended cellular proliferation and continued virus production.11–13 In sum, The central role of TAR in HIV-1 biology underscores its potential as a therapeutic target.14 Thus, inhibition of the Tat-TAR interaction has been pursued as a potential anti-HIV therapeutic strategy. While numerous classes of small molecules have been reported15–17, the lack of anti-HIV drugs targeting TAR in the clinic highlights the challenges in developing small molecule RNA binders. TAR RNA remains both an important anti-viral target and a model system to develop RNA-targeting molecules.
Previously, we utilized a small molecule microarray (SMM) screening platform to rapidly screen immobilized small molecules against nucleic acid targets.18 In one example, we reported the discovery of 1, a thienopyridine compound that interacts with the TAR hairpin.19 However, at that time little was known about the mode of interaction between the compound and RNA. Here, we have developed a chemical synthesis of 1 to generate a focused library of 25 compounds derived from the initial hit structure. Biochemical and biophysical analyses of this series of compounds, including affinity measurements, NMR analysis, and SHAPE, provided information about structure-activity relationships as well as insights into the mode of binding of this class of compounds to HIV-1 TAR.
Results and Discussion
Chemistry
To prepare a focused compound library, we developed a modular and convergent synthetic route to 1 and its analogs. For preparation of 1, 2-chloro-6-methylnicotinonitrile (24) was treated with thiourea in refluxing n-butanol to provide 2-mercapto-6-methylnicotinonitrile (25) in essentially quantitative yield (Scheme 1).20 In parallel, reaction of 3-(trifluoromethyl)aniline (26) with bromoacetyl bromide in the presence of catalytic amounts of 4-N,N-dimethylamino-pyridine (DMAP) provided 2-bromo-N-(3-(trifluoromethyl)phenyl)acetamide (27) in high yield (Scheme 1).21 Reaction of mercaptopyridine 25 with bromoacetamide 27 in refluxing ethanol in the presence of excess sodium ethoxide promoted bromide displacement by the thiol group of the mercaptopyridine intermediate followed by cyclization to afford the thienopyridine ring structure, providing 1 in 58% yield (Scheme 1).22
Scheme 1.

Synthetic route to 1a
a Reagents and conditions: (a) thiourea, n-BuOH, reflux, 3 h, quantitative; (b) bromoacetyl bromide, DMAP, DCM, 0 °C to rt, 12 h, 88%; (c) NaOEt, EtOH, reflux, 1 h, 58%.
We next used this route to design and synthesize several analogs of 1 to evaluate how structural changes would affect affinity for TAR. 2–4, containing a second pyridine ring, were prepared by reaction of the associated aminopyridines (28g-i) with bromoacetyl bromide to provide the bromoacetamide as an HBr salt (29g-i) (Scheme 2A).23 Treatment of bromoacetamides 29g-i with 2-mercapto-6-methylnicotinonitrile (25) in refluxing ethanol in the presence of excess base yielded the assembled thienopyridine ring system (Scheme 2A). 5–9 were prepared via a similar synthetic route to 1 (Scheme 2B). In the case of 10, with a bulkier isopropyl group at the 6-position of the pyridine core, reaction of 2-chloro-6-isopropylnicotinonitrile (30c) with thiourea failed to yield the desired mercaptopyridine. Therefore, Lawesson’s reagent was used to synthesize the desired mercaptopyridine (31c) in modest yield.24 The reaction of mercaptopyridine 31c with the desired bromoacetamide (27) proceeded as described for 1 to provide 10 (Scheme 2B). Preparation of 11, lacking an amine, was achieved by a modified Sandmeyer reaction, employing NaNO2/H2SO4/Cu2O with hypophosphorous acid (Scheme 3).25 Additionally, reaction of 1 with triphosgene at room temperature provided cyclized 12 (Scheme 3).26 Analogs containing a sulfonamide linker were synthesized by the reaction of desired anilines (26 and 34) and chloromethanesulfonyl chloride in the presence of triethylamine followed by a basic workup27 to provide the sulfonamide intermediate (35–36) (Scheme 3). Sulfonamides 35–36 were treated with the desired mercaptopyridine (25) in the presence of excess base in N,N-dimethylformaide (DMF) at 130 °C to afford 13 and 14 (Scheme 3).27
Scheme 2.

Synthetic routes to (A) pyridinyl analogs 2–4 (B) 5–10a
a Reagents and conditions: (a) bromoacetylbromide, DCM, rt, 3–12 h, 55%-95%; (b) 25, NaOEt, EtOH, reflux, 12 h, 32%-46%; (c) thiourea, n-BuOH, reflux, 3 h, quantitative; (d) Lawesson’s reagent, toluene, reflux, 4 h 30 min, 42%; (e) bromoacetyl bromide, DMAP, DCM, 0 °C to rt, 6–12 h, 62%-94%; (f) NaOEt, EtOH, reflux, 1 h, 23%-78%.
Scheme 3.

Synthetic route to 11–14a
a Reagents and conditions: (a) 50% aq. H3PO2, conc. H2SO4, Cu2O, 1M aq. NaNO2, MeOH, 0 °C to rt, 2 h 30 min, 22%; (b) triphosgene, THF, rt, 1 h, 98%; (c) chloromethylsulfonyl chloride, Et3N, DCM, 0 °C to rt, 12 h then 2N NaOH, rt, 2 h 30 min, 60%-72%; (d) 25, NaOEt, DMF, 130 °C, 1 h, 33%-72%.
We also prepared 15–19, containing oxadiazolyl linkers between the thienopyridine core and the aryl group (Scheme 4). For 15 and 16, which contain 1,3,4-oxadiazol-2,5-yl and 1,2,4-oxadiazol-3,5-yl rings (respectively), commercially available chloromethyl phenyl substituted oxadiazoles (37I-m) were treated with 2-mercapto-6-methylnicotinonitrile (25) in the presence of excess base in DMF at 130 °C, producing the thienopyridine scaffold in good yield. 17–23 were synthesized similar to 16 (Scheme 4 and Scheme S1). In the case of 24–26, the desired chloromethyloxadiazoles were conveniently prepared as reported previously28 and further treated with 25 to yield the desired products (Scheme S2). Collectively, the convergent route reported here furnished a library of 25 analogs of the initial hit structure.
Scheme 4.

Synthetic route to oxadiazole analogs 15–19a
a Reagents and conditions: (a) 25 or 2-mercaptopyridine-3-carbonitrile (38), NaOEt, DMF, 130 °C, 1 h, 31%-76%.
Biochemical evaluation of compound library
a. Affinity measurement
To evaluate the TAR binding properties of analogs described above, we utilized a series of biochemical assays that provide information on both compound affinity for TAR and its ability to displace a Tat-derived peptide from TAR, revealing a clear picture of structure-activity relationship. To probe direct binding of compounds to TAR, we utilized two different 2-aminopurine (2-AP) assays. 2-AP is an environmentally-sensitive fluorophore that reports on the local environment of the base.29 Conformational changes in the RNA upon binding of a ligand result in changes in fluorescence intensity, which can be used to derive accurate affinities. Two distinct constructs were used where either U25 or U23 (both of which are located in the bulge region of TAR hairpin) was replaced with a 2-AP fluorophore (Figure 1A). U25 is a flexible residue30 and its replacement by 2AP has been previously utilized in probing TAR-ligand interactions29. To evaluate larger numbers of compounds, we miniaturized the 2-AP assay to be compatible with plate reader-based analysis. This format required a higher concentration of RNA, but enabled a significantly reduced assay volume of 100 >L per sample in a 96 well plate. Once optimized, this assay generated reproducible apparent binding constants (Kdapp), albeit with slightly higher values than obtained under previous assay conditions.19 Values obtained under these conditions were both internally consistent and comparable to recently published Kds of several TAR-binding small molecules.31
Figure 1.

(A) Secondary structure of 29nt TAR hairpin with the sites of 2AP replacement highlighted. Constructs were designed with either U23 or U25 replaced with 2-AP. Binding curves from (B) 25-2AP and (C) FRET assays for 2 and 15.
Kdapp obtained from 2-AP assays with both constructs were in good agreement (Table 1). Synthetic 1 showed an apparent Kdapp 25-2AP of 47.7 ± 11.6 >M. Next, the trifluoromethylphenyl group was replaced by different pyridine isomers (2–4) which dramatically improved compound solubility, making them usable at high concentrations in NMR experiments (see below). Interestingly, analog 2, containing 4-pyridyl substitution, exhibited a similar binding affinity (Kdapp 25-2AP = 40.0 ± 0.7 >M) to 1 (Figure 1B). Analogs 3, containing 3-pyridyl (Kdapp 25-2AP = 68.0 ± 26.0 >M) and 4 with 2-pyridyl (Kdapp 25-2AP = 50.3 ± 10.3 >M) substituents showed ~2-fold weaker binding affinity compared to 2. Changing the position of the meta-trifluoromethyl group in 1 to para (7, Kdapp 25-2AP = 75.3 ± 9.2 >M) or ortho (8, Kdapp 25-2AP = 90.1 ± 4.7 >M) decreased binding affinity approximately 2-fold. Additionally, extending the meta-trifluoromethyl phenyl by a methylene linker (9, Kdapp 25-2AP = 66.4 ± 0.9 >M) maintained a similar binding affinity to 1.
Table 1.
Affinity measurement and Tat/TAR displacement assay for 1 and analogs
| Compound | 25-2AP TAR Kd app (μM)a | 23-2AP TAR Kd app (μM)a | FRET IC50 (μM)a |
|---|---|---|---|
| 1 | 47.7 ± 11.6 | 54.1 ± 5.8 | >400 |
| 2 | 40.0 ± 0.7 | 51.1 ± 5.5 | >400 |
| 3 | 68.0 ± 26.0 | 89.5 ± 7.3 | >400 |
| 4 | 50.3 ± 10.3 | 64.3 ± 2.8 | >400 |
| 5 | 66.5 ± 4.5 | 94.1 ± 20.6 | >400 |
| 6 | 48.5 ± 5.4 | 55.4 ± 9.6 | >400 |
| 7 | 75.3 ± 9.2 | 84.1 ± 9.6 | 87.0 ± 2.6 |
| 8 | 90.1 ± 4.7 | >400 | 90.4 ± 23.3 |
| 9 | 66.4 ± 0.9 | 69.4 ± 14.2 | >400 |
| 10 | 43.3 ± 13.3 | 107.1 ± 35.4 | >400 |
| 11 | > 400 | >400 | >400 |
| 12 | >400 | >400 | >400 |
| 13 | >400 | >400 | >400 |
| 14 | >400 | >400 | >400 |
| 15 | 49.1 ± 7.8 | 75.0 ± 15.4 | 40.1 ± 5.6 |
| 16 | 47.7 ± 17.8 | 46.0 ± 15.2 | 45.1 ± 25.8 |
| 17 | 44.6 ± 7.9 | 56.9 ± 8.1 | >300 |
| 18 | 42.5 ± 2.9 | 45.6 ± 7.8 | >400 |
| 19 | 50.2 ± 8.4 | 44.1 ± 9.0 | 37.6 ± 13.1 |
Highest concentration evaluated was 400 μM unless otherwise indicated. Values represent the average of three replicates, ± standard deviation.
We further investigated the role of substitution on the pyridyl ring of thienopyridine scaffold. Moving the methyl group to the 5-position (6, Kdapp 25-2AP = 48.5 ± 5.4 >M) maintained a similar binding affinity as 1 while the 4-methyl substituent (5, Kdapp 25-2AP = 66.5 ± 4.5 >M) showed ~2-fold weaker binding affinity. Replacing the methyl substituent with a bulkier isopropyl group in 10 (Kdapp 25-2AP = 43.3 ± 13.3 >M) maintained similar affinity compared to 1. Next, Analog 11, a desamino thienopyridine, showed inferior binding affinity, highlighting the importance of the amino group. Additionally, replacing amide for a sulfonamide in analog 13 and 14 or a cyclized analog, 12, completely abolished binding.
Next, we evaluated the oxadiazole series. Interestingly, isosteric replacement of the amide linker in 1 with 1,3,4-oxadiazol-2,5-yl in 15 (Kdapp 25-2AP = 49.1 ± 7.8 >M) (Figure 1B) as well as 1,2,4-oxadiazol-3,5-yl in 16 (Kdapp 25-2AP = 47.7 ± 17.8 >M) retained binding affinity as observed for 1. Analogs 17–19, containing the 1,3,4-oxadiazol-2,5-yl but with different substitution pattern, retained the binding affinity of 15. However, evaluation of 1,3,4-oxadiazol-2,5-yl analogs 20–26 either decreased solubility or abolished binding affinity (Table S1).
b. Competitive binding assay measurements
In addition to the 2-AP direct binding assays, we utilized an orthogonal FRET-based assay to evaluate disruption of the Tat-TAR interaction.32 In this assay, a 16-mer peptide derived from Tat protein is labeled with fluorescein at its N-terminus and TAMRA at its C-terminus. In solution, the two fluorophores in the free peptide are in close proximity and quench each other. Upon binding to TAR, the peptide adopts an extended conformation, inducing FRET. In this assay, a compound capable of disrupting the Tat-TAR interaction will cause a loss of FRET signal. All synthesized analogs were evaluated in FRET assay. While many, including 2, did not display activity in this assay up to 400 >M, it is notable that oxadiazole analogs 15 (Figure 1C) and 16 (IC50 = 40.1 ± 5.6 >M and IC50 = 45.1 ± 25.8 >M respectively) displaced the Tat peptide with IC50 values similar to their apparent Kds (Table 1). Additionally, while removal of meta-trifluoromethyl group on the phenyl ring in 17 or its replacement with meta-chloro in 18 abolished FRET signal, the absence of 6-methyl substituent in 19 maintained FRET activity similar to 15 (Table 1) suggesting the importance of phenyl substituent on the ability of the oxadiazole analogs to displace the Tat peptide.
Taken together, our thienopyridine analogs displayed mid micromolar binding to the TAR hairpin under the assay conditions. While the majority of the structural changes in this study resulted in subtle effects, isosteric replacement of the amide with oxadiazoles provided analogs which not only maintained binding affinity to TAR but displaced the Tat peptide in the FRET assay at concentrations in the mid micromolar range. It is also noteworthy that the compounds in this series with similar binding affinity show differing behavior in the FRET-based assay. However, these observations are consistent with a report in which peptides with similar TAR binding affinity also exhibited very different IC50 values in displacing a Tat peptide.33 This seemingly contradictory observation suggests that the determinants of binding may differ from those required for disrupting an associated RNA/peptide interaction, at least for some ligands.
NMR studies
To better understand the mode of interaction of the compounds with TAR, we investigated the small molecule-RNA complex by NMR. Many compounds in this series exhibited poor aqueous solubility, which limited high-resolution structure analysis. Compound 2 displayed increased solubility relative to 1 and could be studied at concentrations amenable for ligand-detected NMR experiments and some limited RNA-detected experiments. 2 was first evaluated using line broadening methods (Figure 2A and Figure S1A–C).34 In this experiment, its proton spectra was recorded in the presence and absence of 10μM TAR. Upon binding, the ligand resonances broaden in response to the increase in rotational correlation time while bound to target RNA.34 In the case of 2, the two doublets corresponding to protons on the pyridine ring of the thienopyridine scaffold (protons a and b in Figure 2A) are completely broadened in presence of TAR suggesting the pyridine ring directly interacts with the hairpin while other proton peaks do not change (Figure S1A). In contrast, ligand-detected NMR of a non-binding analog 14 shows no line broadening in neither the aromatic (Figure 2B) nor aliphatic region (Figure S1B). The line-broadening pattern in the 1D-1H NMR experiments is consistent with the 2-AP fluorescent-based affinity measurements indicating mid micromolar binding.
Figure 2.

(A) Ligand-detected 1H NMR spectra of aromatic region of 2 (150 >M) and B) 14 (150 >M) in the presence (red spectra) and absence of TAR hairpin (10 >M) (black spectra).
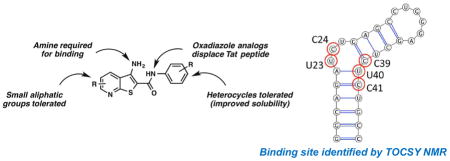
Ligand-detected NMR experiments require lower ligand concentration to measure binding, which is useful for low solubility molecules. However, these methods are also susceptible to nonspecific aggregation effects, which generate false positive results. Therefore, to identify and verify binding of 2, we evaluated ligand-induced chemical shift perturbations in total correlation spectroscopy (TOCSY) experiments. Here, free TAR hairpin was evaluated upon the addition of 2 (Figure 3A–B and Figure S1D–E). Specific changes in the TOCSY spectrum suggest single site binding with the largest changes occurring at bases C39, U40 and C41. These residues are located directly below the -UCU-bulge residues U23, C24 and U25. Resonances within residues U23 and C24 also show significant chemical shift changes. However, unlike many TAR ligands, U25 does not show significant changes in chemical shift, nor do any of the remaining pyrimidines, indicating a new binding mode compared to previously reported TAR-binding ligands such as argininamide and acridine, which disrupt stacking of A22 and U23 and bind to the major groove of TAR similar to Tat-derived peptides.35–37 On the other hand, the solution NMR structure of Neomycin B bound to TAR indicates that this aminoglycoside binds in the minor groove instead.38 However TAR residues G21, U42 and G26 that are shifted in presence of Neomycin B are not the same residues perturbed by 2. Additionally, Hoescht 33258 has been reported to bind to the TAR hairpin, though it binds to the major groove/upper bulge region primarily at and around bases G36-U40.39
Figure 3.

Overlaid 2D TOCSY NMR spectra of pyrimidine residues of TAR (A) with increasing concentrations of 2. Indicated are free TAR (black) and TAR:2 ratio of 1:1 (green), 1:2 (light blue), 1:3 (dark blue), and 1:5 (red). Arrows indicate the direction of chemical shift changes upon addition of 2. (B) Secondary structure of 29nt TAR hairpin with bases perturbed in the presence of 2 highlighted with red circles.
The concentration-dependent chemical shift perturbations of TAR residues induced by 2 suggest that complex formation occurs in the fast exchange regime of the NMR time scale. However, residues U40 and C41 also experience broadening in addition to chemical shift changes specific to these residues. These results are entirely consistent with binding in the mid micromolar range, in agreement with binding affinity determined by biochemical assays. The perturbed residues are concentrated in the bulge and lower bulge region of the TAR hairpin, suggesting that 2 may be a lower bulge binder.
In sum, the NMR studies indicate that 2 binds to TAR and is likely a lower bulge/minor groove binder. This binding mode does not perturb many residues in the Tat binding site itself, which provides insight into the varying ability of compounds in this series to displace the Tat peptide. Attempts to derive a full structure of TAR bound to 2 by intermolecular NOESY experiments were hampered by the limited solubility of the ligand under the required NMR conditions.
SHAPE
Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) can be used to establish RNA secondary structure and identify sites of interaction of RNA with other molecules by examining backbone flexibility.40 Electrophilic reagents, e.g. 1M7 (1-methyl-7-nitroisatoic anhydride) modify the 2′-OH of less-structured RNA nucleotides, which causes reverse transcription (RT) to halt one nucleotide before the modification. Modification frequencies are then used to estimate a ‘reactivity profile’ that quantifies the propensity of each nucleotide to be modified by the chemical probe. High reactivity reflects nucleotides that are flexible and unstructured, while low reactivity provides structural constraining, such as formation of base pairing, stacking or RNA–ligand interactions.
We applied SHAPE to probe RNA conformational changes that would be diagnostic of compound binding in the context of an in vitro-synthesized 365-nt HIV-1 5′ UTR RNA. Changes in 1M7 reactivity at each nucleotide of the HIV-1 5′ UTR RNA in the presence of 2 and 15 are presented in Figure 4 (relative to the analogous experiment performed in its absence). The decrease in nucleotide reactivity upon analog addition is indicated in gradients of blue, while increase in reactivity is represented in gradients of red; unchanged nucleotide reactivity is in gray. While the global topology of the 5′ UTR was unaffected upon incubation with 2 and 15, substantial changes in chemical reactivity were observed mostly within TAR (Figure 4A–B). In particular, addition of 2 caused a decrease in 1M7 reactivity for residue U25 within the TAR bulge and U31, G33 and A35 within the apical loop (Figure 4A). Reactivity changes observed in the apical loop residues for 2 could reflect ligand binding to the lower bulge region (as discussed above) causing disruption of a direct interaction between the apical loop and the bulge as recently demonstrated by Krawczyk et al.41 Addition of 15 to RNA showed a similar 1M7 reactivity reduction throughout TAR compared to 2, and in particular with residues U13–14, C19 and G32 (Figure 4B). Taken together, reduced 1M7 reactivity suggests that analog binding causes structural perturbations to TAR due to a direct or, for loop residues, indirect interaction with 2 and 15. Furthermore, these results are consistent with our observations for 1, which we showed also bound selectively to the TAR hairpin.19 Although the global topology of the 5′ UTR was unchanged upon compound binding, subtle changes in chemical reactivity were observed downstream of the TAR hairpin. These “off target” effects may reflect the influence of compound binding on long-range tertiary interactions42, 43 and/or indirect structural rearrangements in the 5′ UTR.44 In conclusion, the SHAPE data suggest specific binding of both 2 and 15 to the TAR region of HIV-1 UTR in the presence of other structured motifs.
Figure 4.

SHAPE analysis of the HIV-1 5′ UTR in the presence of (A) 2 and (B) 15, respectively. top: nucleotides are color-coded according to normalized SHAPE reactivity values. 1M7 reactivity of the extreme 5′ and 3′ terminal nucleotides (represented in white) are not assigned due to limited resolution in the immediate vicinity of the primer and the final extension product. Nucleotide 1 is vector derived. bottom: step plots for quantitative comparison of 1M7 reactivity values obtained from the HIV-1 RNA 5′ UTR probed in the presence (black) or absence (red) of tested analog. Boxed region corresponds to nucleotides involved in formation of the 5′ TAR hairpin (nts G2–C58).
Conclusions
We have characterized the interaction of a series of thienopyridine compounds to the HIV-1 TAR RNA hairpin. The initial structure, 1, was discovered in a screen of immobilized small molecules, which identified several structures binding to TAR specifically over other RNA stem-loops. Compound 1 and a focused library of 25 analogs were synthesized using a concise and modular synthetic route. These compounds were then evaluated in several biochemical assays for binding to the TAR hairpin as well as to disrupt the Tat-TAR interaction. A clear structure-activity relationship was observed in the binding assays. A number of analogs, but not all those that bound to TAR, were able to displace a Tat-derived peptide from the RNA. Replacing the amide linker with either of two oxadiazolyl groups yielded analogs with similar affinity to the original hit that were also able to disrupt the Tat/TAR interaction. The most potent analog, 15, showed a similar binding affinity to 1 and exhibited an IC50 of 40 >M for displacement of Tat-derived peptide. SHAPE studies showed that both 2 and 15 retained global specificity for binding to the TAR motif within the 365-nt HIV-1 5′ UTR RNA. NMR studies further confirmed that 2 binds to TAR and causes chemical shift perturbations exclusively within and below the bulge region. These results suggest that 2 binds to the lower bulge region, possibly within the minor groove. The perturbed residues differ from those interacting with a Tat-derived peptide, neomycin, Hoechst 33258 or argininamide, which could provide a basis for the varying ability of compounds in this class to displace the peptide. Future efforts will focus on improving solubility of analogs for cell-based studies and further development of a bifunctional molecule to tether our lower bulge binder with an apical loop binder such as 5-(N,N)-dimethyl amiloride31 to achieve both increased specificity and enhanced affinity through synergistic binding.
Experimental Section
Biochemical assays
Nucleic acids were purchased from Dharmacon with the following sequences: TAR RNA: 5′-GGCAGAUCUGAGCCUGGGAGCUCUCUGCC-3′ and 25-2AP TAR: 5′-GGCAGAUC(2AP)GAGCCUGGGAGCUCUCUGCC-3 and 23-2AP TAR: 5′-GGCAGA(2AP)CUGAGCCUGGGAGCUCUCUGCC-3. TAR and 2AP TAR constructs were reconstituted in 1x PBS pH 7.4 to a concentration of ~100 >M and annealed by heating to 95 °C for 3 min. The constructs were allowed to cool to room temperature for at least 30 min. Tat FRET peptide, a 16-mer peptide labeled at its N-terminus by FAM and C-terminus by TAMRA, was purchased from AnaSpec (catalog #: AS-64346-05). A fluorescence-based assay employing TAR constructs labeled with 2-AP either at bulge residues U23 or U25 was used. 2-AP TAR constructs were used at 1 >M. 5 >L of the tested compound in DMSO and 95 >L of 2-AP TAR in the assay buffer (1× PBS pH 7.4) were added to assay plate (Nunc black plates) and incubated at room temperature with rocking for 30 min. Fluorescence values were measured at an excitation wavelength of 310 ± 9 nm and an emission wavelength of 365 ± 9 nm using a microplate reader and values were normalized to free 2-AP TAR. Most analogs were not fluorescent at this wavelength, and in the case of fluorescent compounds, the inherent fluorescence was subtracted from that of the complex (TAR and compound) and then normalized to free 2-AP TAR. Apparent Kd values were determined by non-linear regression by fitting the measured fluorescence (GraphPad Prism 6.0 software). FRET studies were performed as previously reported32 with modification. Unlabeled TAR was used at 100 nM and FAM-Tat-TAMRA at 12 nM. 5 μL of the tested compound (in DMSO) and 95 μL of unlabeled RNA/peptide complex in assay buffer (50 mM Tris pH 7.4; 20 mM KCl; 0.01% Triton X-100) were added to Nunc black assay plates and incubated at room temperature for 30 min with rocking. It should be noted that each plate contained four wells for negative control (equivalent to 0% inhibition and highest fluorescent/FRET) which included 100 nM unlabeled TAR and 12 nM FAM-Tat-TAMRA in presence of 5% DMSO. Additionally, three wells were assigned for the positive control (equivalent to 100% inhibition and lowest fluorescence/FRET) which included 12 nM of FAM-Tat-TAMRA in presence of 5% DMSO. Fluorescence values were measured at an excitation wavelength of 485 ± 20 nm and an emission wavelength at 590 ± 35 nm using a microplate reader. Most analogs were not fluorescent at this wavelength; however for analogs 2, 3, 4, 16, 23 and 25, the fluorescence of complex (unlabeled TAR/labeled Tat) in presence of compound exceeded the maximum fluorescence as indicated by the negative control wells. Therefore, the inherent fluorescence of compound was subtracted from that of the complex plus compounds. IC50 values were determined by non-linear regression fitting of the competition curves.
NMR
HIV-1 TAR RNA hairpin was transcribed and purified as described previously.45–47 Ligand detected NMR experiments were collected on a Bruker DRX 500MHz equipped with TCI cryoprobe or Bruker Avance III 600 MHz spectrometer equipped with TCI cryoprobe. NMR screening buffer consisted of 20 mM d19-Bis-Tris at pH6.5, 10 mM sodium chloride, and 11.1 >M DSA as chemical shift reference (integrates to 100 >M)48, prepared in 99.99% D2O or 95%H2O/5%D2O. Each ligand was first dissolved in DMSO to a 10 mM stock concentration in d6-DMSO. Free ligand screening samples were prepared at 150 >M in NMR screening buffer with final DMSO concentration of 15%. We used this concentration as the safe upper limit beyond which the RNA structure might be affected. For RNA detected experiments, the lyophilized RNA pellets were dissolved in DMSO-free NMR screening buffer and heated for 4 min at 90°C, then snapped cooled for 5 min at −20 °C. 1D-1H experiments for both ligand and RNA were collected using the Bruker “zgesgp” excitation sculpting pulse sequence to suppress background water signal. Data was collected with 64 scans and 16K data points. Data was processed using 16K zero points, Fourier transformed followed by manual phase and baseline correction. 2D-1H TOCSY experiments were collected using the Bruker “mlevesgpph” pulse sequence with excitation sculpting water suppression, data collected with 8 scans, 2K × 512 data points, a recycle delay of 1.2 s and spin-lock mixing time of 80ms. Data was processed using NMRPipe and visualized using SPARKY.
SHAPE
RNA preparation for SHAPE analysis
DNA template for in vitro transcription were generated by PCR amplification of the HIV-1 molecular clone pNL4-3 using the following primers: T7L 5′-TAATACGACTCACTATAGGTCTCTCTG -3′ (containing T7 promoter, underlined) 369R 5′-GCTTAATACCGACGCTCTCGC -3′
All PCR experiments were performed using Invitrogen Platinum® Taq DNA polymerase High Fidelity. Transcripts were synthesized with the MEGAscript T7 transcription kit (Thermo Fisher Scientific) following the manufacturer’s protocol, and RNAs were purified from denaturing 8 M urea/6% polyacrylamide gels, followed by electro-elution and ethanol precipitation. Purified RNAs were dissolved in sterile water and stored at −20 ºC.
Renaturation and 1M7 treatment of RNA
5 pmoles of RNA were heated to 90 ºC for 3 min, and gradually cooled to 4 ºC (0.1 ºC/sec). The volume was adjusted to 10 >L in a final buffer of 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 5 mM MgCl2. Samples were then divided into 5 μl experimental (+) and control (−) aliquots (2.5 pmoles each) and incubated at 37 ºC for 15 min. Analog 2 and 15 were added to both (+) and (−) RNA solutions to a final concentration of 5 >M and 0.5 >M respectively, and incubated for 10 min at room temperature. Chemical modification was initiated by addition of 1 μL of 1M7 (in anhydrous DMSO) to the (+) RNA solution to a final concentration of 3.5 mM, or DMSO alone to the (−) RNA reaction, followed by incubation at 37 ºC for 5 min. Completed reactions were precipitated at −20 ºC with 10 ng/>L glycogen, 0.3M sodium acetate pH 5.2 and 2.5 volumes of cold ethanol. RNA was collected by centrifugation, and resuspended in 10 >L of water. A parallel experiment lacking analogs was performed as a control.
Primer extension
For detection of 2′-O-adducts, 2.5 pmoles of modified and unmodified RNAs were mixed, respectively, with 5 pmoles of Cy5-labelled (for 1M7-modified samples) or 6 pmoles of Cy5.5-labelled (for unmodified samples) DNA primer complementary to their 3′ end. Samples were incubated at 85 ºC for 1 min, 60 ºC for 5 min, 35 ºC for 5 min and 50 ºC for 2 min to anneal primers to initiate reverse transcription. RNA was reverse transcribed at 50 ºC for 50 min with 100U RT (Invitrogen Superscript III), 1^p RT buffer (Invitrogen), 5 mM DTT and 500 mM dNTPs (Promega). RNA was hydrolyzed with 200 mM NaOH for 5 min at 95 ºC and samples were neutralized with an equivalent volume of HCl. Sequencing ladders were prepared using the Epicentre cycle sequencing kit according to the manufacturer’s instructions and primers labeled with WELLRed D2 and LICOR IR-800 dyes. Modified and control samples were mixed with the sequencing ladders, precipitated as above, dried and resuspended in 30 >L deionized formamide. Primer extension products were analyzed on a Beckman CEQ8000 Genetic Analysis System programmed with the separation method described previously.49
SHAPE data analysis
Electropherograms were processed using the program SHAPEfinder program, following the software developer’s protocol and included the required pre-calibration for matrixing and mobility shift for each set of primers as described.40, 50 The area under each negative peak was subtracted from that of the corresponding positive peak. The resulting peak area difference at each nucleotide position was then divided by the average of the highest 8% of peak area differences, calculated after discounting any results greater than the 3rd quartile plus 1.5× the interquartile range. RNAstructure software version 5.7 was used to predict RNA secondary structure(s) on the basis of pseudo-free energy constraints derived from SHAPE reactivity values. All reactivity data used in 2D structure analysis was averaged from three independent experiments. Varna (ver. 3–9), the visualization applet for RNA secondary structure, was used to produce high-quality images.51
Chemistry
Chemicals were purchased from commercial sources and used without further purification unless otherwise noted. Anhydrous solvents were prepared by passage over activated alumina. Thin-layer chromatography was conducted with E. Merck silica gel 60 F254 precoated plates (0.25 mm) and visualized by exposure to UV light (254 nm). Flash column chromatography was performed using normal phase or reverse phase on a CombiFlash® Rf 200i (Teledyne Isco Inc). 1H NMR spectra were recorded at 400 or 500 MHz, and are reported relative to deuterated solvent signals. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz), and integration. 13C NMR spectra were recorded at 100 or 125 MHz. Data for 13C NMR spectra are reported in terms of chemical shift. All NMR spectra were standardized to the NMR solvent signal as reported previously.52 Analytical LC/MS was performed using a Shimadzu LCMS-2020 Single Quadrupole utilizing a Kinetex 2.6 μm C18 100 A (2.1 × 50 mm) column obtained from Phenomenex Inc. Runs employed a gradient of 0→90% ACN/0.1% aqueous formic acid over 4 minutes at a flow rate of 0.2 mL/min. Purities of final compounds were assessed by analytical reversephase HPLC using Agilent Proshell 120 EC-C18 (4.6 mm × 50 mm; particle size 2.7 μm). Runs employed a gradient of 10% ACN/water (1 min), 10–90% ACN/water (6 min), and 90% ACN/water (2 min) flow = 1 mL/min. High-resolution LC/MS analyses acquired on an Agilent 6520 Accurate-Mass Q-TOF LC/MS System, (Agilent Technologies, Inc., Santa Clara, CA) equipped with a dual electrospray source, operated in the positive-ion mode. Separation was performed on Zorbax 300SB-C18 Poroshell column (2.1 mm × 150 mm; particle size 5 μm). All final compounds were purified to >95% purity unless otherwise stated.
Representative procedure for thienopyridine carboxamide preparation
3-Amino-6-methyl-N-(3-(trifluoromethyl)phenyl)thieno[2,3-b]pyridine-2-carboxamide (1)
Synthesized using reported procedure with modification.22 To a suspension of 25 (51 mg, 0.34 mmol) in anhydrous EtOH (2 mL) was added 27 (92 mg, 0.32 mmol) and sodium ethoxide (54 mg, 0.75 mmol). The mixture was heated to reflux for 1 h 15 min. The mixture was diluted with EtOAc (10 mL) and washed with H2O (10 mL × 2) and brine (10 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. Crude was purified by flash column chromatography (hexanes/EtOAc) on silica gel to give 1 (65 mg, 58%) as a light yellow solid. Purity 100%.1H NMR (500 MHz, DMSO-d6) δ 9.70 (s, 1H), 8.41 (d, J = 8.3 Hz, 1H), 8.23 (s, 1H), 7.99 (d, J = 8.3 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.47 (s, 2H), 7.41 (d, J = 7.7 Hz, 1H), 7.35 (d, J = 8.3 Hz, 1H), 2.61 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 164.23, 159.90, 158.54, 147.83, 140.00, 131.15, 129.58, 129.14 (q, J = 31.5 Hz), 125.31 (q, J = 272.4 Hz), 124.20, 123.60, 119.61, 119.41 (q, J = 3.8 Hz), 116.81 (q, J = 4.0 Hz), 94.75, 24.29. ESI HRMS: m/z 352.07332 (M+H)+.
Representative procedure for pyridinylthienopyridine carboxamide preparation
3-Amino-6-methyl-N-(pyridin-4-yl)thieno[2,3-b]pyridine-2-carboxamide (2)
Synthesized using a similar procedure used to prepare 1 except using 29g. Mixture was heated to reflux overnight. Upon cooling to room temperature and addition of H2O, yellow solid precipitated. Solid was filtered and washed with cold EtOH. Crude was purified by flash column chromatography (hexanes/EtOAc) on silica gel to give 2 (18 mg, 22% over two steps) as a yellow solid. Purity 100%.1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.44 – 8.38 (m, 3H), 7.76 (d, J = 6.3 Hz, 2H), 7.53 (s, 2H), 7.35 (d, J = 8.3 Hz, 1H), 2.60 (s, 3H).13C NMR (100 MHz, DMSO-d6) δ 164.49, 160.15, 158.63, 149.99, 148.33, 146.11, 131.24, 123.44, 119.65, 114.19, 94.63, 24.29. ESI HRMS: 285.08048 (M+H)+.
6-Methyl-2-(pyridin-3-ylcarbamoyl)thieno[2,3-b]pyridin-3-aminium 2,2,2-trifluoroacetate (3)
Synthesized using a similar procedure used to prepare 1 except using 29h. Mixture was heated to reflux overnight. Upon cooling to room temperature and addition of H2O, yellow solid precipitated. Solid was filtered and washed with H2O to give the title compound (26 mg, 46%) as a light yellow solid. Purity 98%.1H NMR (400 MHz, DMSO-d6) δ 9.60 (s, 1H), 8.88 (d, J = 2.2 Hz, 1H), 8.39 (d, J = 8.3 Hz, 1H), 8.27 (dd, J = 4.7, 1.4 Hz, 1H), 8.11 – 8.06 (m, 1H), 7.43 (s, 2H), 7.38 – 7.32 (m, 2H), 2.60 (s, 3H).13C NMR (100 MHz, DMSO-d6) δ 164.21, 159.84, 158.52, 147.61, 144.13, 142.59, 135.73, 131.12, 127.95, 123.60, 123.27, 119.59, 94.87, 24.27. ESI HRMS: 285.08043 (M+H)+.
3-Amino-6-methyl-N-(pyridin-2-yl)thieno[2,3-b]pyridine-2-carboxamide 2,2,2-trifluoroacetate (4)
Synthesized using a similar procedure used to prepare 1 except using 29i. Mixture was heated to reflux overnight. Upon cooling to room temperature and addition of H2O, yellow solid precipitated. Solid was filtered and washed with cold EtOH. Crude was purified by flash column chromatography (hexanes/EtOAc) on silica gel to give 4 (18 mg, 32%) as a yellow solid. Purity 100%.1H NMR (400 MHz, DMSO-d6) δ 9.54 (s, 1H), 8.39 (d, J = 8.3 Hz, 1H), 8.35 (d, J = 4.4 Hz, 1H), 8.05 (d, J = 8.3 Hz, 1H), 7.83 – 7.77 (m, 1H), 7.42 (s, 2H), 7.34 (d, J = 8.3 Hz, 1H), 7.12 (dd, J = 6.8, 5.2 Hz, 1H), 2.60 (s, 3H).13C NMR (100 MHz, DMSO-d6) δ 163.99, 159.86, 158.62, 151.96, 147.88, 147.62, 137.86, 131.15, 123.66, 119.51, 119.35, 114.70, 95.67, 24.28. ESI HRMS: 285.08095 (M+H)+.
3-Amino-4-methyl-N-(3-(trifluoromethyl)phenyl)thieno[2,3-b]pyridine-2-carboxamide (5)
Synthesized using a similar procedure used to prepare 1 except using 31a. Crude was purified by flash column chromatography (hexanes/EtOAc) on silica gel to give 5 (44 mg, 37%) as a light yellow solid. Purity 96%. 1H NMR (500 MHz, DMSO-d6) δ 9.78 (s, 1H), 8.49 (d, J = 4.7 Hz, 1H), 8.17 (s, 1H), 8.00 (d, J = 8.3 Hz, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.43 (d, J = 7.8 Hz, 1H), 7.21 (d, J = 5.4 Hz, 1H), 7.08 (s, 2H), 2.81 (s, 3H).13C NMR (125 MHz, DMSO-d6) δ 164.40, 159.28, 150.00, 149.70, 145.16, 139.76, 129.64, 129.19 (q, J = 31.0 Hz), 125.28, 124.93, 124.49, 123.12, 122.01, 119.69, 117.08, 96.93, 20.00. ESI HRMS 352.07318 (M+H)+.
3-Amino-5-methyl-N-(3-(trifluoromethyl)phenyl)thieno[2,3-b]pyridine-2-carboxamide (6)
Synthesized using a similar procedure used to prepare 1 except using 31b. Crude was purified by silica gel chromatography (hexanes/EtOAc) to give 6 (36 mg, 30%) as a light yellow solid. Purity 99%. 1H NMR (500 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.55 (s, 1H), 8.36 (s, 1H), 8.21 (s, 1H), 7.99 (d, J = 8.1 Hz, 1H), 7.56 (t, J = 7.9 Hz, 1H), 7.46 – 7.39 (m, 3H), 2.43 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 164.17, 156.13, 151.54, 147.34, 139.93, 130.86, 129.60, 129.16 (q, J = 31.4 Hz), 128.89, 125.80, 124.24, 124.22 (q, J = 272.2 Hz), 119.49 (d, J = 3.5 Hz), 116.85 (q, J = 3.8 Hz), 96.12, 17.96. ESI HRMS: 352.07272 (M+H)+.
3-amino-6-methyl-N-(4-(trifluoromethyl)phenyl)thieno[2,3-b]pyridine-2-carboxamide (7)
Synthesized using a similar procedure used to prepare 1 except using 33d. Crude was purified by silica gel chromatography (hexanes/EtOAc) to give 7 (28 mg, 23%) as a light yellow solid. Purity 97%. 1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.40 (d, J = 8.3 Hz, 1H), 7.96 (d, J = 8.5 Hz, 2H), 7.67 (d, J = 8.6 Hz, 2H), 7.47 (s, 2H), 7.35 (d, J = 8.3 Hz, 1H), 2.60 (s, 3H).13C NMR (100 MHz, DMSO-d6) δ 164.23, 159.94, 158.57, 147.91, 142.90, 131.14, 125.62 (q, J = 3.5 Hz), 123.55, 123.21, 120.48, 119.61, 94.91, 24.27. ESI HRMS: 352.07336 (M+H)+.
3-Amino-6-methyl-N-(2-(trifluoromethyl)phenyl)thieno[2,3-b]pyridine-2-carboxamide (8)
Synthesized using a similar procedure used to prepare 1 except using 33e. Crude was purified by silica gel chromatography (hexanes/EtOAc) to give 8 (32 mg, 38%) as a light yellow solid. Purity 96%. 1H NMR (500 MHz, DMSO-d6) δ 9.20 (s, 1H), 8.36 (d, J = 8.3 Hz, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.71 (t, J = 8.0 Hz, 1H), 7.57 (d, J = 7.9 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.34 (d, J = 8.3 Hz, 1H), 7.26 (s, 2H), 2.60 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 164.60, 159.64, 158.42, 146.95, 135.82, 132.89, 131.09, 130.77, 126.92, 126.40 (q, J = 4.9 Hz), 125.90 (q, J = 29.2 Hz), 123.74, 123.71 (q, J = 273.3 Hz), 119.56, 95.28, 24.29. ESI HRMS: 352.07291 (M+H)+.
3-Amino-6-methyl-N-(3-(trifluoromethyl)benzyl)thieno[2,3-b]pyridine-2-carboxamide (9)
Synthesized using a similar procedure used to prepare 1 except using 33f. Crude was purified by silica gel chromatography (hexanes/EtOAc) to give 9 (106 mg, 60%) as a light yellow solid. Purity 100%. 1H NMR (500 MHz, DMSO-d6) δ 8.35 – 8.30 (m, 2H), 7.66 (s, 1H), 7.64 – 7.62 (m, 1H), 7.61 – 7.55 (m, 2H), 7.31 (d, J = 8.3 Hz, 1H), 7.18 (s, 2H), 4.48 (d, J = 5.9 Hz, 2H), 2.58 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 165.27, 159.50, 158.22, 145.89, 141.75, 131.65, 131.07, 129.53, 129.11 (q, J = 31.5 Hz), 124.47 (q, J = 272.2 Hz), 124.06, 123.90 (q, J = 3.7 Hz), 123.63 (q, J = 3.8 Hz), 119.69, 95.79, 42.03, 24.35. ESI HRMS: 366.08889 (M+H)+.
3-Amino-6-isopropyl-N-(3-(trifluoromethyl)phenyl)thieno[2,3-b]pyridine-2-carboxamide (10)
Synthesized using a similar procedure used to prepare 1 except using 31c. Crude was purified by flash column chromatography (hexanes/EtOAc) on silica gel to give 10 (44 mg, 78%) as a light yellow oil which solidified upon standing. Purity 93%. 1H NMR (500 MHz, DMSO-d6) δ 9.68 (s, 1H), 8.44 (d, J = 8.4 Hz, 1H), 8.22 (s, 1H), 7.99 (d, J = 8.2 Hz, 1H), 7.56 (t, J = 7.9 Hz, 1H), 7.47 (s, 2H), 7.43 – 7.38 (m, 2H), 3.15 (p, J = 6.9 Hz, 1H), 1.29 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, DMSO-d6) δ 168.50, 164.23, 158.51, 147.78, 139.97, 131.47, 129.58, 129.15 (q, J = 31.7 Hz), 124.27, 124.23 (q, J = 272.8 Hz), 124.03, 119.43 (q, J = 3.8 Hz), 117.37, 116.86 (q, J = 4.0 Hz), 94.92, 39.85, 35.91, 22.41. ESI HRMS: 380.10396 (M+H)+.
6-Methyl-N-(3-(trifluoromethyl)phenyl)thieno[2,3-b]pyridine-2-carboxamide (11)
Synthesized using reported procedure with modification.25 To a solution of 1 (56 mg, 0.16 mmol) in MeOH (6 mL) was added a solution of 50% H3PO2 (259 >L, 2.4 mmol) at 0 °C. To the reaction mixture was added conc. H2SO4 (655 >L), Cu2O (14 mg, 0.10 mmol),1 M NaNO2 aqueous solution (239 >L) and the reaction mixture was stirred for 30 min at 0 °C. After stirring for 2 h and 30 min at room temperature, the reaction mixture was diluted with H2O (30 mL) and the aqueous layer was extracted with EtOAc (20 mL –). The combined organic layer was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. Crude was purified by flash column chromatography (hexanes/EtOAc) on silica gel to give 11 (12 mg, 22%) as a white solid. Purity 98%.1H NMR (500 MHz, DMSO-d6) δ 10.83 (s, 1H), 8.36 – 8.32 (m, 2H), 8.21 (s, 1H), 8.05 (d, J = 8.2 Hz, 1H), 7.63 (t, J = 8.0 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 2.63 (s, 3H).13C NMR (125 MHz, DMSO-d6) δ 161.21, 160.54, 158.36, 139.31, 137.39, 133.71, 130.21, 130.09, 129.46 (q, J = 31.7 Hz), 124.53, 124.09 (q, J = 272.3 Hz), 123.78, 120.96, 120.34 (q, J = 3.7 Hz), 116.35 (q, J = 4.0 Hz), 24.29. ESI HRMS: 337.06096 (M+H)+.
7-Methyl-3-(3-(trifluoromethyl)phenyl)pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine-2,4(1H,3H)-dione (12)
Synthesized using reported procedure with modification.26 Triphosgene (13 mg, 0.043 mmol) was added to a stirred solution of 1 (30 mg, 0.084 mmol) in THF (2.5 mL) at room temperature. The resulting mixture was heated under reflux for 1 h. The mixture was then cooled to room temperature and diluted with THF (5 mL). The solid was collected by filtration and rinsed with cold THF to give a white solid (28 mg, 90%). Purity 98%. 1H NMR (500 MHz, DMSO-d6) δ 12.81 (s, 1H), 8.65 (d, J = 8.4 Hz, 1H), 7.87 (s, 1H), 7.83 (d, J = 7.4 Hz, 1H), 7.78 – 7.72 (m, 2H), 7.55 (d, J = 8.4 Hz, 1H), 2.67 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 161.09, 160.73, 159.08, 151.33, 138.79, 136.39, 133.62, 131.96, 130.16, 129.73 (q, J = 32.1 Hz), 126.28 (q, J = 3.9 Hz), 125.24 (q, J = 3.7 Hz), 124.98, 122.82, 121.07, 120.37, 107.68, 24.46.. ESI HRMS: 378.05243 (M+H)+.
Representative procedure for thienopyridine sulfonamide preparation
3-Amino-6-methyl-N-(3-(trifluoromethyl)phenyl)thieno[2,3-b]pyridine-2-sulfonamide (13)
Synthesized using reported procedure with modification.27 To a solution of 25 (32 mg, 0.21 mmol) in anhydrous DMF (1.5 mL) was added 35 (63 mg, 0.22 mmol) and sodium ethoxide (33 mg, 0.46 mmol). The mixture was heated to 130 °C for 1 h. The mixture was diluted with H2O (10 mL) and extracted with EtOAc (10 mL × 2) and finally washed with brine (10 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. Crude was purified by silica gel chromatography (hexanes/EtOAc) to give 13 (27 mg, 33%) as a light yellow solid. Purity 96%.1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.35 (d, J = 8.3 Hz, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.45 – 7.38 (m, 2H), 7.37 – 7.30 (m, 2H), 6.83 (s, 2H), 2.55 (s, 3H).13C NMR (126 MHz, DMSO) δ 160.74, 158.96, 144.69, 139.28, 132.25, 130.93, 130.25 (q, J = 31.5 Hz), 125.34, 123.57, 123.16, 120.51, 115.43, 97.64, 24.68. ESI HRMS: 388.04044 (M+H)+.
3-Amino-N-(3-fluorophenyl)-6-methylthieno[2,3-b]pyridine-2-sulfonamide (14)
Synthesized using a similar procedure used to prepare 13 except using 36. Crude was purified by silica gel chromatography (hexanes/EtOAc) to give 14 (96 mg, 72%) as an oil which was solidified to a beige solid upon standing. Purity 100%. 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1H), 8.33 (d, J = 8.3 Hz, 1H), 7.33 (d, J = 8.4 Hz, 1H), 7.26 (td, J = 8.4, 6.9 Hz, 1H), 6.97 – 6.88 (m, 2H), 6.85 – 6.80 (m, 1H), 6.80 – 6.76 (m, 2H), 2.54 (s, 3H).13C NMR (100 MHz, DMSO-d6) δ 162.21 (d, J = 242.8 Hz), 160.13, 158.48, 143.89, 139.73 (d, J = 10.7 Hz), 131.72, 130.82 (d, J = 9.5 Hz), 123.13, 119.98, 114.88 (d, J = 2.7 Hz), 110.08 (d, J = 21.0 Hz), 105.70 (d, J = 25.4 Hz), 97.72, 24.17. ESI HRMS: 338.04340 (M+H)+.
Representative procedure for oxadiazolyl thienopyridine preparation
6-Methyl-2-(5-(3-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thieno[2,3-b]pyridin-3-amine (15)
To a stirring solution of 25 (42 mg, 0.28 mmol) in anhydrous DMF (2 mL) was added commercially available 5-(chloromethyl)-3-[3-(trifluoromethyl)phenyl]-1,3,4-oxadiazole, 37I, (66 mg, 0.24 mmol) and sodium ethoxide (50 mg, 0.63 mmol). The mixture was heated to 130 °C for 1 h. The mixture was then cooled to room temperature and solid precipitated at the bottom of the flask. The solid was collected by filtration and rinsed with cold MeOH and dried in vacuo to give a yellow solid (28 mg, 31% yield). Purity 96%. 1H NMR (500 MHz, DMSO-d6) δ 8.50 (d, J = 8.3 Hz, 1H), 8.40 (d, J = 7.9 Hz, 1H), 8.35 (s, 1H), 8.01 (d, J = 7.9 Hz, 1H), 7.87 (t, J = 7.8 Hz, 1H), 7.39 (d, J = 8.3 Hz, 1H), 7.26 (s, 2H), 2.61 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 162.36, 160.47, 159.33, 159.28, 143.73, 131.25, 130.80, 130.50, 130.18, 129.92, 128.06 (q, J = 3.3 Hz), 124.80, 124.45, 123.38, 122.77 (q, J = 3.7 Hz), 122.63, 119.88, 85.96, 24.21. ESI HRMS: 377.06832 (M+H)+.
6-Methyl-2-(3-(3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)thieno[2,3-b]pyridin-3-amine (16)
Synthesized using a similar procedure used to prepare 15 except using commercially available 5-(chloromethyl)-3-[3-(trifluoromethyl)phenyl]-1,3,4-oxadiazole, 37m. Precipitated solid was filtered, rinsed with cold MeOH, and dried in vacuo to give the title compound (31 mg, 67% yield) as a red solid. Purity 99%. 1H NMR (500 MHz, DMSO-d6) δ 8.55 (d, J = 8.3 Hz, 1H), 8.48 (d, J = 7.7 Hz, 1H), 8.43 (s, 1H), 8.00 (d, J = 7.6 Hz, 1H), 7.84 (t, J = 7.7 Hz, 1H), 7.63 (s, 2H), 7.41 (d, J = 8.3 Hz, 1H), 2.61 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.46, 166.38, 160.44, 159.78, 146.38, 131.88, 131.35, 130.51, 129.92 (q, J = 32.1 Hz), 128.09 (d, J = 3.3 Hz), 127.46, 123.64 (q, J = 3.7 Hz), 122.93, 122.76 (q, J = 272 Hz), 119.99, 85.79, 24.28.ESI HRMS: 377.06809 (M+H)+.
6-Methyl-2-(5-phenyl-1,3,4-oxadiazol-2-yl)thieno[2,3-b]pyridin-3-amine (17)
Synthesized using a similar procedure used to prepare 15 except using 25 and commercially available 2-(chloromethyl)-5-phenyl-1,3,4-oxadiazole, 37n. Precipitated solid was filtered, rinsed with cold MeOH, and dried in vacuo to give the title compound (24 mg, 65% yield) as a yellow solid. Purity 96%. 1H NMR (500 MHz, DMSOd6) δ 8.46 (d, J = 8.3 Hz, 1H), 8.11 – 8.08 (m, 2H), 7.65 – 7.61 (m, 3H), 7.40 (d, J = 8.3 Hz, 1H), 7.18 (s, 2H), 2.61 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 161.93, 161.45, 159.19, 159.17, 143.37, 131.70, 131.12, 129.41, 126.50, 123.40, 123.28, 119.87, 86.23, 24.21.ESI HRMS: 309.08079 (M+H)+.
2-(5-(3-Chlorophenyl)-1,3,4-oxadiazol-2-yl)-6-methylthieno[2,3-b]pyridin-3-amine (18)
Synthesized using a similar procedure used to prepare 15 except using 25 and commercially available 2-(chloromethyl)-5-(3-chlorophenyl)-1,3,4-oxadiazole, 37o. Precipitated solid was filtered, rinsed with cold MeOH, and dried in vacuo to give the title compound (26 mg, 42% yield) as a yellow solid. Purity 96%. 1H NMR (500 MHz, DMSO-d6) δ 8.46 (d, J = 8.3 Hz, 1H), 8.11 (t, J = 1.7 Hz, 1H), 8.06 (dt, J = 7.6, 1.2 Hz, 1H), 7.70 (ddd, J = 8.1, 2.0, 1.1 Hz, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.39 (d, J = 8.3 Hz, 1H), 7.20 (s, 2H), 2.61 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 162.22, 160.43, 159.29, 159.27, 143.58, 134.03, 131.44, 131.41, 131.16, 125.93, 125.25, 125.20, 123.38, 119.87, 86.08, 24.21. ESI HRMS: 343.04216 (M+H)+.
2-(5-(3-(Trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thieno[2,3-b]pyridin-3-amine (19)
Synthesized using a similar procedure used to prepare 15 except using commercially available 2-Mercaptopyridine-3-carbonitrile, 38, and 37m. Precipitated solid was filtered, rinsed with cold MeOH, and dried in vacuo to give the title compound (55 mg, 76% yield) as a yellow solid. Purity 99%. 1H NMR (500 MHz, DMSO-d6) δ 8.69 (dd, J = 4.6, 1.6 Hz, 1H), 8.61 (dd, J = 8.2, 1.6 Hz, 1H), 8.41 (d, J = 7.9 Hz, 1H), 8.36 (s, 1H), 8.02 (d, J = 7.9 Hz, 1H), 7.88 (t, J = 7.8 Hz, 1H), 7.54 (dd, J = 8.1, 4.6 Hz, 1H), 7.29 (s, 2H). 13C NMR (125 MHz, DMSO-d6) δ 162.22, 160.66, 159.56, 149.93, 143.49, 131.17, 130.80, 130.55, 129.94 (q, J = 32 Hz), 128.14 (q, J = 3.3 Hz), 125.69, 124.79, 124.41, 122.84 (q, J = 3.9 Hz), 122.62, 119.86, 87.02. ESI HRMS: 363.05232 (M+H)+.
Representative procedure for 2-mercaptopyridine preparation
2-Mercapto-6-methylnicotinonitrile (25)
Synthesized using reported procedure with modification.20 A mixture of 2-chloro-6-methylnicotinonitrile, 24, (222 mg, 1.44 mmol) and thiourea (348 mg, 4.57 mmol) was heated at reflux (118 °C) in n-butanol for 3 h. A yellow suspension was obtained. After cooling to room temperature, the yellow solid was precipitated. The solids were collected by filtration, rinsed with cold n-butanol, and dried in vacuo to give the title compound (217 mg, quantitative) as a light yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 14.07 (s, 1H), 7.99 (d, J = 7.7 Hz, 1H), 6.73 (d, J = 7.7 Hz, 1H), 2.39 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 177.70, 155.00, 145.03, 117.20, 113.40, 112.75, 19.14. ESI MS: m/z 149.1 (M-H)−.
Representative procedure for acetamide preparation
2-Bromo-N-(3-(trifluoromethyl)phenyl)acetamide (27)
Synthesized using reported procedure with modification.21 A stirred solution of 3-(trifluoromethyl)aniline, 26, (170 >L, 1.36 mmol) and 4-(dimethylamino)pyridine (70 mg, 42 mol%) in CH2Cl2 (8 mL) was treated dropwise with bromoacetyl bromide (140 >L, 1.61 mmol) at 0 °C. The reaction mixture was allowed to warm up to room temperature with good stirring overnight. The mixture was diluted with H2O (30 mL) and extracted with EtOAc (20 mL × 2). The organic layer was dried (MgSO4), filtered, and concentrated in vacuo. Crude was purified by flash column chromatography (hexanes/EtOAc) on silica gel to give 27 (339 mg, 88%) as a light brown oil which solidified upon standing. 1H NMR (500 MHz, CDCl3) δ 8.23 (s, 1H), 7.83 (s, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.45 – 7.41 (m, 1H), 4.05 (s, 2H).13C NMR (125 MHz, CDCl3) δ 163.70, 137.55, 131.74 (q, J = 32.7 Hz), 129.87, 123.83 (q, J = 272.4 Hz), 123.18, 121.95 (q, J = 3.8 Hz), 116.88 (q, J = 3.9 Hz), 29.40. ESI MS: m/z 281.55, 283.50 (M+H)+.
Representative procedure for pyridinium acetamido bromide preparation
4-(2-Bromoacetamido)pyridin-1-ium bromide (29g)
Synthesized using reported procedure with modification.23 To a stirred solution of 4-aminopyridine, 28g, (201 mg, 2.14 mmol) in dry CH2Cl2 (5 mL) at 0 °C was added bromoacetyl bromide (195 >L, 2.24 mmol) dropwise. As soon as bromoacetyl bromide was added, the reaction mixture turned cloudy and a white precipitate formed. The mixture was allowed to warm to room temperature with good stirring overnight. Mixture was filtered and the solid was washed with cold CH2Cl2 to give 29g as a white solid which was used in the next step without further purification. ESI MS: m/z 214.45, 216.45 (M+H)+.
3-(2-Bromoacetamido)pyridin-1-ium bromide (29h)
Synthesized using a similar procedure used to prepare 29g using 3-aminopyridine, 28h. The mixture was allowed to warm to room temperature with good stirring over 3 h. Mixture was filtered and the solid was washed with cold CH2Cl2 to give the title compound (609 mg, 95%) as a beige solid which was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ 11.42 (s, 1H), 9.20 (d, J = 2.3 Hz, 1H), 8.65 (d, J = 5.4 Hz, 1H), 8.50 (d, J = 8.6 Hz, 1H), 7.99 (dd, J = 8.5, 5.5 Hz, 1H), 4.19 (s, 2H).13C NMR (100 MHz, DMSO-d6) δ 166.22, 137.81, 137.58, 134.35, 132.52, 127.48, 29.53. ESI MS: m/z 214.45, 216.40 (M+H)+.
2-Bromo-N-(pyridin-2-yl)acetamide hydrobromide (29i)
Synthesized using a similar procedure used to prepare 29g except using 2-aminopyridine, 28i. The mixture was allowed to warm to room temperature with good stirring over 3 h. Mixture was filtered and the solid was washed with cold CH2Cl2 to give the title compound (342 mg, 55%) as a light purple solid which was used in the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 8.81 (s, 1H), 8.32 (d, J = 3.7 Hz, 1H), 8.17 (s, 1H), 7.74 (t, J = 7.6 Hz, 1H), 7.13 – 7.06 (m, 1H), 4.01 (s, 2H).13C NMR (125 MHz, CDCl3) δ 164.23, 150.72, 148.15, 138.66, 120.72, 114.02, 29.20. ESI MS: m/z 214.45, 216.45 (M+H)+.
2-Mercapto-4-methylnicotinonitrile (31a)
Synthesized using a similar procedure used to prepare 25 except using 2-chloro-4-methylnicotinonitrile, 30a. Precipitated solid was filtered, rinsed with cold n-butanol, and dried in vacuo to give the title compound (237 mg, quantitative) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 7.81 (d, J = 6.4 Hz, 1H), 6.82 (d, J = 6.4 Hz, 1H), 2.40 (s, 3H).13C NMR (100 MHz, DMSO-d6) δ 177.05, 157.57, 140.65, 116.31, 115.93, 114.61, 20.99. ESI MS: m/z 149.1 (MH) −.
2-Mercapto-5-methylnicotinonitrile (31b)
Synthesized using a similar procedure used to prepare 25 except using 2-chloro-5-methylnicotinonitrile, 30b. Precipitated solid was filtered, rinsed with cold n-butanol, and dried in vacuo to give the title compound (121 mg, quantitative) as a light yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 14.10 (s, 1H), 8.04 (d, J = 1.9 Hz, 1H), 7.83 (s, 1H), 2.10 (s, 3H). 13C NMR (126 MHz, DMSO) δ 174.77, 147.03, 141.45, 122.24, 116.90, 116.08, 16.06. ESI MS: m/z 149.1 (M-H) −.
6-Isopropyl-2-mercaptonicotinonitrile (31c)
Synthesized using reported procedure with modification.24 A suspension of 2-chloro-6-isopropylnicotinonitrile, 30c, (53 mg, 0.29 mmol) and Lawesson’s reagent (135 mg, >90% purity, 0.30 mmol) in toluene (2.5 mL) was refluxed at 110 °C for 4 h 30 min. The reaction mixture was cooled down to room temperature and filtered through a Celite plug to remove any unreacted Lawesson’s reagent. The solvent was evaporated and crude was purified by silica gel chromatography (hexanes/EtOAc) to give 31c (22 mg, 42% yield) as a yellow sticky solid. 1H NMR (400 MHz, DMSO-d6) δ 14.04 (s, 1H), 8.04 (d, J = 7.8 Hz, 1H), 6.78 (d, J = 7.8 Hz, 1H), 3.06 (p, J = 6.9 Hz, 1H), 1.20 (d, J = 6.9 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 177.78, 163.81, 145.44, 117.06, 113.98, 109.04, 31.42, 21.17. ESI MS: m/z 179.1 (M+H)+, 177.1 (M-H) −.
2-Bromo-N-(4-(trifluoromethyl)phenyl)acetamide (33d)
Synthesized using a similar procedure used to prepare 27 except using 4-(trifluoromethyl)aniline, 32d. The reaction mixture was allowed to warm up to room temperature with good stirring over 8 h. The mixture was diluted with H2O (30 mL) and extracted with EtOAc (20 mL × 2). The organic layer was dried (MgSO4), filtered, and concentrated in vacuo. Crude (355 mg, 94%), a white solid, was used in the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 8.24 (s, 1H), 7.68 (d, J = 8.5 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 4.05 (s, 2H).13C NMR (125 MHz, CDCl3) δ 163.69, 140.05, 127.18 (q, J = 33.0 Hz), 126.56 (q, J = 3.8 Hz), 124.04 (q, J = 271.8 Hz), 119.73, 29.44. ESI MS: m/z 281.50, 283.55 (M+H)+.
2-Bromo-N-(2-(trifluoromethyl)phenyl)acetamide (33e)
Synthesized using a similar procedure used to prepare 27 except using 2-(trifluoromethyl)aniline, 32e, and letting the reaction to stir overnight at room temperature. Crude (397 mg, 89%), a beige solid, was used in the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 8.60 (s, 1H), 8.19 (d, J = 8.3 Hz, 1H), 7.65 (d, J = 7.9 Hz, 1H), 7.59 (t, J = 7.9 Hz, 1H), 7.29 (t, J = 7.7 Hz, 1H), 4.07 (s, 2H). 13C NMR (125 MHz, CDCl3) δ 163.89, 134.59, 133.11, 126.38 (q, J = 5.3 Hz), 125.40, 124.17, 123.98 (q, J = 273.1 Hz), 120.79 (q, J = 29.9 Hz), 29.43. ESI MS: m/z 281.50, 283.50 (M+H)+.
2-Bromo-N-(3-(trifluoromethyl)benzyl)acetamide (33f)
Synthesized using a similar procedure used to prepare 27 except using 3-(trifluoromethyl)benzylamine, 32f, and letting the reaction to stir for 6h at room temperature. Crude was purified by silica gel chromatography (hexanes/EtOAc) to give 33f (316 mg, 62%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.58 – 7.55 (m, 1H), 7.54 (s, 1H), 7.51 – 7.45 (m, 2H), 6.85 (s, 1H), 4.54 (d, J = 6.0 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 165.68, 138.54, 131.31 (q, J = 33.5 Hz), 131.18, 129.49, 124.79, 124.54, 124.06 (q, J = 272.1 Hz), 43.80, 29.16. ESI MS: m/z 295.50, 297.55 (M+H)+.
Representative procedure for chloromethanesulfonamide preparation
1-Chloro-N-(3-(trifluoromethyl)phenyl)methanesulfonamide (35)
Synthesized using reported procedure with modification.27 To a stirring solution of 3-(trifluoromethyl)aniline, 26, (388 >L, 3.11 mmol) in CH2Cl2 (6 mL) was added Et3N (865 >L, 6.20 mmol) at room temperature. The reaction mixture was cool down to 0 °C and chloromethanesulfonyl chloride (626 >L, 6.20 mmol) in CH2Cl2 (3 mL) was added at 0 °C. The reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated on vacuo then 2 N NaOH (15 mL) was added and the mixture was stirred at room temperature for 2 h 30 min. The reaction mixture was neutralized by 1N HCl. The mixture was diluted with H2O (10 mL) and extracted with EtOAc (20 mL × 2) and finally washed with brine (10 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. Crude was purified by silica gel chromatography (hexanes/EtOAc) to give 35 (614 mg, 72%) as a beige solid. 1H NMR (400 MHz, DMSO-d6) δ 10.77 (s, 1H), 7.62 – 7.56 (m, 1H), 7.56 – 7.51 (m, 2H), 7.49 (d, J = 7.6 Hz, 1H), 5.13 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 138.25, 130.64, 129.96 (q, J = 31.8 Hz), 125.17, 123.64, 122.46, 120.71 (q, J = 3.6 Hz), 116.08 (q, J = 4.1 Hz), 54.87. ESI MS: m/z 272.0 (M-H) −.
1-Chloro-N-(3-fluorophenyl)methanesulfonamide (36)
Synthesized using a similar procedure used to prepare 35 except using 3-fluoroaniline, 34. Crude was purified by silica gel column chromatography (hexanes/EtOAc) to give 36 (347 mg, 60%) as a yellow oil which was solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 7.35 (td, J = 8.2, 6.3 Hz, 1H), 7.11 – 7.03 (m, 2H), 6.97 (tdd, J = 8.3, 2.4, 0.8 Hz, 1H), 6.90 (s, 1H), 4.51 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 163.33 (d, J = 248.3 Hz), 136.96 (d, J = 10.2 Hz), 131.22 (d, J = 9.3 Hz), 117.68 (d, J = 3.2 Hz), 113.62 (d, J = 21.2 Hz), 109.71 (d, J = 25.2 Hz), 53.58. ESI MS: m/z 222.00 (M-H) −.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, the National Cancer Institute (NCI), and the IATAP program, National Institutes of Health. Work at the University of Washington was supported by NIH-NIGMS GM110569. M. Shortridge was supported by an American Cancer Society fellowship (PF-13-056-01-RMC). We thank the Biophysics Resource in the Structural Biophysics Laboratory, NCI at Frederick for assistance with Q-TOF LC-MS studies.
Abbreviations Used
- TAR
transactivation response element
- 5′-UTR
5′-untranslated region
- SMM
small molecule microarray
- 2AP
2-aminopurine
- FRET
Forster resonance energy transfer
- SHAPE
selective 2′-hydroxyl acylation analyzed by primer extension
Solvent and reagent abbreviation used
- 1M7
1-methyl-7-nitroisatoic anhydride
- MgCl2
magnesium chloride
- ACN
acetonitrile
- MeOH
methanol
- EtOH
ethanol
- EtOAc
ethylacetate
- Na2SO4
sodium sulfate
- NaOEt
sodium ethoxide
- H2SO4
sulfuric acid
- H3PO2
hypophosphorous acid
- NaNO2
sodium nitrite
- Cu2O
copper (I) oxide
- Et3N
trimethylamine
Footnotes
Notes
The authors declare no competing financial interest.
Molecular formula strings and associated biochemical data, procedure for the synthesis of 20–26 and biochemical data, ligand-detected full 1H NMR spectra of 2 and 14, full TOCSY spectra of free TAR and in presence of different concentration of 2, 1H NMR and 13C NMR spectra for all compounds.
References
- 1.Shortridge MD, Varani G. Structure based approaches for targeting non-coding RNAs with small molecules. Curr Opin Struct Biol. 2015;30:79–88. doi: 10.1016/j.sbi.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dingwall C, Ernberg I, Gait MJ, Green SM, Heaphy S, Karn J, Lowe AD, Singh M, Skinner MA, Valerio R. Human immunodeficiency virus-1 Tat protein binds trans-activation-responsive region (TAR) RNA in vitro. Proc Natl Acad Sci USA. 1989;86(18):6925–6929. doi: 10.1073/pnas.86.18.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dingwall C, Ernberg I, Gait MJ, Green SM, Heaphy S, Karn J, Lowe AD, Singh M, Skinner MA. HIV-1 Tat protein stimulates transcription by binding to a U-rich bulge in the stem of the TAR RNA structure. EMBO J. 1990;9(12):4145–4153. doi: 10.1002/j.1460-2075.1990.tb07637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berkhout B, Jeang KT. Trans activation of human immunodeficiency virus type 1 is sequence specific for both the single-stranded bulge and loop of the trans-acting-responsive hairpin: a quantitative analysis. J Virol. 1989;63(12):5501–5504. doi: 10.1128/jvi.63.12.5501-5504.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berkhout B, Silverman RH, Jeang KT. Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell. 1989;59(2):273–282. doi: 10.1016/0092-8674(89)90289-4. [DOI] [PubMed] [Google Scholar]
- 6.Berkhout B. Structural features in TAR RNA of human and simian immunodeficiency viruses: a phylogenetic analysis. Nucleic Acids Res. 1992;20(1):27–31. doi: 10.1093/nar/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huthoff H, Girard F, Wijmenga SS, Berkhout B. Evidence for a base triple in the free HIV-1 TAR RNA. RNA. 2004;10(3):412–423. doi: 10.1261/rna.5161304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrich D, Ulich C, Gaynor RB. A critical role for the TAR element in promoting efficient human immunodeficiency virus type 1 reverse transcription. J Virol. 1996;70(6):4017–4027. doi: 10.1128/jvi.70.6.4017-4027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harrich D, Ulich C, Garcia-Martinez LF, Gaynor RB. Tat is required for efficient HIV-1 reverse transcription. EMBO J. 1997;16(6):1224–1235. doi: 10.1093/emboj/16.6.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lalonde MS, Lobritz MA, Ratcliff A, Chamanian M, Athanassiou Z, Tyagi M, Wong J, Robinson JA, Karn J, Varani G, Arts EJ. Inhibition of both HIV-1 reverse transcription and gene expression by a cyclic peptide that binds the Tat-transactivating response element (TAR) RNA. PLoS Pathog. 2011;7(5):e1002038. doi: 10.1371/journal.ppat.1002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klase Z, Kale P, Winograd R, Gupta MV, Heydarian M, Berro R, McCaffrey T, Kashanchi F. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol Biol. 2007;8:63. doi: 10.1186/1471-2199-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klase Z, Winograd R, Davis J, Carpio L, Hildreth R, Heydarian M, Fu S, McCaffrey T, Meiri E, Ayash-Rashkovsky M, Gilad S, Bentwich Z, Kashanchi F. HIV-1 TAR miRNA protects against apoptosis by altering cellular gene expression. Retrovirology. 2009;6:18. doi: 10.1186/1742-4690-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouellet DL, Vigneault-Edwards J, Letourneau K, Gobeil LA, Plante I, Burnett JC, Rossi JJ, Provost P. Regulation of host gene expression by HIV-1 TAR microRNAs. Retrovirology. 2013;10:86. doi: 10.1186/1742-4690-10-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Grice SFJ. Targeting the HIV RNA genome: high-hanging fruit only needs a longer ladder. In: Torbett BE, Goodsell DS, Richman DD, editors. future of HIV-1 therapeutics: resistance is futile? Vol. 389. Springer International Publishing; Cham, Switzerland: 2015. pp. 147–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem Rev. 2008;108(4):1171–1224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 16.Guan LR, Disney MD. Recent advances in developing small molecules targeting RNA. ACS Chem Biol. 2012;7(1):73–86. doi: 10.1021/cb200447r. [DOI] [PubMed] [Google Scholar]
- 17.Blond A, Ennifar E, Tisne C, Micouin L. The design of RNA binders: targeting the HIV replication cycle as a case study. ChemMedChem. 2014;9(9):1982–1996. doi: 10.1002/cmdc.201402259. [DOI] [PubMed] [Google Scholar]
- 18.Abulwerdi FA, Schneekloth JS., Jr Microarray-based technologies for the discovery of selective, RNA-binding molecules. Methods. 2016;103:188–195. doi: 10.1016/j.ymeth.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sztuba-Solinska J, Shenoy SR, Gareiss P, Krumpe LRH, Le Grice SFJ, O’Keefe BR, Schneekloth JS. Identification of biologically active, HIV TAR RNA-binding small molecules using small molecule microarrays. J Am Chem Soc. 2014;136(23):8402–8410. doi: 10.1021/ja502754f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang Z, Hu C, Fang J, Yeh M, Zhang Z, Ye M, Chang ZF, Fang JM, Hu CM, Yeh MT. Composition for inhibiting thymidylate kinase and for sensitizing cancer cells to radiation therapy, chemotherapy or immunomodulatory therapy, comprises benzo/pyrido-thiazolone or phthalimide derivative. WO2012072019-A1. 2012
- 21.Kirubakaran S, Gorla SK, Sharling L, Zhang MJ, Liu XP, Ray SS, MacPherson IS, Striepen B, Hedstrom L, Cuny GD. Structure-activity relationship study of selective benzimidazole-based inhibitors of Cryptosporidium parvum IMPDH. Bioorg Med Chem Lett. 2012;22(5):1985–1988. doi: 10.1016/j.bmcl.2012.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai D, Burgeson JR, Tyavanagimatt SRB, Chelsea M, Hruby DE. 20130129677. Thienopyridine derivatives for the treatment and prevention of dengue virus infections. 2013
- 23.Xie H, Ng D, Savinov SN, Dey B, Kwong PD, Wyatt R, Smith AB, Hendrickson WA. Structure-activity relationships in the binding of chemically derivatized CD4 to gp120 from human immunodeficiency virus. J Med Chem. 2007;50(20):4898–4908. doi: 10.1021/jm070564e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tseng CH, Lin RW, Chen YL, Wang GJ, Ho ML, Tzeng CC. Discovery of indeno[1,2-c]quinoline derivatives as inhibitors of osteoclastogenesis induced by receptor activator of NF-kappa B ligand (RANKL) J Med Chem. 2011;54(8):3103–3107. doi: 10.1021/jm1016494. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki T, Miyajima Y, Suzuki K, Iwakiri K, Koshimizu M, Hirai G, Sodeoka M, Kobayashi S. Unexpected diels-alder/carbonyl-ene cascade toward the biomimetic synthesis of chloropupukeananin. Org Lett. 2013;15(7):1748–1751. doi: 10.1021/ol400549q. [DOI] [PubMed] [Google Scholar]
- 26.Jiang M, Li BBX, Xie FC, Delaney F, Xiao XS. Design, synthesis, and biological evaluation of conformationally constrained analogues of naphthol AS-E as inhibitors of CREB-mediated gene transcription. J Med Chem. 2012;55(8):4020–4024. doi: 10.1021/jm300043c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nogradi K, Wagner G, Keserue G, Bielik A, Gati T, Hada V, Koti J, Gal K, Vastag M, Bobok AA, Bobok A, Wagner GA, Keseru G, Keserli G, Krisztina VM. New substituted thienopyridine compounds are metabotropic glutamate receptor 1 inhibitors, useful for treating e.g. gastrointestinal reflux disease, irritable bowel syndrome, neuropathic pain and Alzheimer’s disease. WO2007072095-A1. 2007
- 28.Zhang MZ, Mulholland N, Beattie D, Irwin D, Gu YC, Chen Q, Yang GF, Clough J. Synthesis and antifungal activity of 3-(1,3,4-oxadiazol-5-yl)-indoles and 3-(1,3,4-oxadiazol-5-yl)methylindoles. Eur J Med Chem. 2013;63:22–32. doi: 10.1016/j.ejmech.2013.01.038. [DOI] [PubMed] [Google Scholar]
- 29.Bradrick TD, Marino JP. Ligand-induced changes in 2-aminopurine fluorescence as a probe for small molecule binding to HIV-1 TAR RNA. RNA. 2004;10(9):1459–1468. doi: 10.1261/rna.7620304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bardaro MF, Jr, Shajani Z, Patora-Komisarska K, Robinson JA, Varani G. How binding of small molecule and peptide ligands to HIV-1 TAR alters the RNA motional landscape. Nucleic Acids Res. 2009;37(5):1529–1540. doi: 10.1093/nar/gkn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM, Al-Hashimi HM. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat Chem Biol. 2011;7(8):553–559. doi: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsumoto C, Hamasaki K, Mihara H, Ueno A. A high-throughput screening utilizing intramolecular fluorescence resonance energy transfer for the discovery of the molecules that bind HIV-1 TAR RNA specifically. Bioorg Med Chem Lett. 2000;10(16):1857–1861. doi: 10.1016/s0960-894x(00)00359-0. [DOI] [PubMed] [Google Scholar]
- 33.Pascale L, Azoulay S, Di Giorgio A, Zenacker L, Gaysinski M, Clayette P, Patino N. Thermodynamic studies of a series of homologous HIV-1 TAR RNA ligands reveal that loose binders are stronger Tat competitors than tight ones. Nucleic Acids Res. 2013;41(11):5851–5863. doi: 10.1093/nar/gkt237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shortridge MD, Hage DS, Harbison GS, Powers R. Estimating protein-ligand binding affinity using high-throughput screening by NMR. J Comb Chem. 2008;10(6):948–958. doi: 10.1021/cc800122m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puglisi JD, Tan RY, Calnan BJ, Frankel AD, Williamson JR. Conformation of the TAR RNA-arginine complex by NMR-spectroscopy. Science. 1992;257(5066):76–80. doi: 10.1126/science.1621097. [DOI] [PubMed] [Google Scholar]
- 36.Aboulela F, Karn J, Varani G. The structure of the human-immunodeficiency-virus type-1 Tar RNA reveals principles of RNA recognition by Tat protein. J Mol Biol. 1995;253(2):313–332. doi: 10.1006/jmbi.1995.0555. [DOI] [PubMed] [Google Scholar]
- 37.Hamy F, Brondani V, Florsheimer A, Stark W, Blommers MJJ, Klimkait T. A new class of HIV-1 Tat antagonist acting through Tat-TAR inhibition. Biochemistry. 1998;37(15):5086–5095. doi: 10.1021/bi972947s. [DOI] [PubMed] [Google Scholar]
- 38.Faber C, Sticht H, Schweimer K, Rosch P. Structural rearrangements of HIV-1 Tatresponsive RNA upon binding of neomycin B. J Biol Chem. 2000;275(27):20660–20666. doi: 10.1074/jbc.M000920200. [DOI] [PubMed] [Google Scholar]
- 39.Dassonneville L, Hamy F, Colson P, Houssier C, Bailly C. Binding of hoechst 33258 to the TAR RNA of HIV-1. recognition of a pyrimidine bulge-dependent structure. Nucleic Acids Res. 1997;25(22):4487–4492. doi: 10.1093/nar/25.22.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 2008;6(4):883–899. doi: 10.1371/journal.pbio.0060096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krawczyk K, Sim AY, Knapp B, Deane CM, Minary P. Tertiary element interaction in HIV-1 TAR. J Chem Inf Model. 2016;56(9):1746–1754. doi: 10.1021/acs.jcim.6b00152. [DOI] [PubMed] [Google Scholar]
- 42.Damgaard CK, Andersen ES, Knudsen B, Gorodkin J, Kjems J. RNA interactions in the 5 ′ region of the HIV-1 genome. J Mol Biol. 2004;336(2):369–379. doi: 10.1016/j.jmb.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 43.Stephenson JD, Li HT, Kenyon JC, Symmons M, Klenerman D, Lever AML. Three-dimensional RNA structure of the major HIV-1 packaging signal region. Structure. 2013;21(6):951–962. doi: 10.1016/j.str.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huthoff H, Berkhout B. Mutations in the TAR hairpin affect the equilibrium between alternative conformations of the HIV-1 leader RNA. Nucleic Acids Res. 2001;29(12):2594–2600. doi: 10.1093/nar/29.12.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bardaro MF, Shajani Z, Patora-Komisarska K, Robinson JA, Varani G. How binding of small molecule and peptide ligands to HIV-1 TAR alters the RNA motional landscape. Nucleic Acids Res. 2009;37(5):1529–1540. doi: 10.1093/nar/gkn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davidson A, Leeper TC, Athanassiou Z, Patora-Komisarska K, Karn J, Robinson JA, Varani G. Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. Proc Natl Acad Sci USA. 2009;106(29):11931–11936. doi: 10.1073/pnas.0900629106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leeper TC, Athanassiou Z, Dias RLA, Robinson JA, Varani G. TAR RNA recognition by a cyclic peptidomimetic of Tat protein. Biochemistry. 2005;44(37):12362–12372. doi: 10.1021/bi0510532. [DOI] [PubMed] [Google Scholar]
- 48.Nowick JS, Khakshoor O, Hashemzadeh M, Brower JO. DSA: A new internal standard for NMR studies in aqueous solution. Org Lett. 2003;5(19):3511–3513. doi: 10.1021/ol035347w. [DOI] [PubMed] [Google Scholar]
- 49.Mitra S, Shcherbakova IV, Altman RB, Brenowitz M, Laederach A. High-throughput single-nucleotide structural mapping by capillary automated footprinting analysis. Nucleic Acids Res. 2008;36(11):e63. doi: 10.1093/nar/gkn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Badorrek CS, Weeks KM. Architecture of a gamma retroviral genomic RNA dimer. Biochemistry. 2006;45(42):12664–12672. doi: 10.1021/bi060521k. [DOI] [PubMed] [Google Scholar]
- 51.Darty K, Denise A, Ponty Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25(15):1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fulmer GR, Miller AJM, Sherden NH, Gottlieb HE, Nudelman A, Stoltz BM, Bercaw JE, Goldberg KI. NMR chemical shifts of trace impurities: common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics. 2010;29(9):2176–2179. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.