

Abstract
Disulfide-rich peptides isolated from cone snails are of great interest as drug leads due to their high specificity and potency toward therapeutically relevant ion channels and receptors. They commonly contain the inhibitor cystine knot (ICK) motif comprising three disulfide bonds forming a knotted core. Here we report the successful enzymatic backbone cyclization of an ICK-containing peptide κ-PVIIA, a 27-amino acid conopeptide from Conus purpurascens, using a mutated version of the bacterial transpeptidase, sortase A. Although a slight loss of activity was observed compared to native κ-PVIIA, cyclic κ-PVIIA is a functional peptide that inhibits the Shaker voltage-gated potassium (Kv) channel. Molecular modeling suggests that the decrease in potency may be related to the loss of crucial, but previously unidentified electrostatic interactions between the N-terminus of the peptide and the Shaker channel. This hypothesis was confirmed by testing an N-terminally acetylated κ-PVIIA, which shows a similar decrease in activity. We also investigated the conformational dynamics and hydrogen bond network of cyc-PVIIA, both of which are important factors to be considered for successful cyclization of peptides. We found that cyc-PVIIA has the same conformational dynamics, but different hydrogen bond network compared to those of κ-PVIIA. The ability to efficiently cyclize ICK peptides using sortase A will enable future protein engineering for this class of peptides and may help in the development of novel therapeutic molecules.
Keywords: κ-PVIIA, inhibitory cystine knot, cyclization, sortase A
Introduction
Peptides are undergoing an exciting revival of interest due to recent technological advances in protein and peptide engineering (Fosgerau and Hoffmann, 2015). These advances have led to an increase in interest from the pharmaceutical industry in using peptides in a number of therapeutic applications because unlike small molecules, peptides tend to be highly selective and typically very potent for a specific target. However, a drawback often associated with peptides is a short half-life due to a lack of proteolytic stability. One way to improve the stability of a peptide is through backbone or side chain cyclization, as this reduces susceptibility to proteases and, in some cases, improves activity (Clark et al., 2010) and potentiates orally delivered activity (Clark et al., 2010; Nielsen et al., 2015; Wang et al., 2014). One class of peptides that has gained particular interest is venom-derived disulfide-rich peptides due to their inherent stability (brought about by the high number of cysteine residues) and ability to selectively and potently inhibit voltage-gated potassium (KV), calcium (CaV), and sodium (NaV) channels. This inhibitory activity is particularly relevant given the involvement of these channels in numerous pathologies, including pain, stroke, epilepsy, and multiple sclerosis (Barton et al., 2004; Cushman and Ondetti, 1999; Koo et al., 1997; Miljanich, 2004).
κ-PVIIA is a 27-amino acid peptide isolated from the venom of the purple cone snail, Conus purpurascens (Terlau et al., 1996). It has been reported to block the conductance of the Shaker Kv channel of Drosophila melanogaster with an IC50 of 60 ± 3 nM (Jacobsen et al., 2000; Terlau et al., 1996). Three-dimensional structural analysis and a complete alanine scan suggested that the peptide inhibits the channel by physically occluding the pore (Jacobsen et al., 2000; Savarin et al., 1998; Scanlon et al., 1997). NMR structural analyses showed that κ-PVIIA has an inhibitor cystine knot (ICK) (Savarin et al., 1998; Scanlon et al., 1997)—a structural motif characterized by a triple-stranded anti-parallel β-sheet connected by three disulfide bonds, forming a knotted core. The ICK significantly contributes to the stability of peptides (Craik et al., 2001; Pallaghy et al., 1994). The disulfide connectivity of the ICK motif is CI-CIV, CII-CV, and CIII-CVI, and a common sequence pattern is: CX3–7CX3–6CX0–5CX1–4CX4–13C, (X being any amino acid) (Norton and Pallaghy, 1998).
The ICK motif is found in a broad spectrum of phyla, including animals, plants, and fungi, with a particularly large prevalence in the venoms of cone snails, spiders, and scorpions (Zhu et al., 2003). Interestingly, the ICK is widespread in venoms regardless of the pharmacological target, mechanism of action, or the species from which the peptides are extracted. κ-PVIIA is a member of the O1 conotoxin gene superfamily (Kaas et al., 2010, 2012; Terlau et al., 1996), and contains an ICK motif with a CX6-CX6-CCX3-CX5-C cysteine spacing. In addition to κ-PVIIA, this family comprises a diverse series of conotoxins with an ICK motif including, ω-conotoxin MVIIA (Nielsen et al., 1996), μ-conotoxin GS (Hill et al., 1997), δ-conotoxin PVIA (Shon et al., 1995), and μO-conotoxin MrVIB (Fainzilber et al., 1995), which influence CaV and NaV channels (Fig. 1a) (Heinemann and Leipold, 2007; Kalia et al., 2015).
Figure 1.
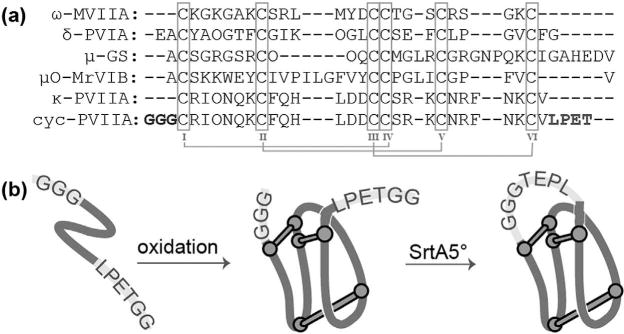
(a) Sequence alignment of 01 gene superfamily conotoxins displaying an ICK motif. (b) Schematic representation of the experimental procedures used to cyclize κ-PVIIA.
One class of peptides that is structurally related to the O1 conotoxin superfamily is the cyclotides (Daly and Craik, 2011). This family of plant-derived peptides comprises a cystine knot that is embedded within a cyclic backbone, a motif referred to as a cyclic cystine knot (CCK) (Craik et al., 2001). Six loops protrude from the core and are thought to direct the activity of cyclotides, which have been referred to as a natural combinatorial template (Craik and Conibear, 2011; Ireland et al., 2006). The combination of the cystine knot coupled with the cyclized backbone makes this family of peptides ultrastable and able to withstand harsh chemical, enzymatic or thermal conditions (Colgrave and Craik, 2004). In addition, their use in African folk medicinal teas suggests that they have a degree of oral bioavailability (Gran, 1970).
Cyclization using chemical methods has been used in a few cases to improve the pharmaceutical properties of ICK peptides by making them more cyclotide-like (Akcan et al., 2015; Hemu et al., 2013). Cyclotides are typically sequentially assembled using standard Boc solid-phase peptide synthesis and cyclized using native chemical ligation (Blanco-Canosa et al., 2015; Daly et al., 1999; Dawson et al., 1994; Tam et al., 1999). Recent reports also describe the production of cyclotides using Fmoc compatible methods (Blanco-Canosa et al., 2015; Cheneval et al., 2014). Furthermore, we recently showed that cyclization of disulfide-rich peptides, including cyclotide kalata B1, sunflower trypsin inhibitor-I (SFTI-I), and cyclic Vc1.1, could be achieved using the bacterial transpeptidase, sortase A (SrtA), as an alternative to chemical ligation methods (Jia et al., 2014). Another recent study also demonstrated successful backbone cyclization of a recombinant precursor of the cyclotide MCoTI-II using a mutant SrtA (P94S/D160N/D165A/K196T) (Stanger et al., 2014).
SrtA, originally isolated from Staphylococcus aureus, recognizes the penta-amino acid LPXTG motif (X = any amino acid) and cleaves the peptide bond between Thr and Gly, forming a thioacyl enzyme intermediate. The intermediate then undergoes nucleophilic attack by Glyn, which results in the formation of a new peptide bond between Thr and Glyn (Ton-That et al., 1999). SrtA has been widely used in protein engineering (Popp and Ploegh, 2011; Witte et al., 2012), and efforts to improve its catalytic activity were recently successful; specifically, through the use of a directed evolution strategy leading to the modified SrtA (P94R/D160N/D165A/K190E/K196T), here after referred to as SrtA5°, with significantly enhanced ligation activity (Chen et al., 2011).
Little is known about the subtype selectivity of κ-PVIIA across KV channels and with increasing interest in the potential treatment of multiple sclerosis by KV1.3 channel inhibition (Rangaraju et al., 2009), we chose κ-PVIIA as a model system not only to enzymatically cyclize an ICK peptide, but also to search for potential targets in addition to Shaker. Following the successful cyclization of kalata B1, SFTI-I and cyclic Vc1.1 using SrtA, we investigated whether cyclization of κ-PVIIA could be achieved by a semi-enzymatic approach using SrtA5° and determined how it would affect the activity and the stability of the peptide. This study describes the synthesis, oxidation and cyclization using SrtA5° of linear κ-PVIIA (Fig. 1b) and reports its serum stability. Structural analysis using nuclear magnetic resonance (NMR) allowed us to compare the structures of cyclic κ-PVIIA (cyc-PVIIA) with κ-PVIIA; electrophysiological studies and molecular dynamics simulations provided insights into differences in interactions between cyc-PVIIA, κ-PVIIA and Shaker, and allowed us to define the κ-PVIIA KV channel selectivity profile.
Materials and Methods
Peptide Synthesis
Linear peptides were synthesized by solid-phase peptide synthesis using an automatic peptide synthesizer (Symphony®, Protein Technologies, Inc., Tucson, AZ) following standard Fmoc chemistry protocols. 2-Chlorotrityl chloride resin was used for [GGG] PVIIA[LPETGG], whereas rink amide resin was used for linear κ-PVIIA to achieve C-terminal amidation. κ-PVIIA with N-terminal acetylation was produced by treating the resin with 6% acetic acid/0.2 M DIPEA in DMF following completion of elongation of the peptide. Peptides were released from the resins using trifluoroacetic acid (TFA)/triisopropylsilane/water (95:2.5:2.5) at room temperature for 2 h. TFA was removed using a rotary evaporator, and peptides were precipitated with ice-cold diethyl ether, dissolved in 50% v/v acetonitrile (ACN) containing 0.05% v/v TFA and lyophilized. Peptides were purified using reversed-phase high performance liquid chromatography (RP-HPLC) and the mass was confirmed using mass spectrometry.
Oxidative Folding of Peptides
κ-PVIIA and GGG-PVIIA-LPETGG were oxidized in 50 mM Tris pH 7.5, 1 mM GSH, 0.5 mM GSSG at 4 °C for 4–5 days. The progress of oxidation was monitored by analytical RP-HPLC with a gradient from 0–50% v/v ACN (0.05% v/v TFA) over 50 min with a flow rate of 0.3 mL/min using an analytical C18 column (Agilent ZORBAX 300SB-C18, 5 μm, 2.1×150 mm). The oxidation yield was calculated based on HPLC profile. The mixture was purified by RP-HPLC using a 0.5%/min gradient, and the molecular weight was confirmed using mass spectrometry.
SrtA5° Expression
SrtA5° (Chen et al., 2011) was expressed as previously described (Popp et al., 2009). Briefly, an overnight culture of E.coli BL21 (DE3) transformed with SrtA5° plasmid was cultured in the presence of 50 μg/mL kanamycin until OD600 = 0.7. Protein expression was induced by addition of isopropyl-β-D-1-thiogalactopyranoside (IPTG) to a concentration of 1 mM for 3 h at 37 °C. Cells were harvested by centrifugation and resuspended in lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 10 mM imidazole, 10% v/v glycerol, 20 μg/mL DNase I). Cells were sonicated, the supernatant was collected and the protein was purified using Ni-Sepharose (HisTrap FF, GE Healthcare). Fractions containing SrtA5° were combined and the buffer was exchanged using a PD-10 desalting column (GE Healthcare).
Cyclization of Peptides Using SrtA5°
For cyclization, 50 μM of the peptide was incubated with 50 μM of SrtA5° in reaction buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 10 mM CaCl2) at 37 °C. Cyclization progress was monitored using RP-HPLC with a gradient of 0–50% v/v ACN (0.05% v/v TFA) over 50 min with a flow rate of 1 mL/min using an analytical C18 column (GraceSmart™ RP18, 5 μm, 4.6 × 150 mm). When the majority of linear peptide was deemed to be cyclic, the peptide was separated from SrtA5° using a centrifugal concentrator (10 kDa), the solution containing the cyclic peptide was loaded onto a semipreparative C18 column (Phenomenex, Jupiter 5UC18, 300 Å, 250 × 10.0 nm, 5 μm) and eluted with a gradient of 36–45% ACN (0.05% TFA) over 30 min with a flow rate of 3 mL/min. The molecular mass of purified cyclic-[GGG]PVIIA[LPET] was confirmed by matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometry. Fractions showing the correct mass were lyophilized for further assays.
NMR Spectroscopy and Structure Calculations
Cyc-PVIIA was dissolved in 90% H2O, 10% D2O (v/v), or 99.96% D2O at a concentration of 1 mM and ~pH 3.5. Two-dimensional NMR spectra were acquired at 298 K on a Bruker Avance 600 MHz NMR spectrometer equipped with a cryogenically cooled probe (Bruker, Karlsruhe, Germany). Spectra were recorded with 4096 data points in the F2 and 512 increments in the F1 dimension for TOCSY, NOESY, E.COSY, and DQF-COSY; 2,048 data points and 256 increments for 1H-13C HSQC; and 2,048 data points and 128 increments for 1H-15N HSQC. Sequential assignment and structure calculations were performed as previously described (Conibear et al., 2012). Briefly, peaks in the NOESY spectrum with 100 ms mixing time were picked manually, intraresidual and sequential NOEs were assigned, and a list of interproton distances was generated using the AUTO and CALC function in CYANA (Guntert, 2004). Following structural refinement within CNS (Brunger et al., 1998), a set of 20 structures with the lowest energies, no NOE violations greater than 0.2 Å and no dihedral violations greater than 2.0°, was chosen for MolProbity analysis (Chen et al., 2010). PyMOL and MOLMOL (Koradi et al., 1996) were used for generating figures and final superimposition.
Two-Electrode Voltage-Clamp Recording From Xenopus oocytes
Shaker-IR (IR for inactivation removed) (Hoshi et al., 1990; Tempel et al., 1987), rKV2.1Δ7 (Frech et al., 1989; Li-Smerin and Swartz, 1998), hKV1.3 (Stuhmer et al., 1989), rKV1.6 (Grupe et al., 1990; Swanson et al., 1990), rNaV1.4 (Trimmer et al., 1989), and hNaV1.6 (Burgess et al., 1995; Schaller et al., 1995) were used in this study. The rKV2.1Δ7 construct contains seven point mutations in the outer vestibule, rendering the channel sensitive to agitoxin-2 (Garcia et al., 1994). cRNA of all constructs and mutants was synthesized using T7/SP6 polymerase (Message Machine kit, Life Technologies, Carlsbad, CA) after linearizing the sequenced DNA with appropriate restriction enzymes. Channel constructs were expressed in Xenopus laevis oocytes and studied following 1–2 days incubation after manual cRNA injection as described before (Gilchrist et al., 2013). Leak and background conductances, identified by blocking the channel with agitoxin-2, were subtracted for all KV channel currents shown. Voltage-activation relationships were obtained by measuring tail currents for KV channels and a single Boltzmann function was fitted to the data. Voltage-activation relationships were recorded in the absence and presence of different toxin concentrations. The ratio of currents (I/I0) recorded in the presence (I) and absence (I0) of toxin was calculated for various strength depolarizations. The value of I/I0 measured in the linear phase at 0 mV was taken as Fraction unblocked. The IC50 of the toxin for KV channels was calculated using the Hill equation. Off-line data analysis was performed using Clampfit 10 (Molecular Devices, Sunnyvale, CA) and Origin 7.5 (Originlab, Northampton, MA).
Molecular Modeling
The molecular model used for the studies of interaction between the Shaker K+ channel and linear κ-PVIIA was kindly provided by Mhadavi and Kuyucak (Mahdavi and Kuyucak, 2013). A model of the interaction between cyc-PVIIA and the Shaker K+ channel was built by homology with Modeller 9v13 (Sali and Blundell, 1993) using the model of Mhadavi and Kuyucak and the solution structure of cyc-PVIIA as templates. The homology model was then embedded in a pre-equilibrated 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer; the Shaker channel was placed in the bilayer and POPC residues that overlap with the channel were removed. SPC water molecules were added to the system and sodium and chloride ions were added to simulate a physiological concentration of 150 mM sodium chloride. The simulated system contained 100 POPC molecules, ~9,000 water molecules, 23 sodium ions and 26 chloride ions. After 10,000 steps of steepest descent minimization, the system was equilibrated for 10 ns while progressively releasing position restraints on the channel and toxin. The system was then equilibrated for 100 ns without restraint. All simulations used the Gromos 54a7 force field (Schmid et al., 2011), the v-rescale thermostat (Bussi et al., 2007), the Parrinello-Rahman barostat (Parrinello and Rahman, 1981), and the reaction field method (Heinz et al., 2001) to simulate the effect of long range dipole-dipole interactions. The electrostatic potential generated by the Shaker K+ channel was computed using APBS software (Baker et al., 2001). Molecular dynamics simulations of cyc-PVIIA and κ-PVIIA individually in solution were carried out using the Amber 99SB-ILDN force field (Lindorff-Larsen et al., 2010). Each peptide was embedded in a water box of ~2,600 SPC water molecules, containing sodium and chloride ions at 150 mM concentrations. All molecular dynamics simulations were carried out using the Gromacs 5.1 molecular dynamics engine (Pronk et al., 2013).
Serum Stability Assay
The stability of native and cyc-PVIIA in human serum was tested at a peptide concentration of 20 μM using a method previously described (Chan et al., 2011).
Results and Discussion
κ-PVIIA Precursor Synthesis, Oxidation, and Cyclization
κ-PVIIA and the linear precursor [GGG]PVIIA[LPETGG] were synthesized by Fmoc chemistry. Oxidative folding was monitored by analytical HPLC over time, and both peptides showed a similar profile, with one prominent peak after 4 days (Fig. 2a). For cyclization, we used a mutated S. aureus SrtA which has improved catalytic activity compared to wild-type SrtA (Chen et al., 2011). Among the mutant SrtAs described by Chen et al. we tested the two most active: (i) P94S/D160N/D165A/K196T and (ii) P94R/D160N/D165A/K190E/K196T (SrtA5°) using GFP-LPETGG as the substrate. In our hands, SrtA5° showed slightly better catalytic activity compared to the tetramutant, and consequently was used in subsequent experiments. The linear oxidized [GGG]PVIIA-[LPETGG] was incubated with SrtA5° for cyclization, and progress was monitored by RP-HPLC. Based on calculations of peak area derived from HPLC, 46% and 57% of conversion from linear [GGG] PVIIA[LPETGG] to cyclic-[GGG]PVIIA[LPET] was observed after 3.5 h and 8 h, respectively, (Fig. 2b, c). The peak eluting before cyc-PVIIA contained a peptide with the same mass of cyclic peptide and was assumed to be an isomer, but did not yield sufficient material for further analysis. An attempt to synthesize κ-PVIIA by Boc-chemistry using an automatic synthesizer was unsuccessful; therefore, cyclization by native chemical ligation was not achieved.
Figure 2.
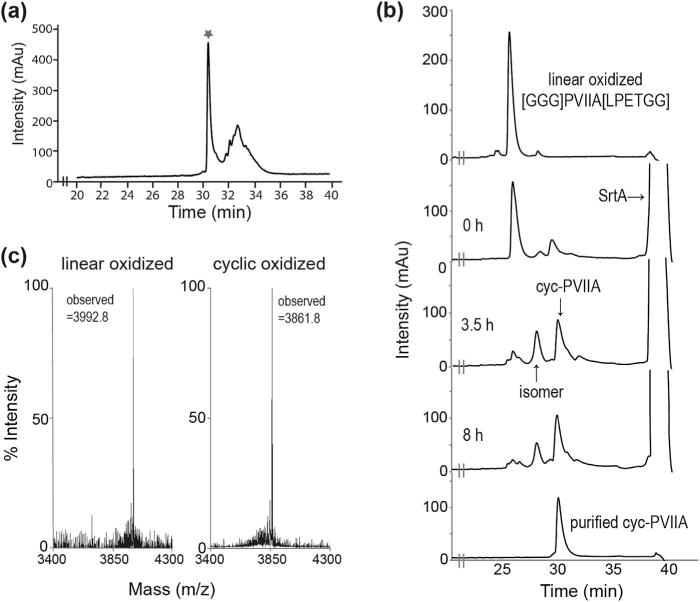
Oxidation and cyclization of [GGG]PVIIA[LPETGG]. (a) RP-HPLC profile of [GGG]PVIIA[LPETGG] oxidation. The asterisk represents the major peak of linear oxidized product. (b) Cyclization progress of [GGG]PVIIA[LPETGG] over time using SrtA5° and (c) MALDI-TOF analysis of linear oxidized (calculated monoisotopic mass: 3992.7 Da) and cyclic oxidized PVIIA (calculated monoisotopic mass: 3860.6 Da).
NMR Solution Structure of Cyc-PVIIA
Linear oxidized [GGG]PVIIA[LPETGG] and cyc-PVIIA were analyzed by two-dimensional 1H NMR. For both peptides the peaks in the amide proton region of the NMR spectra were well dispersed and all protons were assigned by the sequential assignment procedure using TOCSYand NOESY spectra (Wüthrich, 1986). Secondary Ha chemical shift comparison confirmed them to be similar to native κ-PVIIA (PDB identifier 1AV3) (Scanlon et al., 1997) and the positive secondary shifts for residues Q6-C8, K19-C20 and K25-V27 indicated the presence of the characteristic β-sheet secondary structure (Fig. 3a). Backbone cyclization was confirmed by the presence of a αHi-NHi+1 NOE between Thr31 and Gly32, indicating peptide bond formation linking the C-terminal LPETand N-terminal GGG motif of cyc-PVIIA.
Figure 3.
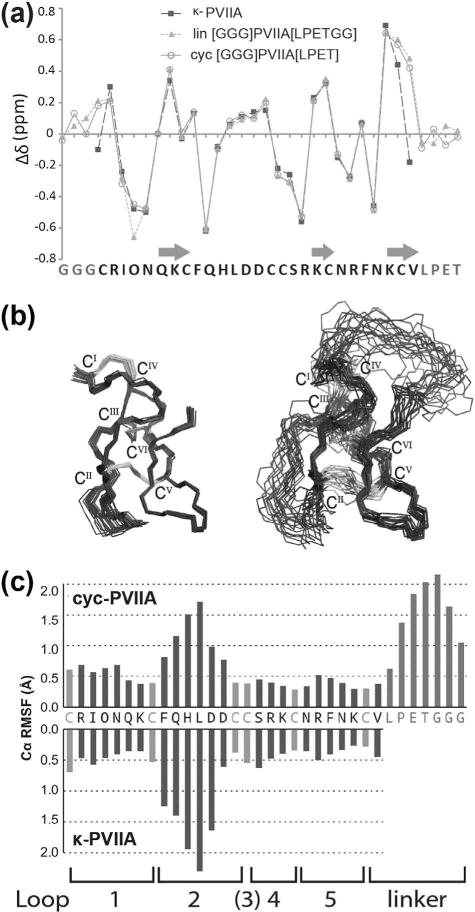
(a) Secondary Ha chemical shift comparison of κ-PVIIA and cyc-PVIIA. Residues forming β-sheets are indicated with arrows. (b) The 20 lowest-energy structres of κ-PVIIA (PDB identifier 1AV3, left) (Scanlon et al., 1997) and cyc-PVIIA (PDB identifier 2N8E, right) superimposed over the backbone residues 2–7 and 15–26. Disulfide bonds are shown in gray with Roman numerals. (c) Root-mean-square fluctuation (RMSF) highlighting the flexibility of each loop of native and cyc-PVIIA.
The three-dimensional structure of cyc-PVIIA was calculated using 384 distance restraints, together with 17 φ, 17 ψ, four χ1 dihedral angle restraints and four hydrogen bonds derived from a D2O exchange experiment. The structure of cyc-PVIIA calculated using CYANA (Guntert, 2004) was refined further in a water shell using CNS (Brunger et al., 1998). The 20 conformers with the lowest energy were chosen for structural statistical analysis and showed a root mean square deviation of 0.91±0.25 Å across the backbone, excluding the flexible loop 2 (residues F9-D14) and the disordered linker (residues L28-G34) (Table I). Cyc-PVIIA showed high similarity to κ-PVIIA (PDB identifier 1AV3) (Scanlon et al., 1997) across the overall structure, except in the linker region (Fig. 3b). As seen in other members of the O1 conotoxin superfamily, including MVIIA (Nielsen et al., 1996) and MrVIB (Daly et al., 2004), loop 2 of PVIIA is the most flexible region (not including the linker) based on RMSD comparison of each loop of NMR structures. To determine if cyclization introduced any changes in backbone flexibility of the structure, the Cα root-mean-square fluctuation (RMSF) of κ-PVIIA and cyc-PVIIA were measured from molecular dynamics simulations. Overall, both peptides showed similar RMSF profiles (Fig. 3c). Loop 2 of cyc-PVIIA had slightly less flexibility than that of native PVIIA, which is consistent with cyclization being generally thought to introduce structural rigidity and prevent disulfide bond shuffling (Clark et al., 2010).
Table I.
Energies and structural statistics for the family of 20 lowest energy cyc-PVIIA structures.
| Energies (kcal/mol) | |
| Overall | −1119.8 ± 73.9 |
| Bonds | 14.1 ± 1.5 |
| Angles | 44.5 ± 5.2 |
| Improper | 18.3 ± 4.9 |
| Dihedral | 158.5 ± 3.3 |
| Van der Waals | −125.0 ± 3.3 |
| Electrostatic | −1230.3 ± 77.7 |
| NOE | 0.1 ± 0.0 |
| cDih | 0.1 ± 0.1 |
| MolProbity statistics | |
| Clashes (>0.4Å/1,000 atoms) | 7.5 ± 2.5 |
| Poor rotamers | 1.0 ± 1.0 |
| Ramachandran outliers (%) | 0.0 ± 0.0 |
| Ramachandran favored (%)a | 93.3 ± 4.1 |
| RMSDb from mean structure (Å) | |
| Mean global backbone (residues 3–8, 15–27) | 0.91 ± 0.25 |
| Mean global heavy (residues 3–8, 15–27) | 1.78 ± 0.26 |
| Mean global backbone (residues 1–34) | 1.65 ± 0.31 |
| Mean global heavy (residues 1–34) | 2.42 ± 0.30 |
| Number of distance restraints | |
| Intraresidue (i–j = 0) | 139 |
| Sequential (|i–j| = 1) | 149 |
| Medium range (|i–j| < 5) | 46 |
| Long range (|i–j| ≥5) | 43 |
| Hydrogen bonds | 4 |
| Disulfide bonds | 3 |
| Total | 384 |
| Number of dihedral angle restraints | |
| φ | 17 |
| ψ | 17 |
| χ1 | 4 |
| Total | 38 |
| Violations from experimental restraints | |
| Total NOE violations exceeding 0.2 Å | 0 |
| Total dihedral violations exceeding 2.0° | 0 |
Data are presented as mean ±S.D.
The remaining (6.7%) were found in the allowed regions of the Ramachandran plot.
Mean RMSD was calculated using MOLMOL (Koradi et al., 1996).
Native and Cyclic κ-PVIIA Inhibit Shaker
To evaluate the effect of cyclization on toxin activity, κ-PVIIA and cyc-PVIIA were tested on the Shaker KV channel expressed in Xenopus oocytes. κ-PVIIA completely blocked Shaker currents at 1 μM over a wide range of voltages, whereas the same concentration of the cyclic peptide only partially blocked the channel (Fig. 4a). Fitting the concentration-dependence of Shaker block with the Hill equation revealed an IC50 value for native κ-PVIIA of 80±5 nM, which is comparable to that previously published, 60±3 nM (Jacobsen et al., 2000; Terlau et al., 1996). In contrast, an IC50 value 10-fold higher was observed for cyc-PVIIA (824±60 nM). Both κ-PVIIA and cyc-PVIIA displayed a slope of 1.1±0.1, suggesting a toxin:channel ratio of 1:1, which is commonly observed for pore-blocking toxins (Fig. 4b). As the ion-conducting pore of KV channels is known to be conserved (Lu et al., 2001), both native and cyc-PVIIA were tested on other KV channels, including rKV2.1Δ7, hKV1.3, rKV1.6. Of the tested channels, 1 μM κ-PVIIA only slightly blocked rKV1.6, whereas cyc-PVIIA had no effect (Fig. 4c). Although Shaker has been used as a model for studying other KV channels due to the high degree of homology, our results suggest that a ligand of Shaker does not necessarily bind to other KV channels. Similar experimental or modeling results were also obtained previously using KV1.1 and KV1.2, (Boccaccio et al., 2004; Mahdavi and Kuyucak, 2013; Shon et al., 1998). Finally, neither peptide is active on rNaV1.4 or hNaV1.6.
Figure 4.
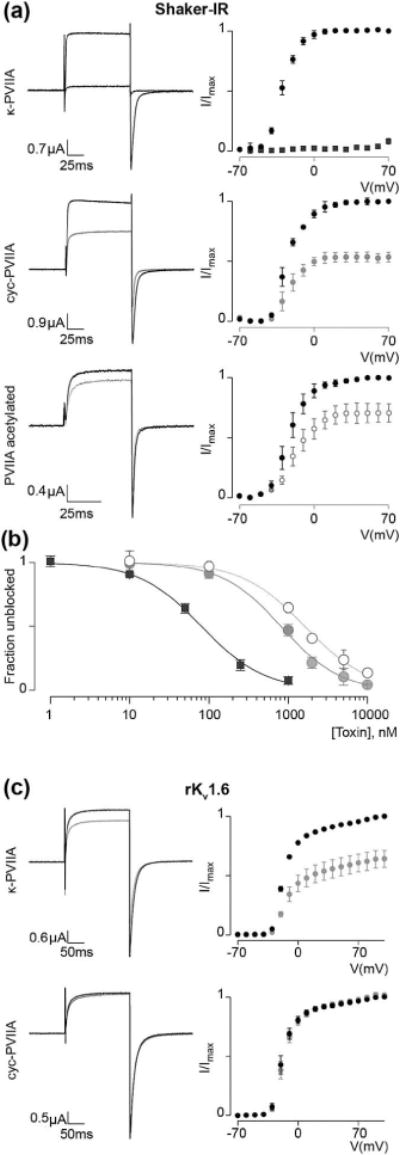
Inhibitory toxin activity on Drosophila Shaker and rKV1.6 (a) 1 μM of κ-PVIIA completely blocks Shaker-IR (inactivation removed) currents over a wide voltage range whereas the same concentration of the cyc-PVIIA and N-terminally acetylated κ-PVIIA only partially block the channel. Representative example traces shown (left) were selected from n = 5 recordings under each condition at a voltage of 20 mV (100 ms) followed by a voltage-step to −70 mV (100 ms) to evoke tail currents (holding potential was −90 mV). Voltage-activation relationships (right) were obtained by plotting tail currents versus applied voltage step. Black lines and markers represent control currents and gray lines and markers show the response on currents following application of toxin. (b) Fitting the concentration-dependence for toxin, block of Shaker-IR with the Hill equation reveals an IC50 value for the κ-PVIIA of 80 ± 5 nM (black squares), cyc-PVIIA is 824±60 nM (gray filled circles) and that of N-terminally acetylated κ-PVIIA is 1616±98 nM (gray empty circles). κ-PVIIA, cyc-PVIIA, and acetylated κ-PVIIA fits display a slope of ~1.1±0.1, suggesting a toxin:channel ratio of 1:1. The data is presented as mean ±s.e.m. (c) 1 μM κ-PVIIA weakly blocks rKV1.6 whereas cyc-PVIIA has no effect (n = 5). Example traces shown on the left were evoked by depolarizing the channel to 0 mV followed by −50 mV (tail) from a holding potential of −90 mV. Black lines and markers represent control currents and gray lines and markers show the response on currents following application of toxin.
The N-terminus of κ-PVIIA is Required for Electrostatic Interaction With Shaker
Previous molecular modeling studies predicted how κ-PVIIA binds to Shaker based on structural analyses and mutagenesis studies (Huang et al., 2005; Jacobsen et al., 2000; Mahdavi and Kuyucak, 2013; Savarin et al., 1998). Initially, recognition of κ-PVIIA by Shaker takes place via electrostatic interactions between positively charged residues on the surface of the peptide and negatively charged residues on the extracellular side of the channel (Huang et al., 2005). The charged residues play an important role in adjusting the conformation of the peptide in the binding process until Lys7 of κ-PVIIA inserts into the pore of the channel and blocks the flow of potassium ions. In addition to Lys7, several residues were found to have a critical role in binding; for example, Arg2 of κ-PVIIA interacts strongly with Asp447 of the channel and Phe9 of κ-PVIIA and Phe425 make hydrophobic interactions (Huang et al., 2005; Mahdavi and Kuyucak, 2013).
We developed a model of cyc-PVIIA bound to Shaker using a recent model of the channel (Mahdavi and Kuyucak, 2013). In our model, the binding mode of cyc-PVIIA is predicted to be generally unchanged compared with that of κ-PVIIA (Fig. 5a–c). The stability of the cyc-PVIIA/Shaker complex was examined in a 100 ns molecular dynamics simulation, analyzed using the backbone RMSD of Shaker, cyc-PVIIA, and the position of cyc-PVIIA on Shaker (Fig. 5d–g). The transmembrane helices of Shaker were rigid and did not deviate significantly from the starting conformation, with a backbone RMSD of ~1 Å during the simulation. The backbone RMSD of the entire channel, including transmembrane helices and loops, was on average 2.2 Å, indicating the loops are more flexible than the helices. The conformation of cyc-PVIIA was also very stable as its backbone RMSD was ~1 Å, (excluding the cyclization linker) (Fig. 5g). The change in position of cyc-PVIIA on Shaker was evaluated by computing the backbone RMSD of the peptide (excluding the linker) after fitting on to Shaker transmembrane helices. The average RMSD of the cyc-PVIIA position (~2.5 Å) is comparable to the backbone RMSD of Shaker, which is mostly accounted for by the flexibility of the loops, thus suggesting that the binding mode of cyc-PVIIA to the channel is preserved. This is further supported by an overlay of the starting and final binding modes of the simulation (Fig. 5d–f), which shows excellent conservation of the relative position of Lys7 and of the interface.
Figure 5.
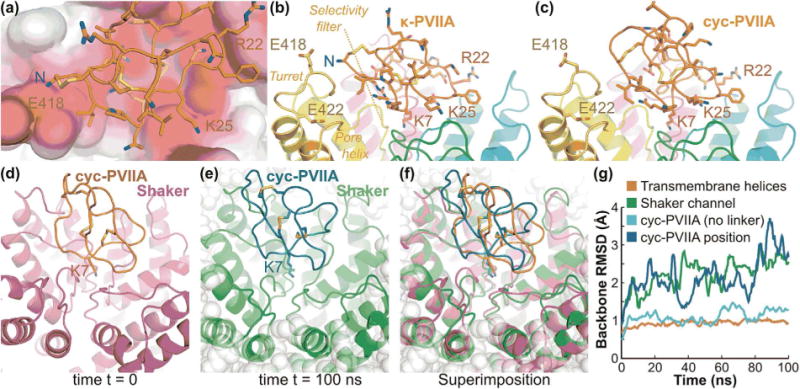
Model of interactions of κ-PVIIA and cyc-PVIIA with Shaker, (a) View of the extracellular entrance of the channel in the molecular model of κ-PVIIA in complex with Shaker (Mahdavi and Kuyucak, 2013). The solvent-accessible surface of the channel is colored according to the Poisson-Boltzmann electrostatic potential it generates, as computed by the APBS software (Baker et al., 2001), with a scale ranging from −4 kT/e (red) to +4 kT/e (blue). The extracellular loop of each of the four subunits of the channel creates an electronegative potential with which the positively charged residues R22 and K25 and N-terminus of κ-PVIIA interact on three different sides of the peptide. (b) Side view of the κ-PVIIA/Shaker complex highlighting two glutamines located on the Shaker turret, E418 and E422 that contribute to generate the electronegative potential surrounding the N-terminus of κ-PVIIA. The side chain of K7 of κ-PVIIA is also shown deeply buried in the ion channel selectivity filter. (c) Molecular model of the complex between cyc-PVIIA and Shaker, which suggests that the binding mode is globally similar to that of the linear peptide apart from the absence of electrostatic interactions between the N-terminus and Shaker. (d) Complex between cyc-PVIIA and Shaker at the start of the molecular dynamics simulation carried out in a POPC lipid membrane environment and explicit solvent. Cyc-PVIIA is in orange and Shaker is in purple; K7 is shown using stick representation. (e) Complex between cyc-PVIIA and Shaker at the end of the molecular dynamics simulation. Cyc-PVIIA is in dark green and Shaker is in light green; K7 is shown using stick representation; POPC lipids are shown as gray balls. (f) Superimposition of first and final frame of the simulation as represented in (d) and (e). (g) Evolution of backbone RMSD from the initial conformation during the 100 ns molecular dynamics simulation of cyc-PVIIA/Shaker embedded in a POPC lipid membrane. The backbone RMSD values were computed using the transmembrane helices of Shaker only (red), all Shaker channel residues (green), cyc-PVIIA residues (light blue) and the position of cyc-PVIIA (blue), which is the backbone RMSD of cyc-PVIIA computed after fitting on the structure of the transmembrane helices.
Despite the unchanged binding mode after cyclization, cyc-PVIIA showed decreased activity (Fig. 4a), and this was hypothesized to be due to loss of electrostatic interaction between the electronegative potential generated by Glu418 and Glu422 on the turret of Shaker and the positively charged N-terminus of κ-PVIIA (Fig. 5a–c). To test this hypothesis, N-terminally acetylated PVIIA was synthesized and indeed found to have ~20-fold lower activity (IC50, 1616 ± 98 nM) than κ-PVIIA on Shaker (Fig. 4a and b) confirming that the N-terminus of κ-PVIIA is required for full inhibition of channel conductivity. We expect that the loss of electrostatic interaction could possibly be compensated by incorporation of a positively charged residue in the linker in future studies.
The Conformations of κ-PVIIA and cyc-PVIIA Are Similar Upon Binding to Shaker, But the Hydrogen Bond Networks Differ
Although the overall backbone conformations of cyc-PVIIA and native κ-PVIIA in the NMR solution structures are very similar, Cys1 and Arg2, which are adjacent to the cyclization linker, are in different spatial locations in the cyclized and native peptides (Fig. 6b and c). By contrast, molecular models suggest that these two residues have similar conformations and interactions at the interface with Shaker. We thus investigated if the difference in affinity between the cyclized and linear form could be explained by differences in the unbound state. An overlay of the unbound and bound peptide conformations suggests that Cys1 is displaced by 3.0 Å for κ-PVIIA and 2.5 Å for cyc-PVIIA. In contrast, the hydrogen bond networks of Arg2 for the native and cyclic peptides are quite different when they are bound and unbound to Shaker (Fig. 6). The changes in the hydrogen bond networks were studied using 100 ns molecular dynamics simulations. Arg2 of κ-PVIIA forms stable hydrogen bonds with Lys7 and Cys16 and transient hydrogen bonds with Asp14. On the other hand, Arg2 of cyc-PVIIA lacks hydrogen bonds with Lys7 or Cys16 but does form a transient hydrogen bond with Asp14. The models suggest that the hydrogen bond network of unbound κ-PVIIA is more similar to the bound state than that of cyc-PVIIA. Although it is not certain whether this difference contributes to the decreased activity of cyc-PVIIA, it appears likely that the two hydrogen bonds between Arg2 and Lys7, and Arg2 and Cys16 stabilize the conformation of κ-PVIIA upon binding to Shaker.
Figure 6.
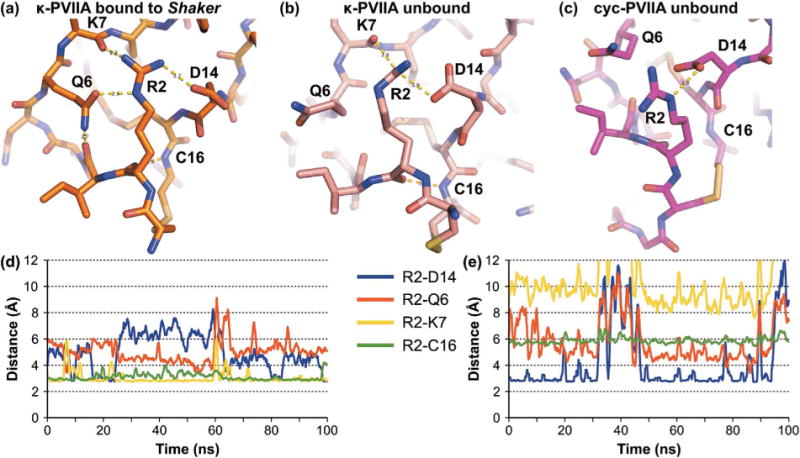
Hydrogen bond network established by Arg2 of κ-PVIIA and cyc-PVIIA. Hydrogen bond network of (a) Arg2 of κ-PVIIA when bound to Shaker in the model (Mahdavi and Kuyucak, 2013), (b) Arg2 of κ-PVIIA Arg2 in the NMR solution structure (PDB identifier 1AV3) (Scanlon et al., 1997), and (c) Arg2 in the molecular model of unbound cyc-PVIIA. The evolution of the Arg2 hydrogen bond network of κ-PVIIA (d) and cyc-PVIIA (e) was studied using 100 ns molecular dynamics simulations. In these simulations, Arg2 of κ-PVIIA formed stable hydrogen bonds with Lys7 and Cys16 and transiently established a hydrogen bond with Asp14. In contrast to the linear peptide, Arg2 of cyc-PVIIA did not form hydrogen bonds with Lys7 and Cys16 but it established a hydrogen bond more frequently with Asp14 than Arg2 of the linear peptide.
κ-PVIIA and cyc-PVIIA Serum Stability
To determine the effect of cyclization on stability, native and cyc-PVIIA were incubated in human serum. The majority of native κ-PVIIA was still detectable after 12 h; this was expected given that the ICK motif is known to contribute to peptide stability. Indeed, a high percentage of acyclic kalata B1, which is topologically similar to κ-PVIIA because of its knotted conformation, remains in serum even after 24 h (Jia et al., 2014). Surprisingly, only ~50% of cyc-PVIIA was detectable in serum after 12 h (Fig. 7). One interpretation of this finding is that the additional constraint introduced by cyclization has made it more susceptible to proteolysis, thus highlighting the importance of optimizing the length and sequence of a linker in cyclization studies.
Figure 7.
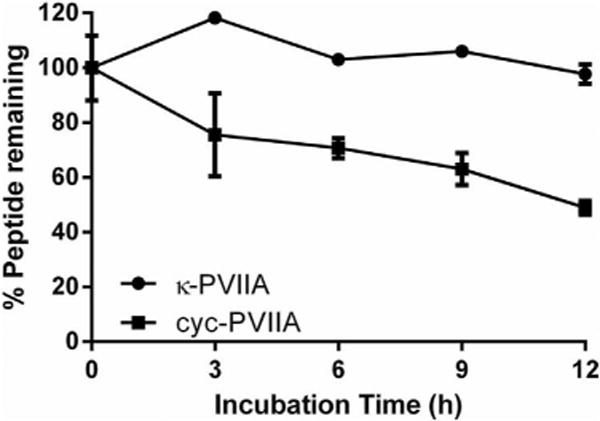
Serum stability assay on native and cyc-PVIIA. Percentage of remaining peptide following 0, 3, 6, 9, and 12 h incubation in human serum is presented as mean ± s.e.m of triplicates.
Conclusion
The ICK motif is found in many venom peptides that have therapeutic potential, but chemical and protein engineering is typically required to achieve suitable pharmaceutical properties for this motif. This study demonstrates that cyclization of an ICK motif-containing conotoxin is achievable via transpeptidation using SrtA. The linear oxidized peptide precursor of κ-PVIIA was efficiently cyclized, producing cyc-PVIIA with a head-to-tail cyclic backbone. The overall backbone structure and conformational dynamics of cyc-PVIIA is similar to native κ-PVIIA; however, following cyclization the peptide has partially reduced activity on the Shaker KV channel. The decrease in activity appears to be due to the removal of previously unrecognized electrostatic interactions between the N-terminus of the peptide and negatively charged residues within the channel. Overall, the results of this study suggests that criteria which need to be considered in the head-to-tail backbone cyclization of ICK peptides include the effect of terminal mutations on activity, as well as the intramolecular hydrogen bond network and the conformational dynamics of the peptides. The study reveals that SrtA is an efficient tool for cyclizing disulfide-rich peptides that are inaccessible to cyclization using traditional synthetic methods.
Acknowledgments
We thank David Liu for providing the SrtA5° expression plasmid, and K. J. Swartz, C. Deutsch, O. Pongs, and B. Chanda for sharing Shaker, rKV2.1Δ7, hKV1.3, rKV1.6, and rNaV1.4, respectively, and C. A. Wang for testing the catalytic activity of SrtA5°. This work was funded by a National Health and Medical Research Council (NHMRC) Project Grant to DJC and CIS (APP1047857) and by the National Institute of Neurological Disorders And Stroke (NINDS) of the National Institutes of Health (NIH) under Award Number 1R01NS091352 to FB. SK is a recipient of an Australian Research Council (ARC) Australian Postgraduate Award, ACC was supported by a University of Queensland International PhD scholarship, CIS in an Institute for Molecular Bioscience Industry Fellow, DJC is an ARC Australian Laureate Fellow (FL150100146) and KJR is an ARC Future Fellow (FT130100890).
Contract grant sponsor: National Health and Medical Research Council (NHMRC) Contract grant number: APP1047857
Contract grant sponsor: National Institute of Neurological Disorders and Stroke (NINDS)
Contract grant number: 1R01NS091352
Contract grant sponsor: Australian Research Council (ARC) Australian Postgraduate Award
Contract grant sponsor: University of Queensland International PhD
Contract grant sponsor: Australian Laureate Fellow
Contract grant number: FL150100146
Contract grant sponsor: ARC Future Fellow
Contract grant number: FT130100890
Footnotes
The structure of cyclized PVIIA has been deposited to the Protein Data Bank (code 2N8E) and 15 the Biological Magnetic Resonance Bank (accession number 25847).
References
- Akcan M, Clark RJ, Daly NL, Conibear AC, de Faoite A, Heghinian MD, Sahil T, Adams DJ, Mari F, Craik DJ. Transforming conotoxins into cyclotides: Backbone cyclization of P-superfamily conotoxins. Biopolymers. 2015;104:682–692. doi: 10.1002/bip.22699. [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ME, White HS, Wilcox KS. The effect of CGX-1007 and CI-1041, novel NMDA receptor antagonists, on NMDA receptor-mediated EPSCs. Epilepsy Res. 2004;59:13–24. doi: 10.1016/j.eplepsyres.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Blanco-Canosa JB, Nardone B, Albericio F, Dawson PE. Chemical protein synthesis using a second-generation N-acylurea linker for the preparation of peptide-thioester precursors. J Am Chem Soc. 2015;137:7197–7209. doi: 10.1021/jacs.5b03504. [DOI] [PubMed] [Google Scholar]
- Boccaccio A, Conti F, Olivera BM, Terlau H. Binding of kappa-conotoxin PVIIA to Shaker K+ channels reveals different K+ and Rb+ occupancies within the ion channel pore. J Gen Physiol. 2004;124:71–81. doi: 10.1085/jgp.200409048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nat Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Chan LY, Gunasekera S, Henriques ST, Worth NF, Le SJ, Clark RJ, Campbell JH, Craik DJ, Daly NL. Engineering pro-angiogenic peptides using stable, disulfide-rich cyclic scaffolds. Blood. 2011;118:6709–6717. doi: 10.1182/blood-2011-06-359141. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Dorr BM, Liu DR. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc Natl Acad Sci USA. 2011;108:11399–11404. doi: 10.1073/pnas.1101046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheneval O, Schroeder CI, Durek T, Walsh P, Huang YH, Liras S, Price DA, Craik DJ. Fmoc-based synthesis of disulfide-rich cyclic peptides. J Org Chem. 2014;79:5538–5544. doi: 10.1021/jo500699m. [DOI] [PubMed] [Google Scholar]
- Clark RJ, Jensen J, Nevin ST, Callaghan BP, Adams DJ, Craik DJ. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew Chem Int Ed Engl. 2010;49:6545–6548. doi: 10.1002/anie.201000620. [DOI] [PubMed] [Google Scholar]
- Colgrave ML, Craik DJ. Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: The importance of the cyclic cystine knot. Biochemistry. 2004;43:5965–5975. doi: 10.1021/bi049711q. [DOI] [PubMed] [Google Scholar]
- Conibear AC, Rosengren KJ, Harvey PJ, Craik DJ. Structural characterization of the cyclic cystine ladder motif of theta-defensins. Biochemistry. 2012;51:9718–9726. doi: 10.1021/bi301363a. [DOI] [PubMed] [Google Scholar]
- Craik DJ, Conibear AC. The chemistry of cyclotides. J Org Chem. 2011;76:4805–4817. doi: 10.1021/jo200520v. [DOI] [PubMed] [Google Scholar]
- Craik DJ, Daly NL, Waine C. The cystine knot motif in toxins and implications for drug design. Toxicon. 2001;39:43–60. doi: 10.1016/s0041-0101(00)00160-4. [DOI] [PubMed] [Google Scholar]
- Cushman DW, Ondetti MA. Design of angiotensin converting enzyme inhibitors. Nat Med. 1999;5:1110–1113. doi: 10.1038/13423. [DOI] [PubMed] [Google Scholar]
- Daly NL, Craik DJ. Bioactive cystine knot proteins. Curr Opin Chem Biol. 2011;15:362–368. doi: 10.1016/j.cbpa.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Daly NL, Love S, Alewood PF, Craik DJ. Chemical synthesis and folding pathways of large cyclic polypeptide: Studies of the cystine knot polypeptide kalata B1. Biochemistry. 1999;38:10606–10614. doi: 10.1021/bi990605b. [DOI] [PubMed] [Google Scholar]
- Daly NL, Ekberg JA, Thomas L, Adams DJ, Lewis RJ, Craik DJ. Structures of muO-conotoxins from Conus marmoreus. Inhibitors of tetrodotoxin (TTX)-sensitive and TTX-resistant sodium channels in mammalian sensory neurons. J Biol Chem. 2004;279:25774–25782. doi: 10.1074/jbc.M313002200. [DOI] [PubMed] [Google Scholar]
- Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- Fainzilber M, van der Schors R, Lodder JC, Li KW, Geraerts WP, Kits KS. New sodium channel-blocking conotoxins also affect calcium currents in Lymnaea neurons. Biochemistry. 1995;34:5364–5371. doi: 10.1021/bi00016a007. [DOI] [PubMed] [Google Scholar]
- Fosgerau K, Hoffmann T. Peptide therapeutics: Current status and future directions. Drug Discov Today. 2015;20:122–128. doi: 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Frech GC, VanDongen AM, Schuster G, Brown AM, Joho RH. A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature. 1989;340:642–645. doi: 10.1038/340642a0. [DOI] [PubMed] [Google Scholar]
- Garcia ML, Garcia-Calvo M, Hidalgo P, Lee A, MacKinnon R. Purification and characterization of three inhibitors of voltage-dependent K+ channels from Leiurus quinquestriatus var. hebraeus venom. Biochemistry. 1994;33:6834–6839. doi: 10.1021/bi00188a012. [DOI] [PubMed] [Google Scholar]
- Gilchrist J, Das S, Van Petegem F, Bosmans F. Crystallographic insights into sodium-channel modulation by the beta4 subunit. Proc Natl Acad Sci USA. 2013;110:E5016–E5024. doi: 10.1073/pnas.1314557110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gran L. An oxytocic principle found in Oldenlandia affinis DC. Medd Nor Farm Selsk. 1970;12:173–180. [Google Scholar]
- Grupe A, Schroter KH, Ruppersberg JP, Stocker M, Drewes T, Beckh S, Pongs O. Cloning and expression of a human voltage-gated potassium channel. A novel member of the RCK potassium channel family. EMBO J. 1990;9:1749–1756. doi: 10.1002/j.1460-2075.1990.tb08299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- Heinemann SH, Leipold E. Conotoxins of the O-superfamily affecting voltage-gated sodium channels. Cell Mol Life Sci. 2007;64:1329–1340. doi: 10.1007/s00018-007-6565-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz TN, van Gunsteren WF, Hunenberger PH. Comparison of four methods to compute the dielectric permittivity of liquids from molecular dynamics simulations. J Chem Phys. 2001;115:1125–1136. [Google Scholar]
- Hemu X, Taichi M, Qiu Y, Liu DX, Tam JP. Biomimetic synthesis of cyclic peptides using novel thioester surrogates. Biopolymers. 2013;100:492–501. doi: 10.1002/bip.22308. [DOI] [PubMed] [Google Scholar]
- Hill JM, Alewood PF, Craik DJ. Solution structure of the sodium channel antagonist conotoxin GS: A new molecular caliper for probing sodium channel geometry. Structure. 1997;5:571–583. doi: 10.1016/s0969-2126(97)00212-8. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Huang X, Dong F, Zhou HX. Electrostatic recognition and induced fit in the kappa-PVIIA toxin binding to Shaker potassium channel. J Am Chem Soc. 2005;127:6836–6849. doi: 10.1021/ja042641q. [DOI] [PubMed] [Google Scholar]
- Ireland DC, Colgrave ML, Craik DJ. A novel suite of cyclotides from Viola odorata: Sequence variation and the implications for structure, function and stability. Biochem J. 2006;400:1–12. doi: 10.1042/BJ20060627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen RB, Koch ED, Lange-Malecki B, Stocker M, Verhey J, Van Wagoner RM, Vyazovkina A, Olivera BM, Terlau H. Single amino acid substitutions in kappa-conotoxin PVIIA disrupt interaction with the shaker K+ channel. J Biol Chem. 2000;275:24639–24644. doi: 10.1074/jbc.C900990199. [DOI] [PubMed] [Google Scholar]
- Jia X, Kwon S, Wang CI, Huang YH, Chan LY, Tan CC, Rosengren KJ, Mulvenna JP, Schroeder CI, Craik DJ. Semienzymatic cyclization of disulfide-rich peptides using Sortase A. J Biol Chem. 2014;289:6627–6638. doi: 10.1074/jbc.M113.539262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas Q, Westermann JC, Craik DJ. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon. 2010;55:1491–1509. doi: 10.1016/j.toxicon.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Kaas Q, Yu R, Jin AH, Dutertre S, Craik DJ. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012;40:D325–D330. doi: 10.1093/nar/gkr886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia J, Milescu M, Salvatierra J, Wagner J, Klint JK, King GF, Olivera BM, Bosmans F. From foe to friend: Using animal toxins to investigate ion channel function. J Mol Biol. 2015;427:158–175. doi: 10.1016/j.jmb.2014.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo GC, Blake JT, Talento A, Nguyen M, Lin S, Sirotina A, Shah K, Mulvany K, Hora D, Jr, Cunningham P, Wunderler DL, McManus OB, Slaughter R, Bugianesi R, Felix J, Garcia M, Williamson J, Kaczorowski G, Sigal NH, Springer MS, Feeney W. Blockade of the voltage-gated potassium channel Kv1.3 inhibits immune responses in vivo. J Immunol. 1997;158:5120–5128. [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Li-Smerin Y, Swartz KJ. Gating modifier toxins reveal a conserved structural motif in voltage-gated Ca2+ and K+ channels. Proc Natl Acad Sci USA. 1998;95:8585–8589. doi: 10.1073/pnas.95.15.8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Klem AM, Ramu Y. Ion conduction pore is conserved among potassium channels. Nature. 2001;413:809–813. doi: 10.1038/35101535. [DOI] [PubMed] [Google Scholar]
- Mahdavi S, Kuyucak S. Why the Drosophila Shaker K+ channel is not a good model for ligand binding to voltage-gated Kv1 channels. Biochemistry. 2013;52:1631–1640. doi: 10.1021/bi301257p. [DOI] [PubMed] [Google Scholar]
- Miljanich GP. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr Med Chem. 2004;11:3029–3040. doi: 10.2174/0929867043363884. [DOI] [PubMed] [Google Scholar]
- Nielsen KJ, Thomas L, Lewis RJ, Alewood PF, Craik DJ. A consensus structure for omega-conotoxins with different selectivities for voltage-sensitive calcium channel subtypes: Comparison of MVIIA SVIB and SNX-202. J Mol Biol. 1996;263:297–310. doi: 10.1006/jmbi.1996.0576. [DOI] [PubMed] [Google Scholar]
- Nielsen DS, Lohman RJ, Hoang HN, Hill TA, Jones A, Lucke AJ, Fairlie DP. Flexibility versus rigidity for orally bioavailable cyclic hexapeptides. Chembiochem. 2015;16:2289–2293. doi: 10.1002/cbic.201500441. [DOI] [PubMed] [Google Scholar]
- Norton RS, Pallaghy PK. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon. 1998;36:1573–1583. doi: 10.1016/s0041-0101(98)00149-4. [DOI] [PubMed] [Google Scholar]
- Pallaghy PK, Nielsen KJ, Craik DJ, Norton RS. A common structural motif incorporating a cystine knot and a triple-stranded beta-sheet in toxic and inhibitory polypeptides. Protein Sci. 1994;3:1833–1839. doi: 10.1002/pro.5560031022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M, Rahman A. Polymorphic transitions in single-crystals—A new molecular-dynamics method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- Popp MW, Ploegh HL. Making and breaking peptide bonds: Protein engineering using sortase. Angew Chem Int Ed Engl. 2011;50:5024–5032. doi: 10.1002/anie.201008267. [DOI] [PubMed] [Google Scholar]
- Popp MW, Antos JM, Ploegh HL. Site-specific protein labeling via sortase-mediated transpeptidation. Curr Protoc Protein Sci. 2009;56:15.3.1–15.3.9. doi: 10.1002/0471140864.ps1503s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju S, Chi V, Pennington MW, Chandy KG. Kv1.3 potassium channels as a therapeutic target in multiple sclerosis. Expert Opin Ther Targets. 2009;13:909–924. doi: 10.1517/14728220903018957. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Savarin P, Guenneugues M, Gilquin B, Lamthanh H, Gasparini S, Zinn-Justin S, Menez A. Three-dimensional structure of kappa-conotoxin PVIIA, a novel potassium channel-blocking toxin from cone snails. Biochemistry. 1998;37:5407–5416. doi: 10.1021/bi9730341. [DOI] [PubMed] [Google Scholar]
- Scanlon MJ, Naranjo D, Thomas L, Alewood PF, Lewis RJ, Craik DJ. Solution structure and proposed binding mechanism of a novel potassium channel toxin kappa-conotoxin PVIIA. Structure. 1997;5:1585–1597. doi: 10.1016/s0969-2126(97)00307-9. [DOI] [PubMed] [Google Scholar]
- Schaller KL, Krzemien DM, Yarowsky PJ, Krueger BK, Caldwell JH. A novel, abundant sodium channel expressed in neurons and glia. J Neurosci. 1995;15:3231–3242. doi: 10.1523/JNEUROSCI.15-05-03231.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, van Gunsteren WF. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophys J. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Shon KJ, Grilley MM, Marsh M, Yoshikami D, Hall AR, Kurz B, Gray WR, Imperial JS, Hillyard DR, Olivera BM. Purification, characterization, synthesis, and cloning of the lockjaw peptide from Conus purpurascens venom. Biochemistry. 1995;34:4913–4918. doi: 10.1021/bi00015a002. [DOI] [PubMed] [Google Scholar]
- Shon KJ, Stocker M, Terlau H, Stuhmer W, Jacobsen R, Walker C, Grilley M, Watkins M, Hillyard DR, Gray WR, Olivera BM. Kappa-Conotoxin PVIIA is a peptide inhibiting the shaker K+ channel. J Biol Chem. 1998;273:33–38. doi: 10.1074/jbc.273.1.33. [DOI] [PubMed] [Google Scholar]
- Stanger K, Maurer T, Kaluarachchi H, Coons M, Franke Y, Hannoush RN. Backbone cyclization of a recombinant cystine-knot peptide by engineered Sortase A. FEBS Lett. 2014;588:4487–4496. doi: 10.1016/j.febslet.2014.10.020. [DOI] [PubMed] [Google Scholar]
- Stuhmer W, Ruppersberg JP, Schroter KH, Sakmann B, Stocker M, Giese KP, Perschke A, Baumann A, Pongs O. Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. EMBO J. 1989;8:3235–3244. doi: 10.1002/j.1460-2075.1989.tb08483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson R, Marshall J, Smith JS, Williams JB, Boyle MB, Folander K, Luneau CJ, Antanavage J, Oliva C, Buhrow SA, Bennet C, Stein RB, Kaczmarek LK. Cloning and expression of cDNA and genomic clones encoding three delayed rectifier potassium channels in rat brain. Neuron. 1990;4:929–939. doi: 10.1016/0896-6273(90)90146-7. [DOI] [PubMed] [Google Scholar]
- Tam JP, Lu YA, Yang JL, Chiu KW. An unusual structural motif of antimicrobial peptides containing end-to-end macrocycle and cystine-knot disulfides. Proc Natl Acad Sci USA. 1999;96:8913–8918. doi: 10.1073/pnas.96.16.8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempel BL, Papazian DM, Schwarz TL, Jan YN, Jan LY. Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science. 1987;237:770–775. doi: 10.1126/science.2441471. [DOI] [PubMed] [Google Scholar]
- Terlau H, Shon KJ, Grilley M, Stocker M, Stuhmer W, Olivera BM. Strategy for rapid immobilization of prey by a fish-hunting marine snail. Nature. 1996;381:148–151. doi: 10.1038/381148a0. [DOI] [PubMed] [Google Scholar]
- Ton-That H, Liu G, Mazmanian SK, Faull KF, Schneewind O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc Natl Acad Sci USA. 1999;96:12424–12429. doi: 10.1073/pnas.96.22.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS, Cooperman SS, Tomiko SA, Zhou JY, Crean SM, Boyle MB, Kallen RG, Sheng ZH, Barchi RL, Sigworth FJ, Goodman RH, Agnew WS, Mandel G. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron. 1989;3:33–49. doi: 10.1016/0896-6273(89)90113-x. [DOI] [PubMed] [Google Scholar]
- Wüthrich K. NMR of proteins and nucleic acids. New York: Wiley-Interscience; 1986. [Google Scholar]
- Wang CK, Northfield SE, Colless B, Chaousis S, Hamernig I, Lohman RJ, Nielsen DS, Schroeder CI, Liras S, Price DA, Fairlie DP, Craik DJ. Rational design and synthesis of an orally bioavailable peptide guided by NMR amide temperature coefficients. Proc Natl Acad Sci USA. 2014;111:17504–17509. doi: 10.1073/pnas.1417611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte MD, Cragnolini JJ, Dougan SK, Yoder NC, Popp MW, Ploegh HL. Preparation of unnatural N-to-N and C-to-C protein fusions. Proc Natl Acad Sci USA. 2012;109:11993–11998. doi: 10.1073/pnas.1205427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu SY, Darbon H, Dyason K, Verdonck F, Tytgat J. Evolutionary origin of inhibitor cystine knot peptides. FASEB J. 2003;17:1765–1767. doi: 10.1096/fj.02-1044fje. [DOI] [PubMed] [Google Scholar]