

Abstract
A major product of oxidative DNA damage is 8-oxoguanine. In humans, 8-oxoguanine DNA Glycosylase (hOGG1) facilitates removal of these lesions, producing an abasic (AP) site in the DNA that is subsequently incised by AP-endonuclease 1 (APE1). APE1 stimulates turnover of several glycosylases by accelerating rate-limiting product release. However, there have been conflicting accounts of whether hOGG1 follows a similar mechanism. In pre-steady state kinetic measurements, we found that addition of APE1 had no effect on the rapid burst phase of 8-oxoguanine excision by hOGG1, but accelerated steady-state turnover (kcat) by ~10-fold. The stimulation by APE1 required divalent cations, was detectable under multiple turnover conditions using limiting concentrations of APE1, did not require flanking DNA surrounding the hOGG1 lesion site, and occurred efficiently even when the first 49 residues of APE1’s N-terminus was deleted. Stimulation by APE1 does not involve relief from product inhibition because thymine DNA glycosylase (TDG), an enzyme that binds more tightly to AP-sites than hOGG1, could not effectively substitute for APE1. A stimulation mechanism involving stable protein-protein interactions between free APE1 and hOGG1, or the DNA bound forms, was excluded using protein crosslinking assays. The combined results indicate a mechanism where dynamic excursions of hOGG1 from the AP-site allow APE1 to invade the site and rapidly incise the phosphate backbone. This mechanism, which allows APE1 to access the AP-site without forming specific interactions with the glycosylase, is a simple and elegant solution to passing along unstable intermediates in base excision repair.
Graphical abstract
8-oxoguanine (8-oxoG) is an abundant form of oxidative damage to DNA that occurs with a frequency of 104–105 per cell per day1. Human 8-oxoguanine glycosylase (hOGG1) initiates the excision of 8-oxoG from oxidized G/C base pairs producing an abasic (AP) site as the major product, which is subsequently removed and filled in by subsequent enzymes in the base excision repair pathway2. In addition to this major activity, hOGG1 also possesses a minor AP lyase activity arising from the formation of a Schiff base linkage between a conserved active site lysine and the C1′ of the damaged site3 The lyase activity eliminates the 3′ phosphate of the AP site and yields a single strand break, but this activity is thought be minor under physiological conditions 4–8. The timely removal of abundant 8-oxoG lesions is imperative in order to prevent G/C→T/A transversion mutations in the genome6,9.
A common kinetic barrier for several human DNA glycosylases is release from their AP-site products10,11. For several of these glycosylases (including hOGG1), the subsequent enzyme in the repair pathway AP-endonuclease 1 (APE1), has been found to stimulate steady-state turnover by promoting enzyme release from the product complex6,12–18. Although the mechanistic basis for APE1 stimulated turnover has been reasonably well-characterized for several of these enzymes2,12,19, mechanistic ambiguity remains in the case of hOGG1. One earlier study of hOGG1 suggested that the endonuclease activity of APE1 was not important for stimulation of turnover, and suggested that the high AP-site binding affinity of APE1 led to displacement of hOGG114. A second study has reported that the AP-endonuclease activity of APE1 was essential for stimulation, and further, that direct interactions between APE1 and hOGG1 were required for facilitated displacement of hOGG117.
To explore this question further we tested the viability of five potential mechanisms for APE1-mediated stimulation of hOGG1 turnover (Fig. 1). These mechanisms fall into three general categories. The first involves stable protein-protein interactions between the free or DNA bound enzymes that promote rapid AP-site release by hOGG1 (Fig. 1A, 1B). The second category involves kinetic trapping of the AP-site by APE1 (Fig. 1C, 1D). A requirement of a trapping mechanism is that hOGG1 must transiently depart the AP-site by microscopic dissociation and/or translocation along the DNA to allow APE1 to gain access to the site. Rapid microscopic dissociation or short-range translocation away from the site are reversible steps that most often lead to reassociation with the AP-site (retrograde binding), which can then be interrupted by APE1. A second requirement for this trapping mechanism is that APE1 must be at a sufficiently high concentration to allow efficient trapping of the site in the lifetime that it is exposed (which may occur by DNA translocation or 3D diffusion through bulk solution). Finally, a simple competitive mechanism involving binding of APE1 to the AP site is tested (Fig. 1E). In this mechanism, APE1 binding to the product increases the concentration of free hOGG1 at later stages in the reaction as product inhibition becomes significant. The efficiency of this mechanism is dictated by the relative binding affinities of each protein to the AP-site and their concentrations. All of the above mechanisms can be influenced by APE1 enzymatic activity.
Figure 1. Possible mechanisms for displacement of hOGG1 from an AP site by APE1.

(A) Complexation of free proteins. In this mechanism, APE1 induces a conformational change in hOGG1 that facilitates turnover from the AP site. (B) DNA-facilitated protein complexation. In this mechanism, non-specific interactions of each protein with DNA are used to facilitate weak protein-protein interactions that facilitate release from the AP-site. (C) Kinetic trapping of the AP site by pre-association of APE1 with the DNA. (D) Kinetic trapping of the AP site by diffusional encounter with the hOGG1-AP site. (E) Equilibrium competitive binding of APE1 and hOGG1 to the AP product site. The experiments in this paper support the stimulation mechanisms shown in panels 1C and 1D (see Discussion).
We have developed several new assays to test the above mechanistic possibilities. Our combined findings lead us to embrace a kinetic trapping mechanism that does not involve stable protein-protein interactions and does not require flanking DNA around the hOGG1 binding site (Fig. 1C, 1D). Importantly, APE1 endonuclease activity is required to allow rapid catalytic recycling of limiting concentrations of APE1 to stalled hOGG1/AP-site complexes. We establish the validity of this model using kinetic simulations of the experimental data and suggest that the model is kinetically competent in the cell nucleus given the APE1 copy number per cell (104–105)19.
MATERIALS AND METHODS
Expression and purification of proteins
Full-length hOGG120, wild type human APE121, Δ49APE1 and the catalytic domain of hUNG22 were expressed and purified as previously described. TDG was a generous gift of Dr. Alexander Drohat. Protein concentrations were determined by absorbance at 280 nm (εhOGG1 = 68.9 mM−1 cm−1; εAPE1 = 56.8 mM−1 cm−1; εΔ49APE1 = 55.3 mM−1 cm−1; εhUNG = 33.7 mM−1 cm−1; εTDG = 33.7 mM−1 cm−1).
DNA Oligonucleotides
DNA oligonucleotides were purchased from Integrated DNA Technologies (IDT) or Midland Certified Reagent Company and were purified by gel electrophoresis before use. The sequences of the DNA molecules used in this work are listed in the Supplementary Information. 8-oxoG containing oligonucleotides radiolabeled on the 5’ end were generated by reacting 30 pmol of single stranded DNA with 40 units of T4 polynucleotide kinase (New England Biolabs) and 36 µCi γ[32P]-ATP (3000 mCi/mmol, Perkin Elmer) at 37 °C for 45 min. in a buffer containing 7 mM Tris-HCl (pH 7.6), 1 mM MgCl2, and 0.5 mM DTT. Duplexes were formed by the addition of a 1.1-fold molar excess of the unlabeled complementary strand and supplementing the buffer with 150 mM potassium acetate. The solution was heated to 95 °C for 5 min. and slowly cooled to room temperature over a three-hour period on a heat block. Following hybridization, the radiolabeled dsDNA was purified from unreacted nucleotides by gel filtration (p30 micro Bio-spin column; BioRad). To confirm purity and labeling efficiency samples of the unpurified and purified radiolabeled dsDNA were run on a 10% denaturing PAGE gel containing 7 M urea for 25 minutes at 25 W and a phosphorimage was obtained. The radioactive counts from each sample on the gel were quantified using the QuantityOne software. The specific activity in the units, counts/pmol, of the unpurified sample was determined based on the known concentrations of reagents added. The concentration of the final purified DNA was then calculated based on the specific activity of the sample and the volume of the purified DNA.
A DNA substrate containing a single apyrimidinic site (AP31) was prepared by digestion of a 5′-[32P]-labeled uracil-containing double stranded DNA with uracil DNA glycosylase (U31, see Supplemental Information). U31 is identical in sequence to °G31 except that uracil replaces 8-oxoG. The labeled U31 duplex was diluted to 500 nM in buffer B (see below) and incubated at 37 °C for 1h in the presence of 4 nM hUNG2 to generate DNA with the abasic site (AP31). The generated abasic DNA was shown to be stable for at least 1.5 h (on ice).
Pre-steady state kinetic experiments of 8-oxoguanine excision by hOGG1
All kinetic experiments with hOGG1 or apoAPE1 (inactive) used reaction buffer A [100 µg/ml BSA (New England Biolabs), 1 mM potassium-EDTA, 14 mM potassium phosphate pH 7.5 (prepared by titration of potassium monophosphate with potassium hydroxide), 120 mM potassium acetate, 1 mM DTT]. The hOGG1 activity is the same in both buffer A and B (Fig. S1). Buffer A has a total concentration of potassium ions equal to 150 mM. To reduce variability in experiments, a master stock solution of 5x buffer A was prepared and stored in single use aliquots at −20 °C. In experiments with active APE1, EDTA was excluded from buffer A and it was supplemented with 1 mM MgCl2 (buffer B). Due to the sensitivity of hOGG1 to dilution or buffer exchange we optimized its handling: single frozen aliquots of hOGG1 (20 µM) were thawed on ice for 10 min. and then mixed gently by pipetting before dilution into buffer A (final concentrations were in the range 50 nM to 2000 nM depending on the experiment). Diluted enzyme solutions were incubated on ice for 5 min. before gentle mixing by pipetting. We found that serial dilutions of more than 10-fold at a single step consistently reduced the 8-oxoG excision activity. Following this protocol, we could consistently obtain ~75 % enzymatic activity based on pre-steady state burst amplitude measurements. Before use, APE1 was buffer exchanged from its storage buffer to buffer B and its concentration was adjusted to 10x of its final desired concentration in the reaction with hOGG1. APE1 was added to the 8-oxoG DNA substrate solution and equilibrated at 37 °C for 2 min. before initiating the reaction by the addition of hOGG1.
Pre-steady state kinetic measurements were performed using 500 nM 31mer or 15mer 8-oxoG containing duplexes with a 5’ 32P label on the lesion containing strand (°G31 or °G15; see Supplemental Information) and using various concentrations of hOGG1 (25, 50 and 125 nM final concentrations). Reactions were performed at 37 °C in buffer A and were initiated by mixing equal volumes (25 µL) of 2x concentrated solutions of hOGG1 and DNA. At various times, 6 µL was removed and quenched in an equal volume of quench solution (70% formamide, 0.2M NaOH, 1x TBE buffer containing bromophenol blue and xylene cyanol). The samples were centrifuged and heated at 95 °C for 20 min. to cleave the abasic sites generated by hOGG1. The samples were centrifuged once more and 12 µL of 90% formamide, 1x TBE buffer was added to reduce the ionic strength of the sample. Prior to sample loading, a denaturing 10% polyacrylamide gel (7 M urea) was pre-run for 15 min. at 25 W. 10 µL of quenched reaction samples were loaded onto the gel, which was run for 25 min. at 25 W. The gel was transferred to filter paper, dried, exposed to phosphor screen and imaged on a Typhoon 9500 imager. The same procedure was followed for the °G15 DNA substrate, except that a 20% gel was used and electrophoresis was at 20 W power for 45 min. These gels were directly exposed to a phosphor screen at room temperature overnight and imaged.
Radioactive substrate and product bands were quantified by densitometric analysis using QuantityOne Basic software after subtraction of background counts. The fraction reaction at each time point was calculated by normalization to the total band intensity in each lane (eq. 1), where f is the fraction of reaction at a given time point,
| (1) |
Iproduct is intensity of the product band and Isubstrate is the intensity of the unreacted substrate band. We normalized the measured activities to the concentration of active hOGG1 enzyme. The time dependence of product formation was plotted in GraphPad Prism software and fitted to eq. 2, where [P] is the product concentration at a time t,
| (2) |
A is the amplitude of burst phase, kbuest is the apparent single turnover rate constant, t is the time in seconds, and υ is the steady-state reaction rate.
Single-turnover 8-oxoguanine cleavage kinetics
The hOGG1 concentration dependence of cleavage of 8-oxoG from the °G31 substrate under single-turnover conditions was performed at 37 °C in buffer A. The reactions were performed in 0.2 mL PCR strip tubes. Reactions were manually initiated using a micropipette by rapid addition of hOGG1 (15 µL in buffer A) to an equal volume of 5′-[32P]-labeled °G31 substrate DNA. In reactions involving the addition of APE1, final solutions of the substrate DNA prior to hOGG1 addition contained the necessary concentration of APE1. Reactions (30 µL) were rapidly quenched at various time points (0.5 – 30 s) by vigorous addition of an equal volume of formamide loading buffer from a second hand held pipette (80% formamide, 1x TBE, 0.025% bromophenol blue, 0.025% xylene cyanol). A metronome was utilized to achieve accurate timing for manual initiation and quenching of the reactions as previously described 23. Indeed, the maximal single-turnover rate constant for hOGG1 measured here matches that of previous studies where a Kintek instrument was employed 24. Following quenching, the samples were heated at 95 °C for 20 minutes. The discrete DNA fragments generated by heating were resolved by electrophoresis on a 10% denaturing PAGE gel containing 7 M urea. The gels were dried, exposed to a storage phosphor screen overnight, and imaged with a Typhoon 9500 phosphorimager (GE Healthcare). The amount of reaction product formed was determined by quantifying the gel images using QuantityOne (Bio-Rad). Reaction rates were determined by fitting the data to a first-order equation (eq 3.), where Pt is the concentration of the product
| (3) |
at time t, k is the observed rate constant for single turnover cleavage, and Pmax is the maximal concentration of product at the completion of the reaction. The endpoints of the single-turnover reactions were in the range 80 to 90% and were independent of hOGG1 or APE1 protein concentration.
Kinetics of covalent complex (CC) formation between hOGG1 and substrate DNA
The kinetics for formation of the imino-sugar covalent linkage between hOGG1 and the °G31 substrate was initiated by mixing 25 µL volumes of 20 nM hOGG1 and 40 nM of 32P 5’-labeled °G31 at 37 °C (final concentrations were 10 nM hOGG1 and 20 nM °G31). To evaluate the amount of covalent complex present at each time, 6 µL reaction samples were removed and quenched at specified times by adding an equal volume of 200 mM NaBH4. This treatment serves to reduce the imino linkage between hOGG1 and the abasic sugar (reduction is complete is less than 5 s). Fifteen minutes after addition of NaBH4, an equal volume (12 µL) of SDS loading buffer was added and the samples were set on ice. The samples were loaded directly onto a 10% SDS-polyacrylamide gel, dried, and exposed overnight using a phosphorimaging screen. The relative amounts of covalently bound DNA and free DNA were quantified using QuantityOne. Reactions carried out with the addition of APE1 followed the same procedure with the exception that 100 nM APE1 (10-fold molar excess over hOGG1) was added to the DNA solution just prior to initiating the reaction with hOGG1. Samples from these same reactions were also evaluated for the rate of glycosidic bond cleavage by quenching time points 1:4 with 90% (v/v) formamide (no NaBH4 reduction). Samples were then heated at 95 °C for 30 min, loaded onto a 10% polyacrylamide urea denaturing gel, and subjected to electrophoresis to separate substrate and product DNA. Gels were dried and exposed overnight using a phosphorimaging screen. The relative amounts of unreacted and cleaved DNA were quantified using QuantityOne.
Effect of apoAPE1 on hOGG1 activity
Inactive APE1 was prepared by removal of Mg2+ ions. APE1 was first buffer exchanged into buffer A and then treated with Chelex-100 resin that had been pre-equilibrated in buffer A (25 mg of resin per 200 µl of 1 µM APE1 sample). The enzyme was incubated with the resin for 15 min. on ice with periodic gentle mixing. Reconstitution of APE1 activity was done by buffer exchange of apoAPE1 into buffer B. The activity of apoAPE1 and metal-reconstituted APE1 (reAPE1) were tested in an activity assay that utilized a 5′-[32P]-labeled 31mer DNA substrate containing an abasic site (AP31, see above). In this activity assay, 50 pM of wild-type, apo or reconstituted APE1 were reacted at 37 °C with 50 nM AP31 in buffer B (wild-type and reconstituted APE1) or buffer A (apoAPE1). Kinetic assays and analyses were done as described above for hOGG1. The kinetic data from the linear range of the reactions were fit to eq. 4:
| (4) |
Where P(t) is the concentration of product at time t, and v is the linear rate. Dividing the rate by the enzyme concentration yielded the observed velocity vobs values (eq. 5).
| (5) |
Equilibrium DNA binding measurements using fluorescence anisotropy
Binding of hOGG1, APE1, apoAPE1, TDG and hUNG to DNA containing an AP site (AP31FAM) was performed at 20 °C on a SPEX Fluoromax-3 spectro-fluorimeter (λex = 494 nm, λem = 518 nm). Magic angle conditions and G factor corrections for the polarizers were used (excitation polarizer = 0°, emission polarizer = 55°). The abasic site DNA (AP31FAM) was generated by pre-treating U31FAM with a small amount of hUNG1 (150 pM) for 1 hour. The FAM label was attached to the 5-position of the 3′-terminal thymine base instead of the phosphodiester group to prevent its removal by the phosphodiesterase activity of APE125. All binding reactions were performed using buffer A by following the increase in anisotropy of the FAM fluorophore as increasing amounts of each enzyme were added from a concentrated stock. The concentration of the AP31FAM duplex was 10 nM in all the experiments. After correcting for dilutions during the titrations, the anisotropy values were converted to fraction bound (eq. 6), where fBound represents the fraction bound, A0 is the initial anisotropy of the free ligand, Amax is the anisotropy of the ligand at saturation, and Aobs is the observed anisotropy value.
| (6) |
The data were then plotted as the function of total enzyme concentration and fit to a quadratic binding isotherm (eq. 7), where ET is the total enzyme concentration, LT is the total DNA concentration, and KD is the dissociation constant. The anisotropy changes were not corrected for changes in fluorescence intensity because emission spectra confirmed that none of the enzymes significantly changed the quantum yield of the FAM fluorescence upon binding.
| (7) |
Protein assisted dissociation of the hOGG1 from an abasic site
To determine if APE1, TDG, or hUNG facilitated dissociation of hOGG1 from the abasic product of its reaction we designed a chase-trapping experiment. This experiment is based on the idea that a protein chase that facilitates hOGG1 dissociation should reduce the amount of hOGG1 that can be trapped on abasic DNA in the form of a reduced Schiff base linkage after quenching with NaBH4. For these experiments, abasic DNA (AP31) was prepared by digestion of 220 nM 5′-[32P] radiolabeled U31 DNA with 400 pM UNG for 1 hour at 37 °C in buffer A. After conversion to AP31, the solution was supplemented with potassium acetate and MgCl2 to a final concentration of 150 mM to a 1 mM, respectively. The hOGG1 reversible Schiff base covalent complex (CC) was prepared by combining 50 nM of 5′-[32P] AP31 with 50 nM hOGG1 and incubating for 5 minutes at 37 °C (25 µLtotal volume). Following pre-equilibration with hOGG1, 25 µL of chase protein (APE1, TDG, or hUNG) was added to a final concentration of 200 nM. Time points were then taken and quenched at fixed intervals by vigorously mixing an aliquot of the reaction (5 µL) with an equal volume of 50 mM NaBH4. The reducing agent serves to irreversibly trap hOGG1 on the DNA in the form of a reduced Schiff base linkage16. Control experiments established that NaBH4 completely reduces the hOGG1 linkage with DNA in less than 5 s. Two minutes after addition of the NaBH4 quench, 10 µl of SDS-PAGE loading buffer was added. The reaction products were run on a 10 % SDS-PAGE gel for 50 minutes at 150 V. The gels were dried, exposed to a storage phosphor screen overnight, and imaged with a Typhoon 9500 phosphorimager (GE Healthcare). The amount of covalent complex in each lane of the gel was determined by quantifying the band intensities using QuantityOne. The fraction of total band counts trapped in the form of the covalent complex was plotted as a function of time after addition of each protein chase relative to the amount trapped by NaBH4 at time zero.
Glutaraldehyde crosslinking
Freezer stocks of hOGG1 and APE1 were exchanged into buffer B (but omitting BSA) and then concentrated to 5 µM and 20 µM for APE1 and hOGG1, respectively. To test for intramolecular self-interactions between the individual proteins, crosslinking experiments were performed over a range of protein concentrations between 25 to 500 nM as follows. First, freshly buffer exchanged APE1 and hOGG1 concentrations were determined using UV absorbance at 280 nm and dilutions were performed to give 10x concentrated stock solutions of each enzyme in buffer B (no BSA), which were left at room temperature to equilibrate for 5 min. After dilution to give concentrations in the range 25 nM to 500 nM protein (50 µL total volume), crosslinking was initiated by the addition of 1 µL of 2.5 M glutaraldehyde followed by incubation at room temperature for 20 min. The reactions were quenched by addition of 10 µL 1 M Tris-HCl pH 7.5, followed by addition of 15 µL of 5x SDS loading buffer. Samples are then run on pre-cast 4–12% SDS bis-trycine denaturing gel in 1x MES-SDS buffer for 90 min. at 150 V. The gel was silver stained for protein detection as follows. The gel was removed from the gel cassette and rinsed in deionized water and then placed into the fixing solution (50% methanol 12% acetic acid 0.05% formalin) and incubated overnight in the dark with gentle shaking. The next day the gel was washed with 60 mL of 36.5% (v/v) ethanol for 20 min. followed by a 2 min. wash with 100 mL of sensitizing agent (0.02% Na2S2O3). The gel was washed two times in deionized water for one minute each and then silver stained with pre-chilled staining solution (0.2% AgNO3, 0.05% formalin) for 20 min. followed by two deionized water washes (one minute each). The gel was then developed for three minutes at room temperature in developing solution (6% Na2CO3, 0.05% formalin 0.008% Na2S2O3), quickly rinsed with deionized water and staining was halted with stop solution (50% methanol, 12% acetic acid). The gels then were imaged using Bio-Rad Gel Doc system with white light transillumination. To test for intermolecular interactions between hOGG1 and APE1, crosslinking and silver staining was performed in an identical fashion except that a fixed concentration of hOGG1 was used (100 nM, where it shows no self-association) and varied concentrations of APE1 (see Results Fig. 6 and Supplementary Information Fig. 4S.).
Figure 6. Detection of interactions between free and DNA bound hOGG1 and APE1 via gluteraldehide crosslinking.

(A) hOGG1 and APE1 do not strongly interact in the free form. Physiologically relevant concentration of hOGG1 (50 nM) and APE1 (50 – 200 nM) were pre-incubated for 5 min followed by addition of the gluteraldehyde crosslinking agent (50 mM) and a further 20 min incubation. The samples then were resolved using SDS-PAGE with detection by silver staining. No bands corresponding to an APE1-hOGG1 complex were detected. (B) Presence of AP-DNA does not facilitate APE1-hOGG1 interaction. AP-DNA (100 nM) was pre-incubated with hOGG1 (100 nM) for 5 min to form a AP-DNA/hOGG1 complex, followed by addition of APE1 (100 nM). After a 1 min incubation, crosslinking with 50 mM glutaraldehyde was performed for 20 min. The samples were resolved using SDS PAGE silver staining detection. No bands corresponding to APE1-hOGG1 or APE1-hOGG1-APDNA complexes were detected. However, presence of AP-DNA significantly facilitated self-crosslinking of APE1 in the absence of hOGG1. (C) APE1 does not form a stable complex with a reduced hOGG1-APDNA complex. In this experiment designed to test whether APE1 can interact with a highly stable hOGG1-APDNA complex, 100 nM hOGG1 was incubated for 5 min with 100 nM of AP-DNA which was labeled with 32P on the 5’ end to allow formation of the Schiff base intermediate. The intermediate was reduced by the addition of 25 mM NaBH4. The reduced complex was then incubated for 1 min with 100 nM APE1 followed by 20 min crosslinking in the presence of 50 mM gluteraldehyde. The samples were resolved on a 10% native polyacrylamide gel with detection by phosphorimaging the 32P radiolabel. Although a stable hOGG1-APDNA complex was readily detected, no unique bands corresponding to APE1-hOGG1-APDNA complex were observed.
To test for the presence of DNA facilitated protein-protein interactions between hOGG1 and APE1, we performed crosslinking in the presence of AP31 DNA (prepared as described above). We first incubated 110 nM of AP31 with 110 nM of hOGG1 for 5 min. (45 µL reaction volume) before addition of 5 µL of 500 nM APE1 (final concentration of 100 nM). After one minute we added 1 µL of 2.5 M glutaraldehyde and the remaining steps are the same as outlined above for the free proteins. In this experiment, DNA-facilitated protein complexes were detected by silver staining for protein using denaturing gel electrophoresis. We also investigated whether DNA facilitated protein complexes could be detected using 5′-[32P]-labeled AP31 DNA (prepared as described above) under native gel conditions. In this approach we preformed the hOGG1-AP31 DNA complex (125 nM) by incubating for 5 min. at room temperature (40 µL volume) in 60 mM K2PO4 pH 7.0. We then stabilized the hOGG1/AP31 complex by reduction using 25 mM NaBH4 for 5 minutes at room temperature. It is important that the solution of NaBH4 did not significantly alter the pH or ionic strength of the reaction solution as not to inactivate the enzymes. Thus we prepared 250 mM NaBH4 stock in 60 mM K2PO4 (pH 7.0 before addition of NaBH4) and added 4.4 µL of this solution to 40 µL of hOGG1/AP31 giving final ionic strength of 160 mM and pH 7.6. After reduction, 5 µL of 1000 nM APE1 was added giving 50 µL reaction volume with 100 nM concentrations of the reduced hOGG1-APDNA complex and APE1. The final ionic strength and pH was 160 mM and pH 7.6, respectively. This mixture was incubated for one minute at room temperature and then 1 µL of 2.5 M glutaraldehyde was added followed by another 20 minutes of incubation at room temperature to allow protein crosslinking. The reaction was quenched by addition of 10 µL of 1 M Tris-HCl pH 7.5. 15 µL of 5x native loading buffer was added to the reaction (375 mM Tris-HCl pH 6.8, 50% glycerol, 0.05% each bromophenol blue and xylene cyanol). Samples were run on a native gel using a standard Tris-Glycine running buffer at 150 V for 1h. The gel contained a 4% stacking gel, a 10% running gel, and a 20% stopping gel (to retard free radiolabeled DNA from running off the gel). Gel was then subjected to standard silver staining procedure followed by phosphor imaging to detect the radiolabeled DNA.
RESULTS
APE1 enhances the turnover rate of hOGG1
We examined the APE1 concentration dependence of the hOGG1 glycosylase activity using pre-steady state kinetic conditions that allowed the observation of a burst of product formation in the first-turnover as well as subsequent slower rounds of steady-state turnover of hOGG1 (Fig. 2A). In the absence of APE1, 25 nM hOGG1 reacting with 500 nM °G31 duplex showed a single-turnover burst amplitude that approximated one equivalent of enzyme (A = 0.88, kburst = 0.025 s−1), followed by a very slow steady-state rate (ν/[hOGG1] = 0.0012 s−1, see Table 1). This behavior is indicative of a rapid kinetic phase leading to one enzyme equivalent of product, followed by a slower step that results in subsequent turnovers. As supported by the experiments described below, we assigned the fast step to glycosidic bond cleavage and the slow step to abasic product release by hOGG1. The burst and linear steady state rate behavior was also observed using hOGG1 concentrations in the range 25 nM to 125 nM (Table S1, Fig. S1A).
Figure 2. Effect of APE1 on hOGG1 turnover.

(A) APE1 concentration dependence of hOGG1 activity under multiple turnover conditions. The ratios of [APE1]/[hOGG1] are indicated above the representative gel images. The concentrations of hOGG1 and °G31 were 25 nM and 500 nM, respectively. All experiments where APE1 was present were carried out in buffer B, experiments in the absence of APE1 were carried out in buffer A. Control experiments shown in Fig. S1D confirmed that hOGG1 alone has the same steady-state kinetic activity in buffer A and B. (B) Effect of APE1 on the single turnover cleavage of °G31 by hOGG1. Reactions (buffer B) contained 10 nM °G31 and 50 nM hOGG1 in the absence and presence of 200 nM APE1 and were carried out in triplicate. The single-turnover cleavage rate was fit to a single exponential and was independent of hOGG1 concentration in the range 50 to 400 nM (see Fig. S2). (C) Kinetics of glycosidic bond cleavage and formation of the imino-sugar covalent linkage between 20 nM hOGG1 and 40 nM of 32P 5’-labeled °G31 at 37 °C (final concentrations were 10 nM hOGG1 and 20 nM °G31). Reactions were carried out in the absence and presence of 100 nM APE1, which was added to the DNA solution just prior to initiating the reaction with hOGG1. The covalent hOGG1-DNA complex was trapped by reduction with sodium borohydride and quantified by phosphorimaging after electrophoresis through an SDS-polyacrylamide gel. The amount of cleaved product DNA in the same reactions was determined by quenching time points in formamide in the absence of NaBH4 reduction followed by electrophoresis through a 10% polyacrylamide urea denaturing gel to separate substrate and product DNA. (D) hOGG1 has low lyase activity in the presence and absence of magnesium in buffer A. Two separate reactions were performed in which 200 nM hOGG1 was incubated at 37 °C with 50 nM °G15 DNA (5’-labeled with γ32P). One reaction was performed in the presence of buffer A, and the other was performed in the presence of buffer A with 1 mM MgCl2 substituted for EDTA. Time points were taken at 0, 5, and 30 minutes by mixing 5 µL aliquots of the reaction with 20 µL formamide load buffer containing 90% formamide, 1X TBE, bromophenol blue, and xylene cyanol. In order to maintain the integrity of the intact abasic and lyase cleaved products, there was no heating step prior to running the samples on a gel. Each time point was loaded onto a denaturing 20% polyacrylamide gel (7 M Urea) that was run for 1.5 hours at 15 W, exposed to phosphor screen, and imaged on a Typhoon 9500 imager. In the samples that were not heated, hOGG1 forms the stable covalent complex (CC) with the DNA in the presence and absence of magnesium and a product containing an intact abasic site (AP15) that runs slightly lower on the gel than the unreacted 15mer substrate (°G15). There is a low abundance band that migrates at the expected size of the β elimination product generated from the hOGG1 lyase activity (LyP). This band represents ~1% of the total signal from each lane.
Table 1.
Concentration dependence of APE1 stimulation of hOGG1 activity
| Substrate |
|
ν/[hOGG1] (s−1) |
kburstb (s−1) |
kmaxc (s−1) |
|
|---|---|---|---|---|---|
| °G31 | - | 0.0012 ± 0.0002 | 0.025 ± 0.01 | 0.37 ± 0.09 | |
| °G31 | 0.25 | 0.006 ± 0.001 | d | ||
| °G31 | 0.5 | 0.008 ± 0.001 | d | ||
| °G31 | 1 | 0.011 ± 0.002 | d | ||
| °G31 | 2 | 0.013 ± 0.0005 | 0.025 ± 0.016 | ||
| °G31 | 4 | 0.011 ± 0.002 | d | ||
| °G31 | 10 | 0.011 ± 0.002 | d | ||
| °G15 | - | 0.0036 ± 0.0003 | 0.01 ± 0.001 | 0.25 ± 0.02e | |
| °G15 | 10 | 0.011 ± 0.0003 | 0.01 ± 0.004 |
The concentrations used were 25 nM hOGG1 and 500 nM °G31; concentrations of APE1 were varied between 6 nM to 250 nM.
The values reported are based of triplicate measurements for the indicated condition.
Average rate constant for single-turnover cleavage of the °G31 or °G15 substrates (10 nM) using concentrations of hOGG1 in the range 50 to 200 nM.
The kburst value was fixed at the optimized value of 0.025 s−1 to obtain more robust fitted values for ν/[hOGG1]. This approach was justified because kburst becomes more difficult to estimate as the burst amplitude decreases with APE1 concentration. The reported errors in the fitted values for ν/[hOGG1] were taken as the maximum errors (~17%) of the measurements that were made in triplicate.
The standard deviation of two replicate measurements.
Upon addition of 6 to 250 nM APE1 to a reaction containing a fixed concentration of 25 nM hOGG1 and 500 nM °G31, a concentration dependent increase in the steady-state rate was observed (Fig. 2A). The maximal rate increase was about 10-fold (ν/[hOGG1] = 0.013 s−1, Table 1), which was reached at a ratio of [APE1]/[hOGG1] equal to one. At [APE1]/[hOGG1] ratios lower than unity, the rate acceleration decreased, approaching that of hOGG1 alone. The finding that limiting amounts of APE1 can increase the turnover of hOGG1 suggests that APE1 rapidly recycles between multiple hOGG1-product complexes and accelerates their dissociation. This conclusion that APE1 acts catalytically at the product release step of the hOGG1 reaction is supported by the observation that APE1 has no effect on the rate constant for single-turnover cleavage of °G31 by hOGG1 (kmax = 0.37 ± 0.09 s−1, Table 1) (Fig. 2B and Fig. S2).
To further investigate the mechanistic basis for the rate stimulation by APE1, we monitored the rate of formation of the covalent Schiff base linkage between Lys249 of hOGG1 (10 nM) and the abasic sugar derived from substrate °G31 (20 nM) (Fig. 2C). This was achieved by quenching reaction samples in sodium borohydride, which reduces and stabilizes the linkage10. In the absence of APE1, the covalent complex (CC) appeared rapidly in the first 20 s reaching a peak concentration equal to about 30% of the total substrate concentration (Fig. 2C, black squares). The CC then decayed slowly by about 20% of its peak level over the next 300 s, which corresponded closely with the rate of steady-state production of the cleaved DNA product (black circles, Fig. 2C). This behavior is consistent with rapid glycosidic bond cleavage and CC formation, followed by rate-limiting product release. In contrast in the presence of 100 nM APE1, the CC accumulated to only 20% of the total substrate concentration and decayed completely over the next 300 s (red squares, Fig. 2C). This rapid decay of the CC in the presence of APE1 also correlated with the rapid appearance of the cleaved product (red circles, Fig. 2C). These findings strongly support the conclusion that APE1 provides stimulation by acting at a step after glycosidic bond cleavage and CC formation.
Metal ions are required for APE1 stimulation of hOGG1
Previous studies have suggested that metal free APE1 (apoAPE1) was capable of efficient stimulation of hOGG1 turnover and that the high binding affinity of APE1 for the abasic site of the hOGG1 reaction was sufficient to relieve severe product inhibition13. We investigated this possibility by stringent removal of divalent cations from APE1 using dialysis and metal chelating resin to generate metal free apoAPE1. In contrast with the previous report, this procedure completely ablated the activation that we observed with active APE1 (Fig. 3A; for comparison, the dashed lines show the theoretical curves for hOGG1 activity in the absence and presence of APE1). The lack of stimulatory activity by apoAPE1 cannot arise from non-specific irreversible inactivation of APE1 during metal removal, because full activity was restored after adding back the metal (Fig. S1C). The previous findings likely resulted from incomplete removal of enzyme bound metals through the addition of EDTA rather than treatment with chelation resin. Consistent with this explanation, we found that residual APE1 nuclease activity and hOGG1 stimulation persisted upon addition of 5 mM EDTA to a metal free reaction buffer. The mechanistic basis for the lack of stimulation by apoAPE1 could arise from weak AP-site binding activity (see below) and/or its lack of catalytic activity.
Figure 3. APE1 stimulation of hOGG1 turnover requires metal ions but not the unstructured amino terminus of APE1 or flanking DNA adjacent to AP site.

(A) Stripping metal ions from APE1 (apoAPE1) results in complete loss of hOGG1 stimulation. Dashed lines show the theoretical curves in the absence of APE1 or using one equivalent of wild-type APE1. The experiments were carried out in buffer A in triplicate and standard errors are plotted (error bars are smaller than simbol size). The experiments used 500 nM °G31 substrate, 25 nM hOGG1 and a 4-molar excess of apoAPE1. Representative gel images for the assay are shown at the top of the panel. (B) The Δ49APE1 deletion mutant enhances hOGG1 activity under multiple turnover conditions nearly as well as the full-length enzyme. Dashed lines show the theoretical curves for the same experiment in the absence of APE1 or using one equivalent of wild-type APE1 (see Fig. 2A). The experiments were done in buffer B and used 500 nM °G31 substrate, 25 nM hOGG1 and a 1, 4 or 10 hOGG1 molar equivalent of Δ49APE1 (experiments were done in duplicate and standard errors plotted). (C) APE1 stimulates hOGG1 turnover using the °G15 substrate that contains no flanking DNA when hOGG1 is bound to the AP product site. The experiments were done in buffer B and used 500 nM °G15 substrate, 25 nM hOGG1 and a 1, 4 and 10-molar equivalents of APE1 relative to hOGG1. Experiments were performed in triplicate and standard errors are plotted.
N-terminal tail of APE1 is not required for hOGG1 stimulation
APE1 has an unstructured amino terminal tail that is not required for catalytic activity12, but could be involved in transient protein-protein or DNA interactions that stimulate the release of hOGG1 from its product DNA. To evaluate this possible function for the tail, we used a mutant APE1 where the first 49 N-terminal amino acids were deleted (Δ49APE1). We confirmed that Δ49APE1 and the full-length enzyme displayed similar AP-endonuclease activities. We also found that Δ49APE1 was capable of stimulating hOGG1 activity by nearly the same amount as APE1, even when we used the minimal stoichiometric amount of Δ49APE1 relative to hOGG1 (Fig. 3B, Fig. S3; the dashed lines show the theoretical data in the presence and absence of APE1 for comparison purposes). These findings exclude a significant role for the first 49 amino acid residues of APE1 in the stimulation of hOGG1 turnover.
Flanking DNA is not required for APE1 stimulation
We explored whether the presence of flanking DNA on either side of the 8-oxoG site had any effect on the observed stimulation provided by APE1 (Fig. 3C). For this purpose, we performed kinetic experiments with hOGG1 in the presence and absence of APE1, but utilized a substrate where the 8-oxoG was located at the center of a shorter 15bp duplex (°G15). Based on crystal structures of hOGG1 bound to DNA, a short 15bp sequence would leave no room for APE1 to bind adjacent to hOGG115,26. The baseline steady-state rate of hOGG1 with °G15 was 3.5 fold faster than °G31, even though the single-turnover rate for °G15 was slightly slower (0.25 ± 0.02 s−1)(Table 1). The faster steady-state rate may arise from a faster off-rate from the AP-site product in this short duplex context. Nevertheless, upon addition of a 10-fold excess of APE1 to a reaction of hOGG1 with °G15, a 2.5-fold increase in the steady-state rate was observed, producing a kinetic curve that closely overlays that observed with °G31 (compare Fig. 3C with upper dashed curve in Fig. 3B). Thus, the reduced stimulation by APE1 using the 15mer duplex arises from a faster baseline rate rather than a reduced ability of APE1 to facilitate turnover on this substrate. It is significant that the maximal rate increase with °G15 also could be achieved with 1:1 ratio of hOGG1/DNA, just as observed with °G31. These findings indicate that flanking DNA is not required for maximal stimulation of hOGG1 activity by APE1 and that simple diffusional encounter with the hOGG1-product complex is sufficient.
Can proteins that bind competitively to AP-sites stimulate hOGG1 activity?
One of the simplest models for stimulation of hOGG1 involves competitive binding of a protein to the AP site, thereby alleviating severe product inhibition (Fig. 1E). To test this possibility, we used fluorescence anisotropy measurements to assess the binding affinity of thymine DNA glycosylase (TDG), human uracil DNA glycosylase (hUNG) and apoAPE1 for the AP product site of the hOGG1 reaction (Fig. 4). These binding affinity measurements can then be correlated with the abilities of these various proteins to stimulate hOGG1 activity (see below). This experiment first involves the generation of a 5′ FAM-labeled DNA duplex with a single AP-site opposite to a cytosine (AP31FAM, Supplemental Information). This was accomplished by treating a DNA duplex that contained a single U/C mismatch (U31FAM) with hUNG (see Methods). We found that the affinities of these enzymes for the AP site varied by almost four orders of magnitude using our standard buffer A. The enzymes with the highest affinity for the AP site were TDG and hOGG1 with KDAP values of 12 and 20 nM, respectively (Table 2). In contrast, apoAPE1 and hUNG had much weaker affinities (KDAP values of ~1 and 50 µM, respectively). We could not evaluate KDAP for APE1 in the presence of metal ions due to its enzymatic activity, but previous kinetic trapping experiments have suggested its AP-site affinity to be in the low nanomolar range27. Our kinetic simulations of the stimulatory effects of APE1 on hOGG1 activity also require a low nanomolar value for KDAP (see Discussion). These thermodynamic findings suggest that TDG would be the strongest competitive binder to AP-sites and non-specific DNA.
Figure 4. Equilibrium binding affinities of APE1, apoAPE1 hOGG1, TDG, and hUNG to AP-DNA.

Binding data for TDG (black), hOGG1 (red), apoAPE1 (green), and hUNG (purple) for the AP site DNA (AP31FAM; 10 nM) were obtained by fluorescence anisotropy measurements using buffer A at 20°C. Errors were estimated by performing three independent replicates of the titrations. In the case of hUNG, which showed very weak binding to the AP site, the saturating anisotropy value was fixed at a value previously observed in titrations using other salt concentrations where saturation could be achieved20. This value was consistent with the extrapolated value predicted from the fit to the data set. The uncertainty in the weak KD value for hUNG has no impact on any of our interpretations.
Table 2.
Binding affinities of APE1 and DNA glycosylases for an AP-site in DNA
AP31FAM
Includes covalent binding.
Not determined due to catalytic activity with AP sites.
We then explored whether there was a correlation between the binding affinity of a protein to an AP-site and its ability to stimulate hOGG1 release from an AP-site (Fig. 5A). As above with AP31FAM, a 5′ 32P-labeled DNA duplex with a single AP-site opposite to a cytosine (AP31) was generated by hUNG catalyzed uracil excision. The resulting AP-DNA (50 nM) was incubated with hOGG1 (50 nM) for 5 min. to form an equilibrium mixture of non-covalently bound DNA (NC) and the Schiff base covalent complex (CC). Next, a 4-fold molar excess (200 nM) of each AP-site binding enzyme was added. The reactions were quenched at various times by the addition of sodium borohydride to reduce the Schiff base linkage (RCC, Fig. 5A). Thus, if an enzyme accelerated the departure of hOGG1 from the AP-site, less RCC would be detected at each time point after separation of the RCC from free DNA using SDS-PAGE (Fig. 5B). Of the four enzymes tested using this approach, only APE1 gave a rapid and complete time-dependent decrease in the amount of RCC detected (Fig. 5C). TDG provided a ~30% reduction in the RCC at the first time point which remained essentially constant over time (Fig. 5C). This level of decrease is consistent with the high-affinity of TDG for AP-sites and is consistent with an equilibrium being established between TDG and hOGG1 binding to the AP-site prior to addition of the sodium borohydride. We note that pure equilibrium competitive binding by TDG cannot accelerate the process of rate-limiting product release, but can only prevent product inhibition of the hOGG1 reaction (see below).
Figure 5. Effects of AP site binding enzymes on dissociation of hOGG1 from an AP site.

(A) The experimental strategy was to incubate hOGG1 with AP-DNA until a reversible equilibrium was established between the covalent Schiff base complex (CC) and the noncovalent complex (NCC). A chase enzyme (APE1, TDG, hUNG, or no enzyme) was then added and after an aging time, NaBH4 was added to reduce the imino linkage between the DNA and Lys249 of hOGG1 (RCC). The chase enzymes were each present at a 200 nM concentration. (B) The reaction products as a function of aging time. The RCC and free DNA species were resolved by SDS-PAGE. Representative gel images for no chase and each chase enzyme are shown. (C) Disappearance of the RCC over 15 minutes using various chase enzymes. (D) TDG modestly stimulates hOGG1 turnover. A 4-fold molar excess of TDG over hOGG1 was not significantly stimulatory (blue), while a 40-fold molar excess (1000 nM) increased the turnover rate by about 2.5 fold (green). The dashed lines are the theoretical curves for reactions with hOGG1 only and in the presence of one equivalent of APE1 (see Fig. 2A). The concentrations of °G31 substrate and hOGG1 were 500 and 25 nM respectively. Experiments were carried out in triplicates and standard errors were plotted.
TDG also stimulated the activity of hOGG1 in a pre-steady state kinetic assay format, but the stimulation was much weaker than with APE1 (Fig. 5D). In the kinetic assay, one equivalent of TDG was not stimulatory and a 4-fold or 40-fold molar excess of TDG produced only 1.5 and 2.5-fold stimulations. These fold stimulations—and the enzyme concentrations required to achieve them—pale in comparison to the stimulation provided by one-equivalent of APE1 (upper dashed line, Fig. 5D). As expected, the turnover of hOGG1 was not significantly stimulated by inclusion of up to a 40-fold molar excess hUNG in the reaction (Supplemental Table S2 and Fig. S1B). This result is consistent with the weak binding affinity of hUNG to AP-sites (Fig. 4) and its inability to promote release of hOGG1 from AP sites (Fig. 5B). Based on these findings, we conclude that competitive binding of a protein to an AP-site is an inefficient activation mechanism because it requires high concentrations of the competitor to saturate the sites. The observation that TDG accelerates steady-state turnover and hOGG1 dissociation from the AP site (see above) suggests that it must either actively promote hOGG1 dissociation (a formal possibility), or that it can intercept the site as hOGG1 takes microscopic excursions (see Discussion).
APE1 and hOGG1 do not interact in their free or DNA bound forms
The observation that APE1 stimulates hOGG1 turnover even when both proteins are present at low nanomolar concentrations suggested the possibility of significant protein-protein interactions between these enzymes (Fig. 2A). Such interactions could occur between the free proteins (Fig. 1A), or alternatively, be facilitated by DNA interactions (Fig. 1B). To test these possibilities, we used a glutaraldehyde crosslinking method28. In preliminary experiments we first determined the concentrations of the individual proteins where self-crosslinking was observed (Fig. S4). We found that APE1 formed crosslinked dimers and higher order structures when its concentration exceeded 100 nM (a concentration much higher than the nanomolar concentrations needed for activation) and that hOGG1 showed a lesser tendency to self-crosslink. We then tested for the formation of hetero-crosslinked complexes using concentrations of the two proteins that minimized self-crosslinking. The hOGG1 concentration was fixed at 50 nM and increasing concentrations of APE1 were added in the range 50 to 200 nM (Fig. 6A). The crosslinked complexes were separated by SDS-PAGE and then detected by silver staining. Although we easily detected APE1 self-crosslinking at higher concentrations of APE1, there was no detectable crosslinking between the two proteins. We performed the same experiment (using 100 nM APE1 and 100 nM hOGG1) in the presence of 100 nM AP31 DNA and also detected no hetero-crosslinked complexes, although the presence of DNA increased the self-crosslinking of APE1 (Fig. 6B)
To further confirm that APE1-hOGG1 interactions are not promoted by the presence of DNA, we used an alternative approach where the hOGG1 complex with AP31 was first stabilized by reduction with sodium borohydride. We surmised that stabilizing the hOGG1 complex would allow the maximum opportunity to detect a DNA dependent interaction with APE1. We developed a very mild reduction condition for this purpose where the pH was maintained at ≈7.5 and only 25 mM sodium borohydride was used to minimally perturb the ionic strength and pH. Using these conditions 80% of the 5′32P-labeled AP31 DNA was trapped in a covalent complex with hOGG1 after separation of the free DNA and the covalent complex using electrophoresis through a native Tris-Glycine gel (Fig. 6C). There was no change in the amount of the covalent complex in the presence and absence of glutaraldehyde, indicating that self-crosslinking of hOGG1 is undetectable under these conditions. APE1 alone did not form any stable complexes with the DNA under the same conditions, which is not surprising because the enzyme has robust AP-endonuclease activity that led to almost instantaneous processing of the AP-DNA (Fig. 6C). Finally, the addition of APE1 to the hOGG1/AP-31 covalent complex did not result in any higher molecular weight bands that would indicate a hetero-protein complex. We note that it is possible in the above experimental design for residual NaBH4 to reduce the glutaraldehyde crosslinking reagent, thereby preventing the detection of a hetero-protein complex. However, control experiments established that when the reducing equivalents of NaBH4 were much less than the glutaraldehyde concentration, identical results were obtained (i.e. no hetero-protein complex detection) (Fig. S5).
DISCUSSION
APE1 has been implicated in the stimulation of catalytic turnover of many human DNA repair glycosylases including uracil DNA glycosylase (hUNG2)29, thymine DNA glycosylase (TDG)13,30,31, alkyl adenine DNA glycosylase (AAG)2,12, mutY and MYH19,32,33, and hOGG16,14,16,17. The experimental findings have been interpreted in terms of two general mechanisms usually referred to as “passive”13,30 and “active”16 although the exact definition of these two terms is not always rigorously defined. Here we define passive to mean that APE1 does not form a specific interaction with hOGG1 or DNA that leads to an enhancement of the rate-limiting step (or unfavorable equilibrium) leading to irreversible departure of hOGG1 from its product AP site. Accordingly, active means that APE1 forms a specific interaction with hOGG1 or DNA that leads to an enhancement of the rate-limiting step (or unfavorable equilibrium). It should not be underestimated how difficult it is to definitively discern between these two limiting possibilities because transient interactions that exist on the millisecond or smaller timescale are exceedingly difficult to experimentally detect. The first four mechanisms shown in Fig. 1A-1D could be passive or active, while the equilibrium binding model (Fig. 1E) is passive because it only involves abatement of product inhibition (the equilibrium model has limitations on the rate accelerations that it can provide, see below).
APE1 stimulation of hOGG1 does not require a stable ternary complex and requires APE1 endonuclease activity
We have performed experiments that indicate a pathway for APE1 stimulation involving kinetic trapping (Fig. 1C and 1D). The alternative pathways involving complexation of hOGG1 and APE1 are deemed minor or non-existent based on our inability to detect any heterocomplexes of these proteins by crosslinking, even though self-association of both proteins was easily detected (Fig. 6). The AP-site competitive binding model (Fig. 1E) is also not viable because catalytic levels of APE1 are strongly stimulatory (Fig. 2A), acceleration occurs at the earliest extent of reaction and the rate remains linear after product accumulates to a significant extent (Fig. 2A). By stringent removal of metal ions from APE1, we established that AP endonuclease activity is required for the observed stimulation (Fig. 3A) and that the weak AP-site affinity of apoAPE1 is not sufficient to provide stimulation by the alternative equilibrium binding mechanism (Fig. 4, Fig. 5). Although active APE1 is reported to bind AP-sites with low nanomolar affinity27, this mechanism alone would be poorly stimulatory because tight binding means slow off-rates and inefficient recycling of APE1 to stalled hOGG1-AP site complexes. In this regard, we view the endonuclease activity of APE1 as the key mechanistic feature that promotes efficient stimulation even at sub-stoichiometric levels of APE1.
A passive mechanism is kinetically competent to account for the stimulation by APE1 and TDG
In addition to the strong stimulation provided by APE1, we observed that high concentrations of the strong AP-site binder TDG gave rise to a modest acceleration of hOGG1 turnover (Figs. 4 and 5). Although the stimulation by TDG could also follow an active or passive mechanism, an active mechanism seems unlikely given that there is no obvious biological rationale for the interaction of hOGG1 and TDG in the cell nucleus. A simple equilibrium mechanism for TDG stimulation is supported by (i) the time independence of the observed effect (Fig. 5B, 5C), and (ii) the correspondence between the equilibrium binding affinities of hOGG1 and TDG for an AP-site and the level of stimulation (Figs. 4 and 5).
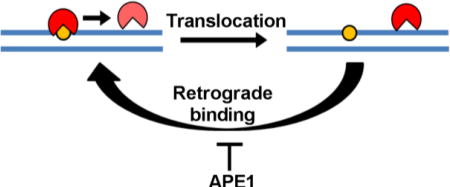
A purely passive stimulation mechanism for both APE1 endonuclease and TDG is apparently not consistent with the kinetic principle that the rate-limiting step leading to the irreversible departure of an enzyme from its product (defined by koff = kcat in this case) cannot be changed by binding of another protein to the site after the first enzyme’s departure. This apparent paradox for the passive mechanism can be resolved if the polymeric nature of DNA is considered along with the microscopic dynamic translocation behavior of these enzymes when interacting with specific and nonspecific DNA chains34,35 In this regard, the macroscopic off rate from a site is determined by the rate of irreversible departure of the enzyme from the site, defined as its equilibration with all other substrate or product molecules in bulk solution. For an enzyme that interacts with DNA, the macroscopic rate also contains the rapid microscopic excursions that an enzyme makes away from a specific site before it irreversibly departs. This in essence sets up a rapid but unfavorable equilibrium that may be heavily biased towards the enzyme rebinding the site rather than departing to bulk solution (“retrograde” AP-site binding; Fig. 7)34. These dynamic excursions of hOGG1 away from the AP-site provide a transient opportunity for APE1 or TDG to invade the site. Invasion by APE1 does not appear to require large segments of flanking DNA because hOGG1 could be efficiently displaced from a short 15mer duplex (Fig. 3C). Once occupying the site, the rapid AP-endonuclease activity of APE1 allows it to irreversibly convert the site to an inert form that allows unimpeded turnover of both enzymes. The viability of a passive mechanism was confirmed using numeric simulations of the data in Fig. 2A (Fig. S6). A similar mechanism has been proposed for activation of AAG by APE1, but these previous studies used excess amounts of apoAPE1 relative to the amount of DNA and AAG that were present12 Our work extends the viability of this mechanism to conditions where catalytic amounts of active APE1 are used.
Figure 7. Model for APE1 stimulation of hOGG1 turnover.

The data are consistent with highly efficient retrograde binding of hOGG1 to the AP product site and its inefficient escape to bulk solution where it can encounter a new substratre molecule. In the presence of sufficient APE1, the AP site is rapidly bound and the DNA phosphate backbone is incised preventing the retrograde binding of hOGG1. The mechanism does not require interactions between the two proteins.
Why can’t APE1 fully stimulate hOGG1 activity?
One subtle aspect of our findings is that addition of APE1, although clearly stimulatory to hOGG1 action, cannot restore the rate to the level seen under single-turnover conditions (Table 1). In addition, the burst phase seen in the pre-steady state experiments is over 10-fold slower than the single-turnover rate constant, suggesting that there is a different rate-limiting step for glycosidic bond cleavage for the two measurements. Such discrepancies between experiments that employ excess enzyme over DNA and the reverse condition of excess DNA have been observed previously with hOGG1 14 and other enzymes 36. The most likely explanation is that with excess DNA binding sites the target search time has become rate-limiting, which is supported by the observation that the pre-steady state burst rate gradually increases as the hOGG1 concentration increases (Fig. S1B, Table S1). Further, our previous studies have indicated that the target search time of hOGG1 can be adversely affected by the presence of nonspecific DNA sequences20. Thus, we infer that a slow step after the formation of the enzyme-DNA complex and before glycosidic bond cleavage is rate-limiting in the first turnover in presence of excess DNA, but in the presence of excess enzyme, the chemical step is rate-limiting. Thus under such conditions, APE1 can only increase the rate of multiple turnovers up to the value of the target search step.
Comparison with APE1 effects on other DNA glycosylase activities
The mechanism for APE1 stimulation of hOGG1 turnover shares some aspects reported in detailed studies of both TDG and AAG12,13,30. Like hOGG1, catalytic sub-stoichiometric levels of APE1 endonuclease were strongly stimulatory for TDG13. Strong APE1 activation of AAG was observed in the absence of metal ions12,18, but high concentrations of APE1 were required to achieve this effect (i.e. exceeding the DNA concentration). Thus, for AAG a non-catalytic AP-site binding mechanism for APE1 was followed that parallels what we observed here using TDG. Consistent with the absence of specific protein-protein interactions between AAG and APE1, a similar AAG activity enhancement was observed when a bacterial endonuclease was used instead of APE1 (EndoIII). Further, for AAG activation by APE1, there was no dependence on whether 3′ or 5′ flanking DNA surrounded the AAG site, suggesting that directional approach by APE1 is not important. However unlike our results with hOGG1, stimulation of AAG by APE1 was less effective when the length of flanking DNA was reduced18. Taken together these studies are broadly consistent with a mechanism that does not involve specific protein-protein interactions. If an active mechanism is involved, it is more likely to involve a disruption in the DNA structure that leads to release of the glycosylase from the site.
Implications
The turnover rate of DNA glycosylases in the cell nucleus is an important question. Although these enzymes are quite plentiful in the nucleus (~105 molecules) and exceed the steady-state load of normal DNA damage, if they are irreversibly bound to AP-product sites or otherwise immobilized in sequestered interactions, the DNA repair capacity of the cell is reduced. An enzyme non-specific hand-off mechanism that involves the dynamic excursion of each glycosylase from its product site followed by the rapid invasion of APE1, is a simple and elegant solution to passing along unstable intermediates in base excision repair. This attractive solution forgoes the complexity of evolving specific interactions for numerous enzymes from different fold families.
Supplementary Material
Acknowledgments
The authors thank Dr. Alex Drohat for generously providing purified TDG for these studies.
Funding
This work was supported by the National Institutes of Health Grants T32 GM8403-23, RO1 GM056834 and National Science Foundation CHE1307275 (to J.T.S). Shannen Cravens was supported in part by NIH F31 GM114994-01A1.
ABBREVIATIONS
- hUNG
human uracil DNA glycosylase
- hOGG1
8-oxoguanine DNA glycosylase
- 8-oxoG
8-oxoguanine
- TDG
thymine DNA glycosylase
- AP-DNA
abasic DNA
- APE1
AP-endonuclease 1
- reAPE1
metal reconstituted APE1
- AP-site
abasic site
- CC
covalent complex
- NCC
noncovalent complex
- RCC
reduced covalent complex
- NS-DNA
nonspecific DNA
- LyP
lyase product
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: to be filled in. DNA sequences, three supporting tables, and six Supporting figures (PDF), one script file and four data files associated with the kinetic simulations in Figure S6.
References
- 1.Lesher DT, Pommier Y, Stewart L, Redinbo MR. 8-Oxoguanine rearranges the active site of human topoisomerase I. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12102–12107. doi: 10.1073/pnas.192282699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brooks SC, Adhikary S, Rubinson EH, Eichman BF. Recent advances in the structural mechanisms of DNA glycosylases. Biochim. Biophys. Acta. 2013;1834:247–271. doi: 10.1016/j.bbapap.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Girard PM, Guibourt N, Boiteux S. The Ogg1 protein of Saccharomyces cerevisiae: A 7,8-dihydro-8-oxoguanine DNA glycosylase AP lyase whose lysine 241 is a critical residue for catalytic activity. Nucleic Acids Res. 1997;25:3204–3211. doi: 10.1093/nar/25.16.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalhus B, Forsbring M, Helle IH, Vik ES, Forstrom RJ, Backe PH, Alseth I, Bjoras M. Separation-of-Function Mutants Unravel the Dual-Reaction Mode of Human 8-Oxoguanine DNA Glycosylase. Structure. 2011;19:117–127. doi: 10.1016/j.str.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 5.Morland I, Luna L, Gustad E, Seeberg E, Bjoras M. Product inhibition and magnesium modulate the dual reaction mode of hOgg1. DNA Repair. 2005;4:381–387. doi: 10.1016/j.dnarep.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Vidal AE, Hickson ID, Boiteux S, Radicella JP. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: bypass of the AP lyase activity step. Nucleic Acids Res. 2001;29:1285–1292. doi: 10.1093/nar/29.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faucher F, Doublie S, Jia ZC. 8-Oxoguanine DNA Glycosylases: One Lesion, Three Subfamilies. Int J Mol Sci. 2012;13:6711–6729. doi: 10.3390/ijms13066711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuznetsov NA, Koval VV, Zharkov DO, Nevinsky GA, Douglas KT, Fedorova OS. Kinetics of substrate recognition and cleavage by human 8-oxoguanine-DNA glycosylase. Nucleic Acids Res. 2005;33:3919–3931. doi: 10.1093/nar/gki694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohno M, Miura T, Furuichi M, Tominaga Y, Tsuchimoto D, Sakumi K, Nakabeppu Y. A genome-wide distribution of 8-oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Res. 2006;16:567–575. doi: 10.1101/gr.4769606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCullough AK, Dodson ML, Lloyd RS. Initiation of base excision repair: Glycosylase mechanisms and structures. Annu. Rev. Biochem. 1999;68:255–285. doi: 10.1146/annurev.biochem.68.1.255. [DOI] [PubMed] [Google Scholar]
- 11.Ide H, Kotera M. Human DNA glycosylases involved in the repair of oxidatively damaged DNA. Biol. Pharm. Bull. 2004;27:480–485. doi: 10.1248/bpb.27.480. [DOI] [PubMed] [Google Scholar]
- 12.Baldwin MR, O’Brien PJ. Human AP Endonuclease 1 Stimulates Multiple-Turnover Base Excision by Alkyladenine DNA Glycosylase. Biochemistry. 2009;48:6022–6033. doi: 10.1021/bi900517y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fitzgerald ME, Drohat AC. Coordinating the initial steps of base excision repair. Apurinic/apyrimidinic endonuclease 1 actively stimulates thymine DNA glycosylase by disrupting the product complex. J. Biol. Chem. 2008;283:32680–32690. doi: 10.1074/jbc.M805504200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruner SD, Norman DPG, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 16.Sidorenko VS, Nevinsky GA, Zharkov DO. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair. 2007;6:317–328. doi: 10.1016/j.dnarep.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 17.Sidorenko VS, Nevinsky GA, Zharkov DO. Specificity of stimulation of human 8-oxoguanine-DNA glycosylase by AP endonuclease. Biochem. Biophys. Res. Commun. 2008;368:175–179. doi: 10.1016/j.bbrc.2008.01.076. [DOI] [PubMed] [Google Scholar]
- 18.Baldwin MR, O’Brien PJ. Nonspecific DNA Binding and Coordination of the First Two Steps of Base Excision Repair. Biochemistry. 2010;49:7879–7891. doi: 10.1021/bi100889r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luncsford PJ, Manvilla BA, Patterson DN, Malik SS, Jin J, Hwang BJ, Gunther R, Kalvakolanu S, Lipinski LJ, Yuan WR, Lu WY, Drohat AC, Lu AL, Toth EA. Coordination of MYH DNA glycosylase and APE1 endonuclease activities via physical interactions. DNA Repair. 2013;12:1043–1052. doi: 10.1016/j.dnarep.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cravens SL, Schonhoft JD, Rowland MM, Rodriguez AA, Anderson BG, Stivers JT. Molecular crowding enhances facilitated diffusion of two human DNA glycosylases. Nucleic Acids Res. 2015;43:4087–4097. doi: 10.1093/nar/gkv301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erzberger JP, Barsky D, Scharer OD, Colvin ME, Wilson DM. Elements in abasic site recognition by the major human and Escherichia coli apurinic/apyrimidinic endonucleases. Nucleic Acids Res. 1998;26:2771–2778. doi: 10.1093/nar/26.11.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cravens SL, Hobson M, Stivers JT. Electrostatic Properties of Complexes along a DNA Glycosylase Damage Search Pathway. Biochemistry. 2014;53:7680–7692. doi: 10.1021/bi501011m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stivers JT, Harris TK, Mildvan AS. Vaccinia DNA topoisomerase I: Evidence supporting a free rotation mechanism for DNA supercoil relaxation. Biochemistry. 1997;36:5212–5222. doi: 10.1021/bi962880t. [DOI] [PubMed] [Google Scholar]
- 24.Leipold MD, Workman H, Muller JG, Burrows CJ, David SS. Recognition and removal of oxidized guanines in duplex DNA by the base excision repair enzymes hOGG1, yOGG1, and yOGG2. Biochemistry. 2003;42:11373–11381. doi: 10.1021/bi034951b. [DOI] [PubMed] [Google Scholar]
- 25.Thakur S, Sarkar B, Cholia RP, Gautam N, Dhiman M, Mantha AK. APE1/Ref-1 as an emerging therapeutic target for various human diseases: phytochemical modulation of its functions. Exp. Mol. Med. 2014;46:e106. doi: 10.1038/emm.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fromme JC, Bruner SD, Yang W, Karplus M, Verdine GL. Product-assisted catalysis in base-excision DNA repair. Nat. Struct. Biol. 2003;10:204–211. doi: 10.1038/nsb902. [DOI] [PubMed] [Google Scholar]
- 27.Strauss PR, Beard WA, Patterson RA, Wilson SH. Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J. Biol. Chem. 1997;272:1302–1307. doi: 10.1074/jbc.272.2.1302. [DOI] [PubMed] [Google Scholar]
- 28.Cheung DT, Nimni ME. Mechanism of Crosslinking of Proteins by Glutaraldehyde .1. Reaction with Model Compounds. Connect. Tissue Res. 1982;10:187–199. doi: 10.3109/03008208209034418. [DOI] [PubMed] [Google Scholar]
- 29.Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 2002;277:39926–39936. doi: 10.1074/jbc.M207107200. [DOI] [PubMed] [Google Scholar]
- 30.Waters TR, Gallinari P, Jiricny J, Swann PF. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem. 1999;274:67–74. doi: 10.1074/jbc.274.1.67. [DOI] [PubMed] [Google Scholar]
- 31.Hardeland U, Steinacher R, Jiricny J, Schar P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002;21:1456–1464. doi: 10.1093/emboj/21.6.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang HJ, Clendenin WM, Wong D, Demple B, Slupska MM, Chiang JH, Miller JH. Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/GO mismatch. Nucleic Acids Res. 2001;29:743–752. doi: 10.1093/nar/29.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pope MA, Porello SL, David SS. Escherichia coli apurinic-apyrimidinic endonucleases enhance the turnover of the adenine glycosylase MutY with G : A substrates. J. Biol. Chem. 2002;277:22605–22615. doi: 10.1074/jbc.M203037200. [DOI] [PubMed] [Google Scholar]
- 34.Rowland MM, Schonhoft JD, McKibbin PL, David SS, Stivers JT. Microscopic mechanism of DNA damage searching by hOGG1. Nucleic Acids Res. 2014;42:9295–9303. doi: 10.1093/nar/gku621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schonhoft JD, Stivers JT. Timing facilitated site transfer of an enzyme on DNA. Nat. Chem. Biol. 2012;8:205–210. doi: 10.1038/nchembio.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson BG, Stivers JT. Variola type IB DNA topoisomerase: DNA binding and supercoil unwinding using engineered DNA minicircles. Biochemistry. 2014;53:4302–4315. doi: 10.1021/bi500571q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.