

Abstract
The combination of gemcitabine (Gem), busulfan (Bu) and melphalan (Mel) is a promising regimen for autologous stem-cell transplantation (SCT) for lymphomas. To further improve the efficacy of [Gem+Bu+Mel], we added PARP inhibitor olaparib (Ola). We hypothesized that Ola would inhibit the repair of damaged DNA caused by [Gem+Bu+Mel]. Exposure of J45.01 and Toledo cell lines to IC10–20 of individual drug inhibited proliferation by 6%–16%; [Gem+Bu+Mel] by 20%–27%; and [Gem+Bu+Mel+Ola] by 61%–67%. The synergistic cytotoxicity of the 4-drug combination may be attributed to activation of the DNA-damage response, inhibition of PARP activity and DNA repair, decreased mitochondrial membrane potential, increased production of reactive oxygen species, and activation of the SAPK/JNK stress signaling pathway, all of which may enhance apoptosis. Similar observations were obtained using mononuclear cells isolated from patients with T-cell lymphocytic leukemia. Our results provide a rationale for undertaking clinical trials of this drug combination for lymphoma patients undergoing SCT.
Keywords: olaparib, gemcitabine, busulfan, melphalan, stem cell transplantation, DNA repair
Introduction
Lymphomas are blood disorders that affect the immune cells or lymphocytes. Treatments may include chemotherapy, radiation therapy, autologous hematopoietic stem cell transplantation (HSCT) and/or immunotherapy. Although there are two major types of lymphomas (Hodgkin’s and non-Hodgkin’s), several subtypes have been described.[1] This heterogeneity of these diseases dictates the need for exploration of more efficacious treatments.
HSCT improves overall and disease-free survival of high-risk lymphoma.[2–4] A critical phase of HSCT is the pre-transplant conditioning therapy where patients receive cytotoxic drugs with or without total body irradiation to destroy their hematopoietic capabilities. The combinations of nucleoside analogs and DNA alkylating agents as part of the pre-transplant regimen have improved the survival of high-risk lymphoma patients. A cohort analysis showed that combined gemcitabine (Gem), busulfan (Bu) and melphalan (Mel) provided better outcomes when compared with BEAM (BCNU, etoposide, ara-C, melphalan) for refractory relapsed Hodgkin’s lymphoma patients who had autologous HSCT.[5,6] The efficacy of this drug combination may be attributed to the ability of Gem to inhibit DNA replication and repair of DNA adducts mediated by Bu and Mel[7] and/or its ability to induce chromatin relaxation and increase access of the DNA alkylators to DNA.[8,9] Addition of epigenetic modifiers to this pre-transplant regimen further improved the survival of patients with refractory relapsed lymphoma,[10] suggesting the relevance of chromatin remodeling to their mechanisms of action.
The success of [Gem+Bu+Mel] in HSCT for lymphomas underscores the importance of mechanism-based chemotherapy and of using drugs with different mechanisms of action to eradicate tumor cells. Knowing that chemotherapy resistance is often related to enhanced DNA damage repair in tumor cells[11,12] and that poly(ADP-ribose) polymerase (PARP) is important in the recognition of damaged DNA and its repair through the base excision repair (BER) and homologous recombination (HR) pathways,[13–15] we hypothesized that addition of a PARP inhibitor to the [Gem+Bu+Mel] regimen would further enhance its cytotoxicity and potentially improve lymphoma patient survival.
PARP proteins catalyze the addition of poly-ADP ribose (PAR) to themselves or other proteins; the resulting PARylation is an important post-translational modification in the assembly of the DNA repair machinery.[16] Inhibition of PARP activity leads to unrepaired DNA breaks which cause cell death. In some circumstances, the ability of PARP inhibitors to trap PARP at damaged DNA sites may be more cytotoxic than unrepaired DNA damage caused by PARP inactivation.[17] Although there are few PARP inhibitors under investigation, olaparib (Ola) is approved by the FDA as a monotherapy for ovarian cancer, and several studies showed its efficacy in solid tumors.[18–22] Although Ola is not yet indicated for lymphoma patients, pre-clinical and clinical studies show its activity as a single agent in lymphoma cell lines and patients.[23,24]
To evaluate the possibility of using [Gem+Bu+Mel+Ola] as a salvage regimen for high-risk lymphoma patients, we determined the cytotoxicity and mechanisms of action of this 4-drug combination in lymphoma cell lines and patient-derived cell samples. Using concentrations close to their IC10-IC20 values, the four-drug combination effectively induced tumor cell death which we attributed to activation of the DNA-damage response, inhibition of PARP activity and DNA repair, and activation of the stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) stress-signaling pathway.
Methods
Cell lines and drugs
The J45.01 and Toledo lymphoma cell lines were obtained from the American Type Culture Collection (Manassas, VA). J45.01 is a human T-cell lymphoblast line that was originally established from a patient with T-cell leukemia and is now widely used as a cellular model for T-cell lymphoma. The Toledo cell line was originally derived from a patient with diffuse large B-cell lymphoma. Cells were cultured in RPMI-1640 (Mediatech, Manassas, VA) supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich, St. Louis, MO) and 100 U/ml penicillin and 100 μg/ml streptomycin (Mediatech) at 37°C in a humidified atmosphere of 5% CO2. Gemcitabine and Ola were obtained from Selleck Chemicals (Houston, TX) and dissolved in phosphate-buffered saline (PBS) or dimethyl sulfoxide (DMSO), respectively. Busulfan and Mel were purchased from Sigma-Aldrich and stock solutions were prepared in DMSO.
Patient samples
Three lymphocytic leukemia cell were isolated from peripheral blood, after obtaining written informed consent, using lymphocyte separation medium (Mediatech), and cultured in RPMI-1640 medium described above. UPN 101 (unique patient number) was a 62-year old Caucasian male with relapsed cutaneous T-cell lymphoma and leukemic blood picture with mixed myeloid and lymphoid phenotype. His circulating leukemic cells were isolated during an aggressive stage. UPN 102 is a 59-year old Hispanic male diagnosed with T-cell prolymphocytic leukemia. Blood samples were taken prior to induction chemotherapy and allogeneic HSCT. UPN 103 is a 37-year old Caucasian male with T-cell prolymphocytic leukemia and the samples used in this study were taken during relapse. The protocol for using patient samples was approved by the Institutional Review Board of the MD Anderson Cancer Center.
Cell proliferation, cell death assays and Western blot analysis
Cell proliferation was determined as previously described using the MTT assay.[25] Cell death was determined by flow-cytometric measurements of phosphatidylserine externalization with Annexin-V-FLUOS (Roche Diagnostics, Indianapolis, IN) and DNA staining with 7 aminoactinomycin D (BD Biosciences, San Jose, CA) using a Muse Cell Analyzer (EMD Millipore, Billerica, MA). Western blot analysis was performed as previously described.[25] The sources of the antibodies and their optimum dilutions are provided in Table 1 under Supplemental Materials.
Table 1.
List of primary antibodies, their sources and dilutions
| Antigen | Source/Cat. # | Clone type* | Dilution** |
|---|---|---|---|
|
| |||
| AIF | Cell Signaling/5318 | mAb | 2500 |
| ATF2 | Cell Signaling/9226 | mAb | 2500 |
| ATM | Santa Cruz Biotech/25921 | mAb | 750 |
| CHK2 | Cell Signaling/2662 | pAb | 2500 |
| Cleaved CASPASE 3 | Cell Signaling/9661 | pAb | 2500 |
| Cytochrome c | BD Bioscience #556433 | mAb | 2000 |
| DNA LIGASE I | Gene Tex/GTX102936 | pAb | 3500 |
| DNA LIGASE III | Gene Tex/GTX102888 | pAb | 3000 |
| FANCD2 | Santa Cruz Biotech/20022 | mAb | 700 |
| Histone 3 | Abcam//1791 | pAb | 3000 |
| KAP1 | Bethyl Lab/A300-275 | pAb | 3000 |
| MRE11 | Active&Motif/39220 | pAb | 2500 |
| NBS1 | Cell Signaling/3002 | pAb | 2500 |
| NOXA | Calbiochem/OP180 | mAb | 1500 |
| Nucleoporin | Santa Cruz Biotech/48373 | mAb | 700 |
| PAR | Trevigen/#4336-BPC-100 | pAb | 3500 |
| PARP1 | Santa Cruz Biotech/8007 | mAb | 1000 |
| PNK | Gene Tex/GTX107488 | pAb | 2000 |
| RAD50 | Cell Signaling/3427 | pAb | 2500 |
| RAD51 | Santa Cruz Biotech/6862 | pAb | 1000 |
| RAD51AP1 | Gene Tex/106411 | pAb | 3000 |
| RAD52 | Cell Signaling/3425 | pAb | 2500 |
| P-ATF2 (T71) | Cell Signaling/5112 | mAb | 2500 |
| P-ATM (S1981) | Rockland/200-301-400 | mAb | 2000 |
| P-CHK2 (S19) | Cell Signaling/2666 | pAb | 2500 |
| P-KAP1 (S824) | Cell Signaling/4127 | pAb | 2000 |
| P-MRE11 (S676) | Cell Signaling/4859 | pAb | 2000 |
| P-NBS1 (S343) | Cell Signaling/3001 | pAb | 2000 |
| P-RAD50 (S635) | Cell Signaling/14223 | pAb | 2000 |
| P-SAPK/JNK (Thr183/Tyr185) | Cell Signaling/4668 | pAb | 2000 |
| P-SEK1/MKK4 (S257) | Cell Signaling/4514 | mAb | 2000 |
| SAPK/JNK | Cell Signaling/9258 | pAb | 2500 |
| SEK1/MKK4 | Cell Signaling/9152 | pAb | 2000 |
| Ubiquitin | Cell Signaling/3936 | mAb | 2500 |
| VDAC1 | Gene Tex/GTX114187 | pAb | 3500 |
| XRCC1 | Gene Tex/GTX111712 | pAb | 2500 |
| β-ACTIN | Sigma/A5316 | mAb | 6000 |
| γ-H2AX | EMD Millipore/05-636 | mAb | 3000 |
pAb: polyclonal antibody; used anti-rabbit IgG (or anti-goat as indicated - G) for secondary antibody from Bio-Rad Lab
mAb: monoclonal antibody; used anti-mouse IgG for secondary antibody from Bio-Rad Lab
Fold dilution in PBS with 0.05% Tween 20
PARP enzyme assay
Preparation of total cell extracts and determination of the PARP enzymatic activity were performed using a Universal Colorimetric PARP Assay kit from Trevigen, Inc. (Gaithersburg, MD). Cells were exposed to drugs for 48 hrs and the protein concentrations in cell extracts were determined using a BCA Protein Assay kit (ThermoFisher Scientific); 50 μg protein was used to analyze the PARP enzymatic activity according to the manufacturer.
Preparation of cytoplasmic and nuclear extracts
Cells were exposed to drugs, and cytoplasmic and nuclear extracts were prepared using a Thermo Scientific Subcellular Protein Fractionation kit for cultured cells (ThermoFisher Scientific). The protein concentrations were determined as described above and 25 μg protein was analyzed by Western blotting.
Genomic DNA isolation and analysis
Genomic DNA was isolated from cells exposed to drugs for 24 hrs and 48 hrs using a Wizard Genomic DNA Purification kit from Promega (Madison, WI). The DNA concentrations were determined using a spectrophotometer and 15 μg DNA was loaded onto 0.8% agarose gel/Tris-acetate buffer and electrophoresis was conducted at 100 volts. The resolved DNA was stained with ethidium bromide and visualized with a G:Box EF2 gel documentation system from Syngene (Frederick, MD).
Analysis of mitochondrial membrane potential (MMP) and reactive oxygen species (ROS)
Cells were exposed to drugs for 48 hrs and analyzed for changes in MMP using the JC-1 probe contained in a detection kit (Cayman Chemical Co., Ann Arbor, MI) as previously described.[26] Flow cytometry was performed using a Gallios Flow Cytometer (Beckman Coulter, Inc., Brea, CA). Production of ROS was determined by exposing cells to drugs for 48 hrs followed by incubation with CM-H2DCFDA for 1 hr, as previously described.[26]
Statistical Analysis
Results are presented as the average ± standard deviation of at least three independent experiments and statistical analysis was performed using a Student’s paired t-test with a two-tailed distribution.
Results
Combination of Gem, Bu, Mel and Ola provides synergistic cytotoxicity toward lymphoma cell lines
Cells were exposed to drugs alone, or in combination, at concentrations equivalent to their calculated IC10-IC20 values (concentrations that caused 10%–20% inhibition of proliferation). Exposure of J45.01 T-cells to 27 nM Gem, 27 μM Bu, 0.66 μM Mel, or 7 μM Ola for 48 hrs inhibited cell proliferation by 6–16%, and to [Gem+Bu+Mel] by ~20% (Figure 1A). Combination of Ola with Gem, Bu or Mel inhibited cell proliferation by 18–36% and addition of Ola to the [Gem+Bu+Mel] combination inhibited cell proliferation by ~61% (P= 0.00005). The results for the proliferation (MTT) assay are consistent with the Annexin V assay, which was used to measure early cell death. Exposure to each individual drug or combination of Ola with Gem, Bu or Mel resulted in 16–21% and 24–40% Ann V-positive cells, respectively (control has 13%). Exposure to [Gem+Bu+Mel] resulted in ~28% Ann V-positive cells, which significantly increased to ~64% when Ola was added (P = 0.00004).
Figure 1.

Cytotoxicity of drugs (alone or in combination) in malignant T- and B-cells. Cells were exposed continuously for 48 hrs to drug(s) prior to MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) and Annexin V (Ann V) assays. Results are average ± SD of at least three independent experiments. Gem or G, gemcitabine; Bu or B, busulfan; Mel or M, melphalan; Ola or O, olaparib
Exposure of Toledo cells to 3.3 nM Gem, 18 μM Bu, 0.6 μM Mel or 7 μM Ola inhibited cell proliferation by 10–16%, and to [Gem+Bu+Mel] by ~27% (Figure 1B).). Combination of Ola with Gem, Bu or Mel inhibited Toledo cell proliferation by 16–30%. When Ola was added to the [Gem+Bu+Mel], cell proliferation was significantly inhibited by ~67% (P = 0.00007). Cell death for each individual drug treatment, [Gem+Ola], [Bu+Ola], and [Mel+Ola] was in the range of 5–33% versus ~3% in control cells. Cell death was ~22% following [Gem+Bu+Mel] and further increased to ~53% when Ola was added to this three-drug combination (P = 0.0001). These results suggest synergistic cytotoxicity when Ola was added to the [Gem+Bu+Mel] combination in J45.01 T-cells and Toledo B-cells.
Early effects of [Gem+Bu+Mel+Ola] combination include activation of the DNA-damage response
Gemcitabine, as a nucleoside analog, stalls replication forks when incorporated into nascent DNA strands during replication and causes DNA strand breaks.[27] When Gem is combined with DNA alkylators like Bu and Mel, the DNA-damage response is strongly activated.[9] The resulting DNA adducts and DNA breaks are expected to be repaired, and we hypothesized that inhibition of DNA repair would aggravate DNA damage. Since Ola is known to inhibit PARP activity with concomitant inhibition of DNA repair, we were prompted to determine if addition of Ola would further enhance [Gem+Bu+Mel]-mediated DNA damage. A widely used indicator of the DNA-damage response is the activation of the ATM pathway.[28,29] Since it is an early event we exposed J45.01 cells for 24 hrs and observed that low concentrations of Gem, Bu, Mel or their combination did not significantly increase the phosphorylation of ATM at S1981; 7 μM Ola alone slightly increased the level of P-ATM (Figure 2A). The [Gem+Bu+Mel+Ola] combination dramatically increased the level of phosphorylated ATM at S1981 (Figure 2A). The phosphorylation of ATM at S1981 is known to activate its kinase activity,[29] consistent with the observed increase in the phosphorylation of its substrates histone 2AX, KAP1 and CHK2 (Figure 2A). Similar results were observed in Toledo cells (Figure 2B), suggesting that Ola enhanced the DNA damage caused by [Gem+Bu+Mel] in both cell lines.
Figure 2.

Activation of the DNA-damage response and inhibition of poly(ADP)ribosylation. Cells were exposed continuously for 24 hrs to drug(s) and analyzed by Western blotting (A, B). Cells were treated with drug(s) for 48 hrs and total cell extracts were assayed for PARP enzymatic activity (C), analyzed by Western blotting for the extent of poly(ADP)ribosylation using total cell extracts (D) or soluble and chromatin-bound nuclear extracts (NE, panel E). Similar cell samples were used to isolate genomic DNA that was analyzed (15 μg) on a 0.8% agarose gel (F). DDR, DNA-damage response; NS, non-specific; other abbreviations are the same as described in Figure 1.
To establish whether Ola inhibits PARP under these conditions, we assayed its enzymatic activity in total extracts from cells exposed to the drug(s). While a low concentration of [Gem+Bu+Mel] did not inhibit PARP activity, cell exposure to Ola alone inhibited PARP activity by ~90% (Figure 2C). Combination of the four drugs had similar degree of inhibition, suggesting that [Gem+Bu+Mel] did not negate the effects of Ola (Figure 2C). The Ola-mediated inhibition of PARP is also shown by Western blotting. While [Gem+Bu+Mel] did not change the level of PARylated proteins, it was significantly decreased by Ola alone, and combination of the four drugs similarly inhibited PARylation of proteins (Figure 2D). Moreover, [Gem+Bu+Mel+Ola] caused cleavage of PARP1 (Figure 2D), suggesting that the combined inhibition of PARP enzymatic activity and its drug-mediated cleavage may explain the observed synergistic cytotoxicity.
To determine the status of chromatin-bound PARP1, we analyzed the soluble and chromatin-bound nuclear extracts by Western blotting. PARP1 was present in the soluble fractions from cells exposed to [Gem+Bu+Mel], Ola alone, and [Gem+Bu+Mel+Ola]. Chromatin-bound PARP1 was also observed in cells exposed to [Gem+Bu+Mel] or Ola alone, but not in cells exposed to [Gem+Bu+Mel+Ola] (Figure 2E). Less chromatin-bound PARylated proteins were also observed in the [Gem+Bu+Mel+Ola] lane, suggesting inefficient PARylation of proteins involved in DNA repair. An increase in the ubiquitinated chromatin-bound proteins was observed in cells exposed to [Gem+Bu+Mel+Ola], which may indicate a possible activation of the ubiquitin-proteasome system leading to protein degradation [30]. These results are consistent with the reported increased ubiquitination of XRCC1, a DNA repair protein, when its PARylation is inhibited.[31]
The overall extent of DNA damage was estimated by isolating genomic DNA and resolving it on an agarose gel. The presence of ethidium bromide-smears suggests DNA fragmentation in cells exposed to [Gem+Bu+Mel+Ola] as early as 24 hrs after drug exposure (Figure 2F), consistent with the activation of the DNA-damage response (Figure 2A) and apoptosis as indicated by the Ann V assay (Figure 1A).
[Gem+Bu+Mel+Ola] combination changes the modifications and levels of proteins involved in DNA repair
After showing the drug-mediated inhibition of PARP and its cellular consequences, we sought to determine the effects of [Gem+Bu+Mel+Ola] on the phosphorylation of MRE11, NBS1, and RAD50, which repair double-strand DNA.[32] [Gem+Bu+Mel+Ola] slightly increased the phosphorylation of MRE11 at S676 in J45.01 cells, and the effect was more dramatic in Toledo cells (Figure 3A and 3B). NBS1 and RAD50 proteins were highly phosphorylated at S343 and S635, respectively, in the 4-drug combination (Figure 3A and 3B). The increased phosphorylations of the MRN components may be due to ATM activation (Figure 2A).[33]
Figure 3.

Analysis of proteins involved in DNA repair. Cells were exposed continuously for 48 hrs to drug(s) and total cell extracts were analyzed by Western blotting for the phosphorylation of the MRN complex components (A, B), and changes in the level of proteins involved in BER (C, D) and HR repairs (E, F). Abbreviations for drug names are the same as in Figure 1.
We also examined the effects of [Gem+Bu+Mel+Ola] on the level of XRCC1, a BER protein which acts as a scaffold protein to coordinate DNA repair by interacting with other repair proteins including PARP1, PNKP, DNA ligase I and DNA ligase III.[34–37] The level of XRCC1 slightly decreased, PARP1 and DNA ligase III were cleaved, and the level of PNKP and DNA ligase I proteins dramatically decreased in J45.01 and Toledo cells exposed to [Gem+Bu+Mel+Ola] (Figure 3C and 3D). Although some of the observed effects might be consequences (rather than causes) of drug-mediated cell death, these results nevertheless suggest that [Gem+Bu+Mel+Ola] combination compromised DNA repair by decreasing the level of proteins involved in BER.
Double-strand breaks are also repaired by HR.[38] We, therefore, determined if [Gem+Bu+Mel+Ola] would decrease the level of proteins involved in this pathway. Western blot analysis shows decreased levels of RAD51, RAD51AP1, RAD52 and FANCD2 proteins in the 4-drug combination (Figure 3E and 3F), suggesting that HR, like BER, was also compromised.
[Gem+Bu+Mel+Ola] combination activates apoptosis
The observed Ann V-positivity, DNA-damage response and inhibition of DNA repair are indicative of genomic injuries that may lead to apoptosis in these cell types. Indeed, exposure of cells to [Gem+Bu+Mel+Ola] resulted in the extensive cleavage of PARP1, activation (by cleavage) of CASPASE 3 and increased levels of the pro-apoptotic protein NOXA (Figure 4A).
Figure 4.
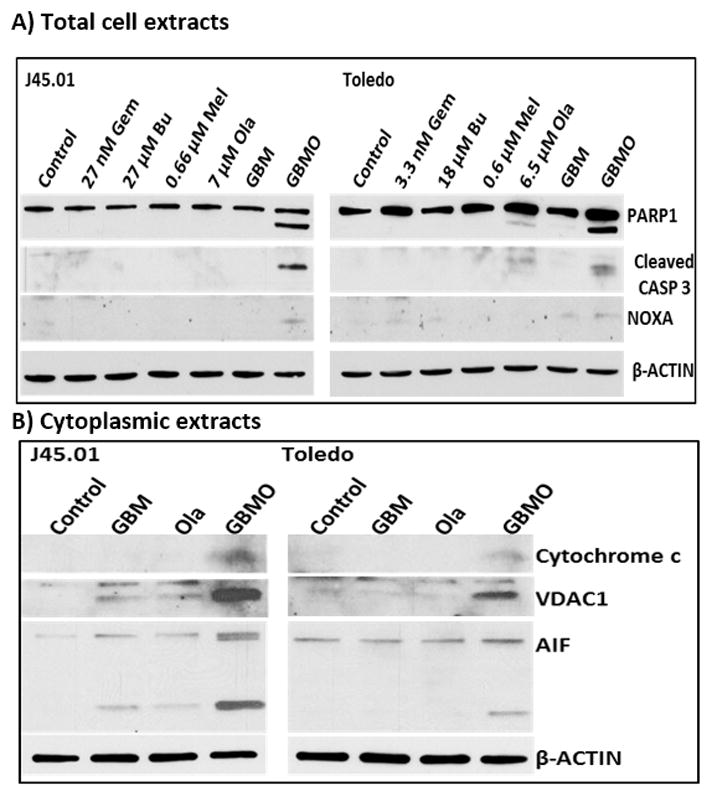
Activation of the apoptosis pathway. Cells were exposed continuously for 48 hrs to drugs (alone or in combination) prior to Western blot analysis of the total cell (A) and cytoplasmic (B) extracts. Abbreviations for drug names are the same as in Figure 1.
To further establish the activation of intrinsic apoptosis, we analyzed cytoplasmic extracts for the release of pro-apoptotic mitochondrial proteins. Indeed, the levels of cytochrome c and voltage-dependent anion channel 1 (VDAC1) increased (Figure 4B). These two proteins are localized to the mitochondrial membrane and upon apoptotic stimulation, cytochrome c is released and induces the activation of pro-apoptotic caspases.[39] The release of cytochrome c is partly dependent on the integrity of VDAC1, an outer mitochondrial membrane protein that controls the mitochondrial permeability.[40] The increased level of VDAC1 in the cytoplasm suggests that the mitochondrial membrane was compromised in cells exposed to [Gem+Bu+Mel+Ola]. Apoptosis-inducing factor (AIF) is another mitochondrial membrane protein which is also released in response to apoptotic stimuli.[41] Similar to cytochrome c and VDAC1, the level of AIF in the cytoplasm increased in the presence of [Gem+Bu+Mel+Ola], and an apparent cleavage of AIF was also observed (Figure 4B).
[Gem+Bu+Mel+Ola] combination activates apoptosis by decreasing the mitochondrial membrane potential and increasing the level of reactive oxygen species
The [Gem+Bu+Mel+Ola]-mediated increase in the cytoplasmic cytochrome c, VDAC1 and AIF suggests increased permeability of the mitochondrial membrane. We therefore analyzed changes in the mitochondrial membrane potential (MMP) using JC-1 reagent as previously described.[42] The aggregated form of JC-1 localizes to the mitochondria which is converted to monomer upon release to the cytoplasm. As a positive control, J45.01 cells were exposed to 1 μM valinomycin, a known ionophore, for 15 min; JC-1 was ~85% monomer and ~15% aggregate. The negative control cells had ~95% aggregated JC-1 and ~5% monomer, which did not significantly change in the presence of [Gem+Bu+Mel] or Ola alone. However, exposure of cells to [Gem+Bu+Mel+Ola] resulted in ~55% aggregate and ~45% monomer (Figure 5A), suggesting decreased MMP. Similar results were obtained in Toledo cells (Figure 5B).
Figure 5.

Changes in the mitochondrial membrane potential (MMP) and level of reactive oxygen species (ROS). Cells were exposed for 48 hrs to drugs (alone or in combination) and analyzed for changes in MMP (A, B) and ROS (C, D) by flow cytometry as described under Methods. Abbreviations for drug names are the same as in Figure 1.
To further understand other cellular consequences of drug-mediated decrease in MMP, we examined the production of ROS, which could be triggered by a reaction of oxygen with leaked electrons from the mitochondrial respiratory chain.[43] An almost 2-fold increase in the level of ROS was observed in cells exposed to [Gem+Bu+Mel+Ola] (Figure 5C and 5D), suggesting that the 4-drug combination has perturbed the mitochondria and increased ROS production.
[Gem+Bu+Mel+Ola] combination activates the SAPK/JNK stress signaling pathway
The observed perturbation of mitochondria in cells exposed to [Gem+Bu+Mel+Ola] may cause drug-mediated activation of stress signaling pathways that promote apoptosis. The SAPK/JNK signal transduction pathway is known to convert stress signaling into apoptotic signaling.[44] This pathway is controlled by the activation of MKK4/7.[45] Initial analysis showed an increased phosphorylation of SEK1/MKK4 at S257, which probably activated SAPK/JNK as suggested by increased phosphorylation at threonine 183 and 185 of SAPK/JNK in cells exposed to [Gem+Bu+Mel+Ola] (Figure 6). The activation of this pathway is further supported by increased phosphorylation of ATF2 at T71, a known substrate of SAPK/JNK.[46]
Figure 6.

Activation of the SAPK/JNK pathway. Cells were exposed continuously for 48 hrs to drugs (alone or in combination) prior to Western blot analysis of the total cell extracts. Abbreviations for drug names are the same as in Figure 1.
Exposure of mononuclear cells from patients with lymphocytic leukemia to [Gem+Bu+Mel+Ola] activates the DNA-damage response and apoptosis
To determine the potential clinical significance of our cell line studies, we isolated mononuclear cells from patients with lymphocytic leukemia, exposed them to individual drugs (or combinations) and analyzed them by Western blotting. Increased phosphorylation of γ-H2AX was observed in the presence of [Gem+Bu+Mel+Ola], indicative of DNA-damage response activation (Figure 7). PARP1 and CASPASE 3 were significantly cleaved in cells exposed to the 4-drug combination, suggesting activation of apoptosis. These results show a synergistic cytotoxicity of [Gem+Bu+Mel+Ola] in cells derived from patients with aggressive T-cell malignancies involving mechanisms analogous to those seen in cultured cell lines.
Figure 7.

Effects of drugs on mononuclear cells isolated from patients with lymphocytic leukemia. Cells were exposed continuously for 48 hrs to drugs (alone or in combination) prior to Western blot analysis of the total cell extracts. Abbreviations for drug names are the same as in Figure 1. UPN: unique patient number
Discussion
Our previous preclinical and clinical studies demonstrated the synergistic cytotoxicity in vitro and clinical efficacy of [Gem+Bu+Mel] as part of the pre-transplant regimen for refractory lymphoma patients undergoing HSCT.[9,47] Our present study now shows that inclusion of the PARP inhibitor Ola further enhances the cytotoxicity of this drug combination in cell lines and patient-derived cell samples.
We used low concentrations of Gem, Bu and Mel in this study to elicit more pronounced effects when Ola was added to the combination. These IC10–20 values of the drugs may or may not be clinically relevant but we wanted to investigate whether these drug combinations would convey a proof of principle as to their synergistic cytotoxicity. In a clinical setting, we will propose a Phase I/II trial in which we hope to identify safe, yet efficacious drug dosages that are synergistic in lymphoma patients.
The [Gem+Bu+Mel] combination caused only 20–27% inhibition of proliferation of cells; addition of Ola increased the inhibition to 62–67% (Figure 1). The observed synergistic cytotoxicity may be attributed to events occurring at the initial DNA damage level as suggested by the activation of the ATM pathway within 24 hrs of drug exposure (Figure 2). During DNA replication, Gem could be incorporated into the nascent DNA strand and stall the replication fork. When DNA adducts formed by Bu and Mel are repaired, Gem could have also been incorporated in place of normal nucleotides. These events are further aggravated by the ability of Gem to potentiate itself; its diphosphorylated form inhibits ribonucleotide reductase, an enzyme that catalyzes the conversion of ribonucleotides into deoxyribonucleotides, depletes the cellular deoxynucleotide pools, and enhances the preferential incorporation of Gem during DNA synthesis and repair.[48] Busulfan and Mel form DNA intra- and inter-strand crosslinks which also stall replication forks and inhibit DNA synthesis. These collective cellular events increased the formation of DNA strand breaks as suggested by the activation of the DNA-damage response and DNA fragmentation (Figure 2). These DNA breaks also activate the DNA repair machinery, aspects of which are inhibited in the presence of Ola. Such inhibition of DNA repair results in the amplification of DNA breaks and the cells concomitantly commit to apoptosis.
Olaparib inhibits DNA repair through inhibition of PARP enzymatic activity. PARP catalyzes poly(ADP) ribosylation which is important for the stability and assembly of proteins involved in DNA repair.[16,49,50] In the presence of Ola, PARP was almost completely inactivated (Figure 2C), and PARylation of proteins was obliterated (Figure 2D). Under certain conditions, [Gem+Bu+Mel] combination also causes cleavage of PARP1,[9] and together with Ola-mediated PARP inhibition may profoundly compromise DNA repair.
The inhibition of DNA repair is consistent with decreased expression of some proteins involved in BER and HR. The levels of XRCC1, PNKP, DNA LIGASE I and III (involved in BER), and of RAD50, RAD51, RAD51AP1, RAD52 and FANCD2 (involved in HR) were all decreased in cells exposed to [Gem+Bu+Mel+Ola] (Figure 3). It remains to be determined if their downregulation is at the transcription level and/or whether their protein stability is compromised by their poly(ADP)ribosylation status. It is possible that decreased PARylation may inhibit protein complex formation and make individual proteins less stable. In fact, ubiquitination of nuclear proteins increased in cells exposed to [Gem+Bu+Mel+Ola] (Figure 2E), which could make them more susceptible to proteasomal-mediated degradation.
The effects of these drugs in the nucleus may transmit signals to the mitochondria as evident by decreased mitochondrial membrane potential and increased ROS production (Figure 5), both of which lead to cell death. Such mitochondrial stress is further aggravated by the activation of the SAPK/JNK signal transduction pathway (Figure 6), probably triggered by DNA damage,[51,52] which also induces apoptosis by phosphorylating mitochondrial proteins involved in the release of cytochrome c and AIF.[44]
Our results are consistent with the notion that Ola is more effective when used in combination regimens as suggested in previous studies on solid tumors.[53–55] Studies on the combination of PARP inhibitor(s) with other drugs to treat lymphomas are very limited. Olaparib and veliparib (ABT-888) radiosensitized non-Hodgkin’s lymphoma cells,[24] and combination of PARP inhibitors with topotecan (topoisomerase-I inhibitor) or temozolomide (DNA alkylating agent) resulted in enhanced killing of lymphoma cells.[56–58] Our present report on the efficacy of [Gem+Bu+Mel+Ola] ] in the J45.01 T-cell line, Toledo B-cell line and patient-derived cell samples (Figure 7) provides a powerful tool to kill tumor cells and justifies its evaluation as part of a pre-transplant regimen for leukemia and lymphoma patients undergoing HSCT.
Acknowledgments
Part of this research was performed in the Flow Cytometry & Cellular Imaging Facility, which is supported in part by the National Institutes of Health through M.D. Anderson’s Cancer Center Support Grant CA016672. This work was also supported by the Stephen L. and Lavinia Boyd Fund for Leukemia Research, and by funds donated by grateful patients.
References
- 1.Mugnaini EN, Ghosh N. Lymphoma. Prim Care: Clinics Office Practice. 2016;43:661–675. doi: 10.1016/j.pop.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Linch DC, Winfield D, Goldstone AH, et al. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin’s disease: results of a BNLI randomised trial. Lancet. 1993;341:1051–1054. doi: 10.1016/0140-6736(93)92411-l. [DOI] [PubMed] [Google Scholar]
- 3.Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. N Engl J Med. 1995;333:1540–1545. doi: 10.1056/NEJM199512073332305. [DOI] [PubMed] [Google Scholar]
- 4.Schmitz N, Pfistner B, Sextro M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet. 2002;359:2065–2071. doi: 10.1016/S0140-6736(02)08938-9. [DOI] [PubMed] [Google Scholar]
- 5.Nieto Y, Popat U, Anderlini P, et al. Autologous stem cell transplantation for refractory or poor-risk relapsed Hodgkin’s lymphoma: effect of the specific high-dose chemotherapy regimen on outcome. Biol Blood Marrow Transplant. 2013;19:410–417. doi: 10.1016/j.bbmt.2012.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nieto Y, Valdez BC, Thall PF, et al. Vorinostat combined with high-dose gemcitabine, busulfan and melphalan with autologous stem-cell transplantation in patients with refractory lymphomas. Biol Blood Marrow Transplant. 2015;21:1914–1920. doi: 10.1016/j.bbmt.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plunkett W, Huang P, Searcy CE, et al. Gemcitabine: preclinical pharmacology and mechanisms of action. Semin Oncol. 1996;23(5 Suppl 10):3–15. [PubMed] [Google Scholar]
- 8.Valdez BC, Andersson BS. Interstrand crosslink inducing agents in pretransplant conditioning therapy for hematologic malignancies. Environ Mol Mutagen. 2010;51:659–668. doi: 10.1002/em.20603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valdez BC, Nieto Y, Murray D, et al. Epigenetic modifiers enhance the synergistic cytotoxicity of combined nucleoside analog-DNA alkylating agents in lymphoma cell lines. Exp Hematol. 2012;40:800–810. doi: 10.1016/j.exphem.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nieto Y, Valdez BC, Thall PF, et al. Double epigenetic modulation of high-dose chemotherapy with azacitidine and vorinostat for patients with refractory or poor-risk relapsed lymphoma. Cancer. 2016;122:2680–2688. doi: 10.1002/cncr.30100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spanswick VJ, Lowe HL, Newton C, et al. Evidence for different mechanisms of ‘unhooking’ for melphalan and cisplatin-induced DNA interstrand cross-links in vitro and in clinical acquired resistant tumour samples. BMC Cancer. 2012;12:436. doi: 10.1186/1471-2407-12-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12:587–598. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 13.Adhikari S, Choudhury S, Mitra PS, et al. Targeting base excision repair for chemosensitization. Anticancer Agents Med Chem. 2008;8:351–357. doi: 10.2174/187152008784220366. [DOI] [PubMed] [Google Scholar]
- 14.David KK, Andrabi SA, Dawson TM, et al. Parthanatos, a messenger of death. Front Biosci (Landmark Ed) 2009;14:1116–1128. doi: 10.2741/3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bryant HE, Petermann E, Schultz N, et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28:2601–2615. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei H, Yu X. Functions of PARylation in DNA Damage Repair Pathways. Genomics Proteomics Bioinformatics. 2016;14:131–139. doi: 10.1016/j.gpb.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP Inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ledermann JA, Harter P, Gourley C, et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016;17:1579–1589. doi: 10.1016/S1470-2045(16)30376-X. [DOI] [PubMed] [Google Scholar]
- 20.Leichman L, Groshen S, O’Neil BH, et al. Phase II study of olaparib (AZD-2281) after standard systemic therapies for disseminated colorectal cancer. Oncologist. 2016;21:172–177. doi: 10.1634/theoncologist.2015-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bang YJ, Im SA, Lee KW, et al. Randomized, double-blind phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J Clin Oncol. 2015;33:3858–3865. doi: 10.1200/JCO.2014.60.0320. [DOI] [PubMed] [Google Scholar]
- 23.Weston VJ, Oldreive CE, Skowronska A, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–4587. doi: 10.1182/blood-2010-01-265769. [DOI] [PubMed] [Google Scholar]
- 24.Schaefer NG, James E, Wahl RL. Poly(ADP-ribose) polymerase inhibitors combined with external beam and radioimmunotherapy to treat aggressive lymphoma. Nucl Med Commun. 2011;32:1046–1051. doi: 10.1097/MNM.0b013e32834a369b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valdez BC, Li Y, Murray D, et al. 5-Aza-2’-deoxycytidine sensitizes busulfan-resistant myeloid leukemia cells by regulating expression of genes involved in cell cycle checkpoint and apoptosis. Leuk Res. 2010;34:364–372. doi: 10.1016/j.leukres.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valdez BC, Zander AR, Song G, et al. Synergistic cytotoxicity of gemcitabine, clofarabine and edelfosine in lymphoma cell lines. Blood Cancer J. 2014;4:e171. doi: 10.1038/bcj.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ewald B, Sampath D, Plunkett W. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene. 2008;27:6522–6537. doi: 10.1038/onc.2008.316. [DOI] [PubMed] [Google Scholar]
- 28.Rogakou EP, Pilch DR, Orr AH, et al. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 29.Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 30.Deshaies RJ. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014;12:94. doi: 10.1186/s12915-014-0094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei L, Nakajima S, Hsieh CL, et al. Damage response of XRCC1 at sites of DNA single strand breaks is regulated by phosphorylation and ubiquitylation after degradation of poly(ADP-ribose) J Cell Sci. 2013;126(Pt 19):4414–4423. doi: 10.1242/jcs.128272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 33.Zhao S, Weng YC, Yuan SS, et al. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature. 2000;405:473–477. doi: 10.1038/35013083. [DOI] [PubMed] [Google Scholar]
- 34.Vidal AE, Boiteux S, Hickson ID, et al. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001;20:6530–6539. doi: 10.1093/emboj/20.22.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitehouse CJ, Taylor RM, Thistlethwaite A, et al. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 36.Caldecott KW, McKeown CK, Tucker JD, et al. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol Cell Biol. 1994;14:68–76. doi: 10.1128/mcb.14.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masson M, Niedergang C, Schreiber V, et al. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol. 1998;18:3563–3571. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 40.Zheng Y, Shi Y, Tian C, et al. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene. 2004;23:1239–1247. doi: 10.1038/sj.onc.1207205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 42.Valdez BC, Wang G, Murray D, et al. Mechanistic studies on the synergistic cytotoxicity of the nucleoside analogs gemcitabine and clofarabine in multiple myeloma: relevance of p53 and its clinical implications. Exp Hematol. 2013;41:719–730. doi: 10.1016/j.exphem.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suski JM, Lebiedzinska M, Bonora M, et al. Relation between mitochondrial membrane potential and ROS formation. Methods Mol Biol. 2012;810:183–205. doi: 10.1007/978-1-61779-382-0_12. [DOI] [PubMed] [Google Scholar]
- 44.Aoki H, Kang PM, Hampe J, et al. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J Biol Chem. 2002;277:10244–10250. doi: 10.1074/jbc.M112355200. [DOI] [PubMed] [Google Scholar]
- 45.Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18:6087–6093. doi: 10.1038/sj.onc.1203129. [DOI] [PubMed] [Google Scholar]
- 46.Lau E, Ronai ZA. ATF2 - at the crossroad of nuclear and cytosolic functions. J Cell Sci. 2012;125:2815–2824. doi: 10.1242/jcs.095000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nieto Y, Thall P, Valdez B, et al. High-dose infusional gemcitabine combined with busulfan and melphalan with autologous stem-cell transplantation in patients with refractory lymphoid malignancies. Biol Blood Marrow Transplant. 2012;18:1677–1686. doi: 10.1016/j.bbmt.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parker WB, Shaddix SC, Chang CH, et al. Effects of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl) adenine on K562 cellular metabolism and the inhibition of human ribonucleotide reductase and DNA polymerases by its 5′-triphosphate. Cancer Res. 1991;51:2386–2394. [PubMed] [Google Scholar]
- 49.D’Amours D, Desnoyers S, D’Silva I, et al. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 50.Zaja R, Mikoč A, Barkauskaite E, et al. Molecular Insights into Poly(ADP-ribose) Recognition and Processing. Biomolecules. 2012;3:1–17. doi: 10.3390/biom3010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hayakawa J, Depatie C, Ohmichi M, et al. The activation of c-Jun NH2-terminal kinase (JNK) by DNA-damaging agents serves to promote drug resistance via activating transcription factor 2 (ATF2)-dependent enhanced DNA repair. J Biol Chem. 2003;278:20582–20592. doi: 10.1074/jbc.M210992200. [DOI] [PubMed] [Google Scholar]
- 52.Bhoumik A, Lopez-Bergami P, Ronai Z. ATF2 on the double- activating transcription factor and DNA damage response protein. Pigment Cell Res. 2007;20:498–506. doi: 10.1111/j.1600-0749.2007.00414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samol J, Ranson M, Scott E, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs. 2012;30:1493–1500. doi: 10.1007/s10637-011-9682-9. [DOI] [PubMed] [Google Scholar]
- 54.Khan OA, Gore M, Lorigan P, et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer. 2011;104:750–755. doi: 10.1038/bjc.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Del Conte G, Sessa C, von Moos R, et al. Phase I study of olaparib in combination with liposomal doxorubicin in patients with advanced solid tumours. Br J Cancer. 2014;111:651–659. doi: 10.1038/bjc.2014.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palma JP, Wang YC, Rodriguez LE, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009;15:7277–7290. doi: 10.1158/1078-0432.CCR-09-1245. [DOI] [PubMed] [Google Scholar]
- 57.Kummar S, Chen A, Ji J, et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011;71:5626–5634. doi: 10.1158/0008-5472.CAN-11-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Golla RM, Li M, Shen Y, et al. Inhibition of poly(ADP-ribose) polymerase (PARP) and ataxia telangiectasia mutated (ATM) on the chemosensitivity of mantle cell lymphoma to agents that induce DNA strand breaks. Hematol Oncol. 2012;30:175–179. doi: 10.1002/hon.1020. [DOI] [PubMed] [Google Scholar]