

Summary
Faithful propagation of functionally distinct chromatin states is crucial for maintaining cellular identity, and its breakdown can lead to diseases such as cancer. Whereas mechanisms that sustain repressed states have been intensely studied, regulatory circuits that protect active chromatin from inactivating signals are not well understood. Here we report a positive feedback loop that preserves the transcription-competent state of RNA polymerase II-transcribed genes. We found that Pdp3 recruits the histone acetyltransferase Mst2 to H3K36me3-marked chromatin. Thereby, Mst2 binds to all transcriptionally active regions genome-wide. Besides acetylating histone H3K14, Mst2 also acetylates Brl1, a component of the histone H2B ubiquitin ligase complex. Brl1 acetylation increases histone H2B ubiquitination, which positively feeds back on transcription and prevents ectopic heterochromatin assembly. Our work uncovers a molecular pathway that secures epigenome integrity and highlights the importance of opposing feedback loops for the partitioning of chromatin into transcriptionally active and inactive states.
Keywords: heterochromatin, RNA interference, epigenetic gene silencing, histone modification, Mst2, Gcn5, Brl1/BRE1, H3K36me3, H2B ubiquitin, acetylomics
Graphical Abstract
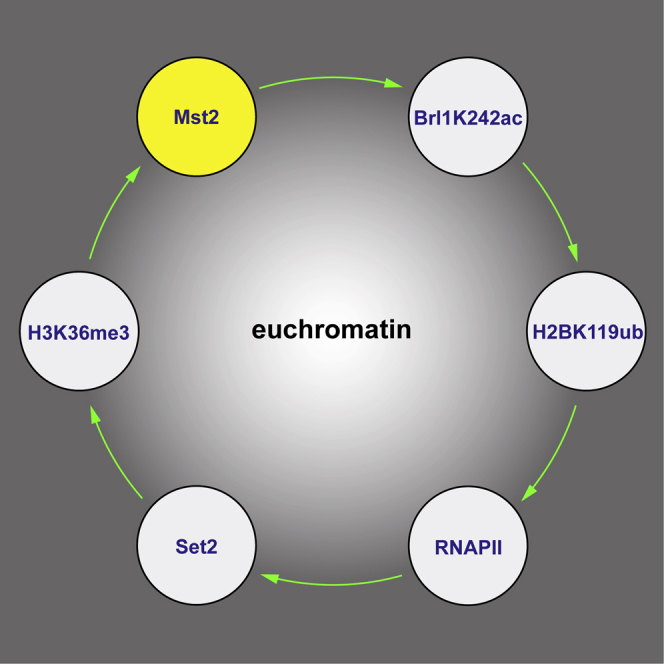
Highlights
-
•
A positive feedback loop maintains euchromatic genes in an active state
-
•
The histone acetyltransferase Mst2 acetylates the ubiquitin ligase Brl1
-
•
Brl1 acetylation inhibits initiation of RNAi-directed heterochromatin formation
-
•
H3K36me3 sequesters Mst2 to prevent illegitimate activation of heterochromatin
The partitioning of distinct chromatin states is crucial for maintaining cellular identity. Flury et al. demonstrate that anchoring an acetyltransferase complex to actively transcribed genes protects euchromatin from transformation into heterochromatin. This mechanism acts through a positive feedback loop that involves acetylation of a non-histone target, which in turn mediates histone ubiquitylation.
Introduction
Chromatin exists in different states that are intimately linked with gene activity. Active chromatin is associated with histone H3 lysine 36 (H3K36) methylation in all eukaryotes. H3K36-specific methyltransferases contain a catalytic SET domain, but they have varying preferences to catalyze mono-, di-, or trimethylation of H3K36. In yeast, the conserved SET domain-containing protein 2 (Set2) performs all H3K36 methylation and is recruited to chromatin co-transcriptionally via direct interaction with RNA polymerase II (Wagner and Carpenter, 2012). Specific reader proteins that interact with histone deacetylases (HDACs) recognize methylated H3K36, which is necessary for HDAC activity within active genes (Drouin et al., 2010, Govind et al., 2010, Nicolas et al., 2007). This compensates for transcription-coupled disruption and hyperacetylation of chromatin, which would otherwise activate cryptic promoters within coding sequences (Carrozza et al., 2005, Nicolas et al., 2007). H3K36me2 is sufficient for localized HDAC activity on protein-coding genes in both Saccharomyces cerevisiae and Schizosaccharomyces pombe (Li et al., 2009, Suzuki et al., 2016), suggesting that H3K36me3 may have a distinct function.
How the active chromatin state is maintained and protected from aberrant inactivation is not well understood. In contrast, we have a detailed understanding of how repressed heterochromatin is maintained due largely to genetic studies in S. pombe and other model organisms. S. pombe shares many of the heterochromatin-specific histone modifications (H3K9 methylation and histone hypoacetylation) and protein components with animals and plants. Constitutive heterochromatin is found at the pericentromeric DNA repeats, telomeres, and the silent mating-type loci in S. pombe. As in all other eukaryotes studied, cis-acting DNA elements have evolved to specify the assembly of these heterochromatic regions (Beisel and Paro, 2011, Moazed, 2011). Additionally, the RNAi pathway is directly involved in the formation of heterochromatin at these loci, and it is indispensable for the stable propagation of pericentromeric heterochromatin (Grewal, 2010). The RNA-induced transcriptional silencing complex (RITS), which includes the RNAi protein Ago1, is directed to chromatin co-transcriptionally via base pairing of the Ago1-bound small RNA with complementary sequences in RNA polymerase II-generated nascent transcripts (Shimada et al., 2016). RITS recruits the sole S. pombe H3K9 methyltransferase Clr4 (Bayne et al., 2010), which methylates H3K9 to form a binding site for heterochromatin protein 1 (HP1) homologs. RITS also helps recruit an RNA-dependent RNA polymerase-containing complex to amplify the process by generating more double-stranded RNA substrate for Dcr1 (Motamedi et al., 2004, Sugiyama et al., 2005). This creates a positive feedback loop on centromeric repeats, guaranteeing high levels of H3K9 methylation and rapid turnover of centromeric RNAs into small interfering RNAs (siRNAs) to maintain a repressive chromatin state.
H3K9 methylation also promotes the recruitment of the class II HDAC Clr3, which deacetylates H3K14 and restricts the access of RNA polymerase II to heterochromatin, thus limiting transcription (Bjerling et al., 2002, Fischer et al., 2009, Motamedi et al., 2008, Sugiyama et al., 2007). In contrast, acetylation of H3K14 is associated with active chromatin, and it is mediated by the Gcn5 and Mst2 histone acetyltransferases (HATs). Interestingly, deletion of the mst2+ gene strengthens heterochromatin silencing at telomeres (Gómez et al., 2005), and it bypasses the requirement of RNAi to maintain centromeric heterochromatin (Reddy et al., 2011). Mst2 also potentiates the phenotype of cells lacking Epe1, which is a putative H3K9 demethylase (Wang et al., 2015). These results indicate that Mst2 antagonizes heterochromatin silencing; however, the underlying mechanisms are unknown.
Whereas we have an advanced understanding of RNAi-mediated maintenance of heterochromatin, relatively little is known about de novo formation of heterochromatin because this is repressed by the RNA polymerase II-associated factor 1 complex (Paf1C). In Paf1C mutant cells, siRNAs initiate gene silencing in an all-or-nothing fashion characteristic of an epigenetic silencing response. Once established, the OFF state is stably propagated even in the absence of the primary siRNAs (Kowalik et al., 2015, Shimada et al., 2016). Yet, the rate at which individual cells initiate silencing is quite low, implying the existence of additional repressive activities (Figure 1A). To isolate factors that are specifically involved in initiating heterochromatin assembly, but not maintenance, we tested previously described chromatin regulators in combination with Paf1C mutants, and we identified the HAT Mst2. We show that Mst2 represses RNAi-mediated heterochromatin formation specifically during the initiation phase of heterochromatin assembly. This is achieved by H3K36me3-dependent sequestration of Mst2 on actively transcribed genes, which is mediated by its interaction partner Pdp3. By restricting Mst2 activity to transcribed protein-coding genes, H3K36me3 maintains those in a euchromatic state. Surprisingly, we discovered that Mst2 acetylates a specific lysine in Brl1, a component of the histone H2B ubiquitin ligase complex (HULC), revealing insights into the mechanism by which Mst2 antagonizes the assembly of ectopic heterochromatin and secures epigenome integrity.
Figure 1.

Mst2 Counteracts siRNA-Directed De Novo Heterochromatin Assembly
(A) Scheme: de novo silencing in trans by siRNAs from a hairpin RNA-producing locus is repressed by Paf1C and other protein complexes.
(B) Experimental setup: white (expressed) or red (silenced) colonies were selected, and their descendants were analyzed for initiation and maintenance of the silenced state, respectively.
(C) Descendants were categorized by color, and the percentage of colonies containing non-white (red) cells was calculated. The p value was calculated using the two-sided, two-sample Student’s t test (n ≥ 3 individual white colonies). Exact numbers are listed in the STAR Methods.
(D) Silencing assays were performed with indicated mutant strains to illustrate the difference between initiation and maintenance of silencing. A representative experiment is shown. Note that quantification shown in (C) was not based on this assay, because individual colonies cannot be clearly distinguished. For (C) and (D), see the STAR Methods for details.
Results
Mst2 Counteracts Small RNA-Directed Initiation of Heterochromatin Assembly
Ectopic heterochromatin formation in S. pombe can be triggered by the temporary expression of trans-acting primary siRNAs in cells harboring mutations in Paf1C subunits; however, the frequency of de novo silencing is low (Kowalik et al., 2015). To identify additional repressors of RNAi-directed heterochromatin assembly, we tested candidate proteins that have previously been implicated in chromatin regulation. We used a reporter strain expressing an RNA hairpin (ade6-hp) complementary to 250 nt of ade6+ and harboring a nonsense mutation in the paf1+ gene (paf1-Q264Stop) (Kowalik et al., 2015). We chose ade6+ as a reporter gene because it allowed us to quantify the initiation of silencing in individual colonies that derived from single cells. When grown on limiting adenine indicator plates, cells with active or inactive ade6+ form white or red colonies, respectively. To determine the frequency of initiation of silencing, we grew single- and double-mutant strains of paf1-Q264Stop alone or in combination with a deleted candidate gene on low-adenine originator plates, selected white colonies, and seeded a given number of (white) cells at single-cell density on low-adenine assay plates (Figure 1B). Subsequent counting of the colonies containing red cells allowed us to calculate the percentage of cells that had initiated ade6+ silencing since they were seeded.
This analysis revealed that deletion of the mst2+ gene, which encodes one of two MYST family HATs (Gómez et al., 2005), dramatically increased the rate at which paf1-Q264Stop cells silenced the ade6+ reporter gene: 11% of paf1-Q264Stop cells formed red colonies (inactive ade6+), whereas more than 90% of the mst2Δ paf1-Q264Stop double-mutant cells inactivated ade6+ (Figure 1C). Importantly, although deletion of mst2+ alone did enable ade6+ silencing (Figure 1C), the repressed state was poorly maintained (Figure 1D). In contrast, ade6+ silencing was stably propagated in mst2Δ paf1-Q264Stop double-mutant cells (Figure 1D).
Therefore, we have identified Mst2 as a repressor of RNAi-mediated heterochromatin formation. Mst2 acts specifically during the initiation phase of heterochromatin assembly as, in contrast to Paf1C, it does not disrupt heterochromatin once it has been established (Figure 1D and results described hereafter).
Mst2 Prevents RNAi-Dependent Spreading of Heterochromatin
The siRNAs trigger de novo formation of heterochromatin by guiding the H3K9 methylation machinery to complementary target sites (Kowalik et al., 2015). Consistent with this, we observed high levels of H3K9me2 at the endogenous ade6+ locus in both paf1-Q264Stop single and mst2Δ paf1-Q264Stop double mutants, but not in wild-type cells (Figure 2A). As reported previously, H3K9me2 was not restricted to ade6+ but spread into neighboring regions (Kowalik et al., 2015, Shimada et al., 2016), resulting in gene repression (Figures 2A and S1A). Remarkably, this spreading was greatly enhanced in the double-mutant strain. Whereas H3K9me2 enrichments dropped to wild-type levels in paf1-Q264Stop cells within a few kilobases (kb) around ade6+, concomitant deletion of mst2+ led to a substantial increase and extensive spreading of H3K9 methylation and gene silencing up to 30 kb downstream of ade6+ (Figure 2A).
Figure 2.

Large Heterochromatin Domains Form upon Removal of mst2+
(A) Upper panel: ChIP analysis of H3K9me2 showing enrichments at the target gene ade6-704 and neighboring regions. Error bars indicate SD (n ≥ 3 independent biological replicates). The y axis is shown in logarithmic scale. Middle and lower panels: siRNAs (middle panel) and RNA (lower panel) reads mapping to the ade6-M210 locus and neighboring regions in wild-type (gray), paf1-Q264Stop (red), and paf1-Q264Stop mst2Δ cells (blue), respectively, are shown. Read counts were normalized to the total read number and are depicted in log2 (middle panel) or linear scale (lower panel).
(B) H3K9me2 enrichments at the right centromere boundary of chromosome 1 (IRC1R). ChIP enrichments are shown relative to the centromeric repeats dg/dh, which was set at 100%. Error bars indicate SD (n = 2 or 3 independent biological replicates; mst2Δ ago1Δ and mst2Δ dcr1Δ or wild-type (WT) and mst2Δ, respectively).
(C) siRNAs mapping to IRC1R and neighboring regions in wild-type (gray), paf1-Q264Stop (red), and paf1-Q264Stop mst2Δ cells (blue). Read counts were normalized to the total read number and are depicted in log2 scale.
See also Figure S1.
Another characteristic of RNAi-induced de novo formation of heterochromatin is the subsequent production of secondary siRNAs complementary to the target locus (Jain et al., 2016, Kowalik et al., 2015, Shimada et al., 2016, Simmer et al., 2010). By sequencing small RNAs, we observed that these secondary siRNAs were also more abundant in paf1-Q264Stop mst2Δ cells compared to the paf1-Q264Stop single mutant, particularly beyond the nucleation site targeted by the trans-acting primary siRNAs (Figure 2A). This suggests that de novo targeting of RITS to the neighboring genes via cis-acting secondary siRNAs mediates H3K9me2 spreading. However, H3K9me2 was still strongly enriched 20 kb downstream of the ade6+ gene in the double-mutant cells, whereas secondary siRNAs mapping to this region were barely detectable. Thus, it is possible that H3K9me2 spreads independently of siRNAs in the absence of Mst2. Alternatively, very low levels of siRNAs might be sufficient to promote spreading of heterochromatin in cis. We could not distinguish between these two possibilities at the ade6+ locus, because RNAi is absolutely necessary to maintain ectopic heterochromatin at this site (Kowalik et al., 2015, Shimada et al., 2016) (Figure S1B). Therefore, we analyzed the effect of mst2+ deletion on the boundary of constitutive heterochromatin at centromere 1.
Similar to genes flanking ade6+, we observed increased H3K9me2 at the centromeric heterochromatin IRC1R boundary and spreading to its proximal genes emc5+ and rad50+ in mst2Δ cells (Figure 2B). Thus, consistent with a previous study (Wang et al., 2015), Mst2 counteracts spreading of H3K9 methylation also at this locus. This is interesting because H3K9me2 levels are low in wild-type cells, despite abundant siRNAs that originate from the IRC1R boundary (Figure 2C) (Keller et al., 2013), indicating that Mst2 counteracts small RNA-directed initiation of H3K9 methylation at this locus as well. Notably, also here we observed H3K9me2 spreading into the more distal emc5+ and rad50+ genes without the concomitant production of high levels of secondary siRNAs (Figure 2C). Because the RNAi machinery is not required to maintain H3K9 methylation at pericentromeric dg/dh repeats in the absence of Mst2 (Reddy et al., 2011), we could delete ago1+ and dcr1+ in the mst2Δ background to test whether a functional RNAi pathway is necessary for the observed cis-spreading of H3K9me2. This revealed that RNAi is indeed essential for H3K9me2 spreading into the adjacent euchromatic regions in the absence of Mst2 (Figures 2B and S1C). Together, our results reveal that Mst2 prevents RNAi-directed de novo assembly and spreading of heterochromatin.
Mst2 Is Excluded from Constitutive Heterochromatin
Because Mst2 prevents spreading of constitutive and synthetic heterochromatin, it is unlikely that Mst2 is recruited to heterochromatin boundaries in a sequence-specific manner. To assess the localization of Mst2 genome-wide in wild-type cells, we used DNA adenine methyltransferase identification (DamID), a sensitive chromatin-profiling technique that we and others have previously adapted for use in S. pombe (Steglich et al., 2012, Woolcock et al., 2011). We generated strains that express Mst2 fused to DNA adenine methylase (Dam), and we assessed GATC methylation, and thereby Mst2 binding, throughout the S. pombe genome using tiling arrays.
Comparing Dam-Mst2-binding profiles with genome-wide H3K9 methylation data (Keller et al., 2013) revealed a striking anti-correlation (Figure 3A). Whereas Mst2 bound throughout the entire genome, it was strongly depleted from constitutive heterochromatin found at centromeres, telomeres, and the silent mating-type locus (Figure 3B). We did not observe specific Mst2 enrichment at the borders of constitutive heterochromatin, consistent with Mst2 not being a bona fide boundary factor (Figure 3C). In contrast, we observed a preferential enrichment of Mst2 on RNA polymerase II-transcribed protein-coding genes (Figure 3D). Of note, regions transcribed by RNA polymerases I and III, such as tRNA, small nucleolar RNAs (snoRNAs), and rRNAs, were depleted of Mst2, similar to heterochromatic regions. To validate these data with an alternative method, we performed chromatin immunoprecipitation (ChIP) with cells expressing C-terminally FLAG-tagged Mst2. Consistent with our DamID results, we observed Mst2 preferentially enriched on transcribed genes, including bub1+, ade6+, and vtc4+ (Figures 3E and 3F), the region targeted for de novo formation of heterochromatin by primary ade6 siRNAs in our initiation of silencing assays. Generally, Mst2 bound weakly to intergenic and promoter proximal regions and most strongly in the body of transcribed genes (Figures 3D–3F).
Figure 3.

Mst2 Is Excluded from Constitutive Heterochromatin and Specifically Associates with Transcriptionally Active Chromatin
(A) Mst2 DamID maps for all three chromosomes (bottom) compared to previously generated H3K9me2 ChIP-sequencing data (top; Keller et al., 2013). The signal of Dam-Mst2 (normalized to Dam only) was averaged over 500 probes and is shown in log2 scale. The x axis shows position on chromosomes.
(B) Enrichment of Dam-Mst2 at different genomic regions. Two independent replicates are shown (scale in log2).
(C) Zoom of DamID map from (A) on centromere of chromosome 1.
(D) Enrichment of Dam-Mst2 at different euchromatic elements. Two independent replicates are shown (scale in log2).
(E and F) ChIP enrichment of Mst2-FLAG (blue) compared to untagged Mst2 (black) over the mto1+/tef3+ locus (E) and the ade6-704 locus (F). ChIP data are shown relative to the mean of the untagged control with the background subtracted (n = 4 ± SEM).
Pdp3 Binding to H3K36me3 Confines Mst2 to Euchromatin
How Mst2 is recruited to active genes is unclear. It is known to physically interact with a number of proteins to form a complex, which includes homologs of S. cerevisiae NuA3 HAT complex subunits (Wang et al., 2012). One of its subunits, Pdp3, contains a PWWP domain (Figure 4A), which binds methylated lysines and arginines (Adams-Cioaba and Min, 2009, Maurer-Stroh et al., 2003, Vermeulen et al., 2010). Interestingly, the S. cerevisae Pdp3 homolog recognizes H3K36me3 (Gilbert et al., 2014), which is enriched in the gene body and the 3′ UTR of active genes (Bell et al., 2007). This pattern is highly reminiscent of Mst2 localization (Figure 3D). Therefore, we speculated that Mst2 is targeted to transcribed genes via Pdp3 binding to H3K36me3. Indeed, we observed a positive correlation between our Dam-Mst2 and published H3K36me3 ChIP on microarray (ChIP-on-chip) data (Wilhelm et al., 2011) (Figure 4B; R2 = 0.25). This positive correlation was completely lost upon deletion of the pdp3+ gene (Figure 4C; R2 = −0.27). Similarly, the enrichment of Mst2 on transcribed genes, including bub1+, ade6+, and vtc4+, was abolished in pdp3Δ cells when interrogated by ChIP (Figures 4D and 4E). These results are consistent with Mst2 recruitment to transcribed genes via Pdp3. This is further supported by ChIP experiments with FLAG-tagged Pdp3, which revealed very similar binding patterns for Mst2 and Pdp3 on transcribed genes (Figures 4D and 4F). Importantly, Mst2 binding at the ade6+ and bub1+ genes was abolished in set2Δ mutants (Figure 4G), demonstrating that methylation of H3K36 is necessary for the recruitment of Mst2 to actively transcribed genes. Moreover, Pdp3 binding to chromatin was lost when we introduced a single point mutation (F109A) in the bona fide recognition site of the PWWP domain (Figure 4F).
Figure 4.

Pdp3 Anchors the Mst2 Complex to Euchromatin via H3K36me3
(A) Scheme of the Mst2 complex and protein domain organization of Pdp3.
(B and C) Correlation between enrichment of Dam-Mst2 and ChIP-on-chip data of H3K36me3 in wild-type (B) and pdp3Δ cells (C). Two independent biological replicates are shown (in log2 scale). ChIP-on-chip data are from Wilhelm et al. (2011).
(D and E) ChIP enrichment of Mst2-FLAG in WT (blue) or pdp3Δ (red) cells compared to an untagged wild-type strain (black) at the mto1+/tef3+ locus (D) or the ade6-704 locus (E).
(F) ChIP enrichment of FLAG-Pdp3 (blue) or FLAG-Pdp3 mutant (pdp3-F109A, red) compared to untagged wild-type strain (black) at the mto1+/tef3+ locus.
(G) ChIP of Mst2-FLAG in WT (blue), set2Δ (purple), and set2-SRIΔ (green) cells at the ade6-704 locus.
(H) ChIP of H3K36me3 in WT (orange), set2Δ (purple), and set2-SRIΔ (green) cells at the ade6-704 locus. ChIP data in (D)–(G) are shown relative to the mean of the untagged control with the background subtracted; ChIP data in (H) are shown relative to the mean of WT over the entire chromatin region examined. All ChIP experiments have been performed with n = 3–4 ± SEM.
(I) Initiation frequencies in paf1-Q264Stop cells additionally deleted for mst2+, pdp3+, or set2+ as in Figure 1D. The p values were calculated using the two-sided, two-sample Student’s t test (n ≥ 4 independent white colonies). Exact numbers are listed in the STAR Methods.
H3K36me3 is deposited co-transcriptionally by the methyltransferase Set2 (Morris et al., 2005). Set2 is recruited to the transcribing RNA polymerase II through its conserved C-terminal Set2-Rpb1 interaction (SRI) domain (Kizer et al., 2005). Intriguingly, mutations in the SRI domain of S. pombe Set2 cause a specific loss of H3K36me3, without affecting H3K36me2 (Suzuki et al., 2016). This enabled us to determine whether Pdp3 is specific to H3K36me3 or whether it would also recognize H3K36me2. Resembling Mst2 binding, H3K36me3 was enriched throughout the gene bodies of ade6+ and bub1+ but depleted from the intergenic region. As expected, H3K36me3 was completely abolished in set2Δ cells and in set2-SRIΔ cells (Figure 4H). Importantly, Mst2 binding at the ade6+ and bub1+ genes was also abolished in both mutants (Figure 4G). Together, these results reveal that Pdp3 specifically recognizes H3K36me3, but not H3K36me2, to recruit Mst2 to transcribed genes.
Thus far we have shown that the HAT Mst2 is recruited to transcribed genes via H3K36me3 and that it acts as a suppressor of RNAi-directed heterochromatin assembly and spreading. To assess whether these functions are connected, we quantified the initiation of ade6+ silencing in mutants unable to recruit Mst2 to transcribed genes (i.e., pdp3Δ and set2Δ cells). Similar to mst2+ deletion (mst2Δ paf1-Q264Stop), deletion of pdp3+ (pdp3Δ paf1-Q264Stop) or set2+ (set2Δ paf1-Q264Stop) also initiated silencing more frequently than the paf1-Q264Stop single mutant (Figure 4I). This demonstrates a functional link between H3K36 modification and RNAi-directed heterochromatin formation. Yet, we note that although phenocopying each other, pdp3Δ and set2Δ did not fully phenocopy an mst2Δ mutant in this assay. This indicates that Mst2 remains partially repressive in the absence of H3K36me3 binding.
H3K36me3 Preserves Euchromatin and Safeguards Heterochromatic Genes from Illegitimate Activation
The foregoing results establish Pdp3 as a reader of the H3K36me3 mark that recruits Mst2 to active genes. Nevertheless, in the absence of Pdp3 and Set2 there was residual Mst2 chromatin association above background (Figures 5A and S2A). Most prominently, Mst2 exclusion from constitutive heterochromatin was largely abolished in both pdp3Δ and set2Δ cells (Figures 5A and S2A). In contrast, deletion of set1+, which encodes the histone methyltransferase that methylates H3K4, had no effect on Mst2 localization (Figure S2B).
Figure 5.

Promiscuous Mst2 Activity in the Absence of Pdp3 Attenuates Heterochromatin Silencing
(A) Mst2 DamID maps for the centromere of chromosome 1 in WT, pdp3Δ, and set2Δ cells. The signal of DamMst2 (normalized to Dam only) was averaged over 500 probes and is shown in log2 scale. The x axis shows position on chromosomes.
(B and D) RNA expression in WT, pdp3Δ, and mst2Δ cells at centromere 1 (B) and telomere 1L (D) assessed by RNA-seq. Relative read counts normalized to total read number (axis scale in log2) are shown.
(C) Relative RNA expression levels of ura4+ (qRT-PCR analysis) at the innermost repeat (imr) in indicated mutants. Shown are the transcript levels relative to WT after normalization to act1+. Data are represented as mean ± SEM from four independent biological experiments.
(E) Scheme of experimental setup for tethering Mst2 at heterochromatin: Four LexA-binding sites were inserted downstream of the ade6+ reporter at the outermost repeat (otr) of chromosome 1 (top). The ade6-M210 allele at the euchromatic endogenous locus of ade6 (bottom) was used as a reference.
(F–H) Expression levels of heterochromatic ade6+ reporter relative to euchromatic ade6-M210 allele in WT (F), pdp3Δ (G), or set2Δ (H) cells. The p values were calculated using the two-sided, two-sample Student’s t test. Error bars indicate SD (n ≥ 3 independent biological replicates).
See also Figure S2.
These results suggest that H3K36me3-mediated recruitment of Mst2 to transcribed genes serves a dual function: it prevents RNAi-mediated heterochromatin formation in euchromatin, and it sequesters Mst2 away from heterochromatin to prevent aberrant activation of heterochromatic genes. In further support of the latter, it was previously reported that deletion of set2+ or pdp3+ alleviated heterochromatin silencing (Braun et al., 2011, Chen et al., 2008, Creamer et al., 2014, Matsuda et al., 2015, Suzuki et al., 2016) (Figure S2C). To test this more directly, we inhibited Mst2 recruitment to active genes using pdp3Δ cells, and we analyzed the effect on silencing of heterochromatic genes. We observed increased expression of centromeric repeats and subtelomeric genes in pdp3Δ cells, but not in mst2Δ cells (Figures 5B–5D). Importantly, these heterochromatin-silencing defects in the pdp3Δ background were rescued by deleting mst2+ (Figures 5C, S2C, and S2D).
To further support our model that Mst2 recruitment to active genes prevents it from aberrantly activating heterochromatic genes, we fused the high-affinity DNA-binding protein LexA to wild-type Mst2 (LexA-Mst2) and catalytically inactive Mst2 (LexA-Mst2∗[E274Q]) (Reddy et al., 2011), both expressed from the endogenous mst2+ locus. In addition, we inserted an ade6+ reporter gene linked to four LexA-binding sites into pericentromeric heterochromatin on chromosome 1 by homologous recombination (Figure 5E). Tethering of LexA-Mst2, but not LexA-Mst2∗, caused a mild silencing defect of the heterochromatic ade6+ reporter (Figure 5F). Because H3K36me3 ought to sequester LexA-Mst2 away from heterochromatin, we would expect a stronger silencing defect in cells lacking Pdp3 or Set2. Indeed, ade6+ silencing was almost completely abolished in pdp3Δ (Figure 5G) and set2Δ cells (Figure 5H). Expression of the euchromatic ade6-M allele remained unaffected in those experiments (Figure S2E).
Therefore, we conclude that the Pdp3 subunit of the Mst2 complex serves two purposes: first, it focuses Mst2 activity on transcribed genes to maintain those in a euchromatic state; and second, it prevents Mst2 from functioning promiscuously and thereby safeguards heterochromatic genes from illegitimate activation.
Mst2 Acetylates Lysine 242 of the E3 Ubiquitin Protein Ligase Brl1
Mst2 is a MYST family HAT that specifically acetylates K14 on histone H3 in vitro and in vivo (Wang et al., 2012). However, H3K14 acetylation levels remained unchanged at centromeres, telomeres, or euchromatic protein-coding genes in mst2Δ cells (Figures S3A–S3C). Notably, Mst2 functions together with another HAT, Gcn5, to regulate global levels of H3K14ac (Wang et al., 2012). Thus, both HATs might interfere with the initiation of heterochromatin assembly through the acetylation of H3K14. However, we observed neither spreading of heterochromatin nor strongly enhanced initiation of heterochromatin assembly triggered by siRNAs in gcn5Δ cells (Figures S3D and S3E). Thus, the repressive effect on de novo formation of heterochromatin is unique to Mst2, and it appears to be mediated by an additional, unknown target of Mst2.
Therefore, we explored whether Mst2 also acetylates non-histone substrates using a liquid chromatography triple-stage mass spectrometry (LC-MS/MS/MS) approach (Aebersold and Mann, 2016, McAlister et al., 2014). Briefly, we extracted total protein from wild-type and HAT mutant cells and digested with Lys-C and Trypsin. The total protein digest was used to quantify global protein changes. Additionally, a fraction of the digest was enriched for peptides containing acetylated lysine using an anti-acetyl lysine-specific antibody. Before the LC-MS/MS/MS analysis on the Orbitrap Fusion Tribrid Mass spectrometer, samples were labeled with tandem mass tags (TMTs) and pooled (Figure 6A). MS-based analysis revealed that the S. pombe proteome remains largely unchanged in mst2Δ and pdp3Δ cells compared to wild-type cells (Figures S4A–S4C). The few proteins that changed in abundance were encoded by genes that reside in the vicinity of telomeric heterochromatin. As expected from our RNA measurements, protein levels of subtelomeric genes decreased in mst2Δ cells and increased in pdp3Δ cells (Figures S4A–S4C). In contrast, deletion of gcn5+ caused substantial proteome-wide changes in protein abundance (Figure S4D). These results consolidate our findings described above and highlight that Gcn5 and Mst2 have distinct roles in controlling genome expression.
Figure 6.

Mst2-Mediated Acetylation of Brl1 Represses Initiation of Heterochromatin Assembly
(A) Scheme: acetylomics workflow. Total peptides or peptides enriched for acetylation were labeled with TMT and subjected to LC-MS/MS/MS. We identified 8,926 acetylated peptides and quantified 3,933 proteins (Table S1). See the STAR Methods for more information.
(B) Volcano plot showing fold changes in pdp3Δ mst2Δ compared to WT cells. Identified acetylated Brl1 peptides are shown in red (n = 3 independent biological replicates). The x axis is shown in log2 scale.
(C) Immunodetection of H2BK119ub in different strains. Dilution series was 1/9, 1/3, and 1/1 of the respective protein extracts. Tubulin served as a loading control. A representative experiment is shown.
(D) Quantification of H2BK119ub (left) and H2B (right) levels normalized to tubulin and relative to WT (brl1+). Multiple independent biological replicates were for H2BK119ub (WT n = 5 and brl1-KR/KQ n = 7) and H2B (WT n = 3 and brl1-KR/KQ n = 6). The p values were calculated using the two-sided, two-sample Student’s t test with equal/unequal variance according to prior evaluation with the F test.
(E) Silencing assays of siRNA-directed de novo heterochromatin assembly (as described in Figure 1) in the strains indicated (close up). All strains contained a paf1+ allele. A representative experiment is shown.
(F and H) Initiation (F) and maintenance (H) frequencies in paf1-Q264Stop cells with additional mutations in mst2+ and brl1+. Assessment of initiation/maintenance frequency was as in Figure 1D. The p values were calculated using two-sided, two-sample Student’s t test (n ≥ 8 individual white colonies). Exact numbers are listed in the STAR Methods.
(G) siRNA reads mapping to the ade6-M210 locus and neighboring regions in brl1-K242R (red) and mst2Δbrl1-K242R (blue) cells. Read counts were normalized to total read number and are depicted in log2 scale.
(I and J) Dilution assays showing gradual loss of ectopic silencing at the trp1+::ade6+ locus in brl1-K242Q mutants, but not in brl1-K242R mutants. Cells were grown exponentially for 1 day (I) or 5 days (J), and equal cell numbers were plated onto yeast extract-nourseothricin (YE-Nat). A representative experiment with independent strains is shown. Different yeast strains are depicted on the right.
See also Figures S3 and S4 and Table S1.
Intriguingly, quantification of the acetyl-lysine-enriched samples revealed widespread acetylome changes in gcn5Δ compared to wild-type cells (Figure S4E). In contrast, only one lysine acetylation site was significantly decreased in mst2Δ cells (Figure 6B). We mapped this Mst2-dependent acetylation site to K242 of the E3 ubiquitin protein ligase Brl1 (Figure S4F). Importantly, Brl1 protein abundance remained unaffected in Mst2 and Pdp3 mutants (Figures S4A–S4C). Furthermore, acetylation of K242 was highly specific, because acetylation levels of other lysine residues in Brl1 (K339 and K138) remained unchanged in the absence of Mst2 (Figure 6B). Thus, in addition to H3K14 (Wang et al., 2012), Mst2 specifically acetylates the non-histone substrate Brl1 at lysine 242.
Brl1 Acetylation Antagonizes RNAi-Directed Heterochromatin Formation
Brl1 is an E3 ligase responsible for the ubiquitination of H2BK119 as part of the HULC complex, which promotes transcription and antagonizes heterochromatin silencing (Tanny et al., 2007, Zofall and Grewal, 2007). This raises the intriguing possibility that Mst2 regulates the activity of the HULC complex via the acetylation of Brl1. To test the role of Brl1 acetylation on H2B monoubiquitination directly, we substituted Brl1-K242 with glutamine (Q) or arginine (R) to mimic acetyl lysine or non-acetylated lysine, respectively. This revealed an overall reduction of H2B ubiquitination in brl1-K242R cells compared to wild-type or brl1-K242Q cells (Figures 6C and 6D). Confirming Brl1 as the sole H2B-ubiquitinating enzyme, H2BK119ub was undetectable in brl1Δ cells (Figure 6C). Moreover, H2BK119ub enrichment at the ade6+ gene was depleted specifically upon expression of ade6 siRNAs in paf1-Q264Stop cells (Figure S4G).
Next, we tested whether Brl1-K242 Q and R mutations would affect RNAi-directed heterochromatin formation. Mimicking an mst2Δ phenotype in the presence of a wild-type paf1+ allele, brl1-K242R cells enabled primary siRNAs to initiate ade6+ silencing. In contrast, brl1-K242Q cells were refractory to ade6 siRNAs (Figure 6E). Remarkably, the brl1-K242Q allele completely disabled the initiation of ade6+ silencing by trans-acting siRNAs in both mst2Δ single- (Figure 6E) and paf1-Q264Stop mst2Δ double-mutant cells (Figure 6F). In contrast, the initiation of siRNA-directed ade6+ silencing in paf1-Q264Stop mst2Δ cells was not affected by the Brl1-K242 to R mutation (Figure 6F). We further observed secondary siRNAs covering the ade6+ gene (Figure 6G), validating that the observed ade6+ silencing was indeed mediated by RNAi.
In line with the abovementioned results that Mst2 does not disrupt heterochromatin once it is established, short-term maintenance of ade6+ silencing remained largely unaffected in both Brl1-K242 Q and R mutants (Figure 6H). However, when cells were cultivated over several days, silencing was gradually lost in Brl1-K242Q, but not Brl1-K242R, mutant cells (Figures 6I and 6J). Hence, long-term maintenance of the silent state is impaired in the Brl1-K242Q mutant. We conclude that the Brl1-K242 Q mutation disables siRNA-directed re-initiation of ade6+ silencing and, thus, affects consolidation of the silent state.
These results show that mimicking acetylation of a single lysine in Brl1 abrogates small RNA-directed initiation of epigenetic gene silencing. This places Brl1 at the center of a regulatory circuit that maintains protein-coding genes in an active state. Conjugation of mono-ubiquitin to H2B has been associated with active transcription by RNA polymerase II (Jason et al., 2002, Tanny, 2014). Thus, the observed stimulatory effect on H2B ubiquitination is expected to feed back positively on transcription. Indeed, we observed that overall H3K4me3 levels were slightly elevated in brl1-K242Q compared to wild-type or brl1-K242R cells, indicating augmented transcription initiation (Figures S4H and S4I). Thus, we have discovered an unexpected positive feedback loop that maintains transcriptionally active regions of the fission yeast genome in a euchromatic state. Because much of the enzymatic machinery involved is conserved from yeast to human, we anticipate that epigenome integrity is secured through similar mechanisms also in other organisms. Although not addressed in our study, it is possible that RNAi-independent pathways that assemble silent chromatin are equally counteracted.
Discussion
Studies in a wide variety of eukaryotic systems have established the cooperation of sequence-dependent specificity factors with existing repressive histone marks to reinforce the silent chromatin state through positive feedback loops as a common principle (Gottschling, 2004, Moazed, 2011). Our study highlights that, like silent chromatin, maintenance of the active chromatin state equally depends on positive feedback. Because faithful propagation of active chromatin states through cell divisions is crucial to maintain cellular identity, we anticipate that multicellular organisms depend on similar mechanisms to shield specific cell types from differentiation signals. Below we discuss the implications of our findings on our understanding of how chromatin is partitioned into silent and active domains.
Reinforcement of the Active Chromatin State through Positive Feedback
Akin to positive feedback loops that reinforce silent chromatin states (Moazed, 2011), we propose a positive feedback system that maintains euchromatic genes in an active state that involves Paf1C and Mst2 (Figure 7A). Both Paf1C and Set2 are recruited to euchromatin co-transcriptionally through interactions with RNA polymerase II, which results in high levels of H3K36 methylation. H3K36me3 is recognized by Pdp3, leading to a high local concentration of Mst2, which acetylates K242 of the HULC subunit Brl1. HULC is required for the ubiquitination of histone H2B at lysine 119 (H2Bub), which is universally linked to active gene transcription, thereby closing the positive feedback loop. Acetylation of a non-histone protein by a HAT is intriguing and highlights that chromatin phenotypes in HAT or HDAC mutants may not necessarily be caused by histone acetylation. Because there is evidence from other systems that histone modifications deposited co-transcriptionally regulate the mechanisms that direct their formation (Tanny, 2014), we believe that consolidation of the active chromatin state through positive feedback is more prevalent than generally assumed.
Figure 7.

Mst2 Constitutes an Activation Barrier for Heterochromatin Assembly
(A) Scheme depicting the regulatory circuit that protects active chromatin from inactivating siRNAs (solid green arrows). Transcription by RNA polymerase II promotes Set2-mediated tri-methylation of H3K36 (Me3, green), which is recognized by Pdp3. This results in high local concentrations of Mst2 on actively transcribed genes. Mst2 acetylates Brl1 at lysine 242, which causes increased H2B ubiquitination and reinforced transcription. Black circles depict opposing feedback loops that maintain the inactive chromatin state (RNAi and CLRC). Relevant residues in the N-terminal tails of H3 and H2B are highlighted in blue. See the text for details.
(B) Model highlighting that a maximal transition state energy warranties stable propagation of euchromatic and heterochromatic states. Note that Mst2 and Paf1C repress distinct steps in the transition from euchromatin (A) to heterochromatin (R). The heterochromatin assembly reaction proceeds the fastest in paf1 mst2 double-mutant cells, where the activation energy (Ea) is the lowest and heterochromatin eventually reaches a low-energy state (R stable). See the text for details.
(C) Mitotic propagation of siRNA-directed silencing of the ade6+ gene monitored by red pigmentation of clones that grew on YE-Nat plates.
High Activation Energy Warrants Stable Propagation of Euchromatic and Heterochromatic States
Our recent discovery of Paf1C as a repressor of siRNA-directed heterochromatin formation (Kowalik et al., 2015) and the results presented in this study demonstrate that siRNAs are sufficient to initiate the formation of heterochromatin independently of genomically encoded silencers, but only if repressive activities in the cell are constrained. Mst2 and Paf1C repress distinct steps in the transition from euchromatin to heterochromatin. This can be best illustrated in analogy to a chemical reaction with high activation energy (Ea) and where reactants (euchromatin) and products (heterochromatin) assume different energetic states (Figure 7B). In the absence of Mst2 or Paf1C, the energy level of the transition state is reduced so that the heterochromatin assembly reaction can proceed; but, only Paf1C mutant cells reach a heterochromatic state that is energetically lower than that of euchromatin, explaining why silencing is not stably maintained in mst2 single mutants. The heterochromatin assembly reaction proceeds the fastest in paf1 mst2 double-mutant cells, where the activation energy is the lowest and heterochromatin eventually reaches a low-energy state. Yet at the same time, Ea for the reverse reaction is smaller in double mutants compared to paf1 single mutants, which predicts less stable inheritance of heterochromatin. Indeed, we observed a variegating silencing phenotype in mst2 paf1 double, but not paf1 single, mutants (Figure 7C).
This model highlights that a maximal transition state energy warranties stable propagation of both euchromatic and heterochromatic states. Thus, it does not seem surprising that previous attempts to induce epigenetically stable gene silencing by using trans-acting siRNAs have failed under wild-type conditions (Bühler et al., 2006, Iida et al., 2008, Sigova et al., 2004, Simmer et al., 2010). It is the aforesaid self-reinforcing feedback regulation that possibly underlies the high activation energy of a heterochromatin assembly reaction. We speculate that Ea might be lowered under certain conditions, enabling the organism to establish facultative heterochromatin and, thus, adapt its gene expression program for optimal fitness. It will be a challenging but exciting task to find the putative enzymes that lower Ea in such a scenario.
Conservation of H3K36 Methylation-Mediated Anti-silencing
Certain post-transcriptional histone modifications are highly correlated with transcription. Yet, it is often unclear whether active chromatin marks are simply a consequence or rather a cause of transcriptional activity. The active H3K36me3 mark is generated by the SET domain containing proteins recruited by RNA polymerase II as a consequence of transcriptional activity. Our results suggest that, in S. pombe, H3K36me3 is also a cause of transcriptional activity. Importantly, it functions to sequester Mst2 on transcriptionally active genes, which serves a dual purpose: (1) it fuels the euchromatic positive feedback loop described above, and (2) it prevents Mst2 from acting promiscuously on constitutive heterochromatin. Thereby, H3K36me3 maintains euchromatic genes in an active state, and it concurrently safeguards constitutive heterochromatin from illegitimate activation by an invasion of Mst2, providing an explanation for the previously reported silencing defects in set2+-deficient cells (Chen et al., 2008, Creamer et al., 2014, Matsuda et al., 2015, Suzuki et al., 2016).
In S. cerevisiae, the methylation of H3K36 by Set2 protects euchromatin from silencing by spreading of the Sir complex from neighboring silent chromatin independently of the Rpd3S HDAC complex (Tompa and Madhani, 2007). This highlights that H3K36 methylation functions through different effector mechanisms that may be conserved. Indeed, SETD2-mediated tri-methylation of H3K36 targets the de novo DNA methyltransferase DNMT3B to transcribed genes in mouse embryonic stem cells (Baubec et al., 2015). Similar to Pdp3-mediated recruitment of Mst2 to active genes in S. pombe, the PWWP domain of DNMT3B is likely to specify recruitment of DNA methylation activity to transcribed genes via interactions with methylated H3K36 (Baubec et al., 2015). The functional relevance of genic DNA methylation in mammalian cells is just being unraveled (Neri et al., 2017). If it had stimulatory effects on transcription, it would also constitute a positive feedback regulatory system. Finally, the polycomb repressive complex 2 (PRC2), reconstituted from humans, flies, mouse, and plants, is directly inhibited by H3K36 methylation (Schmitges et al., 2011, Yuan et al., 2011). Thus, it is tempting to speculate that methylation of H3K36 is an evolutionarily conserved strategy to maintain euchromatin in an active state. In light of this hypothesis, it is intriguing that human H3K36 methyltransferases have been implicated in a wide range of cancers (Papillon-Cavanagh et al., 2017, Schneider et al., 2002, Wagner and Carpenter, 2012). Moreover, the H3K36 to M mutation is seen in 95% of chondroblastomas and promotes sarcomagenesis through altering polycomb-mediated gene silencing (Behjati et al., 2013, Lu et al., 2016). Failure to partition chromatin into silent and active domains as a result of defective H3K36 methylation may, hence, contribute to the initiation and progression of human disease. Therefore, a detailed understanding of the mechanisms that control the active state of chromatin is extremely relevant to human health.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-H3K9me2 | (Kimura et al., 2008) | N/A |
| anti-FLAG | Sigma | Cat# F3165; RRID: AB_259529 |
| anti-H3K14ac | Abcam | Cat# ab52946; RRID: AB_880442 |
| anti-H3K36me3 | Abcam | Cat# ab9050; RRID: AB_306966 |
| anti-H3 (ChIP) | Active Motif | Cat# 61475 |
| anti-H2B | Active Motif | Cat# 39238; RRID: AB_2631110 |
| anti-H2BK119ub | Cell Signaling | Cat# 5546 |
| anti-H3K4me3 | Abcam | Cat# ab8580; RRID: AB_306649 |
| anti-H3 (Western) | Abcam | Cat# ab1791; RRID: AB_302613 |
| anti-tubulin | (Woods et al., 1989) | N/A |
| goat anti-mouse IgG (H + L)-HRP conjugate | Bio-Rad | Cat# 1706516; RRID: AB_11125547 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson Immunoresearch | Cat# 115-035-146; RRID: AB_2307392 |
| Peroxidase AffiniPure Goat Anti-Rabbit IgG (H+L) | Jackson Immunoresearch | Cat# 111-035-144; RRID: AB_2307391 |
| Acetyl-Lysine Motif Kit | Cell Signaling | Cat# 13416S |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-Fluoroorotic Acid (FOA) | US biological, Thermo Fisher | Cat# 207291-8-4 |
| nourseothricin dihydrogen sulfate (NAT) | Fisher or WERNER BioAgents GmbH | Cat# 5029426 or Cat# 5.0 |
| G418 sulfate (Geneticin) | Roche or Invitrogen/Life Technologies | Cat# 04727878001-2 or Cat# 10131027 |
| Hygromycin | Sigma or Invitrogen/Life Technologies | Cat# H7772 or Cat# 10687010 |
| PrimeSTAR GXL DNA Polymerase | Clontech | Cat# R050A |
| Taq DNA Polymerase | NEB | Cat# M0267 |
| Formaldehyde | Sigma or Carl Roth | Cat# F8775 or Cat# 4979 |
| PMSF | Sigma | Cat# P7626 |
| cOmplete Protease Inhibitor Cocktail | Roche | Cat# 11836145001 |
| AEBSF (Pefabloc SC) | Roche | Cat# 11585916001 |
| Dynabeads M-280 Sheep anti-mouse IgG | Thermo Fisher | Cat# 11202D |
| Dynabeads Protein G | Thermo Fisher/ Life Technologies | Cat# 10009D |
| Leupeptin hemisulfate | Carl Roth | Cat# CN33 |
| Proteinase K | Roche | Cat# 3115879001 |
| RNase A | Roche | Cat# 10109169001 |
| SuperScript III | Thermo Fisher/ Life Technologies | Cat# 18080085 |
| Acrylamid/BIS solution (30%) 37.5:1 | Serva | Cat# 10688 |
| Titriplex III (EDTA) | Merck Millipore | Cat# 108418 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | Cat# 172-5274 |
| PowerUp SYBR Green Master Mix | Life Technologies | Cat# A25742 |
| Zymolyase | Fischer | Cat# 6064819 |
| Lysing enzyme from Trichoderma harzianum | Sigma | Cat# L1412 |
| DpnI | NEB | Cat# R0176 |
| DpnII | NEB | Cat# R0543 |
| T4 DNA ligase | Roche | Cat# 10481220001 |
| Lys-C | Wako Chemicals | Cat# 125-05061 |
| Trypsin | Thermo Fisher | Cat# 20233 |
| SEP-PAK | Waters | Cat# WAT036790 |
| YMC Triart C18 0.5 × 250 mm column | YMC Europe GmBH | Cat# TA12S0325J0AU |
| PepMap 100 C18 2 cm trap | Thermo Fisher | Cat# 164946 |
| EASY-Spray C18 column | Thermo Fisher | Cat# ES801 |
| Bolt 4-12% Bis-Tris Plus Gels | Thermo Fisher | Cat# NW04127BOX |
| Immobilon Western Chemiluminescent HRP Substrate | Millipore | Cat# WBKLS0500 |
| Immobilon-P Membran, PVDF, 0,45 μm | Merck Millipore | Cat# IPVH00010 |
| Critical Commercial Assays | ||
| MasterPure Yeast RNA Purification Kit | Epicenter | Cat# MPY03100 |
| Bio-Rad Protein Assay Dye Reagent Concentrate | Bio-Rad | Cat# 500-0006 |
| TruSeq Small RNA library preparation kit | Illumina | Cat# RS-200-0012 |
| TruSeq Stranded mRNA library preparation kit | Illumina | Cat# RS-122-2101 |
| DNeasy Blood and Tissue Kit | QIAGEN | Cat# 69506 |
| RNeasy Midi Kit | QIAGEN | Cat# 75144 |
| GeneChip Hybridization, Wash, and Stain Kit | Affymetrix | Cat# 900720 |
| Turbo DNA free | Thermo Fisher/ Life Technologies | Cat# AM1907 |
| ChIP DNA Clean & Concentrator | Zymo Research | Cat# D5201 |
| Deposited Data | ||
| smallRNA and poly(A) mRNA sequencing data | This study | GEO: GSE93434 |
| DamID data | This study | GEO: GSE93434 |
| Mendeley Data dataset (original unprocessed Western Blot images) | This study | http://dx.doi.org/10.17632/98ywc24xv7.1 |
| Mass spectrometry raw data | This study | ProteomeXchange: PXD005714 |
| H3K36me3 ChIP-chip data | (Wilhelm et al., 2011) | ArrayExpress: E-TABM-946 |
| Experimental Models: Organisms/Strains | ||
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX | (Kowalik et al., 2015) | spb464 |
| h+leu1-32 ura4-D18 ade6-M210 trp1+::ade6+nmt1+::ade6-hp+::natMX | (Kowalik et al., 2015) | spb1788 |
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX paf1-sms8::LEU2 | (Kowalik et al., 2015) | spb2047 |
| h+leu1-32 ura4-D18 ade6-704 or 210 trp1+::ade6+nmt1+::ade6-hp+::natMX Paf1-SMS8::kanMX | (Kowalik et al., 2015) | spb2076 |
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX paf1-sms8::LEU2 mst2Δ::kanMX | This study | spb2151 |
| h+leu1-32 ura4-D18 ade6-M210 or 704 trp1+::ade6+nmt1+::ade6-hp+::natMX mst2Δ::kanMX paf1-sms8::LEU2 | This study | spb2630 |
| h+leu1-32 ura4-D18 ade6-M210 trp1+::ade6+nmt1+::ade6-hp+::natMX mst2Δ::kanMX | This study | spb2094 |
| h+leu1-32 ura4-D18 ade6-M210 trp1+::ade6+nmt1+::ade6-hp+::natMX clr4Δ::hphMX | (Kowalik et al., 2015) | spb1950 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX | (Keller et al., 2013) | spb1591 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA (C. albicans) mst2Δ::hphMX | This study | spb1719 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX clr4Δ::kanMX | This study | spb1754 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 or M216 ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX dcr1Δ::kanMX | This study | spb1776 |
| h? mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 or 216 ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX ago1Δ::kanMX | This study | spb1755 |
| h+otr1R(SphI)::ura4+ura4-DS/E ade6-M210 leu1Δ::nmt1(81x)-dam-myc::kanMX | (Woolcock et al., 2011) | spb492 |
| h+otr1R(SphI)::ura4+ura4-DS/E leu1-32 ade6-M210 leu1Δ::nmt1(81x)-dam-myc-mst2::kanMX | This study | spb2104 |
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+ | (Kowalik et al., 2015) | spb426 (PSB1782) |
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+mst2-CBP-2xFLAG::natMX | This study | PSB1855 |
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+mst2-FLAG::natMX set2Δ::kanMX | This study | PSB1870 |
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+mst2-FLAG::natMX pdp3D::kanMX | This study | PSB1871 |
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+mst2-FLAG::natMX set2-SRIΔ::kanMX | This study | PSB1882 |
| h+imr1L(NcoI)::ura4+otr1R(SphI)::ade6+leu1-32 ura4-DS/E ade6-M210 | (Ekwall et al., 1999) | PSB65 |
| h+imr1L(NcoI)::ura4+otr1R(SphI)::ade6+leu1-32 ura4-DS/E ade6-M210 natMX::CBP-2xFLAG-pdp3 | This study | PSB1696 |
| h+imr1L(NcoI)::ura4+otr1R(SphI)::ade6+leu1-32 ura4-DS/E ade6-M210 pdp3Δ::natMX::CBP-2xFLAG-pdp3_F109A | This study | PSB1698 |
| h+otr1R(SphI)::ura4+ura4-DS/E leu1-32 ade6-M210 leu1Δ::nmt1(81x)-dam-myc-mst2::kanMX pdp3Δ::natMX | This study | spb2212 |
| h? leu1-32 ura4-D18 ade6-704 or ade6M210 trp1+::ade6+ nmt1+::ade6-hp::natMX paf1-sms8::LEU2 pdp3Δ::kanMX | This study | spb2647 |
| h? leu1-32 ura4-D18 ade6-704 or ade6M210 trp1+::ade6+ nmt1+::ade6-hp+::natMX paf1-sms8::LEU2 set2Δ::kanMX | This study | spb2646 |
| h+otr1R(SphI)::ura4+ura4-DS/E leu1-32 ade6-M210 leu1Δ::nmt1(81x)-dam-myc-mst2::kanMX set2Δ::natMX | This study | spb2220 |
| h+otr1R(SphI)::ura4+ura4-DS/E leu1-32 ade6-M210 leu1Δ::nmt1(81x)-dam-myc-mst2::kanMX set1Δ::natMX | This study | spb2239 |
| h+leu1-32 ura4-D18 ade6-M210 trp1+::ade6+nmt1+::ade6-hp+::natMX pdp3Δ::kanMX | This study | spb2319 |
| h-SPSQ (cyhR) SPL42 (cyhS) hphMX::cen1 imr1L(NcoI)::ura4+otr1R(SphI)::ade6+leu1-32 ura4-DS/E ade6-M210 | (Barrales et al., 2016) | PSB582 |
| h-SPSQ (cyhR) SPL42 (cyhS) hphMX::cen1 imr1L(NcoI)::ura4+otr1R(SphI)::ade6+leu1-32 ura4-DS/E ade6-M210 pdp3Δ::natMX | This study | PSB689 |
| h-SPSQ (cyhR) SPL42 (cyhS) hphMX::cen1 imr1L(NcoI)::ura4+otr1R(SphI)::ade6+leu1-32 ura4-DS/E ade6-M210 mst2Δ::natMX | This study | PSB1122 |
| h-SPSQ (cyhR) SPL42 (cyhS) hphMX::cen1 imr1L(NcoI)::ura4+otr1R(SphI)::ade6+leu1-32 ura4-DS/E ade6-M210 pdp3D::natMX mst2Δ::kanMX | This study | PSB2099 |
| h? otr1R(SphI)::ade6-4LexAb.s. (binding site) leu1-32 ura4-D18 or DS/E ade6-M210 | This study | spb2835 |
| h+ otr1R(SphI)::ade6-4LexAb.s. leu1-32 ura4-D18 or DS/E ade6-M210 LexA-mst2 | This study | spb2821 |
| h+ otr1R(SphI)::ade6-4LexAb.s. leu1-32 ura4-D18 or DS/E ade6-M210 LexA-mst2(E274Q) cat.dead | This study | spb2822 |
| h? otr1R(SphI)::ade6-4LexAb.s. leu1-32 ura4-D18 or DS/E ade6-M210 pdp3Δ::kanMX | This study | spb2836 |
| h+ otr1R(SphI)::ade6-4LexAb.s. leu1-32 ura4-D18 or DS/E ade6-M210 LexA-Mst2 pdp3Δ::kanMX | This study | spb2804 |
| h+ otr1R(SphI)::ade6-4LexAb.s. leu1-32 ura4-D18 or DS/E ade6-M210 LexA-mst2(E274Q) cat.dead pdp3Δ::kanMX | This study | spb2852 |
| h- leu1-32 ura4-D18 or DS/E otr1R(SphI)::ade6-4LexAb.s. ade6-M210 set2Δ::kanMX | This study | spb2881 |
| h+ otr1R(SphI)::ade6-4LexAb.s. leu1-32 ura4-D18 or DS/E ade6-M210 set2Δ::kanMX LexA-mst2 | This study | spb2885 |
| h? otr1R(SphI)::ade6-4LexAb.s. leu1-32 ura4-D18 or DS/E ade6-M210 set2Δ::kanMX LexA-mst2(E274Q) cat.dead | This study | spb2894 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA3 (C. albicans) gcn5Δ::kanMX | This study | spb2101 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA3 (C. albicans) pdp3Δ::kanMX | This study | spb2153 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX pdp3Δ::kanMX | This study | spb2115 |
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX brl1-K242R | This study | spb2982 |
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX brl1-K242Q | This study | spb2983 |
| h+leu1-32 ura4-D18 ade6-M210 trp1+::ade6+nmt1+::ade6-hp+::natMX mst2Δ::kanMX brl1-K242Q | This study | spb2984 |
| h+leu1-32 ura4-D18 ade6-M210 or ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX mst2Δ::kanMX brl1-K242R | This study | spb3023 |
| h-leu1-32 ura4-D18 ade6-M210 or ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX mst2Δ::kanMX paf1SMS8::LEU2 brl1-K242R | This study | spb3024 |
| h-leu1-32 ura4-D18 ade6-M210 or ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX paf1-sms8::LEU2 mst2Δ::kanMX brl1-K242Q | This study | spb2996 |
| h+leu1-32 ura4-D18 ade6-M210 trp1+::ade6+nmt1+::ade6-hp+::natMX mst2Δ::kanMX dcr1Δ::hphMX | This study | spb2364 |
| h+leu1-32 ura4-D18 ade6-M210 trp1+::ade6+nmt1+::ade6-hp+::natMX dcr1Δ::hphMX paf1sms8::LEU2 mst2Δ::kanMX | This study | spb2661 |
| h-leu1-32 ura4-D18 ade6-M210 trp1+::ade6+nmt1+::ade6-hp+::natMX dcr1Δ::hphMX paf1sms8::LEU2 mst2Δ::kanMX | This study | spb2662 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX arb2Δ::kanMX | This study | spb1739 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX arb1Δ::kanMX | This study | spb1740 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX cid12Δ::kanMX | This study | spb1741 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX ers1Δ::kanMX | This study | spb1745 |
| h90mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX stc1Δ::kanMX | This study | spb1747 |
| h? mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-? ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX tas3Δ::kanMX | This study | spb1753 |
| h? mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-? ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX chp1Δ::kanMX | This study | spb1756 |
| h? mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-? ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX rik1Δ::kanMX | This study | spb1757 |
| h? mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-? ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMXD hrr1Δ::kanMX | This study | spb1772 |
| h90 mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-M210 ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX rdp1Δ::kanMX | This study | spb1774 |
| h? mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-? ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX raf1Δ::kanMX | This study | spb1802 |
| h? mat3::GFP-natMX (ura4 promoter and adh1 terminator) ura4-DS/E leu1-32 ade6-? ncRNA.95Δ::URA3 (C. albicans) mst2Δ::hphMX raf2Δ::kanMX | This study | spb1803 |
| h+SPSQ (cyhR) hphMX::cen1 imr1L(NcoI)::ura4+otr1R(SphI)::ade6+leu1-32 ura4-DS/E ade6-M210 clr3Δ::kanMX | This study | PSB1524 |
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX gcn5Δ::kanMX | This study | spb2404 |
| h-leu1-32 ura4-D18 ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX gcn5Δ::kanMX paf1SMS8::LEU | This study | spb2443 |
| h+leu1-32 ura4-D18 ade6-M210 or ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX paf1-sms8::LEU2 mst2Δ::kanMX brl1-K242Q | This study | spb3115 |
| h+leu1-32 ura4-D18 ade6-M210 or ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX paf1-sms8::LEU2 mst2Δ::kanMX brl1-K242Q | This study | spb3116 |
| h+leu1-32 ura4-D18 ade6-M210 or ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX paf1-sms8::LEU2 mst2Δ::kanMX brl1-K242Q | This study | spb3117 |
| h+leu1-32 ura4-D18 ade6-M210 or ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX paf1-sms8::LEU2 mst2Δ::kanMX brl1-K242R | This study | spb3118 |
| h+leu1-32 ura4-D18 ade6-M210 or ade6-704 trp1+::ade6+nmt1+::ade6-hp+::natMX paf1-sms8::LEU2 mst2Δ::kanMX brl1-K242R | This study | spb3119 |
| Oligonucleotides | ||
| Primers | This study | Table S2 |
| Recombinant DNA | ||
| pFa6a 81xnmt1 - Dam - mst2 | This study | pMB1436 |
| pJR1 - 3xL - LexA - Mst2 gDNA | This study | pMB1636 |
| pJR1 - 3xL - LexA - Mst2 mut (E274Q)-gDNA | This study | pMB1639 |
| Software and Algorithms | ||
| ImageJ 1.47v | (Schneider et al., 2012) | N/A |
| R | (Team, 2014) | https://www.r-project.org/ |
| QuasR | (Gaidatzis et al., 2015) | N/A |
| Proteome Discoverer 2.1 software | Thermo Fisher | Cat# IQLAAEGAB SFAKJMAUH |
| Other | ||
| Illumina HiSeq2500 | Illumina | N/A |
| GeneChip Scanner 3000 7G System | Affymetrix | Cat# 00-0213 |
| Agilent 1100 system | Agilent | Cat# DE33201061 |
| Orbitrap Fusion Tribrid | Thermo Fisher | Cat# IQLAAEGA APFADBMBCX |
| Easy nLC 1000 system | Thermo Fisher | Cat# LC-010190 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marc Bühler (marc.buehler@fmi.ch).
Experimental Model and Subject Details
Schizosaccharomyces pombe strains used in this study are derivatives of the standard laboratory strain 972 and are listed in the Key Resources Table. Cultures were grown at 30°C in liquid YES media (160 rpm, 12-24 hr) or at 30°C on solid YES agarose plates (for 3 days).
Method Details
Strains and Plasmid Construction
All strains were constructed using the PCR-based protocol (Bähler et al., 1998) or by standard mating and sporulation. Brl1-K242R/Q point mutants were generated by first deleting the ORF with URA3 from Candida albicans and then reinserting the mutated ORF into the same locus by FOA counter-selection. LexA-Mst2 and LexA-Mst2∗ strains were generated by deleting the mst2+ ORF and reinserting mst2(∗) fused LexA from a plasmid by homologous recombination. FLAG-Pdp3, FLAG-Pdp3-F109A point mutant and Mst2-FLAG were similarly generated by first deleting the respective ORF with a kanMX cassette; subsequently the kanMX cassette was replaced by inserting the FLAG-fusion together with a natMX selection marker. In case of Pdp3, the natMX marker is upstream of the 5′ UTR of pdp3+. In the case of Mst2, the selection marker is downstream of the mst2+ ORF (between the FLAG-tag ADH1 terminator and the 3′UTR of mst2+). Western Blots to assess FLAG-tagged protein levels were deposited in Mendeley Data, http://dx.doi.org/10.17632/98ywc24xv7.1.
Silencing Assays
For ade6+ reporter silencing assays, cells were spotted on YES and YE-Nat plates (containing 100 ug/mL nourseothricin) in a ten-fold serial dilution (initial seeded cell number 105 cells) and grown for four days. White colonies were picked to perform dilution assays for initiation of silencing, whereas red colonies were picked to visualize maintenance of silencing.
Serial dilutions of the strains indicated in Figure S1C were plated on PMGc (nonselective, NS) or on PMGc plates containing 2 mg/mL 5-Fluoroorotic Acid (FOA). For ura4+ reporter assays in Figure S2C, cells were plated on EMM (non-selective, NS) or EMM containing 1 mg/mL FOA. The strains were grown at 30°C for three and four days, respectively.
Quantification of silencing frequency
In order to quantify the initiation and maintenance frequency of the silent state in different strains either a single cell-derived white (for initiation) or red (for maintenance) colony was selected. The single colony was resuspended in water and 50 - 500 cells were seeded on YE-Nat plates, which were incubated at 30°C for 5 days. Colonies were counted/categorized after an additional over night incubation at 4°C. Characterization distinguished between white and non-white cells (which could be either red, pink or variegating) and the relative percentage of white cells was used for visualization by boxplots. Multiple individual colonies were quantified for each strain ( = independent biological replicate): n = 3 (wild-type), n = 4 (paf1-Q264Stop,pdp3Δ; paf1-Q264Stop,set2Δ), n = 5 (gcn5Δ), n = 8 (paf1Q-264Stop,brl1-K242R,mst2Δ), n > 10 (mst2Δ; paf1-Q264Stop; paf1-Q264Stop,mst2Δ; paf1-Q264Stop,gcn5Δ; paf1Q-264Stop,brl1K242Q,mst2Δ).
ChIP-qPCR
ChIP experiments with H3K9me2 were performed as described in Bühler et al. (2006), using 2 μg of an anti-H3K9me2 antibody (Kimura et al., 2008). Briefly, 50 mL of exponentially growing cells were harvested at OD = 1.2 and crosslinked with 1% Formaldehyde for 15 min at room temperature. Cell pellets were lysed in ChIP lysis buffer (50 mM HEPES KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 1 mM PMSF, 1x Roche cOMPLETE protease inhibitor cocktail) using a Bead-beater. Lysates were sonicated 13 × 30 s (60 s off) in a Bioruptor and centrifuged for 1x 5 min and 1x 15 min while proceeding with the supernatant. Protein concentration was determined using the Bio-Rad Assay and equal protein amounts were incubated with antibody for 2 hr and with 25 ug Dynabeads (Sheep anti-mouse IgG). Washes were performed three times with lysis buffer, once with wash buffer (10 mM Tris/HCl pH 8, 250 mM LiCl, 0.5% NP40, 0.5% sodium deoxycholate and 1 mM EDTA) and once with TE buffer. Eluates were de-crosslinked in TE and 1% SDS over night at 65°C and subsequently treated with RNase A (0.2 mg/mL) for 1 hr at 37°C and 0.1 mg protease K for 1 hr at 65°C. DNA was purified using phenol/chloroform extraction and real-time qPCR performed on the eluates using the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). Enrichment was calculated by normalization to the adh1+ locus in the clr4Δ mutant that lacks H3K9me2.
ChIP experiments with FLAG-tagged Mst2 or Pdp3, H3K36me3, H3K14ac and H3 were essentially conducted as described in Barrales et al. (2016), using a Q800R1 sonicator (QSonica) for chromatin shearing (30 min, 30 s on /off cycles, 90% amplitude). For each IP, 2 μg of the following antibodies was used (cell lysates corresponding to different amounts of OD600 in brackets): anti-FLAG (Sigma F3165; 30 ODs); anti-H3K14ac (Abcam ab52946, 10 ODs); anti-H3K36me3 (Abcam ab9050, 5 ODs); anti-H3 (Active Motif 61475, 5 ODs). For ChIP experiments with Mst2-FLAG, 4 μg of anti-FLAG antibody and cell lysates corresponding to 50 OD600 were used. DNA was immunoprecipitated with Dynabeads Protein G (Life Technologies) and quantified by qPCR using the PowerUp SYBR Green Master Mix (Life Technologies) and a 7500 Fast Real-Time PCR System (Applied Biosystems). Datasets from each independent experiment (n = 3-4) were standardized using an experimental normalization by defining a global mean value for ChIP efficiency. This global mean value includes all qPCR amplicons (used for each tiling array) from the entire sample pool of strains (wt and mutant strains used in each experiment). For ChIP experiments with FLAG-tagged Mst2 and Pdp3, the raw values were first normalized against mitochondrial DNA as an internal control before applying the same calculations as above. The results are shown with the background subtracted. As background signal, we used for each amplicon the mean value of the untagged strain and the pdp3Δ (or pdp3-F109A) mutant, which significantly reduced the noise in the background-corrected data (as compared to the untagged control only). For ChIP with H3K14ac and H3, the raw values were also normalized against input and mitochondrial DNA; these normalized data are presented relative to the mean value of the wild-type for each amplicon.
small and poly(A)-RNA sequencing
Briefly, total RNA was isolated from exponentially growing cells with the hot phenol method. For small RNA-sequencing, the RNA was fractionated with RNeasy Midi columns (QIAGEN) according to the RNA cleanup protocol provided by the manufacturer. The flow-through fraction was precipitated (‘small-RNA’ fraction). The RNA retained on the column was eluted and ethanol-precipitated (‘large-RNA’ fraction). 25 ug of the small-RNA fraction was separated by 17.5% PAGE and the 15-28 nt fraction excised and purified. Libraries were prepared using the TruSeq Small RNA and TruSeq Stranded mRNA library preparation kits from Illumina for sRNA- and mRNA-Seq, respectively. Following the isolation of the 145-nt to 160-nt population, the libraries were sequenced on an Illumina HiSeq2500. Reads were processed, normalized and analyzed using QuasR (Gaidatzis et al., 2015) with two mismatches allowed.
DamID and Microarray analysis
DamID was performed as described in Woolcock et al. (2011). Briefly, strains expressing either unfused Dam or Dam fusion proteins were grown to OD = 0.4. Approximately 5.3 × 107 cells were harvested, washed once with water and flash frozen in liquid nitrogen. Cells were spheroplasted in 500 μL spheroplast buffer (1.2 M sorbitol, 100 mM KHPO4, pH 7.5, 0.5 mg/ml Zymolyase (Zymo Research), 1 mg/ml lysing enzyme from Trichoderma harzianum (Sigma)). Genomic DNA was isolated using the DNeasy Blood and Tissue Kit (QIAGEN). gDNA was first digested with DpnI (NEB) before ligation of PCR adapters and subsequent DpnII digestion and final PCR amplification. Fragmentation and labeling was done using the GeneChip Whole Transcript Double-Stranded DNA Terminal Labeling Kit (Affymetrix). The fragmented and labeled DNA was hybridized to GeneChip S. pombe Tiling 1.0FR Arrays (Affymetrix). Average enrichment values were calculated for all oligos overlapping the major heterochromatic regions: mating type locus (chromosome 2, 2′114’000-2′137’000), telomeres (chromosome 1, 1-20’000 and 5′571’500-5′579’133; chromosome 2, 4’516’200-4’539’804), centromeres (chromosome 1, 3′753’687-3′789’421, chromosome 2, 1’602′264-1’644’747, chromosome 3, 1’070’904-1’137’003) and subtelomeres (chromosome1, 20’001-35’600 and 5′530’001-5′571’500; chromosome2, 1-15800 and 4497201-4516200). R scripts are available on request.
Microarray data was taken from Wilhelm et al. (2011) (ArrayExpress: E-TABM-946) and processed according to Woolcock et al. (2011), including a pseudocount of 64 to reduce background. R scripts are available upon request.
RT-qPCR
RNA isolation, reverse transcription and RT-qPCR were performed as described previously in Barrales et al. (2016) and Kowalik et al. (2015). For analysis, act1+-normalized datasets were standardized against the mean of a sample pool of strains (wt and mutant strains) from each experiment. These results are shown as relative to the mean value of the wt (which is set to 1). qPCR primers used in this study are listed in Table S2.
Acetylomics
Experimental procedure
Briefly, 400 mL of indicated strains were grown to mid-late growth phase (OD = 1.5), harvested in lysis buffer (50 mM Tris, pH7.5, 150 mM NaCl, 1 mM EDTA, 1x mini complete protease inhibitor cocktail (Roche)) and frozen in liquid N2. Cells were ground in a liquid nitrogen-chilled steel container for 3 × 3 min at 30 Hz using a Retsch MM 400 Ball Mill (Retsch, Haan, Germany) in presence of 1% NP-40 and 0.1% Na-deoxycholate (final concentration). Cell lysates were incubated at 4°C for 15 min and then centrifuged for 15 min at 8’000 g. Proteins in the supernatant were precipitated with > 4 volumes of acetone overnight at −20°C. Protein pellets were resuspended in 8M guanidine hydrochloride and 50 mM HEPES pH 8.5. Approximately 80-120 mg of proteins were reduced, alkylated and digested with Lys-C (Wako Chemicals) and Trypsin (Thermo Fisher) at 37°C. Peptides were purified using a SEP-PAK (Waters) and eluted in 50% acetonitrile in water. 15 mg of peptides were subjected to immunoprecipitation using the PTMScan Acetyl-Lysine Motif Kit from Cell Signaling Kit (13416S). Acetylated peptides were enriched and eluted according to the manufacturer’s instructions. Eluted peptides were labeled with TMT 10plex isobaric labeling reagents (Thermo Fisher) as described in the manufacturer’s instructions. To determine global proteome changes 20 μAc of each sample prior acetyl enrichment was labeled with TMT reagents. TMT labeled peptides were subjected to high pH offline fractionation on a YMC Triart C18 0.5 × 250 mm column (YMC Europe GmbH) using the Agilent 1100 system (Agilent Technologies). 72 fractions were collected for each experiment and concatenated into 24 fractions as previously described (Wang et al., 2011). For each LC-MS analysis, approximately 1 μg of peptides were loaded onto a PepMap 100 C18 2 cm trap (Thermo Fisher) using the Proxeon NanoLC-1000 system (Thermo Fisher). On-line peptide separation was performed on the 15 cm EASY-Spray C18 column (ES801, Thermo Fisher) by applying a linear gradient of increasing ACN concentration at a flow rate of 150 nL/min.
Spectra Acquisition
An Orbitrap Fusion Tribrid (Thermo Fisher) mass spectrometer was operated in a data-dependent mode and TMT reporter ions were quantified using a synchronous precursor selection (SPS)-based MS3 technology, as previously described (McAlister et al., 2014). In brief, the top 10 most intense precursor ions from the Orbitrap survey scan were selected for collision-induced dissociation (CID) fragmentation. The ion-trap analyzer was used to generate the MS2 CID spectrum from which the notches for the MS3 scan were selected. The MS3 spectrum was recorded using the Orbitrap analyzer at a resolution of 60000.
Data processing
Thermo RAW files were processed using Proteome Discoverer 2.1 software (Thermo Fisher), as described in the manufacturer’s instruction. Briefly, the Sequest search engine was used to search the MS2 spectra against the Schizosaccharomyces pombe UniProt database (downloaded on 30/01/2015) supplemented with common contaminating proteins. For total proteome analysis, cysteine carbamidomethylation and TMT tags on lysine and peptide N-termini were set as static modifications, whereas oxidation of methionine residues and acetylation protein N-termini were set as variable modifications. For acetyl-lysine enriched sample analysis, lysine acetylation and lysine TMT tags were set as variable modifications, while other modifications were set the same as for the proteome analysis. The assignments of the MS2 scans were filtered to allow 1% FDR. For reporter quantification, the S/N values were corrected for isotopic impurities of the TMT reagent using the values provided by the manufacturer. The sums across all TMT reporter channels were normalized assuming equal total protein content in each sample for proteome analysis, whereas for acetylome analysis normalization was based on total amount of acetylated peptides. All identified peptides from the proteome and acetylome experiments in this study are listed in Table S1.
Western Blotting
For all proteins examined (unless otherwise indicated), total proteins from exponentially growing cells were extracted using TCA and resuspended in 1M Tris-HCl pH 8.0. Protein concentrations were estimated by Bio-Rad Protein Quantification Assay (Bio-Rad). 5x Laemmli buffer was added to a final concentration of 1x and samples boiled for 5 min at 95°C before separation by SDS-PAGE on a Bolt 4%–12% Bis-Tris gradient gel. Subsequently, proteins were plotted onto a PVDF membrane (Millipore). Antibodies for immunodetection were used at the following concentrations: total H2B (Active Motif, 39238, 1:5′000), H2BK119ubiquitin (Cell Signaling #5546, 1:3′000), H3K4me3 (Abcam, ab8580, 1:2′000), total H3 (Abcam, ab1791, 1:3′000), tubulin (Woods et al., 1989) (1:4’000), HRP-conjugate goat anti-rabbit or goat anti-mouse IgG (Jackson ImmunoResearch, 1:10000). Antibody detection was performed using Millipore Immobilon HRP substrate using the Azure Biosystem c400 Imaging System or the ImageQuant LAS-3000 (GE Healthcare). Equal expression levels of Mst2-FLAG in wt and pdp3Δ cells were validated by quantification using ImageJ. For the F109 point mutant of FLAG-tagged Pdp3, we noticed a two-fold decrease (see original blots in Mendeley Data, http://dx.doi.org/10.17632/98ywc24xv7.1).
For FLAG-tagged proteins, total proteins of exponentially growing cells were extracted by NaOH lysis and TCA precipitation as described (Knop et al., 1999). Samples were resuspended in HU Buffer to a final concentration of 0.1 OD/μl. Samples were boiled for at least 10 min at 65°C prior loading and 0.5 OD per sample was separated by NU-PAGE on a 10% or 12% Bis-Tris gel. Subsequently, proteins were blotted onto an Immobilon-P PVDF membrane (Millipore). Antibodies for immunodetection were used at the following concentrations: anti-FLAG (Sigma, F3165, 1:1’000) and goat anti-mouse IgG (H + L)-HRP conjugate (Bio-Rad, #1706516, 1:10’000). Antibody detection was performed using Millipore Immobilon HRP substrate on a Fusion FX Vilber Lourmat CCD camera. Quantification was done using ImageJ.
Quantification and Statistical Analysis
P values were generated using the two-tailed, two-sample with equal/unequal variance Student’s t test. Error bars are annotated in Figure legends if they show the standard deviation (SD) or standard error of the mean (SEM) and how many replicates were performed. For western blotting, a F-test was performed to assess if the variance between samples is equal or unequal before applying the corresponding Student’s t test (two-tailed, two-sample equal/unequal variance).
Data and Software Availability
The accession number for the sRNA and DamID data reported in this paper is GEO: GSE93434. The accession number for the mass spectrometry raw data is ProteomeXchange: PXD005714. Original Western Blots were deposited in Mendeley Data and are available at http://dx.doi.org/10.17632/98ywc24xv7.1.
Author Contributions
V.F. generated strains; performed H3K9me2-ChIP, western blot, DamID, proteomics, small RNA sequencing (RNA-seq), RNA-seq, and RT-PCR experiments; conducted silencing initiation/maintenance assays; and analyzed the data. P.R.G. generated strains; performed ChIP experiments (FLAG-Pdp3, Mst2-FLAG, H3, H3K36me3, and H3K14Ac), RT-PCR experiments, silencing assays, and western blot; and analyzed the data. V.I. performed proteomics with V.F. and acquired and analyzed data. Y.S. and T.K. generated strains. M.B. and S.B. conceived and supervised the study. S.B. analyzed ChIP data with P.R.G. V.F., P.R.G., S.B., and M.B. designed experiments and prepared figures. V.F. and M.B. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Acknowledgments
We thank F. Mohn and L. Kaaij for discussion and critical feedback on the manuscript and H. Pickersgill (Life Science Editors) for editorial assistance. We thank N. Laschet and Z. Sarkadi for excellent technical assistance. We would like to thank the FMI Functional Genomics facility for assistance in library construction and next-generation sequencing and Y. Murakami for providing a set2ΔSRI strain. This work was supported by funds from the European Research Council (ERC-StG-RNAiGenReg-280410 and ERC-CoG-REpiReg-681213 to M.B.), the European Union Network of Excellence EpiGeneSys (HEALTH-2010-257082 to S.B.), and the German Research Foundation (BR 3511/3-1 to S.B.). The Friedrich Miescher Institute for Biomedical Research is supported by the Novartis Research Foundation.
Published: June 22, 2017
Footnotes
Supplemental Information includes four figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2017.05.026.
Contributor Information
Sigurd Braun, Email: sigurd.braun@bmc.med.lmu.de.
Marc Bühler, Email: marc.buehler@fmi.ch.
Supplemental Information
References
- Adams-Cioaba M.A., Min J. Structure and function of histone methylation binding proteins. Biochem. Cell Biol. 2009;87:93–105. doi: 10.1139/O08-129. [DOI] [PubMed] [Google Scholar]
- Aebersold R., Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- Bähler J., Wu J.Q., Longtine M.S., Shah N.G., McKenzie A., 3rd, Steever A.B., Wach A., Philippsen P., Pringle J.R. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Barrales R.R., Forn M., Georgescu P.R., Sarkadi Z., Braun S. Control of heterochromatin localization and silencing by the nuclear membrane protein Lem2. Genes Dev. 2016;30:133–148. doi: 10.1101/gad.271288.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubec T., Colombo D.F., Wirbelauer C., Schmidt J., Burger L., Krebs A.R., Akalin A., Schübeler D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 2015;520:243–247. doi: 10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- Bayne E.H., White S.A., Kagansky A., Bijos D.A., Sanchez-Pulido L., Hoe K.-L., Kim D.-U., Park H.-O., Ponting C.P., Rappsilber J., Allshire R.C. Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell. 2010;140:666–677. doi: 10.1016/j.cell.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behjati S., Tarpey P.S., Presneau N., Scheipl S., Pillay N., Van Loo P., Wedge D.C., Cooke S.L., Gundem G., Davies H. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013;45:1479–1482. doi: 10.1038/ng.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel C., Paro R. Silencing chromatin: comparing modes and mechanisms. Nat. Rev. Genet. 2011;12:123–135. doi: 10.1038/nrg2932. [DOI] [PubMed] [Google Scholar]
- Bell O., Wirbelauer C., Hild M., Scharf A.N.D., Schwaiger M., MacAlpine D.M., Zilbermann F., van Leeuwen F., Bell S.P., Imhof A. Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J. 2007;26:4974–4984. doi: 10.1038/sj.emboj.7601926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerling P., Silverstein R.A., Thon G., Caudy A., Grewal S., Ekwall K. Functional divergence between histone deacetylases in fission yeast by distinct cellular localization and in vivo specificity. Mol. Cell. Biol. 2002;22:2170–2181. doi: 10.1128/MCB.22.7.2170-2181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S., Garcia J.F., Rowley M., Rougemaille M., Shankar S., Madhani H.D. The Cul4-Ddb1(Cdt)2 ubiquitin ligase inhibits invasion of a boundary-associated antisilencing factor into heterochromatin. Cell. 2011;144:41–54. doi: 10.1016/j.cell.2010.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bühler M., Verdel A., Moazed D. Tethering RITS to a nascent transcript initiates RNAi- and heterochromatin-dependent gene silencing. Cell. 2006;125:873–886. doi: 10.1016/j.cell.2006.04.025. [DOI] [PubMed] [Google Scholar]
- Carrozza M.J., Li B., Florens L., Suganuma T., Swanson S.K., Lee K.K., Shia W.-J., Anderson S., Yates J., Washburn M.P., Workman J.L. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Chen E.S., Zhang K., Nicolas E., Cam H.P., Zofall M., Grewal S.I.S. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature. 2008;451:734–737. doi: 10.1038/nature06561. [DOI] [PubMed] [Google Scholar]
- Creamer K.M., Job G., Shanker S., Neale G.A., Lin Y.-C., Bartholomew B., Partridge J.F. The Mi-2 homolog Mit1 actively positions nucleosomes within heterochromatin to suppress transcription. Mol. Cell. Biol. 2014;34:2046–2061. doi: 10.1128/MCB.01609-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin S., Laramée L., Jacques P.-É., Forest A., Bergeron M., Robert F. DSIF and RNA polymerase II CTD phosphorylation coordinate the recruitment of Rpd3S to actively transcribed genes. PLoS Genet. 2010;6:e1001173. doi: 10.1371/journal.pgen.1001173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekwall K., Cranston G., Allshire R.C. Fission yeast mutants that alleviate transcriptional silencing in centromeric flanking repeats and disrupt chromosome segregation. Genetics. 1999;153:1153–1169. doi: 10.1093/genetics/153.3.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer T., Cui B., Dhakshnamoorthy J., Zhou M., Rubin C., Zofall M., Veenstra T.D., Grewal S.I.S. Diverse roles of HP1 proteins in heterochromatin assembly and functions in fission yeast. Proc. Natl. Acad. Sci. USA. 2009;106:8998–9003. doi: 10.1073/pnas.0813063106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D., Lerch A., Hahne F., Stadler M.B. QuasR: quantification and annotation of short reads in R. Bioinformatics. 2015;31:1130–1132. doi: 10.1093/bioinformatics/btu781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert T.M., McDaniel S.L., Byrum S.D., Cades J.A., Dancy B.C.R., Wade H., Tackett A.J., Strahl B.D., Taverna S.D. A PWWP domain-containing protein targets the NuA3 acetyltransferase complex via histone H3 lysine 36 trimethylation to coordinate transcriptional elongation at coding regions. Mol. Cell. Proteomics. 2014;13:2883–2895. doi: 10.1074/mcp.M114.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez E.B., Espinosa J.M., Forsburg S.L. Schizosaccharomyces pombe mst2+ encodes a MYST family histone acetyltransferase that negatively regulates telomere silencing. Mol. Cell. Biol. 2005;25:8887–8903. doi: 10.1128/MCB.25.20.8887-8903.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschling D.E. Summary: epigenetics—from phenomenon to field. Cold Spring Harb. Symp. Quant. Biol. 2004;69:507–519. doi: 10.1101/sqb.2004.69.507. [DOI] [PubMed] [Google Scholar]
- Govind C.K., Qiu H., Ginsburg D.S., Ruan C., Hofmeyer K., Hu C., Swaminathan V., Workman J.L., Li B., Hinnebusch A.G. Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol. Cell. 2010;39:234–246. doi: 10.1016/j.molcel.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal S.I. RNAi-dependent formation of heterochromatin and its diverse functions. Curr. Opin. Genet. Dev. 2010;20:134–141. doi: 10.1016/j.gde.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida T., Nakayama J., Moazed D. siRNA-mediated heterochromatin establishment requires HP1 and is associated with antisense transcription. Mol. Cell. 2008;31:178–189. doi: 10.1016/j.molcel.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R., Iglesias N., Moazed D. Distinct functions of Argonaute slicer in siRNA maturation and heterochromatin formation. Mol. Cell. 2016;63:191–205. doi: 10.1016/j.molcel.2016.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jason L.J.M., Moore S.C., Lewis J.D., Lindsey G., Ausió J. Histone ubiquitination: a tagging tail unfolds? BioEssays. 2002;24:166–174. doi: 10.1002/bies.10038. [DOI] [PubMed] [Google Scholar]
- Keller C., Kulasegaran-Shylini R., Shimada Y., Hotz H.-R., Bühler M. Noncoding RNAs prevent spreading of a repressive histone mark. Nat. Struct. Mol. Biol. 2013;20:994–1000. doi: 10.1038/nsmb.2619. [DOI] [PubMed] [Google Scholar]
- Kimura H., Hayashi-Takanaka Y., Goto Y., Takizawa N., Nozaki N. The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct. Funct. 2008;33:61–73. doi: 10.1247/csf.07035. [DOI] [PubMed] [Google Scholar]
- Kizer K.O., Phatnani H.P., Shibata Y., Hall H., Greenleaf A.L., Strahl B.D. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell. Biol. 2005;25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M., Siegers K., Pereira G., Zachariae W., Winsor B., Nasmyth K., Schiebel E. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;15(10B):963–972. doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Kowalik K.M., Shimada Y., Flury V., Stadler M.B., Batki J., Bühler M. The Paf1 complex represses small-RNA-mediated epigenetic gene silencing. Nature. 2015;520:248–252. doi: 10.1038/nature14337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Jackson J., Simon M.D., Fleharty B., Gogol M., Seidel C., Workman J.L., Shilatifard A. Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J. Biol. Chem. 2009;284:7970–7976. doi: 10.1074/jbc.M808220200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Jain S.U., Hoelper D., Bechet D., Molden R.C., Ran L., Murphy D., Venneti S., Hameed M., Pawel B.R. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. 2016;352:844–849. doi: 10.1126/science.aac7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda A., Chikashige Y., Ding D.-Q., Ohtsuki C., Mori C., Asakawa H., Kimura H., Haraguchi T., Hiraoka Y. Highly condensed chromatins are formed adjacent to subtelomeric and decondensed silent chromatin in fission yeast. Nat. Commun. 2015;6:7753. doi: 10.1038/ncomms8753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Stroh S., Dickens N.J., Hughes-Davies L., Kouzarides T., Eisenhaber F., Ponting C.P. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem. Sci. 2003;28:69–74. doi: 10.1016/S0968-0004(03)00004-5. [DOI] [PubMed] [Google Scholar]
- McAlister G.C., Nusinow D.P., Jedrychowski M.P., Wühr M., Huttlin E.L., Erickson B.K., Rad R., Haas W., Gygi S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D. Mechanisms for the inheritance of chromatin states. Cell. 2011;146:510–518. doi: 10.1016/j.cell.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S.A., Shibata Y., Noma K., Tsukamoto Y., Warren E., Temple B., Grewal S.I.S., Strahl B.D. Histone H3 K36 methylation is associated with transcription elongation in Schizosaccharomyces pombe. Eukaryot. Cell. 2005;4:1446–1454. doi: 10.1128/EC.4.8.1446-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motamedi M.R., Verdel A., Colmenares S.U., Gerber S.A., Gygi S.P., Moazed D. Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding centromeric RNAs. Cell. 2004;119:789–802. doi: 10.1016/j.cell.2004.11.034. [DOI] [PubMed] [Google Scholar]
- Motamedi M.R., Hong E.-J.E., Li X., Gerber S., Denison C., Gygi S., Moazed D. HP1 proteins form distinct complexes and mediate heterochromatic gene silencing by nonoverlapping mechanisms. Mol. Cell. 2008;32:778–790. doi: 10.1016/j.molcel.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri F., Rapelli S., Krepelova A., Incarnato D., Parlato C., Basile G., Maldotti M., Anselmi F., Oliviero S. Intragenic DNA methylation prevents spurious transcription initiation. Nature. 2017;543:72–77. doi: 10.1038/nature21373. [DOI] [PubMed] [Google Scholar]
- Nicolas E., Yamada T., Cam H.P., Fitzgerald P.C., Kobayashi R., Grewal S.I.S. Distinct roles of HDAC complexes in promoter silencing, antisense suppression and DNA damage protection. Nat. Struct. Mol. Biol. 2007;14:372–380. doi: 10.1038/nsmb1239. [DOI] [PubMed] [Google Scholar]
- Papillon-Cavanagh S., Lu C., Gayden T., Mikael L.G., Bechet D., Karamboulas C., Ailles L., Karamchandani J., Marchione D.M., Garcia B.A. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 2017;49:180–185. doi: 10.1038/ng.3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy B.D., Wang Y., Niu L., Higuchi E.C., Marguerat S.B., Bähler J., Smith G.R., Jia S. Elimination of a specific histone H3K14 acetyltransferase complex bypasses the RNAi pathway to regulate pericentric heterochromatin functions. Genes Dev. 2011;25:214–219. doi: 10.1101/gad.1993611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitges F.W., Prusty A.B., Faty M., Stützer A., Lingaraju G.M., Aiwazian J., Sack R., Hess D., Li L., Zhou S. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell. 2011;42:330–341. doi: 10.1016/j.molcel.2011.03.025. [DOI] [PubMed] [Google Scholar]
- Schneider R., Bannister A.J., Kouzarides T. Unsafe SETs: histone lysine methyltransferases and cancer. Trends Biochem. Sci. 2002;27:396–402. doi: 10.1016/s0968-0004(02)02141-2. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada Y., Mohn F., Bühler M. The RNA-induced transcriptional silencing complex targets chromatin exclusively via interacting with nascent transcripts. Genes Dev. 2016;30:2571–2580. doi: 10.1101/gad.292599.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigova A., Rhind N., Zamore P.D. A single Argonaute protein mediates both transcriptional and posttranscriptional silencing in Schizosaccharomyces pombe. Genes Dev. 2004;18:2359–2367. doi: 10.1101/gad.1218004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmer F., Buscaino A., Kos-Braun I.C., Kagansky A., Boukaba A., Urano T., Kerr A.R.W., Allshire R.C. Hairpin RNA induces secondary small interfering RNA synthesis and silencing in trans in fission yeast. EMBO Rep. 2010;11:112–118. doi: 10.1038/embor.2009.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steglich B., Filion G.J., van Steensel B., Ekwall K. The inner nuclear membrane proteins Man1 and Ima1 link to two different types of chromatin at the nuclear periphery in S. pombe. Nucleus. 2012;3:77–87. doi: 10.4161/nucl.18825. [DOI] [PubMed] [Google Scholar]
- Sugiyama T., Cam H., Verdel A., Moazed D., Grewal S.I.S. RNA-dependent RNA polymerase is an essential component of a self-enforcing loop coupling heterochromatin assembly to siRNA production. Proc. Natl. Acad. Sci. USA. 2005;102:152–157. doi: 10.1073/pnas.0407641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T., Cam H.P., Sugiyama R., Noma K., Zofall M., Kobayashi R., Grewal S.I.S. SHREC, an effector complex for heterochromatic transcriptional silencing. Cell. 2007;128:491–504. doi: 10.1016/j.cell.2006.12.035. [DOI] [PubMed] [Google Scholar]
- Suzuki S., Kato H., Suzuki Y., Chikashige Y., Hiraoka Y., Kimura H., Nagao K., Obuse C., Takahata S., Murakami Y. Histone H3K36 trimethylation is essential for multiple silencing mechanisms in fission yeast. Nucleic Acids Res. 2016;44:4147–4162. doi: 10.1093/nar/gkw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny J.C. Chromatin modification by the RNA polymerase II elongation complex. Transcription. 2014;5:e988093. doi: 10.4161/21541264.2014.988093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny J.C., Erdjument-Bromage H., Tempst P., Allis C.D. Ubiquitylation of histone H2B controls RNA polymerase II transcription elongation independently of histone H3 methylation. Genes Dev. 2007;21:835–847. doi: 10.1101/gad.1516207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R.C. (2014). R: a language and environment for statistical computing. http://www.R-project.org/.
- Tompa R., Madhani H.D. Histone H3 lysine 36 methylation antagonizes silencing in Saccharomyces cerevisiae independently of the Rpd3S histone deacetylase complex. Genetics. 2007;175:585–593. doi: 10.1534/genetics.106.067751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M., Eberl H.C., Matarese F., Marks H., Denissov S., Butter F., Lee K.K., Olsen J.V., Hyman A.A., Stunnenberg H.G., Mann M. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Wagner E.J., Carpenter P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012;13:115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Yang F., Gritsenko M.A., Wang Y., Clauss T., Liu T., Shen Y., Monroe M.E., Lopez-Ferrer D., Reno T. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 2011;11:2019–2026. doi: 10.1002/pmic.201000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Kallgren S.P., Reddy B.D., Kuntz K., López-Maury L., Thompson J., Watt S., Ma C., Hou H., Shi Y. Histone H3 lysine 14 acetylation is required for activation of a DNA damage checkpoint in fission yeast. J. Biol. Chem. 2012;287:4386–4393. doi: 10.1074/jbc.M111.329417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Reddy B.D., Jia S. Rapid epigenetic adaptation to uncontrolled heterochromatin spreading. eLife. 2015;4:80. doi: 10.7554/eLife.06179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm B.T., Marguerat S., Aligianni S., Codlin S., Watt S., Bähler J. Differential patterns of intronic and exonic DNA regions with respect to RNA polymerase II occupancy, nucleosome density and H3K36me3 marking in fission yeast. Genome Biol. 2011;12:R82. doi: 10.1186/gb-2011-12-8-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A., Sherwin T., Sasse R., MacRae T.H., Baines A.J., Gull K. Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J. Cell Sci. 1989;93:491–500. doi: 10.1242/jcs.93.3.491. [DOI] [PubMed] [Google Scholar]
- Woolcock K.J., Gaidatzis D., Punga T., Bühler M. Dicer associates with chromatin to repress genome activity in Schizosaccharomyces pombe. Nat. Struct. Mol. Biol. 2011;18:94–99. doi: 10.1038/nsmb.1935. [DOI] [PubMed] [Google Scholar]
- Yuan W., Xu M., Huang C., Liu N., Chen S., Zhu B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 2011;286:7983–7989. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M., Grewal S.I.S. HULC, a histone H2B ubiquitinating complex, modulates heterochromatin independent of histone methylation in fission yeast. J. Biol. Chem. 2007;282:14065–14072. doi: 10.1074/jbc.M700292200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.