

ABSTRACT
Chronic airway infection and inflammation contribute to the progressive loss of lung function and shortened survival of patients with cystic fibrosis (CF). Rhesus theta defensin-1 (RTD-1) is a macrocyclic host defense peptide with antimicrobial and immunomodulatory activities. Combined with favorable preclinical safety and peptide stability data, RTD-1 warrants investigation to determine its therapeutic potential for treatment of CF lung disease. We sought to evaluate the therapeutic potential of RTD-1 for CF airway infection and inflammation using in vitro, ex vivo, and in vivo models. We evaluated RTD-1's effects on basal and Pseudomonas aeruginosa-induced inflammation in CF sputum leukocytes and CF bronchial epithelial cells. Peptide stability was evaluated by incubation with CF sputum. Airway pharmacokinetics, safety, and tolerance studies were performed in naive mice. Aerosolized RTD-1 treatment effects were assessed by analyzing lung bacterial burdens and airway inflammation using an established model of chronic P. aeruginosa endobronchial infection in CF (ΔF508) mice. RTD-1 directly reduces metalloprotease activity, as well as inflammatory cytokine secretion from CF airway leukocyte and bronchial epithelial cells. Intrapulmonary safety, tolerability, and stability data support the aerosol administration route. RTD-1 reduced the bacterial lung burden, airway neutrophils, and inflammatory cytokines in CF mice with chronic P. aeruginosa lung infection. Collectively, these studies support further development of RTD-1 for treatment of CF airway disease.
KEYWORDS: airway inflammation, Pseudomonas aeruginosa, cystic fibrosis, host defense peptides, theta defensin
INTRODUCTION
Cystic fibrosis (CF) is characterized by a chronic cycle of airway obstruction, infection, and inflammation leading to progressive loss of lung function and eventual respiratory failure (1). Pseudomonas aeruginosa is the most prevalent organism in adults, and chronic infection is associated with lung disease severity and shortened survival (2–5). Susceptibility to infection is attributed in part to a dysfunctional cystic fibrosis transmembrane conductance regulator (CFTR) impairing the primary mucociliary escalator and antimicrobial chemical defenses (6, 7). In particular, accumulation of airway biopolymers and pH acidification reduce the bioactivity of host defense peptides (8–10).
Pathogen-incited inflammation, characterized by excessive neutrophils and proteases (neutrophil elastase [NE]) and matrix metalloproteinase-9 (MMP-9), is strongly associated with matrix breakdown, lung remodeling, and loss of pulmonary function (11–13). In addition, excess proteases degrade host defense peptides and cleave surface receptors on immune cells, leading to further impaired bacterial killing and innate immune responses (14, 15). Clinical trials of anti-inflammatory therapies (i.e., oral prednisone and high-dose ibuprofen) have demonstrated a significant impact on pulmonary disease progression, but serious adverse effects limit their use (16–19). Therefore, new therapies with improved safety profiles are needed to target infection and inflammation, with the goal of slowing the progression of CF lung disease and prolonging survival.
Defensins are small cysteine- and arginine-rich cationic peptides with antimicrobial and immunomodulatory activities (20–22). Uniquely, theta-defensins are backbone cyclized peptides found in Old World primates (23, 24). The prototypical theta-defensin, rhesus theta-defensin-1 (RTD-1), exhibits broad antimicrobial activity, including activity against the known CF pathogens methicillin-resistant Staphylococcus aureus (MRSA) and P. aeruginosa (25–27). Ex vivo, RTD-1 displayed excellent plasma stability and early tumor necrosis factor (TNF) blockade in blood leukocytes (28). In vivo, RTD-1 improved survival in murine bacteremic sepsis and severe acute respiratory syndrome (SARS), likely through immunoregulatory effects (28, 29). One putative mode of anti-inflammatory action is inhibition of MAPK and NF-κB pathways (30).
Previously, we reported in vitro and preliminary in vivo data supporting the antimicrobial activity of RTD-1 against CF P. aeruginosa isolates (25). The goal of the present investigation was to evaluate RTD-1's therapeutic potential in CF. To address this, we utilized in vitro, ex vivo, and murine models of P. aeruginosa-induced CF airway inflammation. Based on the importance of pathogen interactions with resident immune cells and airway epithelia (31, 32), we performed testing in ex vivo CF sputum leukocyte cultures and in vitro with P. aeruginosa-stimulated human bronchial epithelium. In vivo investigations were performed via aerosol administration of RTD-1 in CF mice with chronic P. aeruginosa lung infection.
(This work was presented in part at the North American Cystic Fibrosis Conference, 8 to 10 October 2015.)
RESULTS
Inflammatory gene and protein expression in RTD-1-treated CF bronchial epithelium.
To study RTD-1-associated treatment effects, we quantified soluble-cytokine release in the media from CuFi cells stimulated with P. aeruginosa filtrate in the presence or absence of RTD-1. In addition, we explored the differential expression of genes involved in the host response to the bacterial filtrate stimulus. At the protein level, P. aeruginosa challenge increased release of the cytokines interleukin 1β (IL-1β), TNF, IL-6, and CXCL8 by approximately 8-, 30-, 17-, and 35-fold compared to unconditioned CuFi cells (Fig. 1A to D). With RTD-1 treatment, reductions in IL-1β (P < 0.001), TNF (P < 0.01), CXCL8 (P < 0.01), and IL-6 (P < 0.01) were observed at both 1 and 10 μg/ml compared to untreated P. aeruginosa filtrate-stimulated cells (Fig. 1A to D). At the highest dose tested, IL-1β, IL-6, and CXCL8 were reduced by approximately 1.3-fold, while TNF was observed to be reduced by approximately 2-fold (Fig. 1). Utilizing a PCR array focused on bacterial-infection-associated host response genes, we observed that treatment with RTD-1 at 10 μg/ml resulted in an approximately 2-fold reduction in NLRP3, IL-1β, and CD14 gene expression compared to P. aeruginosa filtrate-induced cells alone (P < 0.05) (see Fig. S1 in the supplemental material). In addition to these inflammasome components, transcript levels of SLC11A1 and CD86 were upregulated approximately 1.5-fold by RTD-1 treatment but did not reach statistical significance (P = 0.06) (see Fig. S1).
FIG 1.
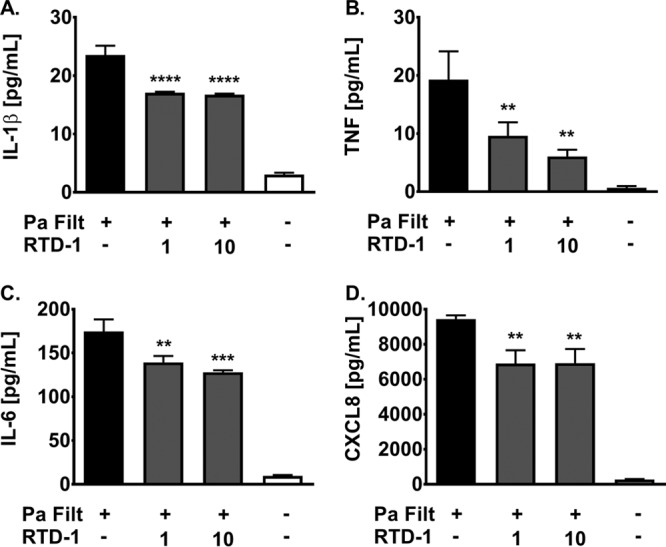
RTD-1 reduces inflammatory cytokines in CF epithelium. CF hBECs were stimulated with P. aeruginosa filtrate (Pa Filt) in the presence or absence of 0, 1, or 10 μg/ml RTD-1 for 24 h. The levels of cytokines IL-1β (A), TNF (B), IL-6 (C), and CXCL8 (D) were quantified by ELISA. Means and SD (n = 3/group) are shown. Treatment differences were analyzed by ANOVA; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 versus P. aeruginosa filtrate alone.
RTD-1 reduces spontaneous inflammatory cytokine secretion in CF sputum leukocytes.
Six patients with CF who were hospitalized for treatment of an acute pulmonary exacerbation (APE) participated in the sputum inflammation study. The median admission demographics were as follows: 28 years of age, normal weight (body mass index [BMI], 23.4), and moderate lung disease (percent predicted forced expiratory volume in 1 s [ppFEV1], 39%). All the patients had chronic P. aeruginosa-positive respiratory cultures and were receiving treatment with azithromycin. Total and differential cell counts in sputum demonstrated neutrophil predominance (see Table S1 in the supplemental material). RTD-1 at 100 μg/ml for 24 h significantly reduced spontaneous secretion of IL-1β (P < 0.001), TNF (P < 0.05), and CXCL8 (P < 0.01), on average by approximately 2.5-, 2-, and 1.3-fold, respectively (Fig. 2). Although IL-6 was also reduced, variability around the point estimate was large and did not reach significance (P = 0.14). Culture media plated on Trypticase soy agar (TSA) demonstrated no bacterial growth (data not shown).
FIG 2.
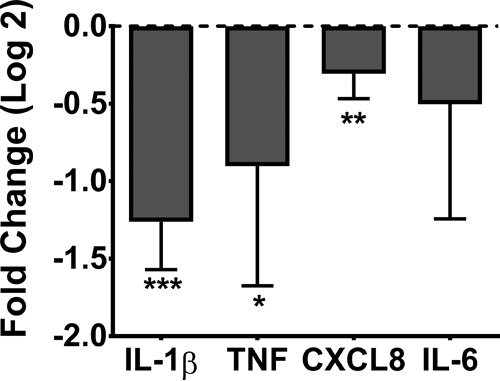
RTD-1 reduces spontaneous inflammatory cytokines in airway leukocytes. Isolated CF airway leukocytes were cultured in the presence or absence of 100 μg/ml RTD-1 for 24 h. Release of cytokines IL-1β, TNF, CXCL8, and IL-6 was quantified by ELISA. Geometric means and 95% CIs (n = 6/group) are shown. Treatment differences (log2) were analyzed by a paired t test after baseline correction (RTD-1/baseline); *, P < 0.05; **, P < 0.01; ***, P < 0.001 versus CF leukocytes alone.
RTD-1 inhibits lung proteases.
RTD-1 inhibited MMP-9 enzyme activity in a dose-dependent manner, with a 50% inhibitory concentration (IC50) of 551 nM (1.1 μg/ml) (Fig. 3A). Based on additional studies performed with varying substrate concentrations, RTD-1 exhibited competitive inhibition, with a Ki of 571 nM (1.2 μg/ml) (95% confidence interval [CI], 513.9 to 628.9; r2 = 0.99) (see Fig. S2 in the supplemental material). Alternatively, RTD-1 at 600 nM (1.25 μg/ml) only slightly inhibited (∼10%) neutrophil elastase activity (Fig. 3C). Increasing the RTD-1 dose 4-fold marginally increased enzyme inhibition (∼18%), suggesting that RTD-1 exhibits minimal NE-inhibitory activity (see Fig. S3 in the supplemental material). In comparison, the well-described NE inhibitor sivelestat completely blocked NE activity at a concentration of 400 nM.
FIG 3.
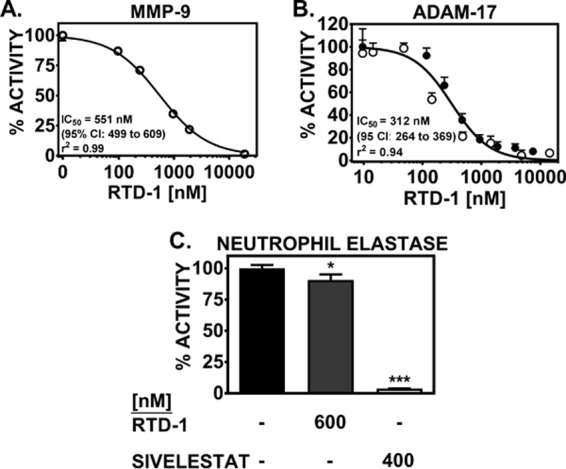
RTD-1 inhibits MMP-9 and ADAM17 metalloproteinases, but not the serine proteinase NE. (A and B) Dose-response curves of recombinant human active MMP-9 (A) and surface ADAM17 activity (B) in CF airway leukocytes (the open and solid circles represent different patients) measured in the presence or absence of RTD-1 at varying concentrations. (C) Alternatively, NE activity in soluble-phase sputum was determined in the presence or absence of either saline, RTD-1, or sivelestat (a known NE inhibitor). Activity was determined by specific fluorogenic substrate reporters. A normalized response sigmoid Emax model was used to determine IC50s. Means and SD (n = 2/group) are shown. Comparative treatment differences for NE inhibition were analyzed by ANOVA; *, P < 0.05; ***, P < 0.001 versus a saline positive control.
In airway leukocytes isolated from CF sputum (n = 2), we observed that RTD-1 inhibited cleavage of an ADAM17 selective exogenous substrate with an IC50 of 312 nM (0.65 μg/ml) (Fig. 3B). Together, the inhibitory activities against both MMP-9 and ADAM17 suggest that RTD-1 shows specificity for the MMPs and may be a biologically relevant inhibitor of metalloproteinase activity.
Sputum pourability (mucoadhesion) is improved by RTD-1.
The effect of RTD-1 on sputum pourability was tested in the six APE participants. Treatment with either 10 μg/ml RTD-1 or 50 μg/ml human recombinant DNase significantly mobilized CF sputum samples compared with no treatment (P < 0.01) (Fig. 4). On average, DNase improved pourability to a greater extent than RTD-1. In addition, on a molar basis, DNase (1.6 μM) appeared to be more potent than RTD-1 (5 μM).
FIG 4.
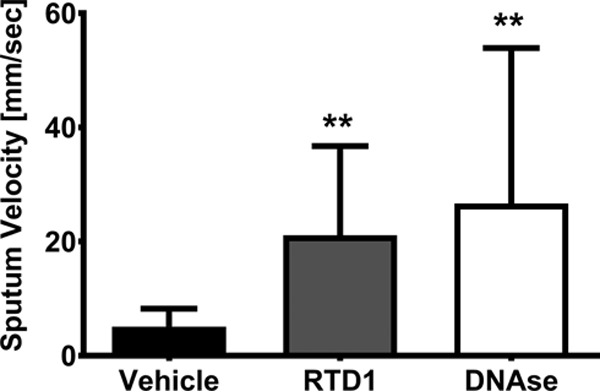
RTD-1 improves CF sputum pourability ex vivo. Sputum pourability as measured by velocity was determined in normal saline, RTD-1, and DNase treatment groups. Geometric means and 95% CIs (n = 6/group) are shown. Treatment differences were analyzed by ANOVA; **, P < 0.01 versus a normal saline control.
RTD-1 retains structural integrity in pooled CF sputa.
Twenty-four-hour peptide stability was evaluated in pooled soluble-phase sputa (SOL) (n = 7 patients). When examined by ultraperformance liquid chromatography (UPLC)-mass spectrometry (MS), RTD-1's signature peak was demonstrated at a retention time (RT) of 4.8 min (see Fig. S4a to c in the supplemental material). The peak area under the concentration-time curve (AUC) for RTD-1 alone compared to the control remained unchanged (P = 0.91). Ionization mass spectrometry confirmed RTD-1's identity (m/z, 521.68). Overall, these data indicate that RTD-1 is a durable peptide able to withstand the high intrapulmonary protease burden observed in CF.
Airway reactivity is unchanged by RTD-1.
Dose-response curves were essentially superimposable for lung resistance (RL) and dynamic compliance (Cdyn) measures (Fig. 5A and B). Reactivity to methacholine (Mch) after topical administration of 3 mg of RTD-1/kg of body weight showed no change in Cdyn or RL compared to sham treatment with phosphate-buffered saline (PBS). Given these findings, RTD-1 does not appear to enhance airway reactive effects, such as narrowing conducting zones or lung unit derecruitment.
FIG 5.
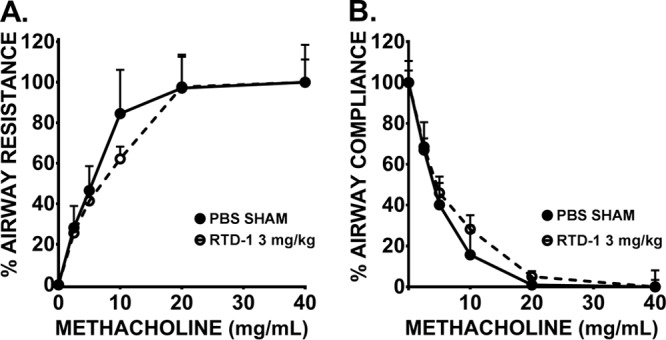
RTD-1 does not induce airway hyperresponsiveness in vivo. Pulmonary mechanics resistance (A) and compliance (B) were evaluated by oscillometry at intranasal doses of 0 and 3 mg/kg RTD-1 under methacholine challenge in BALB/c mice. Means and SD (n = 3/group) are shown.
Aerosol drug delivery.
Delivery of RTD-1 was done through a 16-port nose-only nebulizer system. A 7-stage cascade impactor allowed the characterization of the mass median aerodynamic diameter (MMAD) of nebulized RTD-1 as 0.81 ± 0.12 μM with a geometric standard deviation (SD) of 1.32 ± 0.05 and log-normal distribution (see Fig. S5b to e in the supplemental material). The MMAD did not appear to change significantly across doses. Based on this particle size, and other parameters described in Materials and Methods, the delivered doses of aerosolized RTD-1 were estimated to be 6.8, 49, 167, and 360 μg/kg.
Airway pharmacokinetics (PK) and safety of RTD-1.
The epithelial lung fluid (ELF) concentration-time plot for nebulized RTD-1 after single-dose administration is depicted in Fig. 6. Overall, the observed maximum concentration (Cmax) and exposure defined by the AUC were not proportional to the dose delivered. The half-lives of RTD-1 in the airways across doses were similar, with a median and interquartile range (IQR) of 3.19 ± 0.71 h (Table 1). Lung histology, inflammation scoring, total numbers of airway cells in bronchoalveolar lavage (BAL) fluid (BALF), and body weight changes did not appear to be associated with airway concentrations achieved after a single dose of RTD-1 (see Fig. S6 in the supplemental material). Plasma levels of RTD-1 following aerosol administration were below the lower limit of quantification of 7.8 ng/ml.
FIG 6.
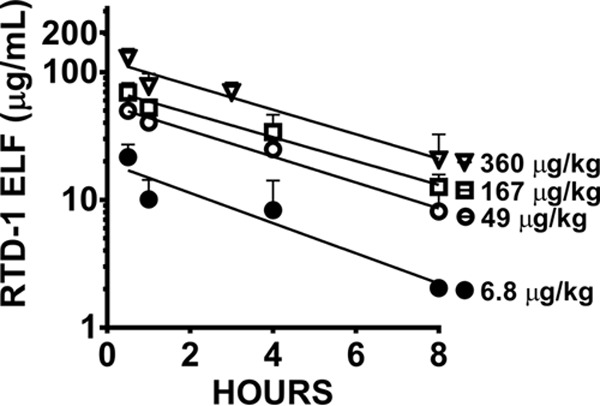
Characterization of aerosolized RTD-1 in vivo. Shown is an epithelial lining fluid concentration-versus-time profile of delivered doses of 360, 167, 49, and 6.8 μg/kg RTD-1. Medians and IQRs (n = 4/group) are shown.
TABLE 1.
RTD-1 noncompartmental PK analysis (NCA) in ELF
| NCA parametera | Valueb for delivered dose (μg/kg) of: |
|||
|---|---|---|---|---|
| 6.8 | 49 | 167 | 360 | |
| AUC (μg · h/ml) | 57.98 (18.88) | 183.15 (8.36) | 257.24 (55.42) | 420.96 (24.24) |
| Cmax (μg/ml) | 20.54 (4.55) | 47.79 (12.90) | 69.17 (11.49) | 137.27 (11.47) |
| Tmax (h) | 0.5 (0) | 0.5 (0) | 0.5 (0) | 0.5 (0) |
| t1/2 (h) | 2.50 (0.10) | 3.07 (0.31) | 3.31 (0.60) | 3.36 (0.76) |
Determined after a single nebulized dose.
The data are presented as median (IQR).
Effect of RTD-1 on lung bacterial burden.
Chronic P. aeruginosa lung infection was achieved in ΔF508 mice with a geometric mean bacterial CFU of 3.98 × 106 in untreated control mice at day 7. RTD-1 treatment reduced the total recovered CFU from both lung tissue and BAL fluid compared with sham-treated controls by approximately 1 log unit at 167 μg/kg (P < 0.01) and 360 μg/kg (P < 0.05) (Fig. 7A). The unbalanced numbers of animals in the treatment groups were the result of spontaneous death during quarantine (n = 6/64) or from surgical complications (n = 12/64), which is consistent with the expected attrition previously described (33, 34). The surviving inoculated mice were assigned to treatment groups. No deaths occurred during the investigational period.
FIG 7.

Antibacterial and anti-inflammatory treatment effects of RTD-1 in vivo. (A to C) Total lung CFU (A), airway leukocytes (B), and cellular differentials (C) were quantified 7 days postinoculation in C57/B6 mice. (D) Weight change from baseline. The data are presented as geometric means and 95% CIs (A and B) or means and SD (C and D). Treatment differences were analyzed by ANOVA; *, P < 0.05; **, P < 0.01; ***, P < 0.001 versus saline-treated infected mice. The graph contains previously published data on the 167-μg/kg dose for comparison (25).
Effect of RTD-1 on airway inflammation.
We observed a bimodal response to daily aerosolized RTD-1 treatment with significantly reduced (∼ 0.5 log unit) airway white blood cells (WBCs) at 49 μg/kg (P < 0.01) and 167 μg/kg (P < 0.05), but not at 360 μg/kg (P > 0.05), compared to untreated infected control mice, despite CFU reductions at the highest RTD-1 doses (Fig. 7B). The differential cell counts indicate that the reduction in airway WBCs reflects a decrease in airway neutrophils (Fig. 7C).
The effects of RTD-1 treatment on proinflammatory cytokine, chemokine, growth factor, and protease levels are summarized in Table 2. In line with the above discussion, we observed a significant reduction in keratinocyte chemoattractant (KC), IL-6, TIMP-1, and total MMP-9 levels at 167 μg/kg (P < 0.05), but no difference at 360 μg/kg RTD-1 (P > 0.05), compared to untreated controls (P < 0.05).
TABLE 2.
Seven-day BALF cytokines in chronic P. aeruginosa lung-infected mice treated with RTD-1
| Variable | Valued at RTD-1 delivered dose (μg/kg) of: |
|||
|---|---|---|---|---|
| 0 (n = 14) | 49 (n = 8) | 167 (n = 12) | 360 (n = 11) | |
| IL-6a | 89.5 (46.7–171.4) | 94.2 (7.3–1,215.0) | 9.7e (2.3–40.5) | 173.5 (36.4–826) |
| TNFa | 16.2 (9.3–28.1) | 17.9 (3.7–86.6) | 4.8 (1.7–14.0) | 15.6 (6.3–38.7) |
| IL-17a | 4.4 (2.9–6.5) | 5.2 (2.0–13.3) | 2.9 (1.2–6.7) | 1.6 (0.9–3.0) |
| KCa | 110.9 (83.0–148.1) | 76.4 (29.5–187.6) | 34.2e (13.6–85.8) | 60.5 (41.3–88.6) |
| MIP2a | 296.4 (200.9–437.4) | 116.4 (15.0–902.6) | 70.6 (16.6–299.9) | 427.9 (101.9–1,797.0) |
| Amphiregulina | 26.7 (8.6–83.5) | 93.9 (29.5–298.9) | 27.8 (14.8–52.2) | 91.6 (41.8–200.7) |
| TIMP-1a | 1,181.0 (501.0–2,784.0) | 918.0 (45.14–18,670) | 54.1e (5.5–529.8) | 1,042.0 (86.4–12,565.0) |
| Total MMP-9b | 11,640.0 (11,130.0–12,160.0) | 8,990.0 (4,390.0–17,850.0) | 6,133.0f (2,328.0–9,938.0) | 10,240.0 (8,086.0–12,390.0) |
| NE activityb,c | 223.40 (66.78–380.10) | 37.5 (−18.5–93.5) | 61.2 (19.4–103.0) | 422.0 (195.0–649.0) |
Results are expressed as geometric mean (95% confidence interval).
Results are expressed as mean (95% confidence interval).
Relative fluorescent units (RFU).
Picograms per milliliter unless otherwise indicated.
P < 0.05.
P < 0.01.
Effect of RTD-1 treatment on weight.
With the exception of the highest dose, RTD-1 treatment reduced weight loss and resulted in a quicker return to baseline weight (Fig. 7D).
DISCUSSION
Persistent infection, inflammation, and mucus-obstructed airways are hallmarks of CF lung disease. Multiple in vivo models of CF, including the piglet and ferret, have demonstrated that infection and inflammation are intimately linked (35). Therefore, strategies to treat lung bacterial infections, inflammation, and obstruction in CF remain important therapeutic targets. Host defense peptides (HDPs) are key components of the innate immune system, providing direct antibacterial activity, as well as modulation of the inflammatory response. As multidrug resistance continues to emerge, novel ways of combating infection must be developed. A promising alternative to conventional antibiotics is HDPs. Their typical net cationic charge and hydrophobic portions allow bacterial cell membrane disruption, translocation, and intracellular targeting. This nonspecific and multiple-site attack reduces the potential for bacterial resistance (36). The dual antibacterial and anti-inflammatory activities of RTD-1 offer a novel approach to treatment of CF lung disease.
We have previously reported that RTD-1 exhibits activity against P. aeruginosa, including multidrug-resistant isolates from patients with CF (25). In addition, RTD-1 has been shown to exhibit anti-inflammatory activity in murine models of peritonitis/sepsis and SARS (28, 29). This activity is thought to be mediated in part by inhibition of NF-κB (30). In the current investigation, we report on a series of in vitro, ex vivo, and in vivo experiments designed to determine the therapeutic potential of RTD-1 for treatment of CF respiratory disease.
Airway inflammation in CF is orchestrated by interactions between invading pathogens and resident immune cells (e.g., macrophages and neutrophils), as well as airway epithelia (31, 32). Therefore, to test the effects of RTD-1 on these distinct cell populations, we performed in vitro evaluations in both human CF bronchial epithelial cells stimulated with P. aeruginosa soluble filtrate and leukocyte cultures from expectorated CF sputum. The data demonstrate diminished release of key inflammatory cytokines and chemokines (e.g., IL-1β and CXCL-8) in both cell populations. The effect of RTD-1 on cytokines from CF sputum leukocytes (30 to 150% reduction) was slightly greater than that reported with azithromycin, prednisone, or roflumilast (25 to 50%) when tested with sputum cells isolated from patients with steroid-naïve chronic obstructive pulmonary disease (COPD) (37).
Transcriptome profiling of the innate and adaptive immune responses in the stimulated CF bronchial cells suggested that RTD-1 may work through Toll-like receptor (TLR)-mediated inflammation pathways, since NLRP3, CD14, and IL-1β transcription levels were reduced approximately 2-fold. Synergy between the TLR2/4 pathway and the NLRP3 inflammasome have been shown to be CD14 dependent and to prime a variety of NLRP3 inflammasome-driven events, such as IL-1β gene expression (38–40). Therefore, reduced chronic pathway activity would reduce noxious proinflammatory reactions, as was observed. In line with our results, published observations demonstrate that RTD-1 reduces NF-κB phosphorylation in leukocytes stimulated with specific TLR agonists (30).
In addition to cytokines/chemokines, published data to date underscore the importance of lung proteases to CF disease progression. In particular, NE and MMP-9 levels are associated with clinical worsening of pulmonary disease (12, 13). Furthermore, proteolytic cleavage by these proteases alters immune regulation/activity (15), augments protease activity (41), and supports chronic neutrophilic signaling (42, 43). While a number of endogenous protease inhibitors (i.e., TIMPs, elafin, and SLPI) are present in the airways, antiproteinase balance is lost because of targeted degradation by the overwhelming proteinase burden (13, 44). Given that host defense peptides, such as LL-37 and serine leukocyte protease inhibitor (SLPI), have demonstrated cysteine and serine antipeptidase activity, respectively (45, 46), we tested the inhibitory activity of RTD-1 against proteinases using in vitro and ex vivo enzyme assays. MMP-9 and NE were chosen, given their strong correlation with disease severity in CF (12, 13). Despite statistical significance, NE activity was largely unaffected in the soluble-phase sputum assay, suggesting minimal clinical relevance. In contrast, RTD-1 concentrations in the high nanomolar range inhibited recombinant active human MMP-9 in a simple buffer system. Given the homology across the catalytic domain of the metalloproteinase family, we hypothesized that RTD-1 would also inhibit ADAM-17. ADAM-17, also known as TACE, is the primary sheddase for TNF, an important proinflammatory cytokine. Using an ex vivo sputum leukocyte assay that measured ADAM-17 ectodomain shedding, we observed that RTD-1 exhibited increased inhibitory activity against ADAM17, with a nearly 2-fold-lower IC50 for ADAM17 than for MMP-9.
Inspissated secretions in CF result in severe airway obstruction. Therapies targeting this pathology are routinely prescribed to mobilize secretions and improve lung function (47). Cationic molecules have demonstrated beneficial effects in compacting sputum DNA and “liquefying” this complex biological matrix (48). We hypothesized that RTD-1, with its high cationic charge cluster, would perform similarly. Therefore, we performed a modified sputum pourability assay, a measure of velocity and mucoadhesiveness (49, 50). Our data demonstrated an increase in the speed of unprocessed sputum traveling down a glass pipette. The magnitude of this effect was less than the 50-μg/ml DNase condition. While speculative, RTD-1 may provide an additive effect via an alternative mechanism for improving mucus clearability. Further studies are warranted to confirm this early observation with definitive rheological assays.
Airway delivery is an attractive option in CF because of the ability to topically deliver large amounts of drug to the afflicted target site while minimizing systemic, whole-body exposure. In preparation for in vivo studies, we conducted a series of experiments designed to characterize RTD-1 stability, pharmacokinetics, safety, and tolerability via aerosol administration. Due to the elevated levels of proteases present in CF sputum, it is important to consider drug stability in preclinical evaluations. Given its unique cyclic backbone, we hypothesized that RTD-1 would be resistant to the soluble proteases present in CF airway sputum. Liquid chromatography (LC)-MS analysis revealed that RTD-1 retains its conformation. This integrity is likely secondary to RTD-1's unique macrocyclic structure, with N terminus to C terminus linkage, which may retard amide bond cleavage by proteases. Single-dose pharmacokinetic studies following aerosol administration demonstrated for all doses that the maximum concentration observed (Cmax) in ELF exceeded the IC50 for MMP-9 and ADAM-17, as well as concentrations used to inhibit inflammation in CF bronchial epithelial cells. The Cmax values relative to the MIC of RP73 (MIC = 4) were estimated to be 5.1, 11.9, 17.3, and 34.3 for 6.8-, 49-, 167-, and 360-μg/kg doses, respectively. These ratios were associated with rapid bactericidal activity in vitro (25). Assuming linear kinetics, the elimination of RTD-1 from the ELF was relatively rapid, with concentrations declining below 10 μg/ml after ∼2.5, 7, 9, and 12.5 hours for 6.8-, 49-, 167-, and 360-μg/kg doses, respectively. While a treatment effect was observed in vivo, these data suggest that increased frequency of administration is warranted to determine if RTD-1's biological effects can be further optimized. Importantly, after aerosol administration of 360 μg/kg, ELF RTD-1 levels achieved concentrations noted to exhibit anti-inflammatory activity in the sputum leukocyte cell culture experiments. Intrapulmonary administration of RTD-1 appeared to be well tolerated based on a lack of drug-induced airway hyperresponsiveness observed in naive mice.
We have shown previously that RTD-1 exhibits in vitro activity against multidrug-resistant isolates of P. aeruginosa from patients with CF (25). We extended our prior findings in this study by testing the efficacy of aerosolized RTD-1 in an established model of chronic endobronchial P. aeruginosa infection in CF (ΔF508/ΔF508) mice (51, 52). Key components of human CF respiratory disease, such as chronic P. aeruginosa lung infection and airway neutrophilia, as well as elevated protease concentrations, were recapitulated in this murine model, as previously described (53) The results demonstrate that RTD-1 exhibits dose-dependent antipseudomonal activity in vivo, with an order of magnitude similar to that of ciprofloxacin (approximately 1 log unit in lung CFU), a widely used antimicrobial against respiratory pathogens in CF (51). RTD-1 treatment exhibited an anti-inflammatory effect, as evidenced by the reduction in airway leukocytes at the 49- and 167-μg/kg doses and in BALF cytokines at the 167-μg/kg dose, though not at the highest dose tested (360 μg/kg). An examination of the between-day controls from each experiment demonstrated no systematic deviations in the lung bacterial burden, indicating that the lack of efficacy at the highest dose was not due to a higher lung bacterial burden. This assertion is supported by others who have reported that establishment of chronic infection and immune cell recruitment are not significantly different across a 1-log10-unit range of infecting inocula (54). Therefore, considering (i) reduced efficacy and (ii) failure to recover lost weight in mice receiving a delivered dose of 360 μg/kg RTD-1, our data may suggest dose-dependent toxicities. Concerns about this cytotoxic potential are commonly cited with host defense peptides (36, 55). RTD-1's minimal amphipathic structure has been shown to interact poorly with zwitterionic lipid membranes of eukaryotes compared with protegrin-1 (56, 57), which supports the lack of fibroblast cytotoxicity and red blood cell hemolysis at concentrations achieved in the ELF in this study (i.e., 100 μg/ml) (27). However, intranasal administration of RTD-1 in uninfected mice resulted in dose-dependent weight loss and lung pathologies. Importantly, at concentrations similar to those reported here, intranasal RTD-1 at 300 μg/kg demonstrated mild perivascular inflammation and airway karyorrhectic/cellular debris (29). This in vitro-in vivo discordance may be the result of assay conditions (i.e., serum proteins). ELF constituents typically have lower concentrations of proteins than serum, and thus, free drug concentrations are likely higher. Additional studies evaluating chronic administration of RTD-1 in uninfected mice are needed to clarify the long-term safety of aerosolized treatment.
In summary, the experimental data reported here demonstrate that aerosol administration of RTD-1 exhibits promising immunomodulatory and antimicrobial effects and warrants further investigation as a potential therapy for CF lung disease.
MATERIALS AND METHODS
Human subjects.
Patients attending the adult CF clinic at the University of Southern California in Los Angeles, CA, were invited to participate in a cross-sectional study investigating lung inflammatory biomarkers in CF. The inclusion criteria were as follows: adults (>18 years old) diagnosed with CF and able to spontaneously expectorate sputum. Eighteen patients provided spontaneously expectorated sputa; 12 were obtained during stable outpatient visits, and 6 were from patients admitted for the treatment of an APE. Age, BMI, ppFEV1, and anti-inflammatory therapy at the time of sampling were recorded (see Tables S1 and S2 in the supplemental material). Leukocyte populations from the APE subset of sputum samples were prepared for cell culture experiments. The study received ethical approval (HS-12-00320) from the institutional review board at the University of Southern California, and all participants provided written informed consent.
Animals.
All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Southern California (protocols 11676, 11956, 20252, and 20157). Male 8- to 10-week-old BALB/c mice (Charles River Laboratories, CA) weighing 20 to 25 g, male and female CftrΔF508/ΔF508 mice with a C57BL/6 background (Case Western Reserve University, Cleveland, OH) 10 to 12 weeks of age weighing 17 to 22 g, and male 10- to 12-week-old C57BL/6N mice (Charles River Laboratories, CA) weighing 24 to 28 g were housed under specific-pathogen-free conditions at a temperature of 22 to 24°C and humidity of 60 to 65%, with 12-h light/dark cycles. The animals were provided standard laboratory chow and water ad libitum.
In vitro and ex vivo models to assess the anti-inflammatory effect and stability of RTD-1. (i) CF bronchial epithelial cells.
CuFi cells (a ΔF508/ΔF508 bronchial epithelial cell line) were maintained at 37°C, 5% CO2, and 100% humidity in bronchial epithelial cell growth medium (BEGM) (Lonza) supplemented with SingleQuots (without gentamicin-amphotericin B; Lonza) and seeded on plates coated with human placental collagen type IV substratum (Sigma). All experiments were conducted between passages 14 and 18. The CuFi cells were stimulated with 20% (vol/vol) P. aeruginosa diffusible material in 24-hour-conditioned BEGM as previously described (58, 59). Briefly, filtrate was obtained from a late-stationary-phase culture of P. aeruginosa isolate RP73 grown in bronchial epithelial cell growth medium containing bronchial epithelial basal medium (catalog no. CC-3171; Lonza) and SingleQuot supplements and growth factors (catalog no. CC-4175; Lonza). The filtrate was clarified, filter sterilized (0.45 μm), and heat inactivated for 10 min at 95°C before use. Filtrate plated on TSA showed no growth. Treatment with 1 or 10 μg/ml RTD-1 was based on previous in vitro work (28, 30). No cytotoxicity was observed over 24 h with or without RTD-1, as determined by lactate dehydrogenase (LDH) release (the biologically relevant threshold was defined as 20%) (58, 59).
(ii) CF sputum leukocyte culture.
Expectorated CF sputum samples (n = 6) were obtained from APE inpatients within 48 h of starting intravenous (i.v.) antibiotics and were processed as previously described (37). Cellular viability after processing was >82%, which is consistent with published data (37, 60). Differential cell counts were determined by placing approximately 5 × 105 cells onto a slide chamber, spinning in a cytocentrifuge (Shandon, Thermo Scientific, Waltham, MA), and staining using a Diff Quick (Polysciences, Warrington, PA) stain kit according to the commercial protocol. Differential cell counts were conducted on 200 cells per slide. The cells were suspended in RPMI medium plus l-glutamine and 10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO) at 2 × 106/ml. A 0.5-ml aliquot of the cell suspension was placed in a 48-well cell plate (Greiner-Bio One, Monroe, NC). The wells were spiked with 100 μg/ml RTD-1 or cell culture medium and incubated for 24 h. This increase in dose relative to the CuFi cell line was based on in-house data suggesting significant protein binding in the presence of serum and previously published safety data demonstrating no hemolytic or cytotoxicity concerns at this dose (27). The clarified cell supernatants were aliquoted and stored at −80°C until analysis (37, 60). No cytotoxicity was observed over 24 h with or without RTD-1, as determined by trypan blue exclusion and LDH release (the biologically relevant threshold was defined as 20%) (37, 61). The sampling of lower airways is supported by <20% squamous epithelial cell contamination (see Table S2 in the supplemental material).
(iii) Human inflammatory gene and protein expression.
RNA was extracted from CuFi human bronchial epithelial cells (hBECs) using an RNeasy minikit according to the manufacturer's instructions with the optional DNase treatment (Qiagen). The quality and quantity of RNA extracted were assessed using UV-visible (Vis) (Nanodrop; Thermo). A260/A280 values of >2.0 and A260/A230 values of >1.8 were achieved for all samples run. cDNA synthesis was performed using the iScript cDNA synthesis kit (Bio-Rad), SSO universal SYBR green supermix (Bio-Rad), and 0.5 μg cDNA/plate. Samples were analyzed for steady-state transcript levels in the host response to bacterial infection using the human innate and adaptive RT2 Profiler PCR array (catalog no. PAHS-052Z; Qiagen) containing 84 paneled genes. Release of IL-1β, TNF, CXCL8, and IL-6 from sputum leukocytes was quantified using an enzyme-linked immunosorbent assay (ELISA) (MSD, Rockville, MD) 24 h post-RTD-1 incubation.
(iv) Antiprotease activity of RTD-1.
In vitro MMP-9 activity was measured using a fluorogenic microtiter substrate assay. A final concentration of 3 μM active recombinant human MMP-9 from Pichia pastoris (Enzo Life Sciences, Farmingdale, NY) was added to increasing concentrations (0.2 to 25 μM) of the fluorogenic MMP substrate 2,4-dinitrophenol (DNP)–Pro–β-cyclohexyl–Ala–Gly–Cys(Me)–His–Ala–Lys(Nma)-NH2 [where Cys(me) is S-methyl-l-cysteinyl and Lys(Nma) is N-epsilon-methylanthranoyl-l-lysine; Enzo Life Sciences] with or without increasing concentrations of RTD-1. Additionally, in pooled expectorated SOL from stable outpatients (n = 7), MMP-9 activity was determined with the above-described fluorogenic-substrate assay. MMP-9 activity was quantified by continuous measurement of fluorescence intensity in a microplate fluorometer (Synergy H1 Hybrid; Bio-Tek Instruments Inc., Winooski, VT) at 37°C and at an excitation wavelength (λex) of 340 nm and an emission wavelength (λem) of 440 nm, in accordance with the manufacturer's suggestions. Data were acquired with Gen5 data analysis software (Bio-Tek Instruments Inc.). The initial velocity was computed and the Ki was estimated by the competitive enzyme inhibition model. Alternatively, the IC50 estimate was calculated using the normalized response sigmoidal maximum-effect (Emax) model with fixed Hill slope.
NE activity was measured using a fluorogenic microtiter substrate assay with MeOSuc-Ala-Ala-Pro-Val-AMC (where MEOSuc is N-methoxysuccinyl and AMC is 7-amido-4-methylcoumarin; InnoZyme; EMD Millipore). SOL samples were assayed at a 200-fold final dilution with or without test compound (RTD-1, sivelestat, or saline), and the reaction was initiated with a 400-μM final substrate concentration in a 110-μl final volume. Activity was measured by continuous measurement of fluorescence intensity in a microplate fluorometer (Synergy H1 Hybrid; Bio-Tek Instruments Inc., Winooski, VT) at 37°C and at a λex of 380 nm and a λem of 460 nm, in accordance with the manufacturer's protocol. Data were acquired with Gen5 data analysis software (Bio-Tek Instruments Inc.). The initial velocity (optical density [OD]/min) was computed, and the percent change from baseline (soluble phase plus saline) was calculated and plotted for comparative analysis.
ADAM17 activity was determined in live airway leukocyte cultures (n = 2). Adult samples were collected from routine clinical visits (patients 25 and 53 years of age with chronic P. aeruginosa lung infections). Both patients were receiving long-term azithromycin treatment and had mild and severe lung disease, respectively (84 and 22 ppFEV1). Sputum leukocytes were processed as described above (CF sputum leukocyte culture method) and then washed and resuspended in assay buffer (Hanks balanced salt solution [HBSS]). Cells were plated in duplicate at 1 × 106 cells/well with 5 μm TACE II fluorogenic substrate (Enzo Life Sciences, Farmingdale, NY). ADAM17 enzymatic activity was quantified by continuous measurement of fluorescence intensity in a microplate fluorometer (Synergy H1 Hybrid; Bio-Tek Instruments Inc., Winooski, VT) at 37°C and at a λex of 485 nm and a λem of 535 nm, in accordance with a protocol described previously (62). Data were acquired with Gen5 data analysis software (Bio-Tek Instruments Inc.). The initial velocity was computed and imported into the dose-response analysis. A normalized response sigmoidal Emax model with variable Hill slope was chosen to estimate the IC50.
(v) Mucoactivity of RTD-1.
Macrorheological analysis was conducted within 3 to 4 h of sputum collection. Samples (n = 6) were stored on wet ice until sputum aliquots (∼0.2 g) were treated for 1 h at 37°C with vehicle (10% [vol/wt] normal saline), recombinant human (rh)-DNase (50 μg/ml) (Qiagen, Valencia, CA), or RTD-1 (10 μg/ml). The RTD-1 concentration was based on previously published in vitro anti-inflammatory work (28). A modified pourability assay was performed as follows. Treated samples were placed into a vertically positioned 2-ml graduated pipette and run under gravity. Quantification of sputum velocity (in millimeters per second) was calculated over a distance of 390 mm (63, 64). This test is an indirect measure of sputum elasticity and adhesion (50, 65).
(vi) RTD-1 stability in CF sputum.
The stability of RTD-1 in sputum was monitored by UPLC (210 nm), and its identity was confirmed by MS (m/z, 521.65). Briefly, pooled CF sputum samples (n = 7) were processed as previously described (66). Undiluted SOL containing extracellular enzymes was spiked with saline or RTD-1 (100 μg/ml; 5% [vol/vol]), vortexed for 5 s, and placed in a 37°C orbital shaker. At 0 and 24 h, 10% acetic acid was added to stop all enzymatic activity. Samples were clarified by centrifugation prior to UPLC-MS and analyzed as described for BALF LC-MS methods.
In vivo models to assess airway tolerance, safety, pharmacokinetics, and anti-inflammatory effects of RTD-1. (i) Airway tolerability.
Airway hyperresponsiveness was assessed by Mch-induced airflow obstruction in naive BALB/c mice exposed to 3 mg/kg RTD-1 via intranasal administration. The dose of RTD-1 administered via this route was based on previous published in vivo work demonstrating efficacy and minimal airway toxicity concerns (29). The route allowed us to test tolerability at concentrations much higher than those achieved with the nebulizer described below. Mice were anesthetized by administration of ketamine (80 mg/kg; Hospira, Lake Forest, IL) and xylazine (10 mg/kg; Lloyd, Shenandoah, IA). An incision was made to access the trachea, and the mice were mechanically ventilated. Lung mechanics were measured by restrained plethysmography (Buxco Electronics, Troy, NY). Aerosolized methacholine was administered in increasing concentrations, and RL and Cdyn were continuously computed by fitting flow, volume, and pressure to an equation of motion.
(ii) Nose-only aerosol drug delivery.
RTD-1 solution (RTD-1 in 0.45% NaCl) was delivered via a syringe pump (flow rate, 0.1 ml/min) to an aerosol inhalation exposure system consisting of a bioaerosol generator (BANG) and a 16-port nose-only inhalation chamber with clear plexiglass nose-only mouse restraint holders. House air was dried by passing it through a desiccator tube prior to entering the BANG nebulizer. A sampling flow rate of 0.5 liter/min (comparable to the operational flow rate of the exposure port) was used in experiments designed to measure aerosol particle size using a 7-stage cascade impactor (Intox, Moriarty, NM) and aerosol concentrations using a Teflon liquid impinger (see Fig. S5a to e in the supplemental material). All items, unless otherwise specified, were purchased from CH Technologies (Westwood, NJ). The estimated delivered dose was based upon the concentration of RTD-1 in the sampled air, the respiratory-minute volume of a 25-g mouse, the duration of exposure, the inhalable fraction, and body weight and was calculated as described previously (67, 68). The operating parameters included the following: exposure times of 1 h for 49-, 167-, and 360-μg/kg delivered doses and 15 min for the 6.8-μg/kg delivered dose and total system flow rates of 2 (6.8 and 49 μg/kg), 4 (167 μg/kg), and 6 (360 μg/kg) liters/min.
(iii) Aerosol pharmacokinetics and safety.
Single-dose pharmacokinetics were determined in C57BL/6NCrl mice following an aerosol dose of 6.8, 49, 167, or 360 μg/kg (see Fig. S7 in the supplemental material). BAL was performed at 0.5, 1, 4, and 8 h postaerosolization (n = 4/time point). RTD-1 concentrations from BALF samples were measured by LC-MS as described below. The urea correction method was utilized to determine RTD-1 concentrations in ELF as previously described (69). Pharmacokinetic parameters were derived from the concentration-time profiles using noncompartmental analysis. The Cmax and time to maximum concentration (Tmax) were obtained from the observed data. The AUC was determined using the linear trapezoidal rule. In addition to respiratory tolerance, total BALF leukocyte counts, and body weight, as described for the infection model, served as surrogates for pulmonary safety. Furthermore, lungs were inflated and infused with 10% formalin-buffered solution (Thermo Fisher Scientific, Waltham, MA) as recommended by the American Thoracic Society (ATS) (70). Hematoxylin-eosin (H&E) staining of 5-μm slices was performed by the University of Southern California (USC) pathology laboratory. Photomicrographs at ×10 magnification using a Nikon Microphot-FXA microscope with a 10× objective lens were captured using a Spot insight 4.0 MP charge-coupled-device (CCD) color digital camera and software. Histopathological evaluation of lung inflammation was performed by a single nonblinded investigator based on neutrophil involvement in the lung and the presence of hyaline membranes, proteinaceous debris, and alveolar wall thickening (70).
(iv) Chronic P. aeruginosa airway infection.
CftrΔF508/ΔF508 mice, 10 to 12 weeks of age, were infected with P. aeruginosa (RP73) by intratracheal instillation exactly as previously described to establish chronic endobronchial infection (54, 71). Briefly, P. aeruginosa entrapped bead preparations were made fresh for each study, and the maximum instillate volume (50 μl) was administered to all mice. The starting inocula were 2.8 × 105, 2.5 × 105, and 5 × 105 for 49-, 167-, and 360-μg/kg delivered doses, respectively. After 24 h of infection, the animals received a 1-hour daily treatment with either aerosolized half-normal saline (n = 15) or RTD-1 (49,167, and 360 μg/kg; n = 8, 12, and 11) for 7 days (see Fig. S8 in the supplemental material). Three independent experiments with treatment and control groups were performed (n = 16/group). The controls were then pooled across experiments. Work regarding the antibacterial effects of aerosolized RTD-1 at 167 μg/kg was previously published (25). These observations were included for comparison across multiple dose groups, as well as to provide new data on the biological effect of each dose on inflammation markers (e.g., cytokines and proteases).
Lungs were washed with 0.8 ml PBS twice on day 7 of infection. Cells were pelleted at 1,000 rpm for 10 min at 4°C. The pelleted cells were resuspended in PBS for analysis of total and differential cell counts. BALF aliquots were stored at −80°C for later analysis of inflammatory cytokines/chemokines and proteases. Total and differential cell counts were performed by diluting cells in Turks blood diluting fluid (Ricca Chemical Company, Arlington, TX, USA). A total cell count was performed using a hemocytometer, while a cell differential count was performed using a commercially available Romanowsky stain variant (Diff-Quick).
Bacterial counts were performed as previously described (54). Briefly, an aliquot of BAL fluid and homogenized lung tissue was serially diluted and plated on TSA plates. The total lung bacterial burden was calculated from the sum of the colony counts in the BAL fluid and lung homogenate.
(v) Murine inflammatory mediators from BALF were quantified using Milliplex MAG (Millipore, Billerica, MA) kits for soluble receptors and inflammatory mediators. Analytes included IL-6, MCP-1, TNF, IL-17, IL-1β, KC, MIP-2, TIMP-1 (total), and amphiregulin. Quantification was performed using Luminex XMap technology and the Luminex 100 platform (Luminex, Austin, TX). All samples were neat and were incubated overnight with the antibodies of interest. Lung proteases were also measured. Total murine MMP-9 (pro-, active, and TIMP complexed) and NE activities were quantified from BALF. The Quantikine Mouse MMP-9 ELISA (R&D, Minneapolis, MN) was used following the manufacturer's protocol to determine total MMP-9. NE activity was measured by a fluorogenic substrate assay using MeOSu-AAPV-AMC (Cayman Chemical, Ann Arbor, MI) as previously described (72). The general clinical response was quantified, in addition to inflammatory markers. Weight and survival were monitored daily. The change from baseline was calculated as follows: [(daily weight/initial weight) − 1] × 100.
(vi) BALF LC-MS.
RTD-1 concentrations from BALF samples were measured by LC-MS as previously described (25). Briefly, samples were acidified with 5% acetic acid, spiked with an internal standard, extracted using C18 SPE columns (Waters, Milford, MA), lyophilized, and resuspended in 100 μl of 10% acetic acid, 5% acetonitrile. Samples were then resolved on a C18 X-Bridge BEH column (Waters) using an Acquity UPLC system (Waters). RTD-1 concentrations were determined from post-photodiode array (PDA) detector eluent using a Micromass Quattro Ultima mass spectrometer under electrospray ionization.
Materials.
The CF P. aeruginosa RP73 clinical strain (multidrug resistant; nonmucoidy phenotype) was a kind gift from Alessandra Bragonzi (71, 73). The hydrochloride salt of RTD-1 (>98%) was synthesized using Fmoc chemistry as described previously (74–76). A stock solution was prepared in sterile water and 0.22-μm-filter sterilized. Working solutions were further prepared in 0.9% sodium chloride for injection (USP; Hospira, Lake Forest, IL) for administration to animals. Turks blood diluting fluid for leukocyte counts was obtained from Ricca Chemical Co. (Arlington, TX). PBS without Ca2+ or Mg2+ was purchased from Lonza (Walkersville, MD). Otherwise, unless specified, materials were purchased from VWR (Radnor, PA).
Statistical methods and analysis.
Statistical and graphical analyses were carried out using GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA). Residual errors of univariate data were inspected for near-normal shape distribution (skewness and kurtosis ± 2; histogram) and central tendency (mean and median) (77). Nonnormal data were subsequently log-transformed and evaluated as described above. The parametric analysis of variance (ANOVA) with post hoc analysis P values were calculated with Bonferroni corrections between P. aeruginosa-treated and untreated groups and reported as either mean and SD or geometric mean and 95% confidence interval. However, multiple-testing adjustment using the Dunn test was performed when unequal sample sizes existed. The ratio paired t test was performed on sputum leukocyte samples as the ratio (treated/control), not the difference, since this was a more consistent measure of effect. Reverse transcription-PCR array analysis was performed using the online Data Analysis Center (Qiagen, Valencia, CA) with multiple internal control normalization genes. The ΔΔCT method with fold expression of each gene treated with RTD-1 compared to that of the control was performed. Changes were log2 normalized, and parametric testing was performed for statistical significance without post hoc adjustments. Significance was determined at a P value of ≤0.05. In regard to model building, the most appropriate model was selected based on the F-test criteria. Noncompartmental analysis of RTD-1 pharmacokinetics was performed by building 4 complete concentration-time profiles and applying the linear trapezoidal rule and statistical-moment theory in Excel for Mac 2016 (Microsoft, Redmond, WA).
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the contribution of the late Niklas Werner to this paper. We also thank Jonathan Lam and Yuzo Suzuki for performing the pulmonary function tests, as well as Diane Da Silva and Joseph Skeate from the Beckman Center for Immune Monitoring for running the multiple-analyte panels.
The research was supported in part by grants from the Cystic Fibrosis Foundation BERING14GO (P.M.B.), the Cystic Fibrosis Research Inc. (P.M.B.), the Webb Foundation (P.M.B.), the National Institute of Dental and Craniofacial Research (5T90DE021982) and National Institute of General Medicine (F31GM109729) (T.J.B.), the National Institutes of Health (RO1AI22931, R01DE021341, and RO1AI125141) (M.E.S.), the Arthritis Foundation (M.E.S.), and the National Cancer Institute (P30CA014089). We also acknowledge the support of the USC Immune Monitoring Core Facility, which is supported in part by National Cancer Institute Cancer Center Shared Grant award P30CA014089.
T.J.B., J.G.J., J.B.S., D.T., O.A., M.E.S., and P.M.B. participated in the experimental design. A.P.R. and J.C.W. evaluated and obtained clinical specimens. T.J.B., J.G.J., J.B.S., D.T., M.S., E.K., and J.M. performed the experiments. T.J.B., J.G.J., M.S., E.K., Niklas Werner, and J.M. analyzed the data. T.J.B., M.E.S., and P.M.B. interpreted results. T.J.B. and P.M.B. prepared the manuscript draft. We all reviewed, edited, and approved the final manuscript.
The content is solely our responsibility and does not necessarily represent the official views of the NIDCR, NIGMS, NCI, or NIH.
M.E.S. has an equity interest in Oryn Therapeutics, LLC, an entity that seeks to commercialize theta-defensins as therapeutics. We declare we have no other conflicts.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00154-17.
REFERENCES
- 1.Chmiel JF, Berger M, Konstan MW. 2002. The role of inflammation in the pathophysiology of CF lung disease. Clin Rev Allergy Immunol 23:5–27. doi: 10.1385/CRIAI:23:1:005. [DOI] [PubMed] [Google Scholar]
- 2.Li Z, Kosorok MR, Farrell PM, Laxova A, West SE, Green CG, Collins J, Rock MJ, Splaingard ML. 2005. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 293:581–588. doi: 10.1001/jama.293.5.581. [DOI] [PubMed] [Google Scholar]
- 3.Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. 2002. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 34:91–100. doi: 10.1002/ppul.10127. [DOI] [PubMed] [Google Scholar]
- 4.Henry RL, Mellis CM, Petrovic L. 1992. Mucoid Pseudomonas aeruginosa is a marker of poor survival in cystic fibrosis. Pediatr Pulmonol 12:158–161. doi: 10.1002/ppul.1950120306. [DOI] [PubMed] [Google Scholar]
- 5.Rosenfeld M, Gibson RL, McNamara S, Emerson J, Burns JL, Castile R, Hiatt P, McCoy K, Wilson CB, Inglis A, Smith A, Martin TR, Ramsey BW. 2001. Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr Pulmonol 32:356–366. doi: 10.1002/ppul.1144. [DOI] [PubMed] [Google Scholar]
- 6.Knowles MR, Boucher RC. 2002. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest 109:571–577. doi: 10.1172/JCI0215217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hiemstra PS, Amatngalim GD, van der Does AM, Taube C. 2016. Antimicrobial peptides and innate lung defenses: role in infectious and noninfectious lung diseases and therapeutic applications. Chest 149:545–551. doi: 10.1378/chest.15-1353. [DOI] [PubMed] [Google Scholar]
- 8.Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, Nelson GA, Chang EH, Taft PJ, Ludwig PS, Estin M, Hornick EE, Launspach JL, Samuel M, Rokhlina T, Karp PH, Ostedgaard LS, Uc A, Starner TD, Horswill AR, Brogden KA, Prather RS, Richter SS, Shilyansky J, McCray PB Jr, Zabner J, Welsh MJ. 2010. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2:29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Banfi B, Horswill AR, Stoltz DA, McCray PB Jr, Welsh MJ, Zabner J. 2012. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487:109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abou Alaiwa MH, Reznikov LR, Gansemer ND, Sheets KA, Horswill AR, Stoltz DA, Zabner J, Welsh MJ. 2014. pH modulates the activity and synergism of the airway surface liquid antimicrobials beta-defensin-3 and LL-37. Proc Natl Acad Sci U S A 111:18703–18708. doi: 10.1073/pnas.1422091112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hilliard TN, Regamey N, Shute JK, Nicholson AG, Alton EW, Bush A, Davies JC. 2007. Airway remodelling in children with cystic fibrosis. Thorax 62:1074–1080. doi: 10.1136/thx.2006.074641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayer-Hamblett N, Aitken ML, Accurso FJ, Kronmal RA, Konstan MW, Burns JL, Sagel SD, Ramsey BW. 2007. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am J Respir Crit Care Med 175:822–828. doi: 10.1164/rccm.200609-1354OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sagel SD, Kapsner RK, Osberg I. 2005. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr Pulmonol 39:224–232. doi: 10.1002/ppul.20165. [DOI] [PubMed] [Google Scholar]
- 14.Voynow JA, Fischer BM, Zheng S. 2008. Proteases and cystic fibrosis. Int J Biochem Cell Biol 40:1238–1245. doi: 10.1016/j.biocel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis SD, Ferkol T. 2013. Identifying the origins of cystic fibrosis lung disease. N Engl J Med 368:2026–2028. doi: 10.1056/NEJMe1303487. [DOI] [PubMed] [Google Scholar]
- 16.Auerbach HS, Williams M, Kirkpatrick JA, Colten HR. 1985. Alternate-day prednisone reduces morbidity and improves pulmonary function in cystic fibrosis. Lancet ii:686–688. [DOI] [PubMed] [Google Scholar]
- 17.Konstan MW, Byard PJ, Hoppel CL, Davis PB. 1995. Effect of high-dose ibuprofen in patients with cystic fibrosis. N Engl J Med 332:848–854. doi: 10.1056/NEJM199503303321303. [DOI] [PubMed] [Google Scholar]
- 18.Konstan MW, Schluchter MD, Xue W, Davis PB. 2007. Clinical use of ibuprofen is associated with slower FEV1 decline in children with cystic fibrosis. Am J Respir Crit Care Med 176:1084–1089. doi: 10.1164/rccm.200702-181OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lands LC, Milner R, Cantin AM, Manson D, Corey M. 2007. High-dose ibuprofen in cystic fibrosis: Canadian safety and effectiveness trial. J Pediatr 151:249–254. doi: 10.1016/j.jpeds.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Ganz T. 2003. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol 3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 21.Mansour SC, Pena OM, Hancock RE. 2014. Host defense peptides: front-line immunomodulators. Trends Immunol 35:443–450. doi: 10.1016/j.it.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Yang D, Liu ZH, Tewary P, Chen Q, de la Rosa G, Oppenheim JJ. 2007. Defensin participation in innate and adaptive immunity. Curr Pharm Des 13:3131–3139. doi: 10.2174/138161207782110453. [DOI] [PubMed] [Google Scholar]
- 23.Selsted ME. 2004. Theta-defensins: cyclic antimicrobial peptides produced by binary ligation of truncated alpha-defensins. Curr Protein Pept Sci 5:365–371. doi: 10.2174/1389203043379459. [DOI] [PubMed] [Google Scholar]
- 24.Lehrer RI, Cole AM, Selsted ME. 2012. Theta-defensins: cyclic peptides with endless potential. J Biol Chem 287:27014–27019. doi: 10.1074/jbc.R112.346098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beringer PM, Bensman TJ, Ho H, Agnello M, Denovel N, Nguyen A, Wong-Beringer A, She R, Tran DQ, Moskowitz SM, Selsted ME. 2016. Rhesus theta-defensin-1 (RTD-1) exhibits in vitro and in vivo activity against cystic fibrosis strains of Pseudomonas aeruginosa. J Antimicrob Chemother 71:181–188. doi: 10.1093/jac/dkv301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tai KP, Kamdar K, Yamaki J, Le VV, Tran D, Tran P, Selsted ME, Ouellette AJ, Wong-Beringer A. 2015. Microbicidal effects of alpha- and theta-defensins against antibiotic-resistant Staphylococcus aureus and Pseudomonas aeruginosa. Innate Immun 21:17–29. doi: 10.1177/1753425913514784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tran D, Tran P, Roberts K, Osapay G, Schaal J, Ouellette A, Selsted ME. 2008. Microbicidal properties and cytocidal selectivity of rhesus macaque theta defensins. Antimicrob Agents Chemother 52:944–953. doi: 10.1128/AAC.01090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaal JB, Tran D, Tran P, Osapay G, Trinh K, Roberts KD, Brasky KM, Tongaonkar P, Ouellette AJ, Selsted ME. 2012. Rhesus macaque theta defensins suppress inflammatory cytokines and enhance survival in mouse models of bacteremic sepsis. PLoS One 7:e51337. doi: 10.1371/journal.pone.0051337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wohlford-Lenane CL, Meyerholz DK, Perlman S, Zhou H, Tran D, Selsted ME, McCray PB Jr. 2009. Rhesus theta-defensin prevents death in a mouse model of severe acute respiratory syndrome coronavirus pulmonary disease. J Virol 83:11385–11390. doi: 10.1128/JVI.01363-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tongaonkar P, Trinh KK, Schaal JB, Tran D, Gulko PS, Ouellette AJ, Selsted ME. 2015. Rhesus macaque theta-defensin RTD-1 inhibits proinflammatory cytokine secretion and gene expression by inhibiting the activation of NF-kappaB and MAPK pathways. J Leukoc Biol 98:1061–1070. doi: 10.1189/jlb.3A0315-102R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, Jung A, Greene C, McElvaney G, Mall M, Doring G. 2012. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros 11:363–382. doi: 10.1016/j.jcf.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 32.Laval J, Ralhan A, Hartl D. 2016. Neutrophils in cystic fibrosis. Biol Chem 397:485–496. doi: 10.1515/hsz-2015-0271. [DOI] [PubMed] [Google Scholar]
- 33.van Heeckeren AM, Schluchter MD. 2002. Murine models of chronic Pseudomonas aeruginosa lung infection. Lab Anim 36:291–312. doi: 10.1258/002367702320162405. [DOI] [PubMed] [Google Scholar]
- 34.van Heeckeren AM, Tscheikuna J, Walenga RW, Konstan MW, Davis PB, Erokwu B, Haxhiu MA, Ferkol TW. 2000. Effect of Pseudomonas infection on weight loss, lung mechanics, and cytokines in mice. Am J Respir Crit Care Med 161:271–279. doi: 10.1164/ajrccm.161.1.9903019. [DOI] [PubMed] [Google Scholar]
- 35.Cohen TS, Prince A. 2012. Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med 18:509–519. doi: 10.1038/nm.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hancock RE, Sahl HG. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol 24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 37.Marjanovic N, Bosnar M, Michielin F, Wille DR, Anic-Milic T, Culic O, Popovic-Grle S, Bogdan M, Parnham MJ, Erakovic Haber V. 2011. Macrolide antibiotics broadly and distinctively inhibit cytokine and chemokine production by COPD sputum cells in vitro. Pharmacol Res 63:389–397. doi: 10.1016/j.phrs.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 38.Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, Bruckner P, Pfeilschifter J, Schaefer RM, Grone HJ, Schaefer L. 2009. Biglycan, a danger signal that activates the NLRP3 inflammasome via Toll-like and P2X receptors. J Biol Chem 284:24035–24048. doi: 10.1074/jbc.M109.014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. 2008. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A 105:8067–8072. doi: 10.1073/pnas.0709684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott P, Ma H, Viriyakosol S, Terkeltaub R, Liu-Bryan R. 2006. Engagement of CD14 mediates the inflammatory potential of monosodium urate crystals. J Immunol 177:6370–6378. doi: 10.4049/jimmunol.177.9.6370. [DOI] [PubMed] [Google Scholar]
- 41.Jackson PL, Xu X, Wilson L, Weathington NM, Clancy JP, Blalock JE, Gaggar A. 2010. Human neutrophil elastase-mediated cleavage sites of MMP-9 and TIMP-1: implications to cystic fibrosis proteolytic dysfunction. Mol Med 16:159–166. doi: 10.2119/molmed.2009.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu X, Jackson PL, Tanner S, Hardison MT, Abdul Roda M, Blalock JE, Gaggar A. 2011. A self-propagating matrix metalloprotease-9 (MMP-9) dependent cycle of chronic neutrophilic inflammation. PLoS One 6:e15781. doi: 10.1371/journal.pone.0015781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hartl D, Latzin P, Hordijk P, Marcos V, Rudolph C, Woischnik M, Krauss-Etschmann S, Koller B, Reinhardt D, Roscher AA, Roos D, Griese M. 2007. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med 13:1423–1430. doi: 10.1038/nm1690. [DOI] [PubMed] [Google Scholar]
- 44.Greene CM, McElvaney NG. 2009. Proteases and antiproteases in chronic neutrophilic lung disease: relevance to drug discovery. Br J Pharmacol 158:1048–1058. doi: 10.1111/j.1476-5381.2009.00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andrault PM, Samsonov SA, Weber G, Coquet L, Nazmi K, Bolscher JG, Lalmanach AC, Jouenne T, Bromme D, Pisabarro MT, Lalmanach G, Lecaille F. 2015. Antimicrobial peptide LL-37 is both a substrate of cathepsins S and K and a selective inhibitor of cathepsin L. Biochemistry 54:2785–2798. doi: 10.1021/acs.biochem.5b00231. [DOI] [PubMed] [Google Scholar]
- 46.Sallenave JM, Si Tahar M, Cox G, Chignard M, Gauldie J. 1997. Secretory leukocyte proteinase inhibitor is a major leukocyte elastase inhibitor in human neutrophils. J Leukoc Biol 61:695–702. [DOI] [PubMed] [Google Scholar]
- 47.Yang C, Chilvers M, Montgomery M, Nolan SJ. 2016. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev 4:CD001127. doi: 10.1002/14651858.CD001127.pub3. [DOI] [PubMed] [Google Scholar]
- 48.Dubois AV, Midoux P, Gras D, Si-Tahar M, Brea D, Attucci S, Khelloufi MK, Ramphal R, Diot P, Gauthier F, Herve V. 2013. Poly-l-lysine compacts DNA, kills bacteria, and improves protease inhibition in cystic fibrosis sputum. Am J Respir Crit Care Med 188:703–709. doi: 10.1164/rccm.201305-0912OC. [DOI] [PubMed] [Google Scholar]
- 49.Keal EE, Reid L. 1972. Neuraminic acid content of sputum in chronic bronchitis. Thorax 27:643–653. doi: 10.1136/thx.27.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubin BK. 1992. Aerosolized recombinant human deoxyribonuclease I in the treatment of cystic fibrosis. N Engl J Med 327:571. [DOI] [PubMed] [Google Scholar]
- 51.Cigana C, Bernardini F, Facchini M, Alcala-Franco B, Riva C, De Fino I, Rossi A, Ranucci S, Misson P, Chevalier E, Brodmann M, Schmitt M, Wach A, Dale GE, Obrecht D, Bragonzi A. 2016. Efficacy of the novel antibiotic POL7001 in preclinical models of Pseudomonas aeruginosa pneumonia. Antimicrob Agents Chemother 60:4991–5000. doi: 10.1128/AAC.00390-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doring G, Bragonzi A, Paroni M, Akturk FF, Cigana C, Schmidt A, Gilpin D, Heyder S, Born T, Smaczny C, Kohlhaufl M, Wagner TO, Loebinger MR, Bilton D, Tunney MM, Elborn JS, Pier GB, Konstan MW, Ulrich M. 2014. BIIL 284 reduces neutrophil numbers but increases P. aeruginosa bacteremia and inflammation in mouse lungs. J Cyst Fibros 13:156–163. doi: 10.1016/j.jcf.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cigana C, Lore NI, Riva C, De Fino I, Spagnuolo L, Sipione B, Rossi G, Nonis A, Cabrini G, Bragonzi A. 2016. Tracking the immunopathological response to Pseudomonas aeruginosa during respiratory infections. Sci Rep 6:21465. doi: 10.1038/srep21465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Facchini M, De Fino I, Riva C, Bragonzi A. 17 March 2014. Long term chronic Pseudomonas aeruginosa airway infection in mice. J Vis Exp doi: 10.3791/51019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Forde E, Humphreys H, Greene CM, Fitzgerald-Hughes D, Devocelle M. 2014. Potential of host defense peptide prodrugs as neutrophil elastase-dependent anti-infective agents for cystic fibrosis. Antimicrob Agents Chemother 58:978–985. doi: 10.1128/AAC.01167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gordon YJ, Romanowski EG, McDermott AM. 2005. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr Eye Res 30:505–515. doi: 10.1080/02713680590968637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knyght I, Clifton L, Saaka Y, Lawrence MJ, Barlow DJ. 2016. Interaction of the antimicrobial peptides rhesus theta-defensin and porcine protegrin-1 with anionic phospholipid monolayers. Langmuir 32:7403–7410. doi: 10.1021/acs.langmuir.6b01688. [DOI] [PubMed] [Google Scholar]
- 58.Berube J, Roussel L, Nattagh L, Rousseau S. 2010. Loss of cystic fibrosis transmembrane conductance regulator function enhances activation of p38 and ERK MAPKs, increasing interleukin-6 synthesis in airway epithelial cells exposed to Pseudomonas aeruginosa. J Biol Chem 285:22299–22307. doi: 10.1074/jbc.M109.098566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu Q, Lu Z, Verghese MW, Randell SH. 2005. Airway epithelial cell tolerance to Pseudomonas aeruginosa. Respir Res 6:26. doi: 10.1186/1465-9921-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Profita M, Chiappara G, Mirabella F, Di Giorgi R, Chimenti L, Costanzo G, Riccobono L, Bellia V, Bousquet J, Vignola AM. 2003. Effect of cilomilast (Ariflo) on TNF-alpha, IL-8, and GM-CSF release by airway cells of patients with COPD. Thorax 58:573–579. doi: 10.1136/thorax.58.7.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scheicher ME, Teixeira MM, Cunha FQ, Teixeira AL Jr, Filho JT, Vianna EO. 2007. Eotaxin-2 in sputum cell culture to evaluate asthma inflammation. Eur Respir J 29:489–495. doi: 10.1183/09031936.00060205. [DOI] [PubMed] [Google Scholar]
- 62.Alvarez-Iglesias M, Wayne G, O'Dea KP, Amour A, Takata M. 2005. Continuous real-time measurement of tumor necrosis factor-alpha converting enzyme activity on live cells. Lab Invest 85:1440–1448. doi: 10.1038/labinvest.3700340. [DOI] [PubMed] [Google Scholar]
- 63.Devereux G, Fraser-Pitt D, Robertson J, Devlin E, Mercer D, O'Neil D. 2015. Cysteamine as a future intervention in cystic fibrosis against current and emerging pathogens: a patient-based ex vivo study confirming its antimicrobial and mucoactive potential in sputum. EBioMedicine 2:1507–1512. doi: 10.1016/j.ebiom.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keal EE. 1971. Biochemistry and rheology of sputum in asthma. Postgrad Med J 47:171–177. doi: 10.1136/pgmj.47.545.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shak S, Capon DJ, Hellmiss R, Marsters SA, Baker CL. 1990. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc Natl Acad Sci U S A 87:9188–9192. doi: 10.1073/pnas.87.23.9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rees DD, Brain JD, Wohl ME, Humes JL, Mumford RA. 1997. Inhibition of neutrophil elastase in CF sputum by L-658,758. J Pharmacol Exp Ther 283:1201–1206. [PubMed] [Google Scholar]
- 67.Alexander DJ, Collins CJ, Coombs DW, Gilkison IS, Hardy CJ, Healey G, Karantabias G, Johnson N, Karlsson A, Kilgour JD, McDonald P. 2008. Association of Inhalation Toxicologists (AIT) working party recommendation for standard delivered dose calculation and expression in non-clinical aerosol inhalation toxicology studies with pharmaceuticals. Inhal Toxicol 20:1179–1189. doi: 10.1080/08958370802207318. [DOI] [PubMed] [Google Scholar]
- 68.Tuttle RS, Sosna WA, Daniels DE, Hamilton SB, Lednicky JA. 2010. Design, assembly, and validation of a nose-only inhalation exposure system for studies of aerosolized viable influenza H5N1 virus in ferrets. Virol J 7:135. doi: 10.1186/1743-422X-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rennard SI, Basset G, Lecossier D, O'Donnell KM, Pinkston P, Martin PG, Crystal RG. 1986. Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution. J Appl Physiol 60:532–538. [DOI] [PubMed] [Google Scholar]
- 70.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM. 2011. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bragonzi A, Wiehlmann L, Klockgether J, Cramer N, Worlitzsch D, Doring G, Tummler B. 2006. Sequence diversity of the mucABD locus in Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology 152:3261–3269. doi: 10.1099/mic.0.29175-0. [DOI] [PubMed] [Google Scholar]
- 72.Kossodo S, Zhang J, Groves K, Cuneo GJ, Handy E, Morin J, Delaney J, Yared W, Rajopadhye M, Peterson JD. 2011. Noninvasive in vivo quantification of neutrophil elastase activity in acute experimental mouse lung injury. Int J Mol Imaging 2011:581406. doi: 10.1155/2011/581406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jeukens J, Boyle B, Bianconi I, Kukavica-Ibrulj I, Tummler B, Bragonzi A, Levesque RC. 2013. Complete genome sequence of persistent cystic fibrosis isolate Pseudomonas aeruginosa strain RP73. Genome Announc 1:e00568–. doi: 10.1128/genomeA.00568-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garcia AE, Osapay G, Tran PA, Yuan J, Selsted ME. 2008. Isolation, synthesis, and antimicrobial activities of naturally occurring theta-defensin isoforms from baboon leukocytes. Infect Immun 76:5883–5891. doi: 10.1128/IAI.01100-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tang YQ, Yuan J, Osapay G, Osapay K, Tran D, Miller CJ, Ouellette AJ, Selsted ME. 1999. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha-defensins. Science 286:498–502. doi: 10.1126/science.286.5439.498. [DOI] [PubMed] [Google Scholar]
- 76.Tran D, Tran PA, Tang YQ, Yuan J, Cole T, Selsted ME. 2002. Homodimeric theta-defensins from rhesus macaque leukocytes: isolation, synthesis, antimicrobial activities, and bacterial binding properties of the cyclic peptides. J Biol Chem 277:3079–3084. doi: 10.1074/jbc.M109117200. [DOI] [PubMed] [Google Scholar]
- 77.Westfall PHK. 2013. Functions of random variables: their distributions and expected values. CRC Press, Boca Raton, FL. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.