

Abstract
We have identified differentially regulated genes in chronic myeloid leukemia (CML) cells upon short treatment with the broad‐spectrum Bcr–Abl inhibitor dasatinib. The highly specific Bcr–Abl inhibitor nilotinib caused a very similar gene expression signature, validating the identified differentially regulated genes as a read‐out of Bcr–Abl activity and implying that Bcr–Abl is the functionally central target of dasatinib in CML cells. Among the strongest downregulated genes, we have further validated the activation marker CD69 and the chemokine interleukin (IL)‐8. Expression of both proteins is upregulated upon Bcr–Abl expression and inhibited by dasatinib and nilotinib. IL‐8 may thus be a useful marker for the monitoring of CML inhibitor efficacy and play a potential pathophysiological role in CML.
Keywords: Bcr–Abl, Chronic myeloid leukemia, Tyrosine kinase inhibitor, Biomarker, Imatinib
1. Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disorder that is characterized by the presence of the translocation (t9;22) (q34;q11) leading to the aberrant expression of the constitutively active Bcr–Abl fusion tyrosine kinase (Wong and Witte, 2004). Targeting Bcr–Abl using the small molecule kinase inhibitor imatinib (Gleevec) has become the front‐line therapy for CML (Deininger et al., 2005; Quintas‐Cardama et al., 2007). Although a majority of CML patients in chronic phase achieve a complete cytogenetic response (CCyR) on imatinib, ∼25% of patients do not achieve such a response by 18months (primary cytogenetic resistance) or loose a previously achieved CCyR due to imatinib resistance caused by point mutations in the kinase domain or other mechanisms (secondary resistance; Druker et al., 2006; O'Hare et al., 2006). Despite the clinical efficacy of the recently approved second‐generation inhibitors nilotinib and dasatinib for the treatment of imatinib resistant or intolerant patients, the general shortcomings of primary and secondary resistance remain problematic, in particular in the advanced and blast crisis phases of CML (Kantarjian et al., 2007; Weisberg et al., 2007; le Coutre et al., 2008).
In general, CML is a well‐studied disease, but the underlying changes in signal transduction networks and gene expression patterns that lead to oncogenic reprogramming of hematopoietic cells by the expression of Bcr–Abl are only poorly understood (Van Etten, 2004; Ren, 2005). With ongoing clinical trials using nilotinib and dasatinib as frontline therapy in CML patients and several “third‐generation” Bcr–Abl inhibitors emerging, diagnostic markers to quickly monitor response to tyrosine kinase inhibitor therapy from peripheral blood may be of great advantage for a rational choice of the appropriate drug, ultimately leading to a personalized treatment of CML patients. At best, such markers could indicate a response before hematologic or cytogenetic remission is achieved or indicate a primary resistance, thus necessitating to switch to a more suited inhibitor, discontinue an expensive ineffective treatment early after diagnosis and opt for alternative therapy.
To shed light on the critical pathways and effectors that are dependent on Bcr–Abl activity and potentially involved in the molecular pathology of CML, we set out to study gene expression signatures induced upon short‐term treatment with dasatinib. We reasoned that, if among the differentially regulated genes we could validate some encoding secreted or membrane‐bound proteins, these could be used conveniently to monitor drug response in patients.
Based on statistical analysis and degree of regulation by dasatinib, we selected 44 genes. A set of 28 genes that were most up‐ or downregulated by dasatinib were validated by using quantitative RT–PCR. Bioinformatic network analysis revealed a particular impact of dasatinib on the JAK–STAT signaling pathway. By employing additional inhibitors and cell lines, we were able to show a very strong dependence of expression of most of the candidate markers on the cellular Bcr–Abl activity status. Two robust candidates, the surface antigen CD69 and the secreted chemokine interleukin (IL)‐8, were found to be prominently downregulated upon inhibitor treatment and are upregulated upon expression of Bcr–Abl, suggesting a potential functional role in the pathogenesis of the disease. Preliminary analysis of a limited number of patient sera is consistent with the possibility of IL‐8 being useful as a biomarker in the disease and may suggest playing a pathogenetic role in CML. Monitoring of IL‐8 levels may thus in future represent an attractive addition to a growing list of other potential early indicators of treatment efficacy in CML.
2. Results
2.1. Microarray analysis identifies gene expression changes induced by dasatinib in K562 cells
In order to monitor the effect of dasatinib on Bcr–Abl dependent signal transduction and gene expression, we performed microarray‐based gene expression analysis from the Bcr–Abl positive cell line K562 that was either treated with 100nM dasatinib for 180min or left untreated in two biological repeats. Under these conditions, Bcr–Abl tyrosine kinase activity was completely inhibited and no Bcr–Abl autophosphorylation or tyrosine phosphorylation of other proteins in K562 cell lysates was detectable, suggesting complete suppression of Bcr–Abl dependent signal transduction (Figure 1 and data not shown). In contrast, cell viability, total cell numbers as well as total RNA yield and quality was not decreased after 180min of dasatinib treatment when compared to untreated control cells (Figure 1 and data not shown). Therefore, differences in the gene expression profiles should enrich for changes caused by loss of Bcr–Abl kinase activity as an oncogenic driver rather than general cell cycle arrest, apoptosis or cell death markers. Statistical analysis of the microarray data revealed 37 regulated genes. This dataset was complemented by manual selection of regulated genes, based on fold‐change and signal intensity, to reach a total of 44 genes. We chose 22 genes that were downregulated at least 3.5‐fold and 6 genes that were upregulated at least 2.5‐fold by dasatinib for further analysis (Table 1 and Supplementary figure S1).
Figure 1.
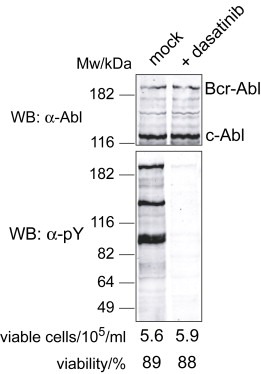
K562 cells treated with dasatinib for 180min show suppression of tyrosine phosphorylation without affecting cell number, viability or Bcr–Abl/c‐Abl stability. K562 cells growing in logarithmic phase were either mock treated with DMSO (left lane) or treated with 100nM dasatinib for 180min (right lane). Cell counts and cell viability was determined prior to harvesting. Proteins were extracted from an aliquot of the cells and were immunoblotted with anti‐Abl and anti‐phosphotyrosine antibodies. Total RNA for microarray hybridizations was extracted from the remaining cells.
Table 1.
Most significantly regulated genes by dasatinib in K562 cells
| Gene name | Fold regulation | qPCR validation |
|---|---|---|
| EGR1 | 0.01 | X |
| CD69 | 0.03 | X |
| IL8 | 0.06 | X |
| IL10RA | 0.06 | X |
| CISH | 0.06 | X |
| SPRY4 | 0.09 | X |
| PHLDA1 | 0.11 | X |
| GDF15 | 0.11 | X |
| MAFF | 0.12 | X |
| HECW2 | 0.12 | X |
| SOCS1 | 0.14 | X |
| MYC | 0.14 | |
| SLC2A3 | 0.14 | X |
| HHEX | 0.15 | X |
| APOL4 | 0.17 | X |
| PIM1 | 0.17 | X |
| CHAC1 | 0.19 | X |
| BCL2L1 | 0.19 | X |
| BHLHB2 | 0.19 | X |
| SOCS3 | 0.20 | X |
| ATF3 | 0.21 | X |
| ETV5 | 0.23 | |
| AMIGO2 | 0.24 | X |
| STC2 | 0.24 | X |
| INHBE | 0.25 | |
| SOCS2 | 0.26 | |
| SPRED1 | 0.26 | |
| ITPR1 | 0.28 | |
| RAB11FIP1 | 0.28 | |
| ETV1 | 0.29 | |
| RHOH | 0.30 | |
| PDE4DIP | 0.32 | |
| FJX1 | 0.33 | |
| IKZF4 | 0.33 | |
| MGC13057 | 0.34 | |
| IER2 | 0.35 | |
| XBP1 | 0.37 | |
| ERMAP | 2.41 | X |
| TMEM56 | 2.53 | X |
| FAM46B | 3.00 | X |
| IFIT1 | 3.29 | X |
| LGR4 | 3.35 | |
| CDKN1C | 3.71 | X |
| CYP3A5 | 5.53 | X |
Based on our statistical analysis, 44 genes were selected. The fold regulation of dasatinib‐treated versus mock‐treated samples in the microarray experiments is indicated. Twenty‐eight genes were included in the qPCR validation.
2.2. Network analysis reveals a major impact of dasatinib on target genes induced by the JAK–STAT pathway
We next performed a detailed bioinformatics analysis of all differentially regulated genes, analyzed the subcellular localization of the protein products as well as common pathways and cellular processes, which were affected by dasatinib. The differentially regulated genes products had a wide range of subcellular distributions (Figure 2). Among the 44 genes included in the analysis, 13 gene products were localized in the nucleus (mostly transcription factors), 12 were membrane proteins, 9 had cytoplasmic localization, 7 were secreted and 2 were localized to the endoplasmic reticulum (Figure 2).
Figure 2.
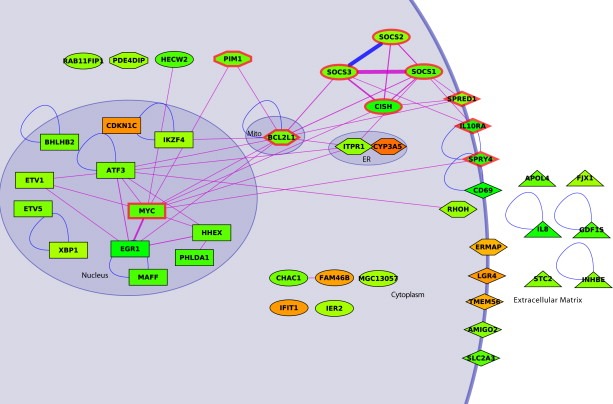
Interaction network and subcellular localization of differentially regulated genes by dasatinib in K562 cells. Regulated genes in response to dasatinib are shown according to their subcellular localization (Mito, mitochondria; ER, endoplasmic reticulum) and reflected in the node shapes (rectangle, nucleus; ellipse, cytoplasm; lozenge, membrane; diamond, Mito or ER; triangle, extracellular). Nodes corresponding to proteins having more than one localization have an intermediary shape. Downregulated genes are indicated in green, upregulated genes in orange. Regulated gene products that interact directly are indicated with blue lines, interactions that can be established over one‐additional partner in‐between are shown by violet lines. Line thickness indicates the number of paths (1–12) linking one protein to another protein. Nine proteins are involved in the JAK–STAT signaling pathway (thick red border).
We next analyzed the differentially regulated genes in the context of a curated list of published molecular interactions and cellular processes using the HPRD and DAVID databases (Dennis et al., 2003; Mishra et al., 2006). By using DAVID, we identified the targets of the JAK–STAT signaling pathway as significantly enriched (10 genes, P=2.1×10−9, Fisher exact test) in selected regulated genes (Supplementary figure S2). More specifically, we identified a multitude of STAT5 target genes (SOCS, PIM‐1, CIS, c‐Myc, Bcl‐XL, Spred, Sprouty) to be downregulated by dasatinib. This is entirely consistent with the transcription factor STAT5 being a critical and established effector of Bcr–Abl action (Shuai et al., 1996; Hoelbl et al., 2006; Ye et al., 2006).
To further elucidate possible functional relations among co‐regulated genes in the process, we considered the corresponding gene products and mined a protein‐protein interaction database (HPRD) using an algorithm that links the regulated genes with the protein–protein interaction dataset (Supplementary figure 2). In one case (SOCS2–SOCS3), we find a direct interaction between regulated gene products. To reveal more functional links among the regulated genes, we considered genes related through one intermediary protein. Therefore, if gene product A (regulated by dasatinib) interacted with protein B (intermediary protein) which interacted with gene product C (also regulated by dasatinib), we linked A to C. Figure 2 displays the network obtained by this procedure. We noticed that a significant part of the JAK–STAT signal transduction pathway was embedded in the computed network, which includes 19 gene products. Supplementary figure S3 shows the network including the intermediary proteins. To further extend the network we applied the same algorithm by accepting linking regulated genes related by two intermediary proteins, resulting in 14 additional genes that were connected to the initial network (Supplementary figure S4).
2.3. Gene expression changes induced by dasatinib are mirrored by nilotinib
In order to validate the findings of the microarray analysis and to provide quantitative information about changes in gene expression induced by dasatinib, we performed quantitative reverse transcriptase–polymerase chain reaction (qRT–PCR) with a panel of 28 genes (22 downregulated, 6 upregulated). In addition to dasatinib and mock‐treated K562 cells, we also treated cells with nilotinib. While dasatinib potently inhibits all major non‐receptor tyrosine kinases, including the Src kinase family and Btk/Tec, and many serine/threonine kinases (Carter et al., 2005; Bantscheff et al., 2007; Hantschel et al., 2007; Rix et al., 2007), nilotinib can be considered the most specific Bcr–Abl inhibitor currently available (Hantschel et al., 2008).
The results of the qRT–PCR analysis showed two major results (Figure 3 and Supplementary table 1): First, there was a very high overlap of the microarray and the qRT–PCR data. With one exception, all of the differentially regulated genes found in the microarray study could be confirmed by qRT–PCR. In addition, all genes showed a very similar level of regulation when comparing the microarray and the qRT–PCR datasets (Figure 3A). Second, all genes that were regulated by dasatinib were also regulated by nilotinib to a similar degree. This is surprising given the difference in target specificity of the two drugs, with dasatinib displaying a very broad specificity, while nilotinib is highly specific. This could be explained by the early time point of analysis, when only the effects on the dominant proliferation driver Bcr–Abl may have manifested. Overall, thesedata argue that the effects observed with the two drugs are indeed valid indicators for the state of activity of Bcr–Abl in K562 cells.
Figure 3.
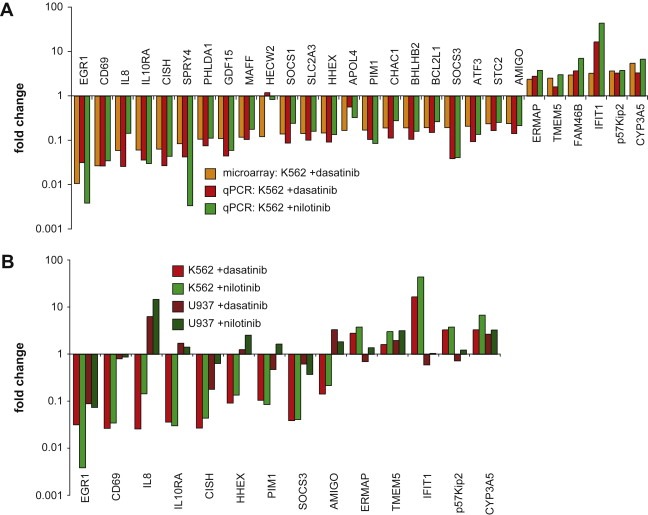
qRT–PCR analysis of differentially regulated genes. (A) Changes in mRNA expression levels detected by microarray after 180min treatment with 100nM dasatinib (black bars) were compared to changes in mRNA expression levels by qRT–PCR after 180min treatment with either 100nM dasatinib (dark grey bars) or 1μM nilotinib (light grey bars). (B) Changes in mRNA expression levels after 180min dasatinib or nilotinib treatment of K562 (black and dark grey bars, respectively) and U937 cells (light grey and white bars, respectively). Both panels show averages of relative fold changes in mRNA expression levels of two independent experiments done in triplicates and. All numerical values and standard deviations are available as Supplementary table 1.
2.4. mRNA expression changes differ between K562 cells and the Bcr–Abl negative lymphoblast cell line U937 upon dasatinib and nilotinib treatment
To test if the observed mRNA expression changes by dasatinib or nilotinib are dependent on the presence of Bcr–Abl, we compared mRNA expression changes induced upon inhibitor treatment in K562 cells with U937 cells. The human lymphoblast cell line U937 does not express Bcr–Abl, but expresses similar surface markers (CD3−, CD13+, CD19−, CD34−) and is morphologically comparable to K562 cells. Exponentially growing K562 and U937 cells were treated with dasatinib, nilotinib or left untreated and changes in mRNA expression levels of 14 genes were measured. For 12 genes, mRNA expression changes in U937 cells were much smaller with both dasatinib and nilotinib when compared to K562 cells (Figure 3B). Eight genes even showed regulation of mRNA expression in the opposite direction in U937 cells when treated with at least one of the two kinase inhibitors.
In summary, these observations suggest that the genes identified in our microarray experiments could be used as markers for Bcr–Abl activity, as nilotinib treatment mirrored the mRNA expression changes induced by dasatinib. Furthermore, the candidate genes were regulated differently or not at all in Bcr–Abl negative U937 cells by either of the two inhibitors.
2.5. CD69 is strongly upregulated by expression of Bcr–Abl and suppressed by inhibition of Bcr–Abl
To determine whether the observed dasatinib‐induced differential regulation of genes on the mRNA level was also reflected in changes in protein expression, we decided to validate selected candidate proteins with emphasis on cell surface receptors and secreted proteins. CD69, a member of the C‐type lectin family with unknown function, was among the strongest downregulated genes upon dasatinib treatment. CD69 is not expressed on resting cells, but is highly upregulated following cellular activation of all hematopoietic cells. Antibody crosslinking of CD69 on platelets and monocytes was shown to activate Erk1/2 and phospholipase C, whereas it was costimulated with the T‐cell receptor to induce proliferation and cytokine production in CD4+ T cells (Sancho et al., 2005).
Both K562 and U937 cells were treated with dasatinib, nilotinib or left untreated and CD69 expression was subsequently determined by FACS staining using a CD69 monoclonal antibody. In line with the results from the microarray and qRT–PCR analysis, 36% of mock‐treated K562 cells showed expression of CD69 (Figure 4, upper panel), which was reduced with treatments of both dasatinib and nilotinib to 11% and 12%, respectively. In contrast, CD69 was found to be expressed in 100% of the U937 cells and remained unchanged with both dasatinib and nilotinib (Figure 4). In addition, we used the GM‐CSF dependent human cell line UT7 that grows in the absence of GM‐CSF when transduced with Bcr–Abl (data not shown). Bcr–Abl expression increased CD69 expression from 26% to 82% when compared to the untransduced controls and was markedly reduced by both drugs. In contrast CD69 expression remained unaffected by dasatinib and nilotinib in untransduced UT7 cells (Figure 4, lower two panels).
Figure 4.
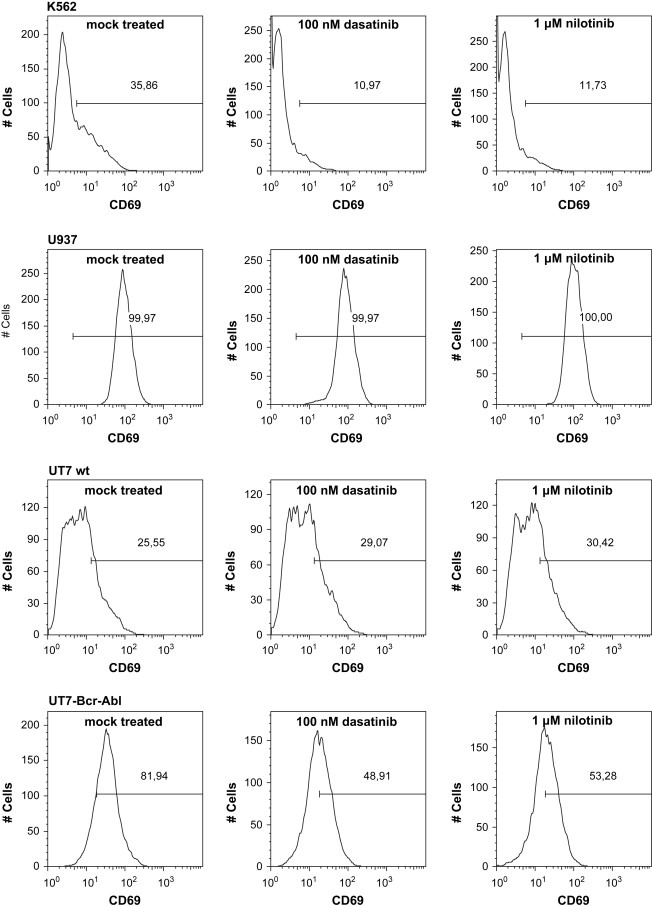
FACS analysis of CD69 expression. FACS analysis of different cell lines showed Bcr–Abl dependent CD69 expression. Cells were either untreated or treated with 100nM Dasatinib and 1μM Nilotinib for 16h. Percentage of CD69‐positive cells is indicated in each histogram. Thirty‐six percent of K562 cells showed CD69 expression, which was reduced upon treatment with Dasatinib and Nilotinib CD69 expression to 11% and 12%, respectively (top panel). U937, a Bcr–Abl negative cell line, did not alter their CD69 expression pattern after treatment with either of the drugs (second panel). 26% of untransduced UT‐7 cells expressed CD69, whereas Bcr–Abl transduction in these cells increased CD69 expression to 82%. After treatment with either Dasatinib or Nilotinib CD69 expression in untransduced UT‐7 remained unchanged, whereas it was decreased in UT7‐Bcr–Abl cells (lower two panels).
2.6. IL‐8 is strongly upregulated by expression of Bcr–Abl and suppressed by inhibition of Bcr–Abl
Another gene that was found to be strongly downregulated by dasatinib was IL‐8. IL‐8 was originally discovered as a neutrophil chemotactic and activating factor and is produced by a wide variety of cell types, including various tumor cell lines, in response to many pro‐inflammatory stimuli, such as exposure to IL‐1, tumor necrosis factor, lipopolysaccharide and viruses. IL‐8 is a member of the alpha (CXC) subfamily of chemokines and is a potent chemoattractant for neutrophils, basophils, T cells and eosinophils. In addition, IL‐8 also has a wide range of other pro‐inflammatory effects and was reported to be pro‐angiogenic (Rot et al., 1996).
First, we determined IL‐8 levels by sandwich ELISA in cell culture supernatants from K562 and U937 cells mock treated, dasatinib or nilotinib treated for 3h, 9h and 24h. Mock treated U937 cells secreted only low levels of IL‐8 and those remained unchanged upon treatment with dasatinib or nilotinib. In contrast, mock treated K562 cells secreted 5–50‐fold higher levels of IL‐8 than U937 cells (Figure 5, upper panel). Dasatinib or nilotinib treatment of K562 cells strongly reduced IL‐8 detectable in the cell culture supernatants. Importantly, UT7 cells transduced with Bcr–Abl also expressed much higher levels of IL‐8 than the untransduced UT7 control cells. As in the K562 cells, dasatinib and nilotinib strongly reduced IL‐8 levels in UT7‐Bcr–Abl cells, but had only little effect on untransduced UT7 cells (Figure 5, lower panel).
Figure 5.

IL‐8 secretion from cell lines. (A) Effects of dasatinib and nilotinib treatment on IL‐8 secretion from K562 and U937 cells. (B) Effects of dasatinib and nilotinib treatment on IL‐8 secretion from UT7 and UT7 cells expressing Bcr–Abl (UT7‐p210). For both panels, cells were either mock treated or treated with 100nM dasatinib or 1μM nilotinib for 3h, 9h or 24h. The supernatants were examined for IL‐8 concentration using an IL‐8 ELISA. The results were normalized to the cell number and supernatant volume in the respective experiment and averages and standard deviation of a representative experiment done in triplicates is shown.
Our results indicate that IL‐8 is upregulated by mechanisms that depend on the expression of Bcr–Abl and that suppression of Bcr–Abl activity leads to downregulation of IL‐8 secretion.
In order to test whether IL‐8 levels are also elevated in the serum of CML patients, we performed a preliminary analysis of serum samples from four patients at diagnosis and at different time points after initiation of imatinib therapy until patients achieved a CCyR. All patients were diagnosed in chronic phase. IL‐8 levels were determined by sandwich ELISA. The IL‐8 levels in the serum from two healthy control individuals were used as control. At diagnosis, IL‐8 levels in our CML patients were much higher than in the healthy controls (Table 2). During the course of imatinib therapy, IL‐8 levels in 3/4 patients decreased to baseline levels, reaching the lowest levels at the latest time points tested. In patient #2, we also observed a drop in IL‐8 levels until month 3, but at 12months, the IL‐8 level was again high despite a complete cytogenetic response. As this patient also did not suffer from an apparent infection, the basis for the increased IL‐8 level at 12months remains uncertain.
Table 2.
IL‐8 in CML patient serum
| Patient | Time point of serum donation | IL‐8 (ng/ml serum) | st.dev. |
|---|---|---|---|
| CML patient 1 | Diagnosis | 3.79 | 0.75 |
| 1month | 0.05 | 0.02 | |
| 3months (CCyR) | 0.10 | 0.05 | |
| CML patient 2 | Diagnosis | 1.79 | 0.11 |
| 1month | 1.58 | 0.01 | |
| 3months | 0.09 | 0.00 | |
| 12months | 1.53 | 0.04 | |
| CML patient 3 | Diagnosis sample 1 | 0.58 | 0.03 |
| Diagnosis sample 2 | 0.36 | 0.01 | |
| 1month | 0.13 | 0.01 | |
| 3months | 0.37 | 0.01 | |
| 24months (CCyR) | 0.13 | 0.01 | |
| CML patient 4 | Diagnosis | 0.54 | 0.03 |
| 1month | 0.18 | 0.01 | |
| 3months (CCyR) | 0.06 | 0.01 | |
| Healthy donor 1 | 0.11 | 0.01 | |
| Healthy donor 2 | 0.06 | 0.00 |
IL‐8 levels in the serum of CML patients at diagnosis and 1–24months after initiation of imatinib therapy. Achievement of a complete cytogenetic response (CCyR) is indicated. Serum samples were examined for IL‐8 concentration using an IL‐8 ELISA. Averages and standard deviations of a representative experiment done in duplicate are shown.
3. Discussion
Our study aimed at identifying transcriptional changes in the signal transduction network in CML cells that are induced by kinase inhibitors like dasatinib. Using global pathway analysis we identified a major impact of dasatinib treatment on the JAK–STAT signaling pathway. We detected a massive downregulation of a large set of canonical STAT5 target genes already after short treatment with dasatinib. Given the requirement of STAT5 signaling for Bcr–Abl dependent oncogenic transformation (Hoelbl et al., 2006; Ye et al., 2006), our results may explain the high efficacy of treatment with dasatinib and other Bcr–Abl inhibitors. According to our data, dasatinib may exert a long‐ranging effect on this Bcr–Abl effector pathway. First, it inhibits tyrosine phosphorylation of STAT5 by Bcr–Abl and its ensuing activation. As a consequence, transcription of the STAT5‐dependent target genes is inhibited.
As the initial microarray experiment in this study was done with the pleiotropic kinase inhibitor dasatinib, we expected to identify a number of differentially regulated genes that can be attributed to inhibition of one of the numerous other targets of dasatinib, besides Bcr–Abl. In the course of our validation strategy, we put particular emphasis on delineating Bcr–Abl dependent changes in mRNA expression from unspecific changes caused by the kinase inhibitor treatment or inhibition of off‐target proteins. To our surprise, we found that the alterations in mRNA expression that we observed with the highly selective Bcr–Abl inhibitor nilotinib mirrored the effects of dasatinib for the vast majority of mRNAs studied. This indicates that Bcr–Abl is the predominant target of dasatinib in K562 cells, which may explain the efficacy of dasatinib in CML. On the other hand, the large number of dasatinib targets that we and others have identified (Bantscheff et al., 2007; Hantschel et al., 2007; Rix et al., 2007) may indicate that adverse off‐target effects of dasatinib may only affect cells of the hematopoietic system that do not express the Bcr–Abl oncoprotein or cells from non‐hematopoietic tissues. Since the target spectrum of imatinib and nilotinib is basically identical (Bantscheff et al., 2007; Hantschel et al., 2007; Rix et al., 2007), it is very likely that the genes differentially regulated by nilotinib are also differentially regulated by imatinib. We preferred to use nilotinib for all in vitro studies presented in this paper, because of the comparable potency of nilotinib and dasatinib that allows an easier comparison of the experiments obtained with the two different drugs. However, for the determination of IL‐8 levels from CML patient sera, only samples from imatinib‐treated patients were available.
We have decided to obtain gene expression profiles of cells after short‐term treatment with dasatinib. In contrast to previous studies that used drug treatments for 16h (Nunoda et al., 2007) and expectedly identified a number of differentially regulated genes related to cell‐cycle control and apoptosis, we have looked at earlier time points, in which Bcr–Abl signaling is suppressed by the drug treatment, but cells are still viable. This strategy enabled us to identify early markers of Bcr–Abl signaling.
The most prominently downregulated gene upon inhibition of Bcr–Abl was the Early growth response protein 1 (Egr1). The mechanism of Abl‐dependent regulation of Egr‐1 expression is quite controversial. Previous studies showed either upregulation induced by c‐Abl expression or suppression of Egr1 expression by Bcr–Abl (Fukada and Tonks, 2001; Stuart et al., 2005). In addition, Egr1 was identified in an overwhelming number of other microarray studies to be highly up‐ or downregulated in very diverse tumor tissues and with various different activators or inhibitors. This argues that the downregulation of Egr1 expression by dasatinib is a rather pleiotropic effect frequently observed in different cancers and not specific to the action of Bcr–Abl. Therefore, Egr‐1 was not followed up further.
We found downregulation of all three members of the Pim kinases by dasatinib (Pim1: 5.8‐fold, Pim2: 2.2‐fold, Pim3: 1.4‐fold). Pim kinases are STAT5 target genes and the activity of Pim kinases was reported to be necessary for conferring growth‐factor independent growth of Bcr–Abl transduced Ba/F3 cells (Adam et al., 2006). Therefore, Pim kinases were proposed as additional therapeutic targets in imatinib resistant cells. One of the best‐described substrates of Pim kinases are the SOCS proteins. We found downregulation of all SOCS proteins by dasatinib, with the exception of SOCS6. SOCS1 was down‐regulated strongest (>7‐fold). As direct transcriptional targets of Stat5, SOCS proteins are negative regulators of the JAK–STAT signaling pathway and phosphorylation of SOCS1 by Pim kinases was shown to stabilize SOCS1 (Chen et al., 2002). In addition to the disruption of the regulatory circuit involving the Pim kinases and SOCS proteins described above, a large number of additional transcriptional targets of STAT5 were identified to be downregulated by dasatinib.
Besides these general insights into the Bcr–Abl signaling network, our study has identified a number of unsuspected proteins, two of which—CD69 and IL‐8—we have validated in more detail. These novel proteins may be of great interest for a more comprehensive understanding of the pathophysiology of CML, but may also be used as suitable markers capable to indicate the state of activity of Bcr–Abl and thus monitor the efficacy of anti‐Bcr–Abl therapies in early stages after initiation if the therapy. This may become of particular importance, as nilotinib, dasatinib and several “third‐generation” Bcr–Abl inhibitors are currently in clinical trials and are expected to dramatically expand the treatment options for CML. Therefore, diagnostic and potentially prognostic marker proteins will become increasingly important to make a rational choice of the drug, identify the most effective drug or drug combination for each individual patient and change the treatment regimen early on once primary or secondary resistance is indicated.
IL‐8 is a secreted chemokine and plays an important role in mediating leukocyte locomotion and activation. It is also a potent angiogenic factor. Various cell types including monocytes express IL‐8. It is interesting to note that K562 cells, although expressing IL‐8, do not express the IL‐8 receptor α or β and expression of both receptors remain unaffected by dasatinib treatment (data not shown). This may indicate that the secreted IL‐8, induced by Bcr–Abl, rather exerts its effects on more mature cells in the CML clone (granulomonocytic cells) rather than on immature blast cells. These data may point to a new interesting concept in which CML cells promote the distribution and expansion of maturing granulomonocytic CML cells in diverse organs not only through increased growth and survival of progenitor cells in the bone marrow, but also by mobilization and locomotion of more mature cells into the tissues through increased IL‐8 production. Indeed, in CML, massive accumulation of leukocytes in the spleen and other tissues is a commonly recognized feature and is often accompanied by clinical symptoms.
Based on the promising results presented in this paper, we now plan to extend our studies on IL‐8 serum measurements to a larger cohort of CML patients treated with imatinib, nilotinib, or dasatinib and controls derived from patients with neoplasms related to CML, in order to learn whether IL‐8 may also be a valuable biomarker and indicator of early disease response in CML.
4. Experimental procedures
4.1. Kinase inhibitors
Dasatinib (Sprycel, BMS‐354825) and nilotinib (Tasigna AMN107) were dissolved in DMSO and used at final concentrations of 100nM for dasatinib and 1μM for nilotinib for all experiments.
4.2. Microarray procedures
1×107 K562 cells were either mock‐treated with DMSO or treated with 100nM dasatinib in two biological repeats. Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany). Preparation of cRNA, hybridization to the human U133 Plus 2.0 GeneChips (Affymetrix, Santa Clara, CA) and scanning of the arrays were carried out according to the manufacturer's protocols (www.affymetrix.com) and as described (Bilban et al., 2006). RMA signal extraction, normalization and filtering (to eliminate genes with extremely low expression) was performed as described (Irizarry et al., 2003; www.bioconductor.org/). The complete microarray dataset has been deposited at Array Express (www.ebi.ac.uk/arrayexpress/) and can be accessed at number E‐MEXP‐1561.
4.3. Data analysis and network analysis
Regulated genes were selected by performing a SAM analysis imposing a 5% false discovery rate (FDR) (Tusher et al., 2001). This yielded 37 genes. All were downregulated by dasatinib treatment. Manual inspection of the fold change of expression levels and signal intensities yielded the selection of another set of 8 genes (7 upregulated, one downregulated), thus giving a total of 44 selected genes. Pathway analysis was performed by using DAVID (Dennis et al., 2003). Integration of selected genes with protein–protein and drug–protein interactions was obtained via an in‐house program. Protein–protein interactions were obtained from HPRD (Mishra et al., 2006).
4.4. Biological material
K562 and U937 cells were obtained from DSMZ (Braunschweig, Germany). UT7 cells (a kind gift of M. Mayerhofer) were retrovirally transduced with p210 Bcr–Abl using standard techniques. Sera from four CML patients and two healthy controls were examined. Serum was obtained from peripheral blood by centrifugation. In CML patients, blood was collected at the time of diagnosis as well as serially during therapy with imatinib (400mg/day per os). Studies performed with serum from CML patients were approved by the institutional review board (Medical University of Vienna). Written informed consent was obtained before blood donation.
4.5. Quantitative RT–PCR
Total RNA was reverse transcribed using oligo(dT)18 primer, 1μM dNTPs and RevertAid M‐MuLV Reverse Transcriptase (Fermentas, Burlington, Canada). Quantitative real‐time PCR was performed with iTaq SYBR Green Supermix with ROX (Bio‐Rad, Hercules, CA) using a Rotor‐Gene 6000 (Corbett) using a standard protocol and GAPDH as a reference gene. Expression changes were calculated by applying the method (Livak and Schmittgen, 2001).
4.6. FACS analysis for CD69
1×105 cells (K562, U937, UT7 or UT7‐Bcr–Abl) were stained with PE‐labeled anti human CD69‐antibody (mouse monoclonal #MHCD6904, Caltag/Invitrogen, Carlsbad, CA), dead cells stained with 7‐AAD (eBioscience, San Diego, CA) and analyzed on a FACSCalibur (BD Biosciences, Franklin Lakes, NJ). For analysis, 7‐AAD negative cells were excluded to compare CD69 expression of non‐treated vs. drug‐treated cells.
4.7. Interleukin‐8 ELISA
The IL‐8 ELISA kit was obtained from BD Biosciences (San Jose, CA) and used according to the instructions of the manufacturer. Concentrated supernatants from cultured cells were used. Serum samples from patients were diluted 1:2 in PBS.
Supporting information
Supplementary data
Acknowledgments
We thank Christian Baumgartner and Harald Herrmann for patient sample collection, and Gerhard Dürnberger for figure S1 and help with SAM. We are deeply grateful to Matthias Mayerhofer for providing the UT7 and UT7‐Bcr–Abl cells. We thank Tilmann Buerckstuemmer and Florian Grebien for the critical reading of the manuscript and all members of the participating labs for help and discussions. This study was supported by the Austrian Academy of Sciences and the FWF (grant #P18737‐B11). J.C. was supported by a APPII‐GenAU Network grant (GZ200.142/1‐VI/I/2006) of the Austrian Ministry of Research and Education.
Supplemental material 1.
1.1.
Supplementary information for this manuscript can be downloaded at doi: 10.1016/j.molonc.2008.07.003.
Hantschel Oliver, Gstoettenbauer Agnes, Colinge Jacques, Kaupe Ines, Bilban Martin, Burkard Thomas R., Valent Peter, Superti-Furga Giulio, (2008), The chemokine interleukin‐8 and the surface activation protein CD69 are markers for Bcr–Abl activity in chronic myeloid leukemia, Molecular Oncology, 2, doi: 10.1016/j.molonc.2008.07.003.
References
- Adam, M. , Pogacic, V. , Bendit, M. , Chappuis, R. , Nawijn, M.C. , Duyster, J. , Fox, C.J. , Thompson, C.B. , Cools, J. , Schwaller, J. , 2006. Targeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABL. Cancer Res. 66, 3828–3835. [DOI] [PubMed] [Google Scholar]
- Bantscheff, M. , Eberhard, D. , Abraham, Y. , Bastuck, S. , Boesche, M. , Hobson, S. , Mathieson, T. , Perrin, J. , Raida, M. , Rau, C. , 2007. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol 25, 1035–1044. [DOI] [PubMed] [Google Scholar]
- Bilban, M. , Heintel, D. , Scharl, T. , Woelfel, T. , Auer, M.M. , Porpaczy, E. , Kainz, B. , Krober, A. , Carey, V.J. , Shehata, M. , 2006. Deregulated expression of fat and muscle genes in B-cell chronic lymphocytic leukemia with high lipoprotein lipase expression. Leukemia 20, 1080–1088. [DOI] [PubMed] [Google Scholar]
- Carter, T.A. , Wodicka, L.M. , Shah, N.P. , Velasco, A.M. , Fabian, M.A. , Treiber, D.K. , Milanov, Z.V. , Atteridge, C.E. , Biggs, W.H. , Edeen, P.T. , 2005. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc. Natl. Acad. Sci. U.S.A. 102, 11011–11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X.P. , Losman, J.A. , Cowan, S. , Donahue, E. , Fay, S. , Vuong, B.Q. , Nawijn, M.C. , Capece, D. , Cohan, V.L. , Rothman, P. , 2002. Pim serine/threonine kinases regulate the stability of Socs-1 protein. Proc. Natl. Acad. Sci. U.S.A. 99, 2175–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Coutre, P. , Ottmann, O.G. , Giles, F. , Kim, D.W. , Cortes, J. , Gattermann, N. , Apperley, J.F. , Larson, R.A. , Abruzzese, E. , O'Brien, S.G. , 2008. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated phase chronic myelogenous leukemia. Blood 111, 1834–1839. [DOI] [PubMed] [Google Scholar]
- Deininger, M. , Buchdunger, E. , Druker, B.J. , 2005. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 105, 2640–2653. [DOI] [PubMed] [Google Scholar]
- Dennis, G. , Sherman, B.T. , Hosack, D.A. , Yang, J. , Gao, W. , Lane, H.C. , Lempicki, R.A. , 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3 [PubMed] [Google Scholar]
- Druker, B.J. , Guilhot, F. , O'Brien, S.G. , Gathmann, I. , Kantarjian, H. , Gattermann, N. , Deininger, M.W. , Silver, R.T. , Goldman, J.M. , Stone, R.M. , 2006. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 355, 2408–2417. [DOI] [PubMed] [Google Scholar]
- Fukada, T. , Tonks, N.K. , 2001. The reciprocal role of Egr-1 and Sp family proteins in regulation of the PTP1B promoter in response to the p210 Bcr–Abl oncoprotein–tyrosine kinase. J. Biol. Chem. 276, 25512–25519. [DOI] [PubMed] [Google Scholar]
- Hantschel, O. , Rix, U. , Schmidt, U. , Burckstummer, T. , Kneidinger, M. , Schutze, G. , Colinge, J. , Bennett, K.L. , Ellmeier, W. , Valent, P. , 2007. The Btk tyrosine kinase is a major target of the Bcr–Abl inhibitor dasatinib. Proc. Natl. Acad. Sci. U.S.A. 104, 13283–13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantschel, O. , Rix, U. , Superti-Furga, G. , 2008. Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leuk. Lymphoma 49, 615–619. [DOI] [PubMed] [Google Scholar]
- Hoelbl, A. , Kovacic, B. , Kerenyi, M.A. , Simma, O. , Warsch, W. , Cui, Y. , Beug, H. , Hennighausen, L. , Moriggl, R. , Sexl, V. , 2006. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood 107, 4898–4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry, R.A. , Bolstad, B.M. , Collin, F. , Cope, L.M. , Hobbs, B. , Speed, T.P. , 2003. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31, e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian, H. , Pasquini, R. , Hamerschlak, N. , Rousselot, P. , Holowiecki, J. , Jootar, S. , Robak, T. , Khoroshko, N. , Masszi, T. , Skotnicki, A. , 2007. Dasatinib or high-dose imatinib for chronic-phase chronic myeloid leukemia after failure of first-line imatinib: a randomized phase-II trial. Blood 109, 5143–5150. [DOI] [PubMed] [Google Scholar]
- Livak, K.J. , Schmittgen, T.D. , 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Mishra, G.R. , Suresh, M. , Kumaran, K. , Kannabiran, N. , Suresh, S. , Bala, P. , Shivakumar, K. , Anuradha, N. , Reddy, R. , Raghavan, T.M. , 2006. Human protein reference database—2006 update. Nucleic Acids Res. 34, D411–D414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunoda, K. , Tauchi, T. , Takaku, T. , Okabe, S. , Akahane, D. , Sashida, G. , Ohyashiki, J.H. , Ohyashiki, K. , 2007. Identification and functional signature of genes regulated by structurally different ABL kinase inhibitors. Oncogene 26, 4179–4188. [DOI] [PubMed] [Google Scholar]
- O'Hare, T. , Corbin, A.S. , Druker, B.J. , 2006. Targeted CML therapy: controlling drug resistance, seeking cure. Curr. Opin. Genet. Dev. 16, 92–99. [DOI] [PubMed] [Google Scholar]
- Quintas-Cardama, A. , Kantarjian, H. , Cortes, J. , 2007. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat. Rev. Drug Discov. 6, 834–848. [DOI] [PubMed] [Google Scholar]
- Ren, R. , 2005. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 5, 172–183. [DOI] [PubMed] [Google Scholar]
- Rix, U. , Hantschel, O. , Durnberger, G. , Remsing Rix, L.L. , Planyavsky, M. , Fernbach, N.V. , Kaupe, I. , Bennett, K.L. , Valent, P. , Colinge, J. , 2007. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib reveal novel kinase and non-kinase targets. Blood 110, 4055–4063. [DOI] [PubMed] [Google Scholar]
- Rot, A. , Hub, E. , Middleton, J. , Pons, F. , Rabeck, C. , Thierer, K. , Wintle, J. , Wolff, B. , Zsak, M. , Dukor, P. , 1996. Some aspects of IL-8 pathophysiology. III: Chemokine interaction with endothelial cells. J. Leukoc. Biol. 59, 39–44. [DOI] [PubMed] [Google Scholar]
- Sancho, D. , Gomez, M. , Sanchez-Madrid, F. , 2005. CD69 is an immunoregulatory molecule induced following activation. Trends Immunol 26, 136–140. [DOI] [PubMed] [Google Scholar]
- Shuai, K. , Halpern, J. , ten Hoeve, J. , Rao, X. , Sawyers, C.L. , 1996. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene 13, 247–254. [PubMed] [Google Scholar]
- Stuart, J.R. , Kawai, H. , Tsai, K.K. , Chuang, E.Y. , Yuan, Z.M. , 2005. c-Abl regulates early growth response protein (EGR1) in response to oxidative stress. Oncogene 24, 8085–8092. [DOI] [PubMed] [Google Scholar]
- Tusher, V.G. , Tibshirani, R. , Chu, G. , 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Etten, R.A. , 2004. Mechanisms of transformation by the BCR-ABL oncogene: new perspectives in the post-imatinib era. Leukemia Res. 28. Suppl. 1, S21–S28. [DOI] [PubMed] [Google Scholar]
- Weisberg, E. , Manley, P.W. , Cowan-Jacob, S.W. , Hochhaus, A. , Griffin, J.D. , 2007. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat. Rev. Cancer 7, 345–356. [DOI] [PubMed] [Google Scholar]
- Wong, S. , Witte, O.N. , 2004. The BCR-ABL Story: Bench to bedside and back. Annu. Rev. Immunol 22, 247–306. [DOI] [PubMed] [Google Scholar]
- Ye, D. , Wolff, N. , Li, L. , Zhang, S. , Ilaria, R.L. , 2006. STAT5 signaling is required for the efficient induction and maintenance of CML in mice. Blood 107, 4917–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data