

Abstract
It is becoming increasingly clear that neurological diseases are multi-factorial involving disruptions in multiple cellular systems. Thus, while each disease has its own initiating mechanisms and pathologies, certain common pathways appear to be involved in most, if not all, neurological diseases described to date. Thus, it is unlikely that modulating only a single factor will be effective at either preventing disease development or slowing disease progression. A better approach is to identify small (< 900 daltons) molecules that have multiple biological activities relevant to the maintenance of brain function. Over the last few years, we have identified an orally active, novel neuroprotective and cognition-enhancing molecule, the flavonoid fisetin. Fisetin not only has direct antioxidant activity but it can also increase the intracellular levels of glutathione, the major intracellular antioxidant. Fisetin can also activate key neurotrophic factor signaling pathways. In addition, it has anti-inflammatory activity against microglial cells and inhibits the activity of lipoxygenases, thereby reducing the production of pro-inflammatory eicosanoids and their by-products. This wide range of actions suggests that fisetin has the ability to reduce the impact of age-related neurological diseases on brain function.
Keywords: oxidative stress, glutathione, neurotrophic factors, memory, microglia, inflammation
2. INTRODUCTION
By far the major risk factor for Alzheimer’s disease (AD) as well as other neurological diseases as well as stroke is old age (1). Currently, there are no disease-modifying drugs for any age-associated neurological diseases or stroke. This is at least in part due to the fact that the vast majority of age-associated neurological diseases arise from a confluence of multiple toxic insults to the brain that accumulate during normal aging and interact with lifestyle, environmental and genetic risk factors with varying degrees of penetrance. For example, although AD is defined in terms of plaque and tangle pathology, it is most frequently associated with other detrimental events such as microvascular damage and inflammation (2). Therefore, it is unlikely that hitting a single target will result in significant benefits to patients with AD (3). This conclusion has been largely born out by the failure of numerous clinical trials for single target drugs in AD (4). Nevertheless, current drug research efforts continue to be almost exclusively focused on single protein targets and the identification of small molecules (low molecular weight (<900 daltons) organic compounds) that can modulate these targets with high affinity (2).
Among the toxic insults that have been identified as contributing to the age-related decline in brain function are increases in oxidative stress, loss of trophic support, accumulation of protein aggregates, dysfunction of the neurovascular system and immune system activation (5). Given this multiplicity of insults and the strong possibility that the relative importance of each of these factors will vary among individuals, it may be necessary to use combinations of drugs directed against different targets. However, this approach is subject to a number of potential problems including pharmacokinetic and bioavailability challenges which in central nervous system (CNS) diseases are exacerbated by the difficulty of getting multiple compounds across the blood brain barrier and the potential for adverse drug-drug interactions. A better approach is to identify small molecules that have multiple biological activities that can impact the multiplicity of insults that are associated with the age-associated toxicity pathways that contribute to disease development and progression.
One excellent source for these small molecules is the original pharmacopeia, plants. The polyphenolic flavonoids are widely distributed in fruits and vegetables and therefore regularly consumed in the human diet (for reviews see 6, 7, 8). Flavonoids were historically characterized on the basis of their antioxidant and free radical scavenging effects. However, more recent studies have shown that flavonoids have a wide range of activities that could make them particularly effective for blocking the age-associated toxicity pathways associated with neurodegenerative diseases. In this article, I will focus on the flavonol fisetin (Fig. 1) which has been found to have therapeutic efficacy in multiple animal neurological disease models including AD, Huntington’s disease (HD), stroke, depression and the neurological complications of diabetes. I will first discuss these and other recent in vivo studies with fisetin in animal models of neurodegenerative diseases and then address the multiple signaling pathways that are targeted by fisetin and are likely to contribute to its efficacy in these disease models.
Figure 1.
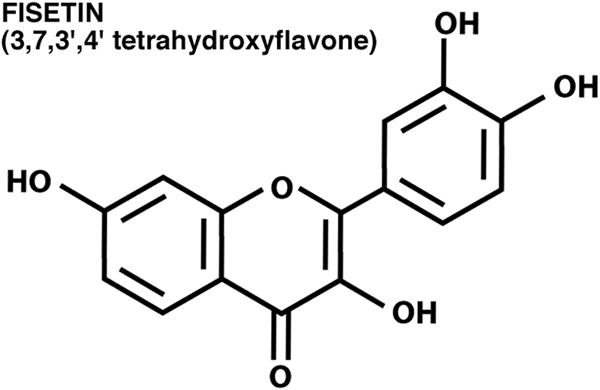
Structure of fisetin.
3. BACKGROUND
Fisetin was originally identified in a screen for compounds that could prevent oxidative stress-induced nerve cell death (9). Of the ~30 flavonoids tested in this study, only two, fisetin and quercetin, were able to maintain GSH levels in the presence of oxidative stress, indicating that this is not a common property of flavonoids. Further studies showed that fisetin also possessed neurotrophic activity, promoting the differentiation of PC12 cells via activation of the Ras-ERK cascade (10). Again, this was a property that distinguished fisetin from almost all of the other ~30 flavonoids tested. Only quercetin, isorhamnetin and luteolin showed some differentiation-inducing activity and they were all much less effective than fisetin. Together, these observations suggested that fisetin had multiple properties that might make it useful for the treatment of age-related neurological diseases.
Unlike many of the better studied flavonoids such as quercetin and luteolin, fisetin is not particularly abundant in fruits and vegetables. The highest levels (160 μg/g) are found in strawberries (11) with 5–10 fold lower levels in apples and persimmons. Small amounts are also found in kiwi fruit, peaches, grapes, tomatoes, onions and cucumbers (12). The bioavailability of fisetin from these sources has not yet been studied.
Fisetin is currently marketed in the US by several nutriceutical companies either alone in 100 mg capsules or in combination with other natural products. The indications for use include the promotion of cognition, the maintenance of brain health and the support of healthy aging.
4. IN VIVO EFFECTS OF FISETIN ON THE CNS
4.1 Fisetin and Alzheimer’s Disease (AD)
Alzheimer’s disease is the most common type of dementia. It is characterized pathologically by the presence of both extracellular neuritic plaques containing amyloid beta (Aβ) peptide and intracellular neurofibrillary tangles containing tau (13). Clinically, AD results in a progressive loss of cognitive ability and eventually daily function activities (14, 15). Current approved therapies are only symptomatic, providing moderate improvements in memory without altering the progression of the disease pathology (16, 17). To test the idea that oral administration of fisetin could prevent the behavioral and pathophysiological changes associated with AD, APPswe/PS1dE9 double transgenic AD mice, a model of the familial form of AD, were used (18). These mice show significant Aβ plaque deposition by 8–9 months of age as well as clear cognitive impairment (19) (20). It was first asked whether fisetin could reduce the behavioral changes associated with AD. Two groups each of wild type and AD mice were used for these studies. Fisetin was fed to one set of wild type and one set of AD mice between the ages of 3 and 12 months in their food at 0.05%, resulting in a daily dose of approximately 25 mg/kg body weight (bw). This dose of fisetin was chosen based on earlier studies on fisetin and cognitive function in mice (21).
Learning and memory were tested using the Morris water maze (MWM) when the mice were 9 months of age and then the memory testing was repeated in a 2 day version of the water maze (WM) at 12 months. The MWM is a test of spatial learning and is strongly correlated with hippocampal synaptic plasticity (22). It can be used to assess both learning and memory deficits. In the standard test, the mice are required to find a submerged platform in a circular pool of opaque liquid (usually water with non-toxic white paint added) by relying on distal visual cues. Typically, they are given 4 trials/day over a period of days and the time required for the mouse to find the platform (latency) is recorded. 24 hr after the end of this acquisition phase, the mice are placed back in the pool but in this case the platform is no longer present. They are given a single, one min trial (probe test) and the amount of time that they spend in the quadrant in which the platform was previously located relative to the time that they spend in the other three quadrants is recorded. At 9 months of age, wild type mice showed decreased times to find the hidden platform over the 5 days of the acquisition phase of the MWM (Fig. 2A). Fisetin had no significant effect on the behavior of the wild type mice in the acquisition task. In contrast, the AD mice showed very little learning over the 5 days of the acquisition task while the fisetin-fed AD mice behaved almost indistinguishably from the wild type mice in this task (Fig. 2A). In the probe test (Fig. 2B), which specifically assays memory, the wild type mice and the wild type mice fed fisetin both showed significant recall of the platform quadrant spending greater than 25% of their time in this quadrant. In contrast, the AD mice showed no indication of memory, spending only 16% of their time in the platform quadrant. Since the platform quadrant is on the opposite side of the tank from where the mice enter, it is not surprising that they spend less than 25% of their time in this quadrant. However, the AD mice fed fisetin showed clear evidence of memory improvement. Fisetin had no effect on swim speeds.
Figure 2.
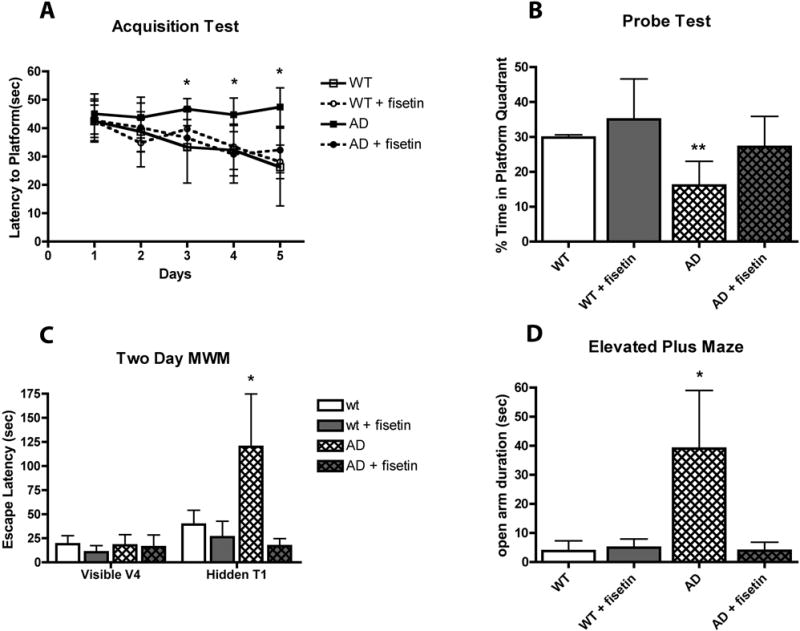
Fisetin maintains learning and memory in AD mice. (A) Performance of the mice during the acquisition phase of the MWM test. 9 month-old AD mice failed to learn the task over the 5-day testing period whereas fisetin-treated AD mice learned to locate the submerged platform. (B) Performance of the mice during the memory phase of the MWM test. AD mice did not spend a greater than random percentage of their time in the quadrant where the submerged platform was previously located whereas fisetin-treated AD mice spent greater than random amounts of time in the platform quadrant. (C) Performance of 12 month old mice during the 2-day MWM test. Although all the mice learned to find the visible platform on day 1, the AD mice did not remember where the platform was located when it was submerged on day 2 whereas fisetin-treated AD mice did remember. (D) 12 month old AD mice spend more time in the open arm of the elevated plus maze, an indicator of disinhibition, as compared with fisetin-treated AD mice. Data represent means ± SD, n = 9–12 per group. Analysis by one-way ANOVA followed by Tukey Kramer multiple comparisons post hoc test. * p < 0.05 and ** p < 0.01 relative to control non-Tg mice. (From 18)
Since AD is a progressive disease, the mice were tested again at 12 months to determine if fisetin continued to reduce memory deficits. Since previous studies in my laboratory had indicated that the MWM could not be repeated on the same set of mice, we used an alternative version that only tests memory and addresses some of the confounding factors that arise when testing older mice in the MWM (23). In the 2 day WM, on day 1 mice are trained to find a visible platform using 4 trials. All of the mice, regardless of genotype or diet, were able to find the visible platform within 30 sec by the last visible trial (Fig. 2C). This result indicated that neither the genotype nor the diet affected the animals’ ability to swim or to locate the visible platform. Thus, any impairment in spatial memory seen with the 2 day WM reflects a cognitive deficit and not a lack of motivation or visual function. 24 hr after the final visible platform trial, the mice were tested in the same tank but with the platform hidden. As shown in Figure 2C, the escape latency of the AD mice was significantly longer than that of the wild type mice, similar to what was reported for triple transgenic AD mice (23). In contrast, the escape latency for the AD mice fed fisetin was indistinguishable from that of the wild type mice.
Another characteristic of AD is social disinhibition which can result in inappropriate behavior in AD patients. The elevated plus maze (EPM), a rodent model of anxiety, is often used as a measure of disinhibition in mice (24). The AD mice used in this study were previously shown to exhibit increased time in the open arms in the EPM which was interpreted as indicative of disinhibition (25). As shown in Figure 2D, the AD mice spent significantly more time in the open arms than the wild type mice. This behavior was reversed in the AD mice fed fisetin. In summary, the fisetin-fed AD mice showed clear improvements in both cognitive and neuropsychiatric behavior.
Cholinergic neurons are among the first to be lost in AD (26, 27) and acetylcholine is the therapeutic target for most FDA approved drugs for AD (28, 29). Scopolamine-induced memory impairment in rodents is a well-established model of memory dysfunction based upon acetylcholine metabolism (30). The available data suggests that reversal of scopolamine-induced cognitive impairment is a suitable model for predicting pharmacodynamic signals of cognitive enhancing compounds in animals (31). The acetylcholinesterase inhibitor, donepezil (Aricept), which improves cognition in AD, reverses the cognitive impairment induced by scopolamine in both humans and animals (32, 33). A recent study (34) tested the ability of fisetin to prevent scopolamine-induced memory impairment in mice. A single oral dose of 40 mg/kg bw of fisetin was as effective as donepezil at rescuing the memory deficit induced by scopolamine as determined using the passive avoidance test. In this test, animals learn to associate an aversive stimulus with a specific environmental context. A two chamber apparatus is used where one chamber is dimly lit and therefore preferable to the mice and the other chamber is brightly lit. During training, the animal is placed in the brightly lit chamber and upon entry into the dimly lit chamber is exposed to a mild foot shock. 24 hr later the mice are returned to the brightly lit chamber and the latency to enter the shock-paired, dimly lit compartment is measured. Animals that remember the task show a much higher latency to enter the shock-paired, dimly lit chamber. These results provide further support for the idea that fisetin could be useful for the treatment of AD.
4.2 Fisetin and Huntington’s Disease (HD)
Huntington’s disease is a late onset, progressive and fatal neurodegenerative disorder for which there is, at present, no cure. It is caused by an expansion of a trinucleotide repeat that encodes an abnormally long polyglutamine tract in the huntingtin protein. The identification of the disease-causing mutation has allowed the development of a number of cellular and animal models of HD and these have been used to elucidate the mechanisms underlying disease development and progression (for reviews see 35, 36–38). Fisetin was tested in several different models of HD including PC12 cells expressing mutant huntingtin exon 1 under the control of an inducible promoter and the R6/2 mouse model of HD (39).
To begin to test for a possible protective role of fisetin in HD, an ecdysone-inducible PC12 cell line (PC12/HttQ103) containing the huntingtin protein exon 1 fused to EGFP (HttQ103 = Httex1-103QP-EGFP) was used (40). Induction of mutant Htt (Httex1-103QP-EGFP) by treatment of PC12/HttQ103 cells with ponasterone resulted in the death of ~45% of the cells within 72 hr (40). Treatment with fisetin at the time of Httex1-103QP induction increased cell survival in a dose-dependent manner with a maximal effect seen between 5 and 10 μM (39). Interestingly, treatment with 10 μM fisetin 24 hr after the induction of Httex1-103QP provided an equal or greater amount of protection indicating that fisetin can reduce the effects of Httex1-103QP already present in cells and that the effect of fisetin was not on suppression of Httex1-103QP synthesis. This latter conclusion was confirmed by fluorescence microscopy where similar numbers of EGFP-tagged Httex1-103QP aggregates could be seen in both the untreated and fisetin-treated cells.
Given the positive results with fisetin in the cell-based assay, it was then tested in a mammalian model of HD, the R6/2 mouse (39). This transgenic mouse line expresses human mutant exon 1 of huntingtin with a highly expanded repeat from the mouse Htt promoter and has been widely used as a model for testing novel therapeutic approaches to the treatment of HD (e.g. 41, 42, 43). Since the overall goal was to determine if oral administration of fisetin could be useful for the treatment of HD, fisetin was fed to genotyped R6/2 mice and their wild type littermates in the food at 0.05% beginning at ~6 weeks of age. The mice were tested on the rotorod from ~7–13 weeks of age and survival was followed. At the time of acquisition of the animals, rotorod performance was already impaired in the R6/2 mice as compared to their wild type littermates (Fig. 3A). However, the performance declined significantly more rapidly in the animals on the control diet as compared to those on the fisetin diet (Fig. 3A). Similarly, as shown in Figure 3B, while the median life span of the R6/2 mice on the control diet was 104 days, that of fisetin-fed mice was increased by ~30% to 139 days. These results indicate that fisetin can reduce the impact of mutant huntingtin in multiple models of HD suggesting that it might be useful for the treatment of the disease.
Figure 3.
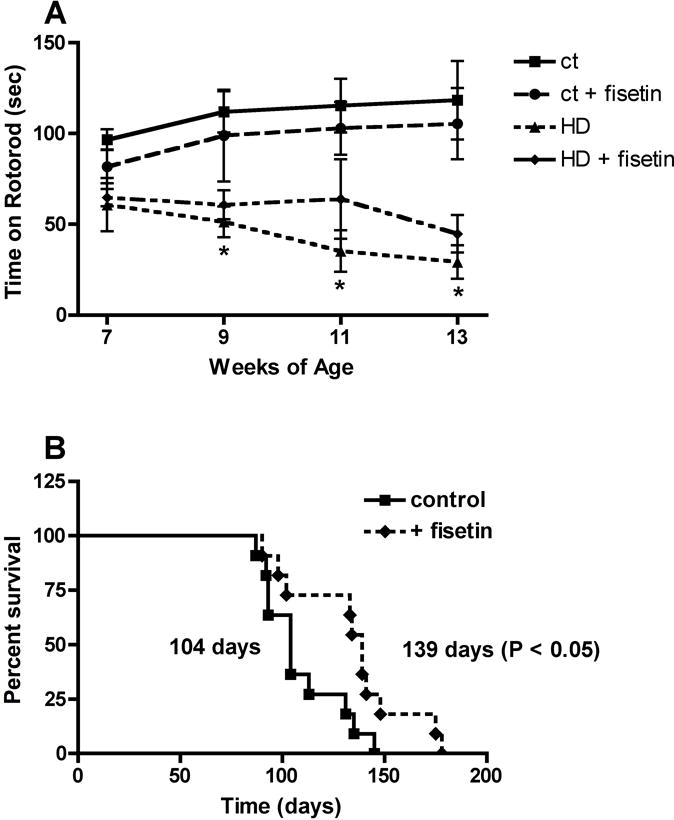
Fisetin treatment decreases motor impairment and mortality in R6/2 mice. R6/2 mice on 0.05% fisetin or control diet (n = 15) were monitored for (A) rotorod performance and (B) mortality. Fisetin improved rotorod performance at 9, 11 and 13 weeks (p < 0.01) and survival (p < 0.05) (Unpaired t test). (From 39)
4.3 Fisetin and Stroke
Ischemic stroke is a devastating disease representing the second leading cause of death in the Western world and the leading cause of disability in adults (44). Ischemic stroke occurs when the normal blood supply to the brain is disrupted, usually due to artery blockage by a blood clot, thereby depriving the brain of oxygen and metabolic substrates and hindering the removal of waste products (for review see 45). Ischemic stroke is believed to evolve in distinct phases. The initial ischemia results in immediate nerve cell death followed by an inflammatory response leading to secondary tissue damage after reperfusion (46). There is good evidence that post-stroke inflammation contributes to secondary tissue damage. The nerve cell damage and death caused by cerebral ischemia results in functional impairment including cognitive impairment or death (47). Fisetin was first tested in an in vitro ischemia model that was developed for the testing of compounds that might have therapeutic value for the treatment of stroke (48). This model utilizes the toxin iodoacetic acid (IAA), a well known, irreversible inhibitor of the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (49), in combination with the mouse HT22 hippocampal nerve cell line. The changes seen following IAA treatment of nerve cells are very similar to changes that have been seen in animal models of ischemic stroke, and include alterations in membrane potential, breakdown of phospholipids, loss of ATP and glutathione (GSH), and an increase in reactive oxygen species (ROS). Fisetin was very effective at protecting the nerve cells from IAA-induced death with an EC50 of 3 μM (48).
Given these positive data, fisetin was then tested in two different animal models of stroke. The rabbit small clot embolism model (SCEM) is a rigorous stroke model that recapitulates the initiating event in most cases of human stroke. For the SCEM, a suspension of small blood clots is injected into the carotid artery of a rabbit. To evaluate the quantitative relationship between clot dose and behavioral deficits, logistic S-shaped quantal analysis curves are fitted to the dose-response data as originally described by Waud (50) and adapted for stroke studies (51–55). A wide range of clot doses is used resulting in behaviorally normal and abnormal animals. In the absence of a neuroprotective treatment regimen, small numbers of microclots cause no grossly apparent neurologic dysfunction and large numbers of microclots invariably cause encephalopathy or death. Using a simple dichotomous rating system, each animal is rated by a naïve observer as either behaviorally normal or abnormal 24 hr post-embolization. Using quantal analysis, it is possible to detect behavioral changes following pharmacological intervention. A separate curve is generated for each treatment condition and a statistically significant increase in the P50 value or the amount of microclots that produce neurologic dysfunction in 50% of a group of animals compared to control is indicative of a behavioral improvement (51–55). To test the protective effect of fisetin in this model, fisetin (50 mg/kg bw) was injected at 5 min after microclot injection and behavioral analysis was conducted 24 hr after treatment. Fisetin significantly (p<0.05) reduced stroke-induced behavioral deficits and increased the P50 value from 1.06 ± 0.15 mg in the control group to 2.53 ± 0.55 mg which directly correlated with an increase in the number of animals that were behaviorally “normal” (48).
Fisetin was also tested in the temporary middle cerebral artery occlusion (tMCAO) stroke model (56). Temporary middle cerebral artery occlusion was induced in mice by the intraluminal filament method and maintained for 60 minutes. Animals were either injected intraperitoneally 20 min before or 180 min after the onset of ischemia with fisetin (25 or 50 mg/kg bw) or placebo. A significant, dose-dependent protective effect of fisetin on stroke size was observed. While animals injected immediately before the onset of ischemia with 25 mg/kg of fisetin showed a trend towards smaller infarcts, animals treated with a higher dose of fisetin (50 mg/kg) had significantly smaller infarcts (placebo 56μl; 25mg/kg fisetin 49μl; 50mg/kg fisetin 30μl; p<0.05). Even when injected three hours after the onset of ischemia, 50 mg/kg fisetin still retained its protective capabilities (placebo 42μl; fisetin 27μl; p<0.05). A strong trend toward earlier recovery in both the low- and high-dose fisetin-treated animals compared to placebo following both the pre- and post-stroke treatment strategies was also seen.
4.4 Fisetin and Depression
Although depression is not directly fatal, it is a major cause of disease-related disability. Depression is associated with both AD (57) and diabetes (58). A recent study looked at the effects of fisetin in two rodent models of depression: the tail suspension test and the forced swim test (59). In both tests, depressive behavior is indicated by the time that the animal remains immobile with classical anti-depressants greatly reducing the immobility time. Oral administration of fisetin showed a dose dependent effect on immobility times in both tests with 20 mg/kg bw being the most effective dose at reducing immobility time. The anti-depressants imipramine and fluoxetine, administered by intraperitoneal injection, were used as positive controls. Further studies showed that the anti-depressant effects of fisetin involved modulation of the serotonergic system by reducing serotonin metabolism possibly through mild inhibition of monoamine oxidase A, the enzyme that metabolizes serotonin (59). Fisetin also appeared to modulate the noradrenergic system which may also contribute to its anti-depressive effects.
4.5 Fisetin and Diabetes
The complications of diabetes have historically been thought to primarily affect the vasculature in tissues such as heart, kidney, retina and peripheral nerves, but may also have a major impact on the microvasculature of the brain. Indeed, there is growing evidence that the complications of diabetes extend to the CNS (60, 61) and that both changes in the flux of glucose through the glycolytic pathway and the glycation of proteins play central roles in these complications (62, 63). There are learning and memory deficits as well as additional CNS pathology associated with diabetes (60, 61) and diabetes is a risk factor for AD (64) and stroke (65).
The Ins2Akita mouse is a model of type 1 diabetes (66). The Akita spontaneous mutation is an autosomal dominant mutation in the insulin II gene. As a consequence, Akita mice develop the pathological characteristics of type 1 diabetes, including diabetic nephropathy (67), neuropathy (68) and elevated anxiety symptoms (69, 70). This model was used to determine if fisetin could reduce the neurological complications of diabetes (71). Four groups of male mice were studied, 2 groups each of wild type and Akita. Fisetin was fed to one set of control and one set of Akita mice between the ages of 6 and 24 weeks in their food at 0.05%, resulting in a daily dose of approximately 25–40 mg/kg bw. Male Akita mice develop hyperglycemia by 4 weeks of age (72). Blood glucose was determined in all animals at 12 weeks and again before sacrificing the animals at 24 weeks. At this time, blood glucose and HbA1c were significantly elevated in the Akita mice. These changes were not affected by the presence of fisetin in the diet. The Akita mice also showed significant weight loss relative to their wild type counterparts and this was also not altered by fisetin in the diet. Fisetin had no effect on blood glucose and HbA1c levels in the wild type mice.
Although cognitive impairment is associated with long-term diabetes in humans (73) and is seen in some mouse models of type 1 diabetes (74, 75), no deficits in behavior in the Akita mice were seen using the MWM, an assay of spatial learning and memory, consistent with an earlier study (68). However, Akita mice displayed significantly decreased locomotor activity in the open field test coupled with a significantly increased time of immobilization (69). These results were interpreted as indicative of anxiety behavior, a CNS complication of many patients with diabetes (70). However, Akita mice fed fisetin showed a significant reduction in these anxiety-related symptoms with both the distance traveled and time ambulatory in the open field test restored to near normal values (71). Fisetin had no effect on the behavior of the wild type mice in the open field test.
Peripheral nerve dysfunction in the form of diabetic neuropathy is a major complication of diabetes that has a significant impact on the quality of life. To assay the effects of fisetin on the development of diabetic neuropathy in the Akita mice both motor nerve conduction velocity and thermal latency, a measure of sensory nerve function, were measured. Akita mice showed a significant decrease in motor nerve conduction velocity that was ameliorated by the presence of fisetin in the diet (Fig. 4A). Fisetin did not have a significant effect on motor nerve conduction velocity in the wild type mice. Akita mice also showed a statistically significant increase in hind paw thermal latency (Fig. 4B) that was prevented by the presence of fisetin in the diet. In contrast, there was no loss of intra-epidermal nerve fibers (IENF) or sub-epidermal nerve plexi (SNP) in the plantar foot skin of Akita mice and fisetin had no effect on these parameters in Akita mice (not shown). Fisetin had no effect on hind paw thermal latency, paw skin IENF or paw skin SNP in wild type mice. Together these results suggest that the elements of diabetic neuropathy measured in the Akita mice result from metabolic and/or neurochemical changes rather than structural damage to neurons. Nevertheless, fisetin was able to reduce the impact of diabetes on peripheral nerve function in this model. Whether it would also be effective in models of diabetic neuropathy which display structural damage is unclear.
Figure 4.
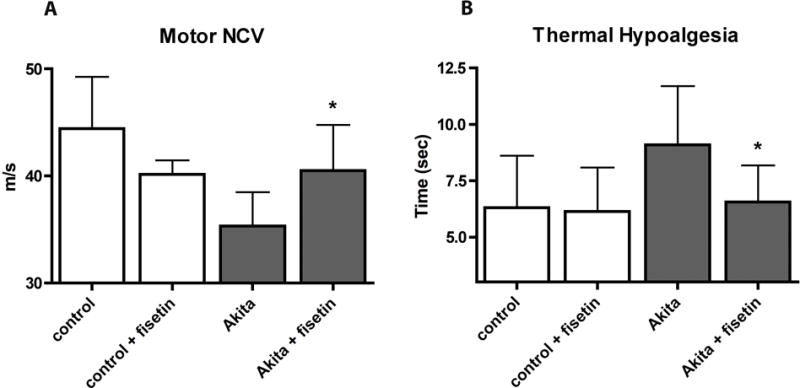
Fisetin reduces the neurological consequences of diabetes in Akita mice. Male Akita mice were fed control diet or fisetin (0.05%) for 18 weeks beginning at 6 weeks of age. (A) large fiber function was tested by measuring motor nerve conduction velocity and (B) small fiber function was measured by paw withdrawal to noxious heat. Diabetes-induced thermal MNCV slowing (A) and hypoalgesia (B) were ameliorated by fisetin treatment. All data are mean ± SD of n = 5–7/group. *(p < 0.05) indicates significantly different from wild type or Akita alone as determined by ANOVA followed by Tukey’s post test.
5. MECHANISMS OF ACTION (Table 1)
Table 1.
Mechanisms of Action of Fisetin and their Potential Relevance to Neurological Diseases
| Mechanism of Action | Potential Disease Relevance |
|---|---|
| Antioxidant and chelating activity | AD; HD; stroke; diabetes |
| Maintenance of GSH | AD; stroke; diabetes |
| Neurotrophic factor signaling pathways | AD; HD; depression |
| Regulation of the innate immune response | AD; stroke; diabetes |
| Modulation of protein aggregation and stability | AD; HD |
5.1 Antioxidant and Chelating Activity
Although it is not entirely clear how relevant the antioxidant activity of flavonoids as measured in test tube assays is to their effects in vivo (76, 77), fisetin is a relatively good antioxidant with a Trolox equivalent antioxidant capacity (TEAC) value of ~3 (9, 78). Although pro-oxidant effects of fisetin have been detected in test tube assays, these are generally seen at high (≥ 50 μM) concentrations (79, 80). In contrast, fisetin was found to be a moderately effective scavenger of superoxide radicals generated by xanthine/xanthine oxidase and Fenton-mediated hydroxyl radicals generated by the combination of ferrous-EDTA and hydrogen peroxide with IC50s in the low (~3–5) micromolar range (81). In addition, fisetin decreased ferrous sulfate/ascorbate induced lipid peroxidation in liver microsomes at low (1.35) micromolar concentrations (82). The latter observations may be related to the efficient chelation of iron by fisetin (83–85). Iron is a transition metal meaning that it exhibits multiple oxidation states and is therefore redox active. While this property is critical to many of its important biological functions, it also means that it can generate ROS such as the highly reactive hydroxyl radical via the Fenton reaction (86). There is growing evidence that iron accumulates in the brain with aging and may contribute to the development and/or progression of age-related neurological diseases such as AD (87–89). For example, a recent MRI study showed that in AD, damage to the hippocampus, the region of the brain that is important for memory, occurs in conjunction with iron accumulation (90). Another MRI study showed that brain iron accumulation was inversely associated with age-related cognitive decline (ie more iron, less memory) (91). Importantly, iron accumulation specifically occurs in the areas of the brain impacted by AD (90). Furthermore, iron chelators have shown beneficial effects in animal models of AD (87–89) as well as some limited positive results in human AD patients (87, 92). How iron accumulation leads to nerve cell damage in AD is not clear at this time. However, the age-related increase in iron found in human, rat and mouse brains (93) was associated with markers of oxidant-mediated damage. Furthermore, iron can promote the accumulation and/or aggregation of Aβ (88, 92, 94) and low levels of iron are a potent catalyst of protein glycation (95). By chelating iron, fisetin could effectively reduce all of these impacts of age-dependent iron accumulation in the brain. Further studies are needed to determine how effective fisetin is at chelating iron in the brain in vivo and the role that this plays in its multiple beneficial effects in age-related neurological diseases.
5.2 Maintenance of GSH
The small molecule antioxidant GSH is a tripeptide consisting of the amino acids glutamate, glycine and cysteine. Cells contain up to millimolar concentrations of GSH. Thus, GSH is one of the most important small molecule antioxidants in somatic cells. GSH and GSH-associated metabolism provide the major line of defense for the protection of cells from oxidative and other forms of toxic stress (for reviews see 96, 97). In addition, as part of the major redox couple in cells, GSH plays a central role in maintaining cellular redox homeostasis (98). Consistent with the idea that higher GSH levels are associated with cell survival, GSH promotes the activity of proteins that maintain cell function such as glutathione peroxidases (99), glyoxalase 1 (100), glutathione S-transferases (101) and Bcl-2 (102) and inhibits the activity of proteins that can promote nerve cell death such as lipoxygenases (LOXs) (103, 104) and cytosolic cytochrome C (105). Thus, maintenance of GSH levels is developing as an important therapeutic target both to reduce the impacts of aging (106) as well as in the treatment of age-associated neurological diseases such as AD and Parkinson’s disease (PD) (107, 108).
A fairly large number of studies have shown age-dependent decreases in total GSH and/or reduced GSH levels in the brain (for reviews see 108, 109, 110). Importantly, a recent study using double-edited 1H MRS to look at GSH levels in the occipital cortex of healthy young (20 yr) and elderly (77 yr) human subjects found that GSH levels were decreased by ~30% in the elderly subjects (111). This finding is of particular relevance with respect to the alterations in cognition with aging and age-related neurological diseases because multiple studies have shown that transient decreases in GSH levels can temporarily impair cognitive function, even in young animals (112–114). Together these studies suggest that acute decreases in GSH levels can impact aspects of both learning and memory. Whether the decreases seen during aging which may occur gradually over a prolonged time period would also have the same effect is not clear but might play a role in the deficits in cognitive function seen with aging and disease.
One of the major transcription factors that regulates GSH metabolism is the ubiquitously expressed protein NF-E2-related factor 2 (Nrf2) (for reviews see 115, 116). Several key enzymes of GSH metabolism are transcriptionally regulated by Nrf2 including the catalytic and regulatory subunits of glutamate cysteine ligase (the rate limiting enzyme for GSH synthesis), GSH synthase, glutathione peroxidase 2, glutathione transferases, xCT (the rate limiting subunit of the glutamate/cystine antiporter system xc−) and glutathione reductase (115, 117). A second transcription factor, activating transcription factor 4 (ATF4), also plays a role in GSH metabolism (118, 119). ATF4 regulates the transcription of a variety of genes involved in amino acid import, GSH biosynthesis and resistance to oxidative stress (118). ATF4 levels are regulated mainly at the translational level by eIF2α phosphorylation via a mechanism that is dependent on the unique 5′ untranslated region of the ATF4 mRNA (120). Recent data (121) indicate that ATF4 plays a key role in regulating basal GSH levels in neuronal and non-neuronal cells, whereas Nrf2 appears to function as a multiplier of ATF4 activity under conditions of oxidative stress.
There is evidence that both Nrf2 and ATF4 decline with age (122–124). Comparing wild type and Nrf2 knockout mice, it was shown that in skeletal muscle, loss of Nrf2 had little impact on ROS or GSH levels or the GSH/GSSG ratio in young mice (2 months) but resulted in a significant increase in ROS and a significant decrease in GSH and GSH/GSSG in elderly mice (>24 months) (125). These changes correlated with an enhancement of myocyte degeneration. Using the reverse approach, it was found that primary skin fibroblasts derived from long-lived Snell dwarf mice have elevated levels of Nrf2 along with increased levels of GSH. Although none of the studies with Nrf2 in aging have looked at brain, similar changes are likely to occur in the brain but further research is needed to confirm this (126). Similar to Nrf2, the levels of ATF4 were found to decline with age (1 month vs > 18 months) in multiple tissues in mice including both cerebral cortex and cerebellum (127). Consistent with the decline in ATF4, eIF2α phosphorylation also decreased with age in the same tissues.
Over the last 15 years, several in vitro models that can be used to identify small molecules that are able to maintain redox homeostasis in the presence of oxidative stress have been developed in my laboratory. Indeed, fisetin was first identified as a potential neuroprotective compound (9) using one of these models, oxidative glutamate toxicity (for review see 128).
How does fisetin maintain GSH levels? Intracellular GSH levels are regulated by a complex series of mechanisms that include substrate (mainly cyst(e)ine) transport and availability, rates of synthesis and regeneration, GSH utilization and GSH efflux to extracellular compartment (96, 129). To date, the most effective approaches for maintaining GSH levels in the CNS include enhancing cyst(e)ine uptake both directly and indirectly via transcriptional upregulation of system xc−, increasing GSH synthesis via transcriptional upregulation of the rate limiting enzyme in GSH biosynthesis, and decreasing GSH utilization (97).
Recently, it was shown that fisetin increases the levels of both Nrf2 and ATF4 in nerve cells in a dose- and time-dependent manner (130). Furthermore, using microarrays, it was found that Nrf2- or ATF4-regulated genes constituted 4 of the top 5 hits of the over 34,000 named mouse genes probed. Under basal conditions, both Nrf2 and ATF4 are very short-lived proteins with reported half-lives of ~15 min (131) and ~10 min (121), respectively. Fisetin was able to increase the half-lives of both of these proteins ~2-fold but via distinct mechanisms (130). Further studies showed that both Nrf2 and ATF4 were essential for the effect of fisetin on GSH levels (130). Treatment of mouse hippocampal HT22 cells with siRNA to ATF4 decreased ATF4 protein levels by ~75%. This resulted in an ~50% reduction in the ability of fisetin to maintain cellular GSH levels under both basal conditions and glutamate-induced oxidative stress conditions (130). Treatment of HT22 cells with siRNA to Nrf2 decreased Nrf2 levels by ~80%. While this reduction had no effect on the ability of fisetin to maintain GSH levels under basal conditions, it reduced by ~30% the ability of fisetin to maintain GSH in the presence of oxidative stress (130). Thus, fisetin induces two distinct transcription factors, ATF4 and Nrf2, that play key roles in regulating GSH levels. Under basal conditions, ATF4 is the more important but the two transcription factors cooperate to increase GSH levels under oxidative stress conditions.
Consistent with these in vitro results, a recent study which looked at the ability of fisetin to protect mice from aluminum chloride toxicity showed that oral administration of fisetin at 15 mg/kg bw could greatly reduce the loss of total GSH from both the cortex and hippocampus (132). Similarly, in the study on scopolamine-induced memory impairment in mice, fisetin was able to prevent the loss of total GSH from the cortex (34).
5.3 Neurotrophic Factor Signaling Pathways
Neurotrophic factors play critical roles in promoting the differentiation, survival and functional maintenance of nerve cells. They are also key players in synaptic plasticity (133), cognition and memory formation (134–136). Changes in the levels of neurotrophic factors and/or their receptors are implicated in the pathophysiology of a variety of neurodegenerative diseases including AD, HD, PD and amyotrophic lateral sclerosis (ALS) (for reviews see 137, 138–140).
A key signaling pathway activated by many neurotrophic factors is the Ras-ERK cascade (141). Previously, it was shown that fisetin can activate the Ras-ERK cascade in nerve cells (10) and that activation of this signaling pathway is associated with the neuroprotective, neurotrophic and cognition enhancing effects of fisetin (21). More recently, it was found that induction of ERK activation by fisetin was required for neuroprotection in the PC12 model of HD (39) (section 4.2). Furthermore, in the AD mice treated with fisetin (section 4.1), increases in ERK phosphorylation in the hippocampus that correlated with improvements in behavior and reductions in neuropathology were seen (18). Thus, fisetin is able to induce ERK activation both in cell culture and in animals and thus may act as at least a partial replacement for the loss of neurotrophic factor signaling that occurs with aging and is exacerbated in neurological diseases.
5.4 Regulation of the Innate Immune Response
Chronic inflammation is a major feature of AD as well as essentially all other neurological disorders (for reviews see 142, 143–145). Microglia (brain macrophages) are the resident immune cell population of the CNS, comprising 5–10% of the total cell population (for reviews see 146, 147, 148). Their presence can have both beneficial and detrimental effects on the brain. Recent studies suggest that activated microglia can exist in at least two states: classically activated M1 microglia and alternatively activated M2 microglia. Alternatively activated M2 microglia play important, protective roles in the CNS such as removing pathogens and promoting tissue regeneration after injury. On the other hand, classically activated M1 microglia are implicated in the pathogenesis of a variety of neurological diseases including AD. M1 microglia produce a wide array of pro-inflammatory and cytotoxic factors including cytokines, free radicals, excitatory neurotransmitters and eicosanoids that may work in concert to promote neurodegeneration. In the context of the AD brain, there are thought to be multiple stimuli that generate an inflammatory response in the microglia including Aβ (145). Thus, inhibiting classically activated M1 microglia is another important therapeutic target for AD as well as other neurological diseases and stroke.
Bacterial lipopolysaccharide (LPS) binds to Toll-like receptor-4 on the surface of microglia and induces classically activated M1 microglia. Using the BV-2 microglial cell line, it was shown that fisetin could reduce LPS-induced microglial activation and neurotoxicity (149). Fisetin blocked LPS-induced nitric oxide production, measured as accumulation of nitrite in the culture medium, as well as LPS-induced increases in the extracellular levels of the pro-inflammatory cytokine tumor necrosis factor-α (TNF-α) and the pro-inflammatory prostaglandin PGE2 and increases in the expression of the inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX2) genes whose protein products regulate nitric oxide and prostaglandin production, respectively. Similar to the results obtained with other types of cells treated with fisetin and LPS (81, 150), the effects of fisetin on microglial activation appeared to be at least partially mediated by the inhibition of LPS-stimulated NF-κB activation. NF-κB is considered a central regulator of inflammation and has been shown to activate more than 500 genes, many of which are involved in inflammation (151). NF-κB normally resides in the cytoplasm of cells in an inactive form comprised of three subunits: p65 and p50 and the inhibitory subunit IκB. Signals that activate NF-κB cause the dissociation of IκB from p50/p65 thereby allowing its translocation to the nucleus where it binds to specific κB DNA consensus sequences in the enhancer region of NF-κB responsive genes. The levels of IκB are in turn regulated by the IκB kinases (IKKs). In the human lung adenocarcinoma cell line H1299, fisetin reduced both NF-κB-dependent reporter gene expression and protein expression through a mechanism that involved inhibition of the activation of the IKKs (150). More recently, it was shown that fisetin can suppress the activity of the NF-κB coactivator CBP/p300 in human monocytes (152). Thus, fisetin may act at multiple levels to block NF-κB activation and NF-κB-induced gene expression.
Other pro-inflammatory signaling pathways that have been shown to be suppressed by fisetin in microglia include the c-jun-N-terminal kinase (JNK) (56) and p38 mitogen activated kinase (p38 MAPK) pathways (149). The activation of the JNK and p38 MAPK pathways contributes to pro-inflammatory cytokine production (153, 154). A recent study showed that in RAW264.7 macrophages, fisetin suppresses both p38 MAPK and JNK activation by increasing the levels of MAPK phosphatase-1 (MKP-1), an enzyme responsible for p38 MAPK and JNK dephosphorylation (155). Fisetin increases MKP-1 levels by a post-translational mechanism involving the inhibition of its ubiquitination and subsequent targeting to the proteasome (155). Interestingly, it has been shown that ERK activation (see section 5.3) can promote MKP-1 stabilization via this pathway (155).
Studies in my laboratory confirmed the effects of fisetin on microglial activation in vitro and then were extended to an analysis of the anti-inflammatory effects of fisetin in the tMCAO model in mice (see section 4.3) (56). Using mouse N9 microglial cells, the effects of fisetin on LPS-induced nitric oxide and TNF-α production were measured. Fisetin caused a significant inhibition of both nitric oxide and TNF-α production compared to placebo (56). To elucidate the underlying mechanism of the anti-inflammatory actions of fisetin, the effects of fisetin on several intracellular signal transduction pathways implicated in inflammation including IκB, JNK and its target cJun were examined. Fisetin treatment of LPS-stimulated murine microglia significantly reduced both IκB and JNK phosphorylation as well as the phosphorylation of the target of JNK, c-Jun, suggesting that fisetin directly inhibits the intracellular signaling pathways induced by pro-inflammatory cytokines. Downstream targets of these transcription factors include pro-inflammatory cytokines but also iNOS. Indeed, following LPS stimulation iNOS expression was significantly reduced 91% ± 14 (p<0.001) in microglial cells treated with 10 μM fisetin.
Next, the amount and composition of the cerebral inflammatory infiltrate at day 3 after stroke were analyzed (56). There were no significant differences in the numbers of microglia in the ipsilesional hemispheres of mice treated with fisetin (50 mg/kg bw) or placebo. In contrast, there was a significant decrease in leukocytes infiltrating the brain in fisetin-treated mice (cells per ischemic hemisphere; mean ± SD; placebo 179,252 ± 23,389; fisetin 93,045 ± 46,963; p=0.045). For further analysis, leukocytes were subdivided into lymphocytes, macrophages and dendritic cells (DC) based on the expression of CD3, CD11b and CD11c, respectively. Since similar absolute numbers of microglia were found in placebo and fisetin-treated mice, the number of microglia was used as an internal reference. Using this approach, a significant decrease in the percentage of macrophages (percentage of macrophages relative to microglia; mean ± SD; placebo 49 ± 5.2; fisetin 12 ± 3.3; p=0.037) and DC (percentage of DC relative to microglia; mean ± SD; placebo 23 ± 0.65; fisetin 14 ± 2.31; p=0.038) as well as lymphocytes (percentage of lymphocytes relative to microglia; mean ± SD; placebo 5.7 ± 0.056; fisetin 3.3 ± 0.207) was observed in the ischemic hemisphere of fisetin-treated animals as compared to placebo.
While the number of cells infiltrating the brain could simply be a function of chemotaxis, which is influenced by the extent of tissue damage, the activation status of these cells likely identifies cells primed to damage additional brain tissue. Therefore, the activation profile of brain resident microglia, infiltrating macrophages and DC was analyzed by flow cytometry of whole brain homogenates three days after stroke. A substantial suppression of intracellular TNF-α production in macrophages in fisetin treated mice at day 3 (placebo 24.6%; fisetin 2.9%) and to a lesser degree at day 7 (placebo 33.8%; fisetin 13.9%) was found. A similar but smaller effect was observed for DC. In addition, a reduction in TNF-α–producing microglia at day 3 which was no longer detectable at day 7 (day 3: placebo 30.4%; fisetin 17.3%; day 7: placebo 31.9%; fisetin 26.8%) was also observed. In contrast, macrophages and DCs isolated from spleen from the same mice did not show any differences in TNF-α production between fisetin-treated and placebo-treated mice either at day 3 or at day 7 post ischemia. Thus, the reduced post-ischemic inflammation was not merely due to a lack of chemotaxis but also to a reduction in the activation of macrophages and microglia in vivo as shown by the decrease in TNF-α. Importantly, residual effects of a single fisetin injection on immune cell activation were still detectable seven days after stroke. Moreover, the observation that fisetin applied three hours after the onset of ischemia is still protective strongly argues in favor of a relevant role for fisetin’s anti-inflammatory effects in vivo.
Further evidence for in vivo anti-inflammatory effects of fisetin was obtained in the studies on fisetin in AD (section 4.1). Both microglia and astrocytes are activated in AD and their activation is implicated in the loss of nerve cell function (156). For example, the activation of astrocytes results not only in the production of pro-inflammatory cytokines but also in a decrease in their normal role as guardians of nerve cell function (157). Increased levels of glial fibrillary acidic protein (GFAP) are a marker of astrocyte activation. While AD had no effect on the number of astrocytes in the hippocampus, it significantly increased both their area and the intensity of GFAP staining and these alterations were largely reversed by fisetin treatment (18). A fisetin-dependent reduction in GFAP levels was also seen in the hippocampus of the AD mice by Western blotting (18).
Together, these results indicate that fisetin has anti-inflammatory activity on microglia and astrocytes both in vitro and in vivo and therefore might be effective in a variety of conditions involving the dysregulation of the immune system in the brain.
To further elucidate the effects of fisetin on inflammation in vivo, a detailed analysis of eicosanoid production in the wild type and AD mice with and without fisetin treatment (section 4.1) was conducted using liquid chromatography tandem mass spectrometry (18). This approach generated global profiles consisting of over 160 individual eicosanoids many of which were altered in AD and after fisetin treatment. Eicosanoids are a class of bioactive lipid mediators derived from the metabolism of polyunsaturated fatty acids, namely arachidonic acid (AA), by COXs, lipoxygenases (LOXs), cytochrome P450s and non-enzymatic pathways (158). They are known to be potent endogenous regulators of the inflammatory response in the periphery but are much less well studied in the brain. AD increased the production of the pro-inflammatory thromboxanes TXB1 and TXB2 and this was partly prevented by fisetin (18). The thromboxane pathway is implicated in platelet aggregation, adhesion and vascular contraction during inflammation (159). This is of relevance given the pro-thrombotic effect of TX’s and the link between thrombosis and vascular pathology in AD (160). On the other hand, fisetin increased production of prostaglandin D2 (PGD2) and its non-enzymatic anti-inflammatory products prostaglandin J2 (PGJ2) and 15-deoxy-PGD2 (15d-PGD2). This is of particular interest because these prostaglandins have potent anti-inflammatory effects in brain microglia (161) including down regulation of iNOS, a known effect of fisetin (56). Historically, PGD2 was regarded as a pro-inflammatory mediator but a potentially anti-inflammatory role is now also recognized (162).
Lipoxygenases (LOXs) metabolize 20-carbon unsaturated fatty acids such as arachidonic acid, which are produced from membrane phospholipids by the action of phospholipases, to eicosanoids, including hydroxyperoxyeicosatetraenoic acids (HPETEs), hydroxyeicosatetraenoic acids (HETEs) and leukotienes. (for review see 163) The LOXs are dioxygenases that incorporate molecular oxygen into specific positions of arachidonic acid and can be distinguished on the basis of the site of oxygen insertion. 5-LOX, 12-LOX and 15-LOX are all expressed in the brain. There is good evidence that both 5-LOX and 12-LOX contribute to the nerve cell loss seen in several neurological disorders. For example, 12-LOX activation plays an important role in the nerve cell death induced by GSH depletion (103, 164–166). 12-LOX was also shown to be increased in AD (167) and 12-LOX inhibition protected cortical neurons from Aβ-induced death (168). In addition, the expression and activity of 5-LOX is specifically increased in the hippocampus with age (169). Using a double transgenic mouse model generated by crossing 5-LOX deficient mice with the Tg2576 mouse model of AD, it was shown that loss of 5-LOX reduced the Aβ burden in the brains of the mice by 64–80% suggesting that inhibition of 5-LOX also might have benefits in AD (170).
In earlier studies, fisetin was found to be an effective inhibitor of 12-LOX activity in the fish gill (171), 5-LOX activity in stimulated peritoneal leukocytes (172) and 15-LOX activity in rabbit reticulocytes (173). As shown in Table 2, these studies were confirmed and extended in my laboratory. Fisetin inhibits 12-LOX from human platelets with an IC50 of 0.14 μM, 5-LOX from human peripheral blood mononuclear leukocytes (PBML) with an IC50 of 0.585 μM and 15-LOX from rabbit reticulocytes with an IC50 of 0.341 μM. In all cases, the IC50 values for fisetin are as good or better than those of known LOX inhibitors. In contrast, fisetin has no effect on COX activity (Table 2). Since LOXs are non-heme iron enzymes (174), at least part of the inhibitory effects of fisetin on LOX activity may be due to its ability to chelate iron. Indeed, other iron chelators have been shown to inhibit LOXs (175). Importantly, in the mouse model of AD, fisetin was found to significantly reduce the levels of pro-inflammatory 5-hydroxyeicosatetraenoic acid (5-HETE) and 12-hydroxyeicosatetraenoic acid (12-HETE), the primary metabolites of 5-LOX and 12-LOX, respectively, in the brains of the AD mice (18). These results clearly indicate that fisetin is able to inhibit both 5-LOX and 12-LOX not only in vitro but in vivo as well, at least in the context of AD.
Table 2.
Fisetin is a Pan-LOX Inhibitor
| Enzyme | Source | Fisetin IC50 |
|---|---|---|
| 5-LOX | Human PBML | 0.58 μM |
| 12-LOX | Human platelets | 0.13 μM |
| 15-LOX | Rabbit reticulocytes | 0.34 μM |
| COX1 | Human platelets | >10 μM |
| COX2 | Human recombinant | >10 μM |
Dose response curves were done with fisetin under standard conditions using the indicated cell types and the IC50 values are presented.
These results suggest that an important activity of fisetin in the brain in the context of age-associated neurological diseases is the inhibition of LOX activity. This action may contribute to both the neuroprotective and anti-inflammatory effects of fisetin and thereby promote the functional maintenance of the CNS.
5.5 Modulation of Protein Aggregation and Stability
Two reports showed that fisetin can inhibit Aβ fibril formation in a cell-free assay system (176, 177). One of these studies (176) also showed that fisetin prevents extracellular Aβ toxicity in the HT22 cells. Since Aβ is thought to play a key role in the nerve cell loss that is the hallmark of AD, these results suggested that fisetin might be able to reduce the burden of Aβ through multiple mechanisms, including inhibition of aggregation and enhancement of degradation.
However, these in vitro results were not supported by the studies on AD mice (18). No significant differences in Aβ plaque loads in sections of brains from control AD and fisetin-fed AD mice were found. Aβ levels were also examined in the radioimmunoprecipitation assay buffer (RIPA) insoluble (100,000 × g pellet) and soluble (RIPA supernatant) fractions of the hippocampi of fisetin-fed and control AD mice. Neither Aβ1–40 nor Aβ1–42 levels, as measured by ELISA, were altered in the RIPA insoluble fraction in the animals fed fisetin relative to untreated animals. However, fisetin treatment did significantly reduce the levels of Aβ1–40 but not Aβ1–42 in the RIPA soluble fraction. Thus, it appears that the major in vivo effects of fisetin do not involve inhibition of Aβ aggregation or accumulation.
The ubiquitin-proteasome pathway mediates the majority of the proteolysis seen in the cytoplasm and nucleus of mammalian cells. As such it plays an important role in the regulation of a variety of physiological and pathophysiological processes (for reviews see 178, 179). Several studies have shown that there is a specific decrease in proteasome activity in the hippocampus, cortex, striatum, globus pallidus and substantia nigra with aging in rodents (180, 181). In contrast, little or no change in proteasome activity is seen in the cerebellum and brain stem. These findings are consistent with studies that have shown that proteasome activity is decreased in a variety of age-associated neurological diseases including AD, PD and ALS (182–184) and may contribute to disease progression. Interestingly, fisetin was shown to increase proteasome activity and this action may also contribute to some of its neuroprotective effects (185).
6. METABOLISM OF FISETIN
Flavonoids are known to be extensively metabolized following oral consumption resulting in glucuronidated, sulfated and methylated metabolites (for reviews see 186, 187). The metabolism of fisetin was first characterized in rats following intravenous and oral administration (188). Following both types of administration, the levels of free fisetin in serum declined within minutes (t1/2= 2.7 min) while the levels of sulfated/glucuronidated fisetin increased. Following oral administration at 50 mg/kg bw, the serum concentration of fisetin sulfates/glucuronides was maintained at ~10 μM for >24 hr. These results are in sharp contrast to those obtained for 5 OH and 7 OH flavones (188) and baicalein (189) where the levels of free and conjugated flavonoid never exceeded 1 μM following oral administration. When tested in an assay for antioxidant activity, the fisetin sulfates/glucuronides showed somewhat reduced but still significant activity as compared to free fisetin (188). This result is consistent with a study on the effects of glucuronidation on the ability of several different flavonoids to protect nerve and lymphoid cells from oxidative stress-induced death (190). Although the flavonoid glucuronides had generally higher EC50s for protection as compared to their unmodified parent, they still showed excellent activity. Furthermore, circulating flavonoid sulfates/glucuronides can be cleaved to the free form in a tissue-specific manner if there is a local release of β-glucuronidases and/or sulfatases (187). In addition, sulfated flavonoids have been shown to have biological activity (191). In the mouse, following intravenous administration of fisetin at 223 mg/kg bw, free fisetin was detected in the serum for several hours (t1/2=3.12 hr) (192). Significant accumulation of fisetin and its metabolites in multiple tissues was also seen. The main metabolites were glucuronidated fisetin, geraldol (3,4′,7-trihydroxy-3′-methoxyflavone) and glucuronidated geraldol. Interestingly, geraldol was as active as fisetin in several biological assays. In contrast, following oral administration of fisetin to mice at 50 mg/kg bw, my laboratory obtained results very similar to those seen with the rat. A very short half-life for free fisetin but a long half-life (>8 hr) for sulfated/glucuronidated fisetin was observed (unpublished results).
There is an ongoing debate about whether flavonoids such as fisetin can reach levels in the brain that are sufficient to affect brain function. As described above (section 4), multiple laboratories have shown that oral administration of fisetin is able to affect brain function in vivo. Whether this is a direct effect of the unmodified fisetin molecule remains to be determined. Importantly, my laboratory recently found that sulfated and/or glucuronidated forms of fisetin reach concentrations of 30 μM in the cerebrospinal fluid with a plasma half life of 8 hr in macaques fed a single oral dose of 25 mg/kg bw (unpublished results).
7. TOXICITY
Off-target effects that can contribute to toxicity are certainly a concern for any potential therapeutic. However, in the case of fisetin, there is no evidence for either short- or long-term toxicity. In a short-term study done by an outside laboratory, mice were orally administered 2000 mg/kg bw of fisetin, examined for 48 hr and sacrificed. No indications of toxicity were observed. The effects of long-term feeding of fisetin on health were also evaluated (18). No significant differences in body weights were seen between control and fisetin-fed animals given fisetin at 0.05% in their diets for 9 months (~25 mg/kg bw). Following sacrifice, multiple tissues (lungs, spleen, liver, kidneys, heart, stomach, intestine, testes and ovary) were examined using standard toxicological pathology criteria and no toxicity was associated with fisetin treatment. Furthermore, fisetin was negative in the Ames test. In addition, fisetin showed no inhibition of hERG activity at concentrations up to 10 μM, the highest concentration tested (unpublished results). It also showed no inhibitory effects on the activities of cytochrome P450s 3A4, 2C9 and 2D6 at concentrations up to 10 μM (unpublished results). Thus, at the present time, there is no indication of fisetin toxicity from either in vitro or in vivo tests.
8. SUMMARY AND PERSPECTIVE
As discussed above, fisetin has positive effects on a number of common pathways associated with age-associated neurological diseases (Fig. 5). While the precise relationships among the pleiotropic effects of fisetin on the cells of the brain are currently under investigation, this combination of actions suggests that fisetin has the potential to maintain brain function even in the presence of the diverse factors that contribute to the development of age-associated neurological diseases (Table 1). Importantly, multiple studies in animal models suggest that fisetin can reduce the impact of age-related neurological diseases, especially those associated with cognitive deficits such as AD. Moreover, many of these diseases do not currently have effective treatments (Table 3). These observations, in combination with the lack of any indication of toxicity, strongly suggest that fisetin might be useful in the clinic. However, because it is a natural product, it is very difficult to obtain the financial support needed to pursue the necessary studies.
Figure 5.
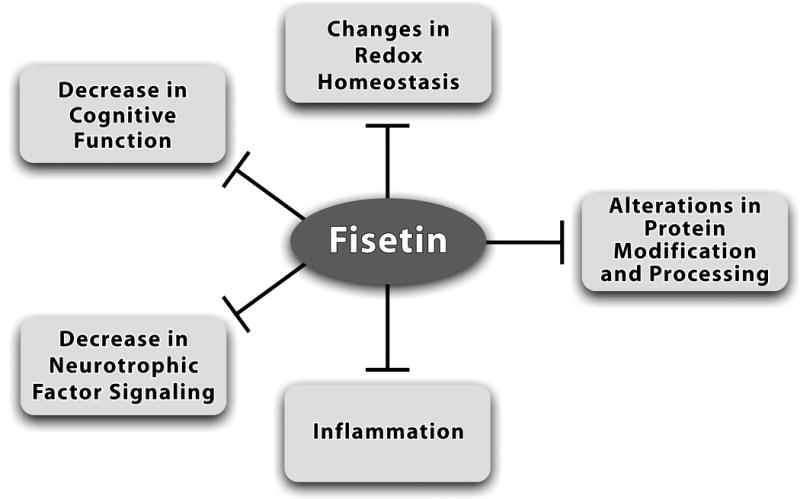
Fisetin affects multiple pathways implicated in brain aging and age-associated diseases. As discussed in this article, fisetin can increase brain cell function and survival through maintenance of redox homeostasis, activating neurotrophic factor signaling pathways and inhibiting inflammatory responses. Fisetin can also enhance cognitive function and in some cases, modulate proteasome activity. Therefore, it has the potential to act as a multi-factorial therapeutic for reducing the decline in brain function associated with age-associated neurological diseases.
Table 3.
Current Treatments for Neurological Diseases for which Fisetin Might Have Clinical Use
| Disease | Current Treatments |
|---|---|
| Alzheimer’s disease | short term symptomatic treatment only: cholinesterase inhibitors and memantine (NMDA receptor antagonist) |
| Huntington’s disease | short term symptomatic treatment only: anti-psychotics; anti-depressants; mood stabilizers |
| Ischemic stroke | tissue plasminogen activator (only useful up to 3 hr after a stroke) |
| Depression | anti-depressants (selective serotonin uptake inhibitors; atypical anti-depressants; tricyclic anti-depressants; monoamine oxidase inhibitors) |
| Diabetic neuropathy | anti-seizure medications; anti-depressants; opioid analgesics |
Acknowledgments
This review was supported by NIH grant R41AI104034 and generous gift from Paul Slavik. I would like to thank Nigel Calcutt (UCSD) for performing the studies on diabetic neuropathy in the Akita mice.
Abbreviations
- Aβ
amyloid beta
- AD
Alzheimer’s disease
- ALS
amyotrophic lateral sclerosis
- AA
arachidonic acid
- ATF4
activating transcription factor 4
- bw
body weight
- CNS
central nervous system
- COX
cyclooxygenase
- DC
dendritic cells
- eIF2α
eukaryotic initiation factor 2α
- EPM
elevated plus maze
- ERK
extracellular signal-regulated kinase
- FDA
US Food and Drug Administration
- GFAP
glial fibrillary acidic protein
- GSH
glutathione
- GSSG
oxidized glutathione
- HD
Huntington’s disease
- Htt
huntingtin protein
- IAA
iodoacetic acid
- IKK
IκB kinase
- JNK
c-jun-N-terminal kinase
- iNOS
inducible nitric oxide synthase
- LOX
lipoxygenase
- LPS
bacterial lipopolysaccharide
- MAPK
mitogen activated protein kinase
- MKP-1
MAP kinase phosphatase 1
- MRI
magnetic resonance imaging
- MWM
Morris water maze
- NF-κB
nuclear factor kappa light chain enhancer of activated B cells
- Nrf2
NF-E2-related factor 2
- PD
Parkinson’s disease
- ROS
reactive oxygen species
- SCEM
small clot embolism model
- TEAC
Trolox equivalent antioxidant capacity
- tMCAO
transient middle cerebral artery occlusion
- TX
thromboxane
- TNF-α
tumor necrosis factor-α
- WM
water maze
References
- 1.Herrup K. Reimagining Alzheimer’s disease-An age-based hypothesis. J Neurosci. 2010;30:16755–16762. doi: 10.1523/JNEUROSCI.4521-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schubert D, Maher P. An alternative approach to drug discovery for Alzheimer’s disease dementia. Future Med Chem. 2012;4:1681–1688. doi: 10.4155/fmc.12.109. [DOI] [PubMed] [Google Scholar]
- 3.Frautschy SA, Cole GM. Why pleiotropic interventions are needed for Alzheimer’s disease. Mol Neurobiol. 2010;41:392–409. doi: 10.1007/s12035-010-8137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Golde TE, Schneider LS, Koo EH. Anti-Abeta therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;69:203–213. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prior M, Chiruta C, Currais A, Goldberg J, Ramsey J, Dargusch R, Maher PA, Schubert D. Back to the future with phenotypic screening. ACS Chem Neurosci. 2014 doi: 10.1021/cn500051h. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Middleton E, Kandaswami C, Theoharides TC. The effects of flavonoids on mammalian cells: implications for inflammation, heart disease and cancer. Pharmacol Rev. 2000;52:673–751. [PubMed] [Google Scholar]
- 7.Heim KE, Tagliaferro AR, Bobilya DJ. Flavonoid antioxidants: chemistry, metabolism and structure-activity relationships. J Nutr Biochem. 2002;13:572–584. doi: 10.1016/s0955-2863(02)00208-5. [DOI] [PubMed] [Google Scholar]
- 8.Ross JA, Kasum CM. Dietary flavonoids: Bioavailability, metabolic effects, and safety. Annu Rev Nutr. 2002;22:19–34. doi: 10.1146/annurev.nutr.22.111401.144957. [DOI] [PubMed] [Google Scholar]
- 9.Ishige K, Schubert D, Sagara Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic Biol Med. 2001;30:433–446. doi: 10.1016/s0891-5849(00)00498-6. [DOI] [PubMed] [Google Scholar]
- 10.Sagara Y, Vahnnasy J, Maher P. Induction of PC12 cell differentiation by flavonoids is dependent upon extracellular signal-regulated kinase activation. J Neurochem. 2004;90:1144–1155. doi: 10.1111/j.1471-4159.2004.02563.x. [DOI] [PubMed] [Google Scholar]
- 11.Arai Y, Watanabe S, Kimira M, Shimoi K, Mochizuki R, Kinae N. Dietary intakes of flavonols, flavones and isoflavones by Japanese women and the inverse correlation between quercetin intake and plasma LDL and cholesterol concentration. J Nutri. 2000;130:2243–2250. doi: 10.1093/jn/130.9.2243. [DOI] [PubMed] [Google Scholar]
- 12.Hendler SS. PDR for Nutritional Supplements. 2nd. Thomson Reuters; 2008. [Google Scholar]
- 13.Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 14.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Humans Services Task Force on Alzheimer’s disease. Neurol. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 15.McKeith I, Cummings JL. Behavioral changes and psychological symptoms in dementia disorders. Lancet Neurol. 2005;4:735–742. doi: 10.1016/S1474-4422(05)70219-2. [DOI] [PubMed] [Google Scholar]
- 16.Haas C. Strategies, development and pitfalls of therapeutic options for Alzheimer’s disease. J Alzheimer’s Dis. 2012;28:241–281. doi: 10.3233/JAD-2011-110986. [DOI] [PubMed] [Google Scholar]
- 17.Rafii MS, Aisen PS. Recent developments in Alzheimer’s disease therapeutics. BMC Med. 2009;7:7. doi: 10.1186/1741-7015-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Currais A, Prior M, Dargusch R, Armando A, Ehren J, Schubert D, Quehenberger O, Maher P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell. 2014;13:379–390. doi: 10.1111/acel.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borshelt DR. Co-Expression of multiple transgenes in mouse CNS: comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- 20.Jankowsky JL, Melnikova T, Fadale DJ, Xu GM, Slunt HH, Gonzales V, Younkin LH, Younkin SG, Borchelt DR, Savonenko AV. Environmental enrichment mitigates cognitive defects in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25:5217–5224. doi: 10.1523/JNEUROSCI.5080-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maher P, Akaishi T, Abe K. Flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc Natl Acad Sci USA. 2006;103:16568–16573. doi: 10.1073/pnas.0607822103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nature Protocols. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gulinello M, Gertner M, Mendoza G, Schoenfeld BP, Oddo S, LaFerla FM, Choi CH, McBride SMJ, Faber DS. Validation of a 2-day water maze protocol in mice. Behav Brain Res. 2009;196:220–227. doi: 10.1016/j.bbr.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walf AA, Frye CA. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nature Protocols. 2007;2:311–328. doi: 10.1038/nprot.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pugh PL, Richardson JC, Bate ST, Upton N, Sunter D. Non-cognitive behaviours in an APP/PS1 transgenic model of Alzheimer’s disease. Behav Brain Res. 2007;178:18–28. doi: 10.1016/j.bbr.2006.11.044. [DOI] [PubMed] [Google Scholar]
- 26.Geula C, Nagykery N, Nicolas A, Wu CK. Cholinergic neuronal and axonal abnormalities are present early in aging and in Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:309–318. doi: 10.1097/NEN.0b013e31816a1df3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schliebs R, Arendt T. The cholinergic system in aging and neuronal degeneration. Behav Brain Res. 2011;221:555–563. doi: 10.1016/j.bbr.2010.11.058. [DOI] [PubMed] [Google Scholar]
- 28.Tricco AC, Vandervaart S, Soobiah C, Lillie E, Perrier L, Chen MH, Hemmelgarn B, Majumdar SR, Straus SE. Efficacy of cognitive enhancers for Alzheimer’s disease: protocol for a systemic review and network meta-analysis. System Rev. 2012;1:31. doi: 10.1186/2046-4053-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanctot KL, Herrmann N, Yau KK, Khan LR, Liu BA, LouLou MM, Einarson TR. Efficacy and safety of cholinesterase inhibitors in Alzheimer’s disease: a meta-analysis. Can Med Assoc J. 2003;169:557–564. [PMC free article] [PubMed] [Google Scholar]
- 30.Klinkenborg I, Blokland A. The validity of scopolamine as a pharmacological model for cognitive impairment: a review of animal behavioral studies. Neurosci Biobehav Rev. 2010;34:1307–1350. doi: 10.1016/j.neubiorev.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Lenz RA, Baker JD, Locke C, Rueter LE, Mohler EG, Wesnes K, Abi-Saab W, Saltarelli MD. The scopolamine model as a pharmacodynamic marker in early drug development. Psychopharmacol. 2012;220:97–107. doi: 10.1007/s00213-011-2456-4. [DOI] [PubMed] [Google Scholar]
- 32.Buccafusco JJ, Terry AV, Webster SJ, Martin D, Hohnadel EJ, Bouchard KA, Warner SE. The scopolamine-reversal paradigm in rats and monkeys: the importance of computer-assisted operant-conditioning memory tasks for screening drug candidates. Psychopharmacol. 2008;199:481–494. doi: 10.1007/s00213-007-0887-8. [DOI] [PubMed] [Google Scholar]
- 33.Snyder PJ, Bednar MM, Cromer JR, Maruff P. Reversal of scopolamine-induced deficits with a single dose of donepezil, an acetylcholinesterase inhibitor. Alzheimer’s & Dementia. 2005;1:126–135. doi: 10.1016/j.jalz.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 34.Cho N, Lee KY, Huh J, Choi JH, Yang H, Jeong EJ, Kim HP, Sung SH. Cognitive-enhancing effects of Rhus verniciflua bark extract and its active flavonoinds with neuroprotective and anti-inflammatory activities. Food Chem Toxicol. 2013;58:355–361. doi: 10.1016/j.fct.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 35.Borrell-Pages M, Zala D, Humbert S, Saudou F. Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci. 2006;63:2642–2660. doi: 10.1007/s00018-006-6242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramaswamy S, Shannon KM, Kordower JH. Huntington’s disease: Pathological mechanisms and therapeutic strategies. Cel Transplant. 2007;16:301–312. doi: 10.3727/000000007783464687. [DOI] [PubMed] [Google Scholar]
- 37.Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C, Krishna G, Davies JE, Ttofi E, Underwood BR, Rubinsztein DC. Huntington’s disease: from pathology and genetics to potential therapeutics. Biochem J. 2008;412:191–209. doi: 10.1042/BJ20071619. [DOI] [PubMed] [Google Scholar]
- 38.Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington’s disease. Eur J Neurosci. 2008;27:2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- 39.Maher P, Dargusch R, Bodai L, Gerard P, Purcell JM, Marsh JL. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum Mol Gen. 2011;20:261–270. doi: 10.1093/hmg/ddq460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs to treat Huntington’s disease. Neurobiol Dis. 2004;16:546–555. doi: 10.1016/j.nbd.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Saydoff JA, Garcia RAG, Browne SE, Liu L, Sheng J, Brenneman D, Hu Z, Cardin S, Gonzalez A, von Bostel RW, Gregorio J, Burr H, Beal MF. Oral uridine pro-drug PN401 is neuroprotective in the R6/2 and N171-82Q mouse models of Huntongton’s disease. Neurobiol Dis. 2006;24:455–465. doi: 10.1016/j.nbd.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 42.Apostol BL, Simmons DA, Zuccato C, Illes K, Pallos J, Casale M, Conforti P, Ramos C, Roarke M, Kathuria S, Cattaneo E, Marsh JL, Thompson LM. CEP–1347 reduces mutant huntingtin-associated neurotoxicity and restores BDNF levels in R6/2 mice. Mol Cell Neurosci. 2008;39:8–20. doi: 10.1016/j.mcn.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 43.Yang L, Calingasan NY, Wille EJ, Cormier K, Smith K, Ferrante RJ, Beal MF. Combination therapy with Coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J Neurochem. 2009;109:1427–1439. doi: 10.1111/j.1471-4159.2009.06074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ingall T. Stroke-incidence, mortality and risk. J Insur Med. 2004;36:143–152. [PubMed] [Google Scholar]
- 45.Lapchak PA, Araujo DM. Advances in ischemic stroke treatment: neuroprotective and combination therapies. Expert Opin Emerg Drugs. 2007;12:97–112. doi: 10.1517/14728214.12.1.97. [DOI] [PubMed] [Google Scholar]
- 46.Gelderblom M, Leypoldt F, Steinback K, Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- 47.Cumming TB, Marshall RS, Lazar RM. Stroke, cognitive deficits and rehabilitation: still an incomplete picture. Int J Stroke. 2013;8:38–45. doi: 10.1111/j.1747-4949.2012.00972.x. [DOI] [PubMed] [Google Scholar]
- 48.Maher P, Salgado KF, Zivin JA, Lapchak PA. A novel approach to screening for new neuroprotective compounds for the treatment of stroke. Brain Res. 2007;1173:117–125. doi: 10.1016/j.brainres.2007.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winkler BS, Sauer MW, Starnes CA. Modulation of the Pasteur effect in retinal cells: implications for understanding compensatory metabolic mechanisms. Exp Eye Res. 2003;76:715–723. doi: 10.1016/s0014-4835(03)00052-6. [DOI] [PubMed] [Google Scholar]
- 50.Waud DR. On biological assays involving quantal responses. J Pharmacol Exper Theory. 1972;183:577–607. [PubMed] [Google Scholar]
- 51.Zivin JA, Waud DR. Quantal bioassay and stroke. Stroke. 1992;23:767–773. doi: 10.1161/01.str.23.5.767. [DOI] [PubMed] [Google Scholar]
- 52.Lapchak PA, Araujo DM, Song D, Wei J, Zivin J. Neuroprotective effects of the spin trap agent disodium-[tert-butylimino)methyl]benzene-1,3-disulfonate N-oxide (generic NXY-059) in a rabbit small clot embolic stroke model: combination studies with the thrombolytic tissue plasminogen activator. Stroke. 2002;33:1411–1415. doi: 10.1161/01.str.0000015346.00054.8b. [DOI] [PubMed] [Google Scholar]
- 53.Lapchak PA, Araujo DM, Zivin JA. Comparison of tenectaplase with alteplase on clinical rating scores following small clot embolic strokes in rabbits. Exp Neurol. 2004;185:154–159. doi: 10.1016/j.expneurol.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 54.Lapchak PA, Song D, Wei J, Zivin JA. Coadministration of NXY-059 and tenecteplase six hours following embolic strokes in rabbits improves clinical rating scores. Exp Neurol. 2004;188:279–285. doi: 10.1016/j.expneurol.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 55.Lapchak PA, Wei J, Zivin J. Transcranial infrared laser therapy improves clinical rating scores after embolic strokes in rabbits. Stroke. 2004;35:1985–1988. doi: 10.1161/01.STR.0000131808.69640.b7. [DOI] [PubMed] [Google Scholar]
- 56.Gelderblom M, Leypoldt F, Lewerenz J, Birkenmayer G, Orozco D, Ludewig P, Thundyil J, Arumugam TV, Gerloff C, Tolosa E, Maher P, Magnus T. The flavonoid fisetin attenuates postischemic immune cell infiltration, activation and infarct size after transient cerebral middle artery occlusion in mice. J Cereb Blood Flow Metab. 2012;32:835–843. doi: 10.1038/jcbfm.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ownby RL, Crocco E, Acevedo A, John V, Loewenstein D. Depression and risk for Alzheimer disease: systemic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry. 2006;63:530–538. doi: 10.1001/archpsyc.63.5.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anderson RJ, Freedland KE, Clouse RE, Lustman PJ. The prevalence of comorbid depression in adults with diabetes. Diabetes Care. 2001;24:1069–1078. doi: 10.2337/diacare.24.6.1069. [DOI] [PubMed] [Google Scholar]
- 59.Zhen L, Zhu JW, Zhao X, Huang W, An Y, Li S, Du X, Lin M, Wang Q, Xu Y, Pan J. The anti-depressant-like effect of fisetin involves serotonergic and noradrenergic system. Behav Brain Res. 2012;228:359–366. doi: 10.1016/j.bbr.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 60.Huber JD. Diabetes, cognitive function and the blood brain barrier. Curr Pharmaceut Des. 2008;14:1594–1600. doi: 10.2174/138161208784705441. [DOI] [PubMed] [Google Scholar]
- 61.Jacobson AM, Ryan CM, Cleary PA, Waberski BH, Weinger K, Musen G, Dahms W, D.E.R. Group Biomedical risk factors for decreased cognitive functioning in type 1 diabetes: an 18 year follow-up of the Diabetes Control and Complications Trial (DCCT) cohort. Diabetologia. 2011;54:245–255. doi: 10.1007/s00125-010-1883-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 63.Meerwaldt R, Links T, Zeebregts C, Tio R, Hillebrands JL, Smit A. The clinical relevance of assessing advanced glycation endproducts accumulation in diabetes. Cardiovasc Diabetol. 2008;7:29–37. doi: 10.1186/1475-2840-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maher PA, Schubert DR. Metabolic links between diabetes and Alzheimer’s disease. Expert Rev Neurother. 2009;9:617–630. doi: 10.1586/ern.09.18. [DOI] [PubMed] [Google Scholar]
- 65.Air EL, Kissels BM. Diabetes, the metabolic syndrome and ischemic stroke. Diabetes Care. 2007;30:3131–3140. doi: 10.2337/dc06-1537. [DOI] [PubMed] [Google Scholar]
- 66.Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997;46:887–894. doi: 10.2337/diab.46.5.887. [DOI] [PubMed] [Google Scholar]
- 67.Gurley SB, Clare SE, Snow KP, Hu A, Meyer TW, Coffman TM. Impact of genetic background on nephropathy in diabetic mice. Am J Physiol Renal Physiol. 2006;290:F214–F222. doi: 10.1152/ajprenal.00204.2005. [DOI] [PubMed] [Google Scholar]
- 68.Choeiri C, Hewitt K, Durkin J, Simard CJ, Renaud JM, Messier C. Longitudinal evaluation of memory performance and peripheral neuropathy in the Ins2C96Y Akita mice. Behav Brain Res. 2005;157:31–38. doi: 10.1016/j.bbr.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 69.Asakawa A, Toyoshima M, Inoue K, Koizumi A. Ins2Akita mice exhibit hyperphagia and anxiety behavior via the melanocortin system. Int J Mol Med. 2007;19:649–652. [PubMed] [Google Scholar]
- 70.Reagan LP, Grillo CA, Piroli GG. The As and Ds of stress: Metabolic, morphological and behavioral consequences. Eur J Pharmacol. 2008;585:64–75. doi: 10.1016/j.ejphar.2008.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maher P, Dargusch R, Ehren JL, Okada S, Sharma K, Schubert D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS ONE. 2011;6:e21226. doi: 10.1371/journal.pone.0021226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barber AJ, Antonetti DA, Kern TS, Reiter CEN, Soans RS, Krady JK, Levison SW, Gardner TW, Bronson SK. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci. 2005;46:2210–2218. doi: 10.1167/iovs.04-1340. [DOI] [PubMed] [Google Scholar]
- 73.Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: a lifespan perspective. Lancet Neurol. 2008;7:184–190. doi: 10.1016/S1474-4422(08)70021-8. [DOI] [PubMed] [Google Scholar]
- 74.Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, Masliah E. Defective insulin signaling pathway and icnreased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. J Neurosci Res. 2008;86:3265–3274. doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burdo JR, Chen Q, Calcutt NA, Schubert D. The pathological interaction between diabetes and presymptomatic Alzheimer’s disease. Neurobiol Aging. 2009;30:1910–1917. doi: 10.1016/j.neurobiolaging.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 76.Schewe T, Steffen Y, Sies H. How do dietary flavanols improve vascular function? A position paper. Arch Biochem Biophys. 2008;476:102–106. doi: 10.1016/j.abb.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 77.Halliwell B. Free radicals and antioxidants-quo vadis? Trends Pharmacol Sci. 2011;32:125–130. doi: 10.1016/j.tips.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 78.Maher P. A comparison of the neurotrophic activities of the flavonoid fisetin and some of its derivatives. Free Rad Res. 2006;40:1105–1111. doi: 10.1080/10715760600672509. [DOI] [PubMed] [Google Scholar]
- 79.Miura YH, Tomita I, Watanabe T, Hirayama T, Fukui S. Active oxygens generation by flavonoids. Biol Pharm Bull. 1998;21:93–96. doi: 10.1248/bpb.21.93. [DOI] [PubMed] [Google Scholar]
- 80.Galati G, Sabzevari O, Wilson JX, O’Brien PJ. Prooxidnat activity and cellular effects of the phenoxyl radicals of dietary flavonoids and other polyphenolics. Toxicol. 2002;177:91–104. doi: 10.1016/s0300-483x(02)00198-1. [DOI] [PubMed] [Google Scholar]
- 81.Wang L, Tu YC, Lian TW, Hung JT, Yen JH, Wu MJ. Distinctive antioxidant and antiinflammatory effects of flavonols. J Agric Food Chem. 2006;54:9798–9804. doi: 10.1021/jf0620719. [DOI] [PubMed] [Google Scholar]
- 82.van Acker FA, Schouten O, Haenen GR, van der Vijgh WJ, Bast A. Flavonoids can replace alpha-tocopherol as an antioxidant. FEBS Lett. 2000;473:145–148. doi: 10.1016/s0014-5793(00)01517-9. [DOI] [PubMed] [Google Scholar]
- 83.van Acker SABE, van den Berg DJ, Tromp MNJL, Griffioen DH, van Bennekom WP, van der Vijgh WJF, Bast A. Structural aspects of antioxidant activity of flavonoids. Free Rad Biol Med. 1996;20:331–342. doi: 10.1016/0891-5849(95)02047-0. [DOI] [PubMed] [Google Scholar]
- 84.Moridani MY, Pourahmad J, Bui H, Siraki A, O’Brien PJ. Dietary flavonoid iron complexes as cytoprotective superoxide radical scavengers. Free Rad Biol Med. 2003;34:243–253. doi: 10.1016/s0891-5849(02)01241-8. [DOI] [PubMed] [Google Scholar]
- 85.Melidou M, Riganakos K, Galaris D. Protection against nuclear DNA damage offered by flavonoids in cells exposed to hydrogen peroxide: The role of iron chelation. Free Rad Biol Med. 2005;39:1591–1600. doi: 10.1016/j.freeradbiomed.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 86.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford, New York: 1993. [Google Scholar]
- 87.Ayton S, Lei P, Bush AI. Metallostasis in Alzheimer’s disease. Free Rad Biol Med. 2013;62:76–89. doi: 10.1016/j.freeradbiomed.2012.10.558. [DOI] [PubMed] [Google Scholar]
- 88.Hare D, Ayton S, Bush AI, Lei P. A delicate balance: iron metabolism and diseases of the brain. Front Aging Neurosci. 2013;5:34. doi: 10.3389/fnagi.2013.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hagemeier J, Geurts JJG, Zivadinov R. Brain iron accumulation in aging and neurodegenerative disorders. Expert Rev Neurother. 2012;12:1467–1480. doi: 10.1586/ern.12.128. [DOI] [PubMed] [Google Scholar]
- 90.Raven EP, Lu PH, Tishler TA, Heydari P, Bartzokis G. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J Alzheimer’s Dis. 2013;37:127–136. doi: 10.3233/JAD-130209. [DOI] [PubMed] [Google Scholar]
- 91.Penke L, Valdes Hernandez MC, Maniega SM, Gow AJ, Murray C, Starr JM, Bastin ME, Deary IJ, Wardlaw JM. Brain iron deposits are associated with general cognitive ability and cognitive aging. Neurobiol Aging. 2012;33:510–517. doi: 10.1016/j.neurobiolaging.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 92.Eskici G, Axelsen PH. Copper and oxidative stress in the pathogenesis of Alzheimer’s disease. Biochemistry. 2012;51:6289–6311. doi: 10.1021/bi3006169. [DOI] [PubMed] [Google Scholar]
- 93.Droge W, Schipper HM. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell. 2007;6:361–370. doi: 10.1111/j.1474-9726.2007.00294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Singh I, Sagare AP, Coma M, Perlmutter D, Gelein R, Bell RD, Deane RJ, Zhong E, Parisi M, Ciszewski J, Kasper RT, Deane R. Low levels of copper disrupt brain amyloid-b homeostasis by altering its production and clearance. Proc Natl Acad Sci U S A. 2013;110:14771–14776. doi: 10.1073/pnas.1302212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nagai R, Murray DB, Metz TO, Baynes JW. Chelation: A fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes. 2012;61:549–559. doi: 10.2337/db11-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meister A, Anderson ME. Glutathione. Ann Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 97.Lewerenz J, Maher P. Control of redox state and redox signaling by neural antioxidant systems. Antioxid Redox Signal. 2011;14:1449–1465. doi: 10.1089/ars.2010.3600. [DOI] [PubMed] [Google Scholar]
- 98.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Rad Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 99.Arthur JR. The glutathione peroxidases. Cell Mol Life Sci. 2000;57:1825–1835. doi: 10.1007/PL00000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Thornalley PJ. Glyoxalase I-structure, function and a critical role in the enzymatic defense against glycation. Biochem Soc Trans. 2003;31:1343–1348. doi: 10.1042/bst0311343. [DOI] [PubMed] [Google Scholar]
- 101.Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003;22:7369–7375. doi: 10.1038/sj.onc.1206940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zimmerman AK, Loucks FA, Schroeder EK, Bouchard RJ, Tyler KL, Linseman DA. Glutathione binding to the Bcl-2 homology-3 domain groove. J Biol Chem. 2007;282:29296–29304. doi: 10.1074/jbc.M702853200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li Y, Maher P, Schubert D. A role of 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- 104.Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metabolism. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 105.Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome C. Nature Cell Biol. 2008;10:1477–1483. doi: 10.1038/ncb1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sohal RS, Orr WC. The redox stress hypothesis of aging. Free Rad Biol Med. 2012;52:539–555. doi: 10.1016/j.freeradbiomed.2011.10.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zeevalk GD, Razmpour R, Bernard LP. Glutathione and Parkinson’s Disease: Is this the elephant in the room? Biomed Pharmacother. 2008;62:236–249. doi: 10.1016/j.biopha.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 108.Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, Hammond CL. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. 2009;390:191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Maher P. The effects of stress and aging on glutathione metabolism. Ageing Res Rev. 2005;4:288–314. doi: 10.1016/j.arr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 110.Currais A, Maher P. Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid Redox Signal. 2013;19:813–822. doi: 10.1089/ars.2012.4996. [DOI] [PubMed] [Google Scholar]
- 111.Emir UE, Raatz S, McPherson S, Hodges JS, Torkelson C, Tawfik P, White T, Terpstra M. Noninvasive quantification of ascorbate and glutathione concentration in the elderly human brain. NMR Biomed. 2011;24:888–894. doi: 10.1002/nbm.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cruz-Aguado R, Almaguer-Melian W, Diaz CM, Lorigados L, Bergado J. Behavioral and biochemical effects of glutathione depletion in the rat brain. Brain Res Bull. 2001;55:327–333. doi: 10.1016/s0361-9230(01)00484-1. [DOI] [PubMed] [Google Scholar]
- 113.Dean O, Bush AI, Berk M, Copolov DL, van den Buuse M. Glutathione depletion in the brain disrupts short-term spatial memory in the Y-maze in rats and mice. Behav Brain Res. 2009;198:258–262. doi: 10.1016/j.bbr.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 114.Kamboj A, Kiran R, Sandhir R. Carbofuran-induced neurochemical and neurobehavioral alterations in rats: attentuation by N-acetylcysteine. Exp Brain Res. 2006;170:567–575. doi: 10.1007/s00221-005-0241-5. [DOI] [PubMed] [Google Scholar]
- 115.Kensler TW, Wakabayashi N, Biswal S. Cell Survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 116.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metabol Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 117.Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas P, Massie A, Smolders I, Methner A, Pergande M, Smith SB, Ganapathy V, Maher P. The cystine/glutamate antiporter system x(c)- in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2012;18:522–555. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 119.Lewerenz J, Albrecht P, Tien ML, Henke N, Karumbayaram S, Kornblum HI, Wiedua-Pazos M, Schubert D, Maher P, Methner A. Induction of Nrf2 and xCT and involved in the action of the neuroprotective antibiotic ceftriazone in vitro. J Neurochem. 2009;111:332–343. doi: 10.1111/j.1471-4159.2009.06347.x. [DOI] [PubMed] [Google Scholar]
- 120.Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lewerenz J, Maher P. Basal levels of eIF2α phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J Biol Chem. 2009;284:1106–1115. doi: 10.1074/jbc.M807325200. doi:PMC Journal-In Process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci USA. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shenvi SV, Smith E, Hagen TM. Identification of age-specific Nrf2 binding to a novel antioxidant response element locus in the Gclc promoter: a compensatory means for the loss of glutathione synthetic capacity in the aging rat liver? Aging Cell. 2012;11:297–304. doi: 10.1111/j.1474-9726.2011.00788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ungvari Z, Bailey DL, Gautam T, Sosnowska D, Wang M, Monticone RE, Telljohann R, Pinto JT, de Cabo R, Sonntag WE, Lakatta EG, Csiszar A. Age-associated vascular oxidative stress, Nrf2 dysfunction and NF-kB activation in the nonhuman primate Macaca mulatta. J Gerentol A Biol Sci Med Sci. 2011;66A:866–875. doi: 10.1093/gerona/glr092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Miller CJ, Gounder SS, Kannan S, Goutam K, Muthusamy VR, Firpo MA, Symons JD, Paine R, Hoidal JR, Rajasekaran NS. Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation pathways in aged skeletal muscle. Biochim Biophys Acta. 2012;1822:1038–1050. doi: 10.1016/j.bbadis.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 126.Leiser SF, Miller RA. Nrf2 signaling, a mechanism for cellular stress resistance in long-lived mice. Mol Cell Biol. 2010;30:871–884. doi: 10.1128/MCB.01145-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hussain SG, Ramaiah KVA. Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem Biophys Res Commun. 2007;355:365–370. doi: 10.1016/j.bbrc.2007.01.156. [DOI] [PubMed] [Google Scholar]
- 128.Tan S, Schubert D, Maher P. Oxytosis: a novel form of programmed cell death. Curr Top Med Chem. 2001;1:497–506. doi: 10.2174/1568026013394741. [DOI] [PubMed] [Google Scholar]
- 129.Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ehren JL, Maher P. Concurrent regulation of the transcription factors Nrf2 and ATF4 mediates the enhancement of glutathione levels by the flavonoid fisetin. Biochem Pharmacol. 2013;85:1816–1826. doi: 10.1016/j.bcp.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 131.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Prakash D, Gopinath K, Sudhandiran G. Fisetin enhances behavioral performances and attenuates reactive gliosis and inflammation during aluminum chloride-induced neurotoxicity. Neuromol Med. 2013;15:192–208. doi: 10.1007/s12017-012-8210-1. [DOI] [PubMed] [Google Scholar]
- 133.Chao MV, Rajagopal R, Lee FS. Neurotrophins signalling in health and disease. Clinical Science. 2006;110:167–173. doi: 10.1042/CS20050163. [DOI] [PubMed] [Google Scholar]
- 134.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 136.Kelly A, Conroy S, Lynch MA. Evidence that nerve growth factor plays a role in long-term potentiation in rat dentate gyrus. Neuropharmacol. 1998;37:561–570. doi: 10.1016/s0028-3908(98)00048-3. [DOI] [PubMed] [Google Scholar]
- 137.Price RD, Milne SA, Sharkey J, Matsuoka N. Advances in small molecules promoting neurotrophic function. Pharmacol Thera. 2007;115:292–306. doi: 10.1016/j.pharmthera.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 138.Chen S, Le W. Neuroprotective therapy in Parkinson disease. Amer J Ther. 2006;13:445–457. doi: 10.1097/01.mjt.0000174353.28012.a7. [DOI] [PubMed] [Google Scholar]
- 139.Levy YS, Gilgun-Sherki Y, Melamed E, Offen D. Therapeutic potential of neurotrophic factors in neurodegenerative diseases. BioDrugs. 2005;19:97–127. doi: 10.2165/00063030-200519020-00003. [DOI] [PubMed] [Google Scholar]
- 140.Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog Neurobiol. 2007;81:294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 141.Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans Soc London B Biol Sci. 2006;36:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.McGeer E, Patrick P. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: A field in its infancy. J Alzheimer’s Dis. 2010;19:355–361. doi: 10.3233/JAD-2010-1219. [DOI] [PubMed] [Google Scholar]
- 143.Lee YJ, Han SB, Nam SY, Oh KW, Hong JT. Inflammation and Alzheimer’s disease. Arch Pharm Res. 2010;33:1539–1556. doi: 10.1007/s12272-010-1006-7. [DOI] [PubMed] [Google Scholar]
- 144.Butchart J, Holmes C. Systemic and central immunity in Alzheimer’s disease: Therapeutic implications. CNS Neurosci Ther. 2012;18:64–76. doi: 10.1111/j.1755-5949.2011.00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wyss-Coray T, Rogers J. Inflammation in Alzheimer’s disease-A brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2:a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rock RB, Peterson PK. Microglia as a pharmacological target in infectious and inflammatory diseases on the brain. J Neuroimmune Pharmacol. 2006;1:117–126. doi: 10.1007/s11481-006-9012-8. [DOI] [PubMed] [Google Scholar]
- 147.Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1:127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- 148.Dringen R. Oxidative and antioxidative potential of brain microglial cells. Antioxid Redox Signal. 2005;7:1223–1233. doi: 10.1089/ars.2005.7.1223. [DOI] [PubMed] [Google Scholar]
- 149.Zheng LT, Ock J, Kwon BM, Suk K. Suppressive effects of flavonoid fisetin on lipopolysaccharide-induced microglial activation and neurotoxicity. Int Immunopharmacol. 2008;8:484–494. doi: 10.1016/j.intimp.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 150.Sung B, Pandey MK, Aggarwal BB. Fisetin, an inhibitor of cyclin-dependent kinase 6, down-regulates nuclear factor-kB-regulated cell proliferation, antiapoptotic and metastatic gene products through the suppression of TAK-1 and receptor-interacting protein-regulated IkBa kinase activation. Mol Pharmacol. 2007;71:1703–1714. doi: 10.1124/mol.107.034512. [DOI] [PubMed] [Google Scholar]
- 151.Gupta SC, Tyagi AK, Deshmukh-Taskar P, Hinojosa M, Prasad S, Aggarwal BB. Downregulation of tumor necrosis factor and other proinflammatory biomarkers by polyphenols. Arch Biochem Biophys. 2014 doi: 10.1016/j.abb.2014.06.006. in press. [DOI] [PubMed] [Google Scholar]
- 152.Kim HJ, Kim SH, Yun J-M. Fisetin inhibits hyperglycemia-induced proinflammatory cytokine production by epigenetic mechanisms. Evidence-based Complemen Alternat Med. 2012:639469. doi: 10.1155/2012/639469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dinarello CA. Anti-inflammatory agents: present and future. Cell. 2010;140:935–950. doi: 10.1016/j.cell.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bachstetter AD, Xing B, de Almeida L, Dimayuga ER, Watterson DM, Van Eldik LJ. Microglial p38alpha MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta amyloid (Abeta) J Neuroinflamm. 2011;8:79. doi: 10.1186/1742-2094-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Leotoing L, Wauquier F, Guicheux J, Miot-Noirault E, Wittrant W, Coxam V. The polyphenol fisetin protects bone by repressing NF-kB and MKP-1-dependent signaling pathways in osteoclasts. PLoS ONE. 2013;8:e68388. doi: 10.1371/journal.pone.0068388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rao JS, Rapoport SI, Kim HW. Altered neuroinflammatory, arachidonic acid cascade and synaptic markers in postmortem Alzheimer’s disease brain. Transl Psychiatry. 2011;1:e31. doi: 10.1038/tp.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 157.Steele ML, Robinson SR. Reactive astrocytes give neurons less support: implications for Alzheimer’s disease. Neurobiol Aging. 2012;33:423.e1–13. doi: 10.1016/j.neurobiolaging.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 158.Buczynski MW, Dumlao DS, Dennis EA. An integrated omics analysis of eicosanoid biology. J Lipid Res. 2009;50:1015–1038. doi: 10.1194/jlr.R900004-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hardwick JP, Eckman K, Lee YK, Abdelmegeed MA, Esterle A, Chilian WM, Chiang JY, Song BJ. Eicosanoids in metabolic syndrome. Adv Pharmacol. 2013;66:157–266. doi: 10.1016/B978-0-12-404717-4.00005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cortes-Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D, Bhuvanendran S, Fenz KM, Strickland S. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer’s disease. Neuron. 2010;66:695–709. doi: 10.1016/j.neuron.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yoon HJ, Jeon SB, Kim IH, Park EJ. Regulation of TLR2 expression by prostaglandins in brain glia. J Immunol. 2008;180:8400–8409. doi: 10.4049/jimmunol.180.12.8400. [DOI] [PubMed] [Google Scholar]
- 162.Scher JU, Pillinger MH. The anti-inflammatory effects of prostaglandins. J Investig Med. 2009;57:703–708. doi: 10.2310/JIM.0b013e31819aaa76. [DOI] [PubMed] [Google Scholar]
- 163.Phillis JW, Horrocks LA, Farooqui AA. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: Their role and involvement in neurological disorders. Brain Res Rev. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 164.Canals S, Casarejos MJ, de Bernardo S, Rodriguez-Martin E, Mena MA. Nitric oxide triggers the toxicity due to glutathione depletion in midbrain cultures through 12-lipoxygenase. J Biol Chem. 2003;278:21542–21549. doi: 10.1074/jbc.M213174200. [DOI] [PubMed] [Google Scholar]
- 165.Kramer BC, Yabut JA, Cheong J, Jnobaptiste R, Robakis T, Olanow CW, Mytilineou C. Toxicity of glutathione depletion in mesencephalic cultures: a role for arachidonic acid and its lipoxygenase metabolites. Eur J Neurosci. 2004;19:280–286. doi: 10.1111/j.1460-9568.2004.03111.x. [DOI] [PubMed] [Google Scholar]
- 166.Wang H, Li J, Follett PL, Zhang Y, Cotanche DA, Jensen FE, Volpe JJ, Rosenberg PA. 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes. Eur J Neurosci. 2004;20:2049–2058. doi: 10.1111/j.1460-9568.2004.03650.x. [DOI] [PubMed] [Google Scholar]
- 167.Pratico D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ, Lee VMY. 12/15-Lipoxygenase is increased in Alzheimer’s disease. Amer J Pathol. 2004;164:1655–1662. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lebeau A, Terro F, Rostene W, Pelaprat D. Blockade of 12-lipoxygenase expression protects cortical neurons from apoptosis induced by beta-amyloid peptide. Cell Death Differ. 2004;11:875–884. doi: 10.1038/sj.cdd.4401395. [DOI] [PubMed] [Google Scholar]
- 169.Chinnici CM, Yao Y, Practico D. The 5-lipoxygenase enzymatic pathway in the mouse brain: Young versus old. Neurobiol Aging. 2007;28:1457–1462. doi: 10.1016/j.neurobiolaging.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 170.Firuzi O, Zhuo J, Chinnici CM, Wisniewski T, Pratico D. 5-Lipoxygenase gene disruption reduces amyloid-beta pathology in a mouse model of Alzheimer’s disease. FASEB J. 2008;22:1169–1178. doi: 10.1096/fj.07-9131.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hsieh RJ, German JB, Kinsella JE. Relative inhibitory potencies of flavonoids on 12-lipoxygenase of fish gill. Lipids. 1988;23:322–326. doi: 10.1007/BF02537342. [DOI] [PubMed] [Google Scholar]
- 172.Laughton MJ, Evans PJ, Moroney MA, Hoult JRS, Halliwell B. Inhibition of mammalian 5-lipoxygenase and cyclo-oxygenase by flavonoids and phenolic dietary additives. Biochem Pharmacol. 1991;42:1673–1681. doi: 10.1016/0006-2952(91)90501-u. [DOI] [PubMed] [Google Scholar]
- 173.Sadik CD, Sies H, Schewe T. Inhibition of 15-lipoxygenases by flavonoids: structure-activity relations and mode of action. Biochem Pharmacol. 2003;65:773–781. doi: 10.1016/s0006-2952(02)01621-0. [DOI] [PubMed] [Google Scholar]
- 174.Schneider C, Pratt DA, Porter NA, Brash AR. Control of oxygenation in lipoxygenase and cyclooxygenase catalysis. Chem Biol. 2007;14:473–488. doi: 10.1016/j.chembiol.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Dailey LA, Imming P. 12-Lipoxygenase: Classification, possible therapeutics benefits from inhibitors and inhibitors. Curr Med Chem. 1999;6:389–399. [PubMed] [Google Scholar]
- 176.Kim H, Park BS, Lee KG, Choi CY, Jang SS, Kim YH, Lee SE. Effects of naturally occurring compounds on fibril formation and oxidative stress of b-amyloid. J Agric Food Chem. 2005;53:8537–8541. doi: 10.1021/jf051985c. [DOI] [PubMed] [Google Scholar]
- 177.Akaishi T, Morimoto T, Shibao M, Watanabe S, Sakai-Kato K, Utsunomiya-Tate N, Abe K. Structural requirement for the flavonoid fisetin in inhibiting fibril formation of amyloid β protein. Neurosci Lett. 2008;444:280–285. doi: 10.1016/j.neulet.2008.08.052. [DOI] [PubMed] [Google Scholar]
- 178.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nature Rev Mol Cell Biol. 2005;6:79–86. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- 179.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 180.Keller JN, Hanni KB, Markesbery WR. Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev. 2000;113:61–70. doi: 10.1016/s0047-6374(99)00101-3. [DOI] [PubMed] [Google Scholar]
- 181.Zeng BY, Medhurst AD, Jackson M, Rose S, Jenner P. Proteasomal activity in brain differs between species and brain regions and changes with age. Mech Ageing Dev. 2005;126:760–766. doi: 10.1016/j.mad.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 182.Chung KKK, Dawson VL, Dawson TM. The role of the ubiquitin-proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders. Trends Neurosci. 2001;24:S7–S14. doi: 10.1016/s0166-2236(00)01998-6. [DOI] [PubMed] [Google Scholar]
- 183.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 184.Betarbet R, Sherer TB, Greenamyre JT. Ubiquitin-proteasome system and Parkinson’s disease. Exper Neurol. 2005;191:S17–S27. doi: 10.1016/j.expneurol.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 185.Maher P. The flavonoid fisetin promotes nerve cell survival from trophic factor withdrawal by enhancement of proteasome activity. Arch Biochem Biophys. 2008;476:139–144. doi: 10.1016/j.abb.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 186.Manach C, Williamson G, Morand C, Scalbert A, Remesy C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Amer J Clin Nutr. 2005;81:230S–242S. doi: 10.1093/ajcn/81.1.230S. [DOI] [PubMed] [Google Scholar]
- 187.Prasain JK, Barnes S. Metabolism and bioavailability of flavonoids in chemoprevention: Current analytical strategies and future prospects. Mol Pharmaceut. 2007;4:846–864. doi: 10.1021/mp700116u. [DOI] [PubMed] [Google Scholar]
- 188.Shia CS, Tsai SY, Kuo SC, Hou YC, Chao PDL. Metabolism and pharmacokinetics of 3,3′,4′,7-tetrahydroxyflavone (fisetin), 5-hydroxyflavone and 7-hydroxyflavone and antihemolysis effects of fisetin and its serum metabolites. J Agric Food Chem. 2009;57:83–89. doi: 10.1021/jf802378q. [DOI] [PubMed] [Google Scholar]
- 189.Lai MY, Hsiu SL, Tsai SY, Hou YC, Chao PDL. Comparison of metabolic pharmacokinetics of baicalin and baicalein in rats. J Pharm Pharmacol. 2003;55:205–209. doi: 10.1211/002235702522. [DOI] [PubMed] [Google Scholar]
- 190.Stevenson DE, Cooney JM, Jensen DJ, Wibisono R, Adaim A, Skinner MA, Zhang J. Comparison of enzymically glucuronidated flavonoids with flavonoid aglycones in an in vitro cellular model of oxidative stress protection. In Vitro Cell Dev Biol Anim. 2008 doi: 10.1007/s11626-007-9072-y. Epub. [DOI] [PubMed] [Google Scholar]
- 191.Correia-da-Silva M, Sousa E, Pinto MMM. Emerging sulfated flavonoids and other polyphenols as drugs: Nature as an inspiration. Medicinal Res Rev. 2014;34:223–279. doi: 10.1002/med.21282. [DOI] [PubMed] [Google Scholar]
- 192.Touil YS, Auzeil N, Boulinguez F, Saighi H, Regazzetti A, Scherman D, Chabot GG. Fisetin deposition and metabolism in mice: Identification of geraldol as an active metabolite. Biochem Pharmacol. 2011;82:1731–1739. doi: 10.1016/j.bcp.2011.07.097. [DOI] [PubMed] [Google Scholar]