

Abstract
Emerging proteomic tools and mass spectrometry play pivotal roles in protein identification, quantification and characterization, even in complex biological samples. The cancer secretome, namely the whole collection of proteins secreted by cancer cells through various secretory pathways, has only recently been shown to have significant potential for diverse applications in oncoproteomics. For example, secreted proteins might represent putative tumor biomarkers or therapeutic targets for various types of cancer. Consequently, many proteomic strategies for secretome analysis have been extensively deployed over the last few years. These efforts generated a large amount of information awaiting deeper mining, better understanding and careful interpretation. Distinct sub‐fields, such as degradomics, exosome proteomics and tumor‐host cell interactions have been developed, in an attempt to provide certain answers to partially elucidated mechanisms of cancer pathobiology. In this review, advances, concerns and challenges in the field of secretome analysis as well as possible clinical applications are discussed.
Keywords: Cancer, Mass spectrometry, Proteomics, Secretome
Non‐standard abbreviations
- ER
Endoplasmic Reticulum
- IL
Interleukin
- FGF
Fibroblast Growth Factor
- MVB
Multivesicular Body
- ILV
Intralumenal Vesicle
- CM
Conditioned Media
- FBS
Fetal Bovine Serum
- GO
Gene Ontology
- HPRD
Human Protein Reference Database
- SIG-Pred
Signal Peptide Prediction
- TMHMM
TransMembrane Hidden Markov Models
- TMpred
Transmembrane Regions and Orientation
- EMT
Epithelial-to-Mesenchymal Transition
- DIGE
Differential Gel Electrophoresis
- MMP
Matrix Metalloproteinase
- KLK
Kallikrein
- TIMP
Tissue Inhibitor of Metalloproteinase
- LC
Liquid Chromatography
- MS
Mass Spectrometry
- MS/MS
Tandem Mass spectrometry
- MALDI
Matrix Assisted Laser Desorption/Ionization
- TOF
Time-of-Flight
- 2DE
2-Dimensional Electrophoresis
- OSCC
Oral Squamous Cell Carcinoma
- OSF2
Osteoblast-Specific-Factor-2
- AXL/UFO
Receptor Tyrosine Kinase
- NGAL
Neutrophil Gelatinase-Associated Lipocalin
- PSA
Prostate Specific Antigen
- uPA
urokinase type Plasminogen Activator
- uPAR
urokinase type Plasminogen Activator Receptor
- MT-MPP
Membrane Type Matrix Metalloproteinase
- ICAT
Isotope-Coded Affinity Tags
- iTRAQ
Isobaric Tag for Relative and Absolute Quantification
- HSP
Heat Shock Protein
- SILAC
Stable Isotope Labeling with Aminoacids in Cell Culture
- ABP
Activity Based Probe
- PI3K
Phosphoinositide-3-Kinase
- Akt
Protein Kinase B
- MAPK
Mitogen Activated Protein Kinase
- EGF
Epidermal Growth Factor
- DEL-1
Developmental Endothelial Locus-1
- ESCRT
Endosomal Sorting Complex Required for Trasport
- CEA
CarcinoEmbryonic Antigen
- HER-2
Human Epidermal Growth Factor Receptor-2
- CAF
Cancer Associated Fibroblast
- TGF-β
Transforming Growth Factor-β
- PDGF
Platelet Derived Growth Factor
- HGF
Hepatocyte Growth Factor
- IGF
Insulin Growth Factor
- VEGF
Vascular Endothelial Growth Factor
- TNF-α
Tumor Necrosis Factor-α
- CAV-1
Caveolin-1
- SPARC
Scalable Processor Architecture
- GPX5
Glutathione Peroxidase-5
- TAM
Tumor Associated Macrophages
- HIF
Hypoxia-Inducible Factor
- LCM
Laser Capture Microdissection
1. Introduction
Novel proteomic tools and mass spectrometry are recently playing a central role in protein research, especially in the simultaneous identification, quantification and characterization of thousands of proteins, even in complex biological samples. The emergence of mass spectrometry‐based proteomics enabled the field of cancer research with a myriad of new opportunities. The new field of “oncoproteomics” deals with the applications of proteomics in clinical and molecular oncology. In the near future, oncoproteomics is expected to play a crucial role in the diagnosis and management of cancer, as well as in the field of personalized medicine for cancer (Jain, 2008). Among the plethora of available tissues/fluids for proteomic analysis, here, we will focus on the cancer secretome. Secretome analysis has only recently been established as a sub‐field of oncoproteomics and the indications thus far point to the fact that this source of proteins is a promising pool of biomarkers and therapeutic targets for various types of cancer. Therefore, there is a reasonable consensus that efforts should continue to comprehensively analyze the cancer secretome. Secreted proteins account for approximately 10–15% of the proteins encoded by the human genome and participate in various physiological processes such as immune defence, blood coagulation, matrix remodelling and cell signalling, but also in pathological conditions including cancer angiogenesis, differentiation, invasion and metastasis. In this review, we intend to discuss some basic biological concepts related to the cancer secretome and secondly, to delineate its applications in oncoproteomics. In brief, we describe how the cancer secretome may serve as a valuable pool of proteins, from which crucial players of cancer development and progression can be identified and serve either as biomarkers or as therapeutic targets.
2. Biology and analysis of the cancer secretome
2.1. Protein secretion pathways
To better understand the nature of secretome analysis, we first provide a brief overview of protein secretion pathways that are responsible for the presence of a large number of extracellular proteins in the microenvironment of normal and/or cancer cells. In eukaryotic cells, soluble proteins are secreted in the extracellular space either by exocytosis of secretory vesicles or by release of secretory/storage granules upon stimulation and activation of intracellular signalling pathways. The secreted proteins are mostly synthesized as protein precursors, which contain N‐terminal signal peptides that direct them to the translocation apparatus of the endoplasmic reticulum (ER). These proteins are transported to the Golgi apparatus and subsequently to the cell surface, where they are liberated into the microenvironment by fusion of the Golgi‐derived vesicles with the plasma membrane. This well‐characterized protein secretion pathway has been termed as the classical secretory pathway (Walter et al., 1984; Mellman and Warren, 2000). Other lines of evidence point out that in addition to this mechanism, proteins can be exported by ER/Golgi‐independent pathways, the so‐called non‐classical secretory pathway. At least four distinct types of non‐classical exports have been distinguished over the years, all of which lack the presence of the classical signal peptide for ER/Golgi‐dependent protein secretion (Nickel, 2003). Certain proteins, such as Interleukin‐1β (IL‐1β), are imported into intracellular vesicles, which are endosomal compartments and through a process called endosomal recycling, they are released in the extracellular space upon fusion of the endosomal vesicle with the plasma membrane (Rubartelli et al., 1990). Other proteins, such as fibroblast growth factor‐1 and ‐2 (FGF‐1 and ‐2), reach the extracellular space by direct translocations across the plasma membrane using distinct transport systems (Mignatti et al., 1992; Trudel et al., 2000). Another proposed mechanism of non‐classical protein secretion involves the direct translocation of the protein to the extracellular space, but it requires that the protein is membrane‐anchored through dual acylation in the N‐terminus, and a flip‐flop mechanism mediates the secretion (Denny et al., 2000). Finally, proteins can also be secreted through exosomes; these vesicles originate from the internalization of activated receptors along with all the scaffolding proteins present therein, followed by traffic through early endosomes. These receptors are further internalized within the endosome, forming the late endosome, which is also referred to as multivesicular body (MVB). These internalized receptors within the late endosomes are referred to as intralumenal vesicles (ILVs), when they are present within the MVBs, but are referred to as exosomes upon fusion of the MVB with the plasma membrane and subsequent secretion (Simpson et al., 2008).
2.2. The cancer secretome
The term “secretome” was introduced by Tjalsma et al. to describe proteins released by a cell, a tissue or organism through the different secretion mechanisms (Tjalsma et al., 2000). Inherent to the description of the various secretory pathways, the cancer secretome has been described as including the extracellular matrix components and all the proteins that are released from a given type of cancer cells, such as growth factors, cytokines, adhesion molecules, shed receptors and proteases, and reflects the functionality of this cell type at a given time point (Kulasingam and Diamandis, 2008). Therefore, the cancer secretome includes proteins released from cancer cells, either with classical or non‐classical secretory pathways, and corresponds to an important class of proteins that can act both locally and systemically (Kulasingam and Diamandis, 2008). Theoretically, the cancer secretome includes all the proteins that can be identified in the interstitial fluid of the tumor mass in vivo (Celis et al., 2005), however it is better conceptualized as the group of proteins identified with mass spectrometry in cancer cell line conditioned media (CM) in in vitro studies (Kulasingam and Diamandis, 2008). Primary tumors are composed of not only cancerous cells but also of a wide diversity of stromal cells, which are recruited as active collaborators, facilitating the development and progression of malignancy. Out of these heterotypic interactions, a great variety of proteins, including growth factors, enzymes such as proteases, smaller protein molecules like chemokines and cytokines, as well as many other proteins are constantly released from all participating cells and act upon others in an autocrine or paracrine fashion, resulting in the acquisition of a favourable milieu for the progression of the malignancy (Mueller and Fusenig, 2004). Therefore, cancer cell‐secreted proteins alone comprise only a subset of the overall microenvironmental proteins. Thus, the investigation of stromal secretomes constitutes a critical strategy for the identification of novel biomarkers or key regulators of carcinogenesis that could be assigned a therapeutic potential. In this context, we propose a wider definition for the term “cancer secretome”, in which it additionally includes secretomes derived from stromal cells in quiescence or as a result of tumor‐host cell interactions, in addition to the cancer cell‐secreted factors. In Figure 1, we provide a schematic overview of the cancer secretome, where all microenvironmental proteins are assigned a possible origin from many types of cells within the neoplastic tissue. Although our main focus is the secreted proteins from cancer cells, we intend to briefly discuss recent proteomic advances in the context of our proposed “heterotypic cancer secretome” model, later in this review.
Figure 1.
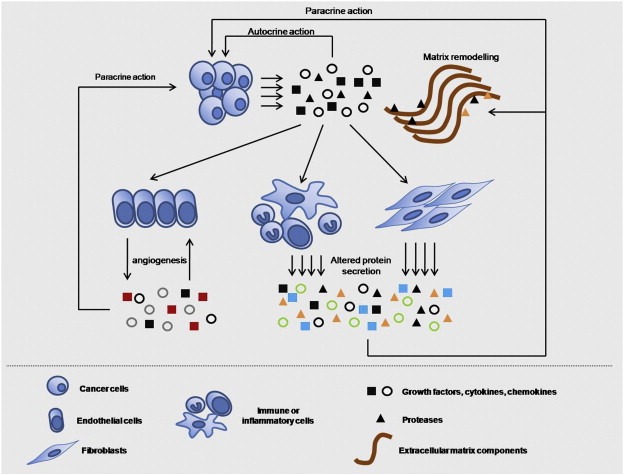
Heterotypic overview of the cancer secretome. All the microenvironmental, secreted proteins may originate either from cancer cells or from associated stromal cells and their secretion may be triggered by paracrine or autocrine actions between them. Proteomic approaches to capture the tumor microenvironment should focus on identifying proteins secreted by all associated cells, not just the cancer cells. Arrows initiating from cells and pointing in molecules represent secretion; the opposite represents the paracrine or autocrine action of the secreted molecules on the cell types.
2.3. Sources of cancer secretomes
Two prominent sources have been utilized in cancer secretome studies: cancer cell line supernatants and proximal biological fluids. The major opposition to tissue culture is the inability to fully replicate the complexity of the tumor microenvironment in vivo; for instance, changes in protein expression may occur because of cell culture stress instead of having certain in vivo relevance. In a relevant study (Celis et al., 1999), the authors found significant changes in protein expression even after short‐term culturing of low‐grade superficial bladder transitional cell carcinomas in vitro. In another relevant study (Ornstein et al., 2000), the authors also noticed significant changes in protein expression between microdissected prostate cancer cells and cell lines developed from the same patient, further demonstrating that culture stress may affect the differential protein potential. To address all these issues, tissue secretomics constitutes an appealing approach to study proteins produced in vivo by the tumor but it has been rather under‐studied, probably due to technical challenges (Celis et al., 2005; Shi et al., 2009; Gromov et al., 2010).
Conditioned media (CM) of cancer cell lines contain secreted or shed proteins released through classical and non‐classical secretion pathways. The limited complexity of CM compared to serum and proximal fluids enhances identification of low abundance proteins. Moreover, as in any in vitro system, experimental conditions can be highly controlled allowing reproducible and quantifiable results. Furthermore, large numbers of cell lines representing various stages and histotypes of a given cancer are readily available; the US National Cancer Institute (NCI)‐60 human tumour cell line anticancer drug screen, developed approximately two decades ago as an in vitro drug discovery tool (Shoemaker, 2006), represents the most notable example of such cell line availability. However, no single cell line can recapitulate the heterogeneity of human tumors; cell lines, for the most part, are deficient from contributions in the host‐tumor microenvironment. In addition, genotypic and phenotypic alterations accumulating over time may give rise to distinct subpopulations in the same cell line.
The presence of serum is important for cell survival and growth under in vitro conditions and frequently conditioned media are supplemented by an exogenous source (e.g. fetal bovine serum; FBS). At the same time, serum starvation has been shown to affect cell survival, proliferation, protein production and secretion patterns (Hasan et al., 1999; Cooper, 2003; Shin et al., 2008; Zander and Bemark, 2008; Levin et al., 2010). However, in the majority of secretome analysis studies, cells are grown in serum‐free media. This approach reduces both sample complexity caused by high protein content of FBS and sample contamination with orthologous proteins that may share amino acid sequences with the proteins of interest. One alternative approach was proposed by Colzani et al. and involved the supplementation of isotopically labelled amino acids in FBS‐containing CM, resulting in the isotopic labelling of proteins that originate from cells (Colzani et al., 2009). Although in this study, labelling made it possible to distinguish between cell‐derived and bovine proteins, additional steps in sample preparation were required to deal with increased sample complexity. A more user friendly approach would be to adapt the cells to serum‐free media by gradually reducing the percentage of serum in the CM prior to proteomic analysis.
An obstacle in the study of actively secreted proteins in the CM is the passive release of proteins into the media caused by cell death. Given that secreted proteins are of low abundance, they can be easily “masked” by highly abundant intracellular proteins. For that reason, frequently, cells are incubated in serum‐free media only for a small period of time such as 24h (Srisomsap et al., 2004; Chung and Yu, 2009; Xue et al., 2010). However, the amount of total protein secreted by the cells in 24h is rather small. In our laboratory, we have established an optimization procedure to maximize protein secretion and minimize cell death. In our workflow, multiple seeding densities and incubation periods are tested and levels of total protein, cell death and protein secretion are monitored for all different conditions. Based on these parameters, optimum conditions are selected. More sophisticated approaches such as hollow fiber culture systems and nanozeolite‐driven enrichment of secretory proteins have also been reported (Cao et al., 2009; Chang et al., 2009).
Fluids proximal to tumors frequently contain cancer cells, in addition to numerous soluble growth factors released by cancer cells and the tumor microenvironment. Many proximal fluids can be obtained with minimally invasive procedures and in large amounts (e.g. ascites fluid from ovarian cancer patients); however, the procedures to obtain such fluids need to be standardized. Since samples are collected from different individuals, the variability caused by behavioural, environmental and genetic differences is unavoidable. Furthermore, contamination by highly abundant serum proteins can increase sample complexity and complicate data interpretation.
2.4. Protein annotation tools
Not all proteins identified in the CM or biological fluids during secretome analysis can be considered per se as actively secreted proteins. Some proteins may be contaminants resulting from cell death or the culture media. Several bioinformatics tools can distinguish between secreted proteins and intracellular contaminants.
One of the most widely used databases for classifying protein subcellular localization is Gene Ontology (GO) (available at http://www.godatabase.org/dev). An advanced understanding of GO structure is critical to interpret the data correctly (Rhee et al., 2008). NCBI PubMed (http://www.ncbi.nlm.nih.gov/), Swiss‐Prot/TrEMBL (http://www.expasy.org/), and Bioinformatic Harvester EMBL (http://harvester.embl.de/) are some additional publicly available databases with protein cellular localization information, based on literature findings. Finally, Human Proteinpedia is a community portal that acts as a reservoir of human protein data and Human Protein Reference Database (HPRD) is used to integrate data deposited in Human Proteinpedia (Mathivanan et al., 2008).
Software tools enable prediction of proteins that are secreted, based on their primary sequence. Certain algorithms screen a target sequence in search of N‐terminal signal sequence or a signal sequence cleavage site. Such proteins are predicted to be secreted by the classical secretion pathway. Protcomp algorithms (http://www.softberry.com) [Softberry ProtComp 6.0 [http://www.softberry.com/berry.phtml?topic=protcompan&group=help&subgroup=proloc]], SignalP (http://www.cbs.dtu.dk/services/SignalP/) (Bendtsen et al., 2004, 2004), web‐based secreted protein database (SPD) (http://spd.cbi.pku.edu.cn) and Signal Peptide Prediction (SIG‐Pred) (http://www.bioinformatics.leeds.ac.uk/prot_analysis/Signal.html) are some of the prediction programs used in secretome analysis studies. Combination of multiple methods may increase predictive accuracy (Klee and Ellis, 2005). As mentioned earlier, protein secretion may occur via a non‐classical pathway. SecretomeP (http://www.cbs.dtu.dk/services/SecretomeP/) is a software tool that predicts mammalian secretory proteins participating in this pathway (Bendtsen et al., 2004, 2004). In addition, given the recent observations on exosomal proteomics, an independent database of proteins secreted through these endocytic‐like vesicles, named ExoCarta, has been generated and is available online (http://exocarta.ludwig.edu.au/index.html) (Mathivanan and Simpson, 2009). Finally, it is also possible that proteins located on the plasma membrane are shed and released to the extracellular space. Therefore, TransMembrane prediction using Hidden Markov Models (TMHMM) (http://www.cbs.dtu.dk/services/TMHMM/) as well as an additional software named Prediction of Transmembrane Regions and Orientation (TMpred) (http://www.ch.embnet.org/software/TMPRED_form.html) are useful tools for predicting transmembrane helices (Moller et al., 2001). The bioinformatic tools for secreted proteins have been incorporated in Figure 2, where the various protein sectretion pathways are also schematically illustrated.
Figure 2.
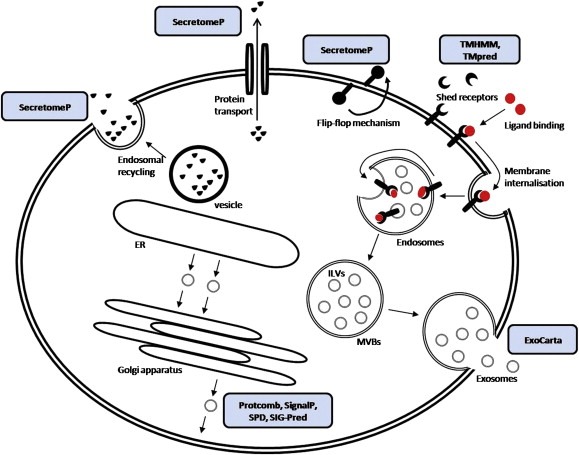
Bioinformatics tools for prediction of protein secretion pathways. The classical protein secretory pathway is ER/Golgi‐dependent and involves the presence of the signal peptide that directs translocation of these proteins to the ER. Protcomb, SignalP, SPD and Sig‐Pred are some of the widely used programs for prediction of proteins secreted through the classical secretory pathway. The non‐classical protein secretion pathway is ER/Golgi‐independent and is associated with absence of signal peptide. SecretomeP has been used for prediction of proteins secreted through non‐classical secretion pathways. Proteolytic events in the extracellular space might also result in shedding of membrane‐bound proteins/particles. Although this is not a protein secretion pathway, but an extracellular proteolytic event, software, such as TMHMM and TMpred is being used for the prediction of membrane and membrane‐bound proteins. Finally, a database of exosome‐secreted proteins, called ExoCarta has been recently generated as a distinct database the proteins secreted as such. ER, endoplasmic reticulum; ILVs, intralumenal vesicles; MVBs, multivesicular bodies; SPD, secreted protein database; SIG‐Pred, signal peptide prediction; TMHMM, transmembrane prediction with hidden Markov models; TMpred, prediction of transmembrane regions and orientation. Arrows indicate protein secretion or vesicle processing/movement. Blue boxes indicate software and their applications.
3. Cancer secretome and cancer pathobiology
Many strategies are constantly employed by the cancer and stromal cells for the acquisition of certain capabilities, which would assist them to overcome the biological limitations of neoplastic development and progression (Hanahan and Weinberg, 2000). New proteomic approaches, including the deep mining of cancer proteomes and secretomes, can provide insights into such mechanisms of carcinogenesis. In particular, the elucidation of key proteins of the metastatic cascade, and to a smaller extent of other cancer hallmarks (e.g. tumor growth and angiogenesis), is in the frontier of proteomic investigations (Everley and Zetter, 2005). The contribution of extracellular proteolysis to tumor invasion and metastasis has been recognized for decades, and new proteomic technologies can identify substrate molecules for many extracellular proteases to elucidate extracellular pathways of cancer progression (Doucet et al., 2008). Other lines of evidence point out that proteins secreted through exosomes might hold hidden but important roles in cancer development and progression, an observation that supports investigations towards the delineation and systematic exploration of the exosomal proteome (Ji et al., 2008; Xiao et al., 2009).
3.1. Cancer secretome analysis and the metastatic cascade
Metastasis consists of a long series of sequential, interrelated steps and is characterized by the activation of specific cell‐biological programs, such as epithelial‐to‐mesenchymal transition (EMT), cell invasion, motility and migration, as well as others (as depicted in Figure 3), which are all orchestrated by diverse extracellular and intracellular protein networks. Below, we briefly discuss how certain proteomic approaches could contribute to the better understanding of the metastatic cascade.
Figure 3.
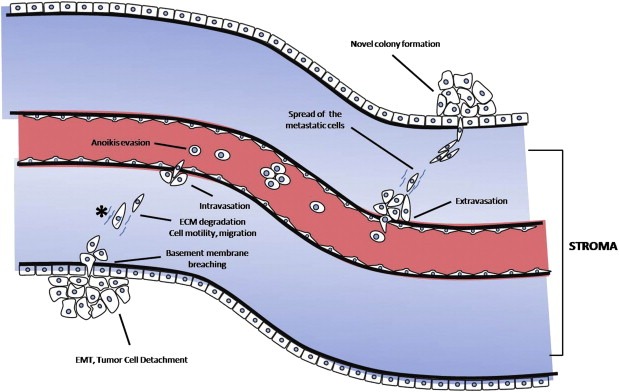
Cell‐biological programs, activated during the metastatic cascade, that could be investigated with mass spectrometry‐based secretome analysis. The metastatic cascade begins with an initial step of localized invasiveness, which enables in situ carcinoma cells that have undergone epithelial‐to‐mesenchymal transition, to breach the basement membrane. Thereafter, they enter into lymphatic or blood microvessels via a process called intravasation. The latter may transport these cancer cells to distant anatomical sites, where they are actually trapped and subsequently they invade into the neighboring tissue via a counter‐related process, called extravasation. This process enables them to form dormant micrometastases, which eventually may acquire the ability to successfully colonize the tissue and form a macroscopic metastasis. Throughout this process, the cancer cells deploy specific cell‐biological programs involving significant alterations in their proteome and secretome profiles to overcome various biological barriers; proteomic investigations have revealed metastasis‐associated proteins with specific roles within the metastatic cascade. EMT, epithelial‐to‐mesenchymal transition; ECM, extracellular matrix.
One of the most interesting cell‐biological programs implicated in metastasis is the EMT, during which, the cancer cells lose their epithelial characteristics, along with the expression of specific epithelial markers, such as E‐Cadherin and cytokeratin and gain a mesenchymal phenotype and fibroblast‐like shape that is partially characterized by expression of other markers, such as N‐cadherin and vimentin (Thiery, 2003; Thiery and Sleeman, 2006; Yilmaz and Christofori, 2009; Zeisberg and Neilson, 2009). Given that specific, but only partially elucidated microenvironmental signals play crucial roles in the initiation and maintenance of EMT, proteomic methodologies have been deployed to analyze the cancer secretomes and shed some light into these phenomena. For instance, in a study by Mathias et al., the well‐established epithelial cell line MDCK underwent EMT after oncogenic Ras transfection; DIGE analysis identified differentially expressed secretome proteins during the transition; some downregulated proteins included clusterin, desmocollin‐2 and collagen XVII, which are known to participate in cell–cell and cell‐matrix adhesion processes, while some upregulated ones included MMP‐1, kallikrein‐6 (KLK6) and TIMP‐1, namely proteases or factors that promote migration and motility in cancer cells (Mathias et al., 2009). The same group repeated these experiments with an LC‐MS/MS approach and also identified numerous potential mediators of EMT. As a proof of concept, they used siRNA‐mediated knockdown of MMP‐1 in the transformed MDCK cells to point out the implication of this protease in cell migration (Mathias et al., 2010). An alternative approach to investigate EMT‐related markers has been proposed by Slany et al. In this study, the authors analyzed the secretome from primary hepatocytes and cells from hepatic tumors, to generate large datasets of secreted proteins; based on the fact that EMT‐related proteins are mainly mesenchymal, they hypothesized that a dataset from the secretome of normal skin fibroblasts could be used as a representative list of proteins for choosing EMT‐associated markers in the hepatic tumor datasets (Slany et al., 2010). This approach enabled the identification of key proteins expressed in hepatocytes and hepatic cancer cells that could potentially serve as markers of EMT.
Other cell‐biological programs, activated during metastasis, which require the cooperation of large intracellular and extracellular protein networks, are the ones implicated in cell invasion, migration and cell motility. One of the most obvious traits of malignant cells is their ability to invade through adjacent cell layers, a process that requires at least two major cellular changes: (a) alteration of their intracellular cytoskeletal rearrangement to acquire an aggressive and motile phenotype and (b) remodelling of the nearby tissue environment by creating passages through the ECM, and pushing aside any stromal cells that stand in their way (Geho et al., 2005). Since the description of cellular proteomes is beyond the purposes of this review, we will only discuss how cancer secretome analysis could assist in the exploration of soluble factors present in the tumor microenvironment that affect cancer cell invasion and migration.
To characterize proteins involved in melanoma dissemination, protein profiles from B16F10 and B16BI6 cells were compared with 2D electrophoresis and MALDI‐TOF mass spectrometric analysis (Rondepierre et al., 2009). Since only the B16BI6 cells were able to generate pulmonary metastases after subcutaneous graft, and their supernatant was able to stimulate in vitro invasion of fibrosarcoma cells, it was hypothesized that these cells should secrete factors that facilitate their metastatic potential. Indeed, the analysis indicated a differential secretome profile in the two cell lines and syntenin was proposed as an invasion modulator (Rondepierre et al., 2009). In a similar study, a 1D SDS‐PAGE and MALDI‐TOF MS strategy was followed to systematically analyze the secretomes of two oral squamous cell carcinoma (OSCC) cell lines and identify key proteins of carcinogenesis. Among others, Mac‐2 was found to be implicated in the regulation of cell growth and motility of OSCC cells (Weng et al., 2008). In another study, it has been hypothesized that the differential expression of key proteins of breast cancer progression could be quantified; to test this, a SILAC‐based strategy was deployed and used to quantify proteins secreted in the supernatants from a pair of one normal and one malignant breast cancer cell line (Liang et al., 2009). This analysis revealed a multitude of potential cancer‐associated soluble factors, among which osteoblast‐specific factor 2 (OSF‐2) has been previously shown to also be overexpressed in the plasma membrane of breast cancer cells (Liang et al., 2006).
Other cell‐biological programs of metastasis, such as tumor cell intravasation (depicted in Figure 3) have been investigated with cell‐surface proteomic analysis of tumor cells (Conn et al., 2008). These studies focused on the cell‐surface proteome, since it has been demonstrated that cell–cell and cell‐matrix adhesion molecules (e.g. selectins, integrins) play significant roles in the efficiency of the intravasation process, although it is generally known that these processes are also mediated by large extracellular protein networks (e.g. matrix metalloproteinases) (Paschos et al., 2009, 2009). We believe that secretome analysis to study tumor intravasation, as well as other metastasis‐associated cell‐biological programs, has been rather unexplored so far, an observation that may point to opportunities in the functional proteomics community.
3.2. Cancer secretome analysis and other aspects of tumor progression
Although a large number of secreted proteins has been shown to be implicated in various aspects of the metastatic process, and proteomic research is effective towards elucidating mechanisms of invasion and metastasis, it is evident that cancer cells and/or stromal cells also liberate a wide variety of growth and survival factors that act in an autocrine or paracrine manner, and mediate other aspects of cancer development and progression, such as tumor cell proliferation, evasion of apoptosis, angiogenesis and resistance to antiproliferative signals and/or chemotherapy. The cancer secretome is almost certainly involved with the acquisition of such hallmarks of carcinogenesis and mass spectrometry‐based analysis can provide novel and insightful evidence for the regulatory pathways therein. For instance, in the previously described study by Weng et al., the identified protein Mac‐2 has been shown to be a strong factor that regulates the growth and survival of OSCC cells (Weng et al., 2008). An interesting approach in secretome analysis for identification of proliferation and/or survival factors has been performed by Hill et al. The authors exposed glioblastoma U87MG cells to a cAMP analog, which is known to lead to a decreased proliferation and invasion potential and then applied a label‐free quantification approach with mass spectrometry to identify key proteins that regulate these processes. A worth‐noticing finding in their analysis is the secretion of the glycoprotein AXL/UFO (Hill et al., 2009), a tyrosine‐protein kinase receptor that has been previously linked to brain tumor growth, prolonging of cell survival and invasion (Vajkoczy et al., 2006). Another interesting approach in cancer secretome analysis for the identification of survival factors has been previously performed by Iannetti et al. The authors generated NF‐kB‐null FRO cells, based on the fact that NF‐kB inhibition causes an increased susceptibility of drug‐induced apoptosis in thyroid carcinoma cells of the anaplastic type and subjected the conditioned media of these cells to differential proteomic analysis. This analysis depicted neutrophil gelatinase‐associated lipocalin (NGAL) protein, an NF‐kB regulated gene, as a potent survival factor of thyroid neoplastic cells (Iannetti et al., 2008).
3.3. Proteomic tools to investigate the extracellular proteolysis of the cancer secretome
Proteolysis affects every protein at some point of its life cycle and it constitutes the major post‐translational modification of secreted proteins. Distorted proteolysis has been considered a pivotal strategy of neoplastic progression, with distinct roles in tumor‐associated inflammation, angiogenesis, invasion and metastasis, as well as regulation and activation of latent forms of growth factors, cytokines and other molecules, which implicates them in tumor growth and proliferation. Therefore, it seems that proteolytic enzyme systems have a significant role in cancer development and progression (Doucet et al., 2008). Proteases do not operate in isolation; they are interconnected in proteolytic pathways and cascades, where the proteolytic information moves in a unidirectional flow or in regulatory feedback loops (Figure 4). It has been articulated that all these pathways and cascades are bridged in more complex and sophisticated networks that have been termed “the protease web” (Overall and Kleifeld, 2006). For instance, in our laboratory, we have been investigating for more than a decade the largest family of extracellular serine proteases, the kallikrein and kallikrein‐related peptidases (KLKs). These serine proteases have been implicated in many aspects of cancer progression, such as proliferation, angiogenesis, invasion and metastasis (Borgono and Diamandis, 2004) and many KLKs might hold promise as putative tumor biomarkers, with KLK3 [also known as the prostate specific antigen (PSA)] being the most prominent and well‐established (Emami and Diamandis, 2008). Current evidence shows that KLKs are implicated in various proteolytic cascades in the extracellular space that also influence other proteases, including matrix metalloproteinases and the uPA/uPAR system (Figure 4) (Borgono and Diamandis, 2004). Although many in vitro and in silico studies have indicated putative KLK substrates and demonstrated specific roles of KLKs based on these findings, it is presumed that a vast proportion of the in vivo targets of KLKs remain mostly unknown.
Figure 4.
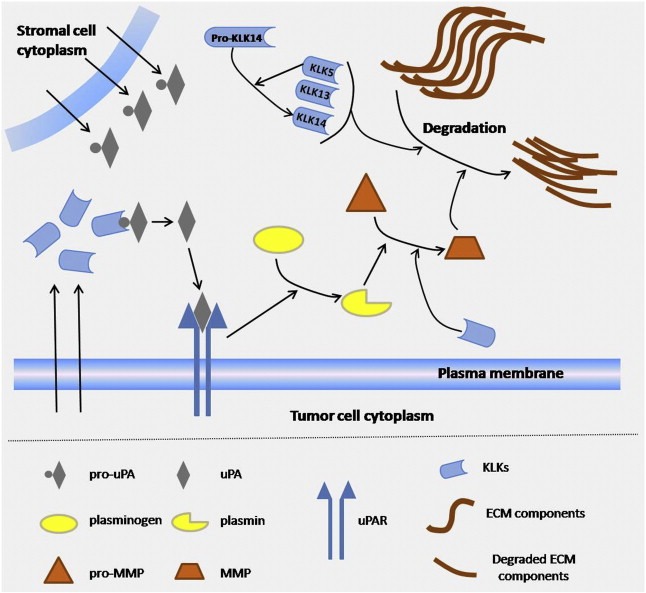
Schematic view of the region represented with an asterisk in Fig. 3, showing a distinct network of proteolytic relationships during cancer cell migration within the stroma. Kallikrein‐related peptidases, many of which are secreted by cancer cells, have been found capable of activating pro‐uPA (produced abundantly by stromal cells) and generate active uPA. In turn, uPA binds to its receptor, uPAR, present in the plasma membrane of the cancer cells, and converts plasminogen into active plasmin. Once plasmin is activated, it may, in turn, proceed to activate several inactive pro‐MMPs and generate active enzymes (MMPs). The latter are mainly responsible for ECM degradation. In addition, KLKs (e.g. KLK1) may be able to directly activate MMPs and also cleave constituents of the ECM themselves. uPA, urokinase‐type plasminogen activator; pro‐uPA, proform of uPA; uPAR, uPA receptor; MMPs, matrix metalloproteinases; pro‐MMPs, proform of MMPs; KLKs, kallikreins; ECM, extracellular matrix. Arrows between two molecules represent activation; arrows initiating from a molecule and pointing out in arrows represent enzymatic interaction; arrows initiating from cell interior and pointing in molecules represent secretion.
Our discussion on KLKs is a good example that the full substrate repertoire of a protease, termed “the protease degradome”, must be deciphered in order to define protease function and to identify possible drug targets. In this direction, degradomics, which involve the utilization of proteomic technologies to investigate protease substrates, have been developed and may facilitate understanding of the role of extracellular proteolysis in cancer (Doucet et al., 2008). Proteomic techniques such as those using multidimensional LC or 2DE and mass spectrometry have highly contributed to the protease substrate discovery platform. The major protease degradomes investigated thus far are those of matrix metalloproteinases, a family of proteases with diverse but distinct roles in cancer. For instance, substrates for MT1‐MPP that were either shed from the plasma membrane or the pericellular microenvironment were identified in the conditioned medium of human breast cancer cell lines transfected with MT1‐MPP, compared with vector or an inactive MT1‐MPP mutant, using ICAT labelling. Out of this analysis, previously unknown substrates for MT1‐MPP have been identified, such as interleukin‐8, death receptor‐6, and secretory leukocyte protease inhibitor (Tam et al., 2004). In the same context, substrates for MMP2 were identified in the secretome of cultured MMP2 (−/−) murine fibroblasts transfected to express low levels of active MMP2 compared to the catalytically inactive MMP2 mutant, using iTRAQ. Novel substrates for MMP2 have been identified, such as CX3CL1 chemokine fractalkine, osteopontin, galectin‐1 and Hsp90a (Dean and Overall, 2007). These analyses point out that quantitative proteomic analyses, like ICAT and iTRAQ, are unbiased techniques and as such, can provide precious insight for specific protease substrate identification. More recently, SILAC approaches have been successfully employed and novel substrates for atrolysin A were identified (Pinto et al., 2007). Taken together, these data show that proteomic approaches could prove to be of considerable assistance to elucidate key proteolytic pathways in cancer.
Another aspect of extracellular proteolysis is the protease activity, not just the amount. For many proteases, the activity status in biological samples is unknown and even in cascades, many proteases are participating with their inactive pro‐forms and are either autoactivated or activated by other proteases at some point. This is also the case for the proteolytic events, involving the KLK family of serine proteases, as depicted in Figure 4. The identification and quantification of active proteases can be achieved by coupling activity‐based probes (ABPs) to mass spectrometric analysis. The ABPs are able to target a specific protease class and irreversibly bind to its active site; upon binding of the ABP to the active site of the protease, the chemically reactive group of the ABP can be either visualized (if the ABP is tagged with a fluorophore or radioactive molecule), or isolated and analyzed through mass spectrometry (if the ABP is tagged with an affinity tag) (Schmidinger et al., 2006). Saghatelian et al. designed ABPs by coupling zinc‐chelating hydroxamate to a benzophenone photocrosslinker, which promoted the selective binding to active matrix metalloproteinases (but not to the inactive zymogens or inhibitor‐bound counterparts) and used these ABPs to identify members of the MMP enzyme class that were upregulated in invasive cancer cells. Their analysis identified a membrane‐bound matrix metalloproteinase that was not reported before to be either increasingly expressed or activated in highly invasive cancers (Saghatelian et al., 2004). The same group designed a biotinylated fluorophosphonate, referred to as FP‐biotin, with which active serine hydrolases could be visualized (Liu et al., 1999). Fluorescent ABPs have also been developed for specific labelling and visualization of the papain family of the lysosomal cysteine cathepsins in live mice. After labelled cathepsins in human breast cancer cell lines were implanted in mice, proteins were extracted from solid tumors and in‐gel digestion coupled to MS/MS was performed for the labelled proteases (Blum et al., 2007). Overall, these data point out that the use of ABPs coupled to visualization or mass spectrometry, may provide further insight about the activity status of extracellular proteases, especially in cancer where proteolysis is disturbed and most of the times, the knowledge on the activity of the key proteases might be obscure.
3.4. Proteomic analysis of tumor‐derived exosomes
Exosomes are 40–100 nm membrane vesicles of endocytic origin, secreted by most cell types in vitro and in vivo. It has been made clear that exosomal proteins participate in life‐preserving processes, supporting the hypothesis that exosomes maintain conserved functions in mammalian tissues. It is also spectacularly evident that exosomes might constitute a highly sophisticated form of cell–cell communication in an autocrine, paracrine or even endocrine fashion. More recent evidence shows that tumor‐derived exosomes may harbour proteins and/or mRNAs with important pathobiological functions in cancer development and progression. For instance, recent work by Jung et al. in a rat model of pancreatic adenocarcinoma, indicated that CD44 protein is responsible for acquiring a soluble matrix in the pre‐metastatic niche, into where tumor‐derived exosomes are able to disseminate and assist in tumor cell embedding and growth. The fact that tumor‐derived exosomes are able to travel to the pre‐metastatic niche might also explain how the long‐distance communication between the cancer‐initiating cells and the niche is achieved (Jung et al., 2009). In addition, gastric cancer‐derived exosomes were able to induce tumor cell proliferation through PI3K/Akt and MAPK/Erk pathways (Qu et al., 2009b), as well as induce apoptosis to Jurkat T‐cells in a dose‐ and time‐dependent manner (Qu et al., 2009a); the latter observation supports the notion that tumor‐derived exosomes might regulate the inflammatory cancer microenviroment and thus have a major impact on tumor progression. Additional evidence that exosomes are implicated in specific communications between cancer and stromal cells have been provided in a study by Hood et al., where the authors indicated that melanoma exosomes were able to interact with, and influence, the morphology of endothelial tube formation and also stimulate the formation of endothelial spheroids and sprouting in a dose‐dependent manner, possibly through growth factors and relevant cytokines (Hood et al., 2009). Overall, all these data provide proof that exosomes are important in cancer development and progression and also provide a quick but accurate explanation as to why proteomic technologies could be preferentially used to systematically analyze the tumor‐derived exosomal proteomes.
An early attempt to characterize the proteome of melanoma‐derived exosomes with mass spectrometry identified several known proteins, as well as novel proteins (e.g. radixin and p120 catenin) that had not been previously documented to be secreted through exosomes (Mears et al., 2004). Also, Ochieng et al. have shown that breast cancer cell uptake of circulating serum exosomes might assist in their anchorage‐independent growth, by activation of the MAPK pathway. Their proteomic analysis identified many cancer–related proteins, that could also serve as exosome markers, including heat shock protein 90, alpha tubulin, galectin‐3 binding protein and chloride intracellular channel protein (Ochieng et al., 2009). In another study, a DIGE‐LC‐MS/MS strategy was performed to compare and contrast the exosomal proteins secreted from a pair of normal and Ras‐transformed murine fibroblasts; it was hypothesized that the frequently disturbed Ras signalling pathway, would be an efficient model to generate a list of exosomal proteins that are differentially expressed in such cancers. Indeed, the analysis showed an up to 10‐fold increase in various proteins, including milk fat globule EGF factor 8, 14‐3‐3 isoforms and collagen a‐1 (VI), confirming their initial hypothesis (Ji et al., 2008).
Given other recent data that exosomes may regulate specific communications between cancer and stromal cells, a proteomic analysis in mesothelioma‐derived exosomes revealed the presence of the angiogenic factor developmental endothelial locus‐1 (DEL‐1) among others, which has been shown to be implicated in vascular development in the tumor stroma (Hegmans et al., 2004). This study suggests that tumor‐endothelial cell (or even other stromal cell) communications could be mediated with the diffusion of exosomes in the extracellular matrix, an observation that is in concordance with a recent model of hypoxia‐triggered exosomal protein secretion with very high angiogenic and metastatic potential in the tumor microenvironment (Park et al., 2010).
The most significant drawback with exosome proteomics is the absence of standardized and well‐characterized methods for isolation and purification of these vesicles. This process is empirical and has been described as “laboratory‐dependent”. Typically, a series of differential centrifugations and ultracentrifugations, followed by further purification steps through flotation in linear sucrose gradients (2.0–0.25M sucrose), are carried out for the isolation of exosomes (Simpson et al., 2008). More recently, antibody‐coated magnetic beads, using antibodies against tumor‐ or cell‐specific proteins have been used to isolate exosomes from supernatants of cancer cell lines; the prerequisite for this processing is the a priori knowledge of at least one exosomal marker, specific to the cancer type under consideration. For instance, an immunoaffinity‐capture method with a colon epithelial cell‐specific A33 antibody was used, in order to purify exosomes derived from the colon cancer cell line LIM1215. Proteomic analysis revealed numerous proteins involved in cytoskeletal rearrangement, signalling, trafficking and exosome biology‐related proteins, such as ESCRT complex proteins (Mathivanan et al., 2009). In addition, this study also revealed that the molecular components of exosomes are cell‐type dependent, since the analysis also identified proteins that are specific to the gastrointestinal tract, such as carcinoembryonic antigen (CEA) (Mathivanan et al., 2009). In a similar manner, a specific antibody against HER‐2 has been used to isolate exosomes from breast cancer cell lines and carry out systematic proteomic analyses in them (Koga et al., 2005).
4. Heterotypic nature of the cancer secretome
There is now abundant evidence that cancer‐associated stromal cells are recruited by cancer cells to allow for more efficient development and progression of the malignancy, and that such recruitment usually causes altered protein expression and secretion profiles, in all participating cell types within the tumor microenviroment. The host cell participation has been termed as (a) ‘desmoplasia’, which involves the implication of fibroblasts and extracellular matrix in the tumorigenic process, (b) inflammation and/or immune response, which is the infiltration of macrophages, neutrophils, mast cells, myeloid cell‐derived suppressor cells and mesenchymal stem cells in the tumorigenic stroma, and (c) angiogenesis, which comprise the further sprouting of blood and lymphatic circulatory systems within the tumor mass (Coussens and Werb, 2002; Ferrara et al., 2003; Mareel and Leroy, 2003; Pugh and Ratcliffe, 2003; Mueller and Fusenig, 2004; Bertout et al., 2008; Joyce and Pollard, 2009). Proteome alterations, regarding the intracellular proteomes of tumor and/or host cells, have been investigated through comprehensive quantitative or non quantitative proteomic approaches, usually in a context of in vitro, co‐culture, or microenvironment alteration experiments (Boraldi et al., 2007; Cancemi et al., 2009) or with the extended use of laser capture microdissection (LCM) in in vivo tissue proteomics studies (Li et al., 2009; Rho et al., 2009). Analysis of such studies is beyond the scope of this review; in contrast, our main focus will be a thought‐provoking discussion over the secretome analysis of tumor and/or host cells, which has not been thoroughly explored yet and warrants further investigation.
4.1. The desmoplasia‐derived secretome
Cancer‐associated fibroblasts (CAFs) play important roles in tumor initiation and progression through specific communications with the cancer cells. Diverse evidence shows that cancer cell‐secreted factors, such as TGF‐β and PDGF are responsible for initiating and maintaining the myofibroblastic phenotype in associated fibroblasts; the latter usually respond to those stimuli with dramatic changes in their protein expression profile, including their intracellular proteome as well as secretome (Kunz‐Schughart and Knuechel, 2002; Kalluri and Zeisberg, 2006; Xing et al., 2010). Certain notable alterations of the CAF secretome include: (a) the induction of an altered extracellular matrix that provides additional oncogenic signals to the tumor by the de novo expression of tenascin‐C (De Wever et al., 2004; Koperek et al., 2007) and matrix metalloproteinases, like, for example, the gelatinases MMP‐2 and MMP‐9 (Saad et al., 2002; Singer et al., 2002), (b) the increased expression of growth factors and cytokines, like insulin‐like growth factor 1 (IGF1) and hepatocyte growth factor (HGF) that promote tumor cell survival and motility, respectively (Aebersold and Mann, 2003; Lewis et al., 2004), (c) the regulation of inflammatory responses at the primary tumor sites by secreting chemotactic, proinflammatory agents, like for example interleukin 1b (IL‐1b) and tumor necrosis factor‐alpha (TNF‐α) (Mueller et al., 2007), and (d) the regulation of angiogenesis by interactions with the local microvasculature, by aberrantly expressing vascular endothelial growth factor (VEGF) (Orimo et al., 2001). In one proteomic study, the authors sought to investigate the mammary cancer‐associated fibroblast secretome, so they induced the myofibroblastic phenotype by generating CAV‐1 (−/−) fibroblasts, based on the hypothesis that since CAV‐1 inhibits TGF‐β signalling, then CAV‐1 (−/−) fibroblasts could maintain a constantly active TGF‐β pathway, which is known to trigger the induction of CAFs. Secretome analysis of CAV‐1 (−/−) fibroblasts indicated the secretion of factors associated with the myofibroblastic phenotype (e.g. Colla1, Colla2 and SPARC), verifying the initial hypothesis (Pavlides et al., 2009). All these studies demonstrate that CAFs are active participants in neoplastic tissues, with an extensively altered secretome, compared to their normal counterparts.
The interactions of cancer cells with their associated fibroblasts have only recently been investigated with proteomic technologies. In a murine model of lung cancer, in which cells were co‐cultured with cancer‐associated fibroblasts and other stromal cells (including endothelial and macrophage cells), SILAC approaches were used to quantitate the differential secretomes of cancer cells and co‐cultured cells (Zhong et al., 2008). The analysis showed that a multitude of extracellular proteins are increased in the co‐cultures, implying their possible participation in various neoplastic phenomena. Among these, the chemokine CXCL1 was abundantly produced in the cocultures and the main source was found to be the cancer cells, probably through paracrine signalling mediated by the stromal cells (Zhong et al., 2008). Another cytokine, found to be increased, was IL‐18 (Zhong et al., 2008) that has already been shown to have contradicting roles in tumorigenesis (inhibiting or promoting) (Park et al., 2007). Various cytokine signalling pathways (HGF, TGF‐β, CXCR) between cancer cells and associated stroma have long been hypothesized to play pivotal roles in the development and progression of cancer (Delany and Canalis, 1998; Tsukinoki et al., 2004; Eck et al., 2009). Remarkably, this study demonstrated that SILAC‐based quantitative proteomic technologies could be a significant tool for investigating cytokine signalling pathways, overcoming a potential limitation that cytokines are secreted in very low amounts, and still are efficiently detected by mass spectrometry. In another study, Paulitschke et al. proposed a model of secretome analysis of associated stromal cells to identify markers for melanoma metastasis, that could either be utilized as biomarkers of disease progression or to study melanoma metastasis. As a proof of concept, they cultured melanoma and associated stromal cells, including melanoma‐associated fibroblasts and normal skin fibroblasts and performed mass spectrometric analysis in cell lysates and supernatants using LC‐MS/MS; their analysis showed many melanoma‐specificic secreted proteins (lumican, Pmel 17), as well as proteins secreted by normal (extracellular matrix proteins) or melanoma‐associated fibroblasts (neuropillin, stanniocalcin‐1, periostin). This strategy provided novel insights into secreted proteins, which have not been previously identified in melanoma or the other cell types, like, for example, GPX5 (Paulitschke et al., 2009).
4.2. The inflammation‐derived secretome
Inflammatory cells are also significant components of neoplastic tissues; for example tumor‐associated macrophages (TAMs) are derived from monocytes and are recruited by monocytic protein chemokines, secreted by the cancer cells. Upon differentiation, TAMs secrete a considerable number of angiogenic and lymphagiogenic growth factors, cytokines and proteases, all of which are mediators of neoplastic development and progression (Schoppmann et al., 2002; Marconi et al., 2008; Sierra et al., 2008). The interactions of TAMs with the cancer cells have been investigated for a long time, but only recently, proteomic technologies have been deployed for studying the altered secretion profiles of these cells. In one such study, the authors performed secretome analysis using LC‐MS/MS on supernatants from a normal monocytic/macrophage cell line, buffy coat monocytes, as well as purified, in vitro‐cultured TAMs, isolated from ovarian cancer ascitic fluid and they noticed the de novo secretion of 14‐3‐3 zeta protein in cancer‐associated macrophages (Kobayashi et al., 2009). Given the previously documented role of 14‐3‐3 zeta protein as adaptor protein in intracellular signalling pathways (Van Der Hoeven et al., 2000; Bialkowska et al., 2003; Birkenfeld et al., 2003), and more recent evidence that this protein can also be secreted in the extracellular space by monocytes/macrophages infected with HIV‐1 virus (Ciborowski et al., 2007), the authors speculated that TAM‐secreted 14‐3‐3 zeta protein may promote neoplastic progression of epithelial ovarian cancer, under conditions which promote its secretion. Remarkably, this was the first mass spectrometry‐based study that demonstrated a novel mechanism of neoplastic progression, mediated by cancer cell‐TAM interactions, using secretome analysis. Given the recent, well‐documented link of chronic inflammatory diseases to cancer incidence (Pages et al., 2010), this paradoxical role of the immune system in cancer progression should be carefully investigated; the interaction of the immune/inflammatory cells, like macrophages, T‐cells, dendritic cells and neutrophils with the cancer cells could be partially elucidated with currently available proteomic technologies.
4.3. The angiogenesis‐derived secretome
Endothelial cells have also been shown to interact with cancer cells, with the most notable mechanism involving the increased secretion of angiogenic growth factors, like, for example, VEGF and angiopoietin, that mostly act in an autocrine manner in endothelial cells, a process that allows the neoformation of blood vessels. This altered secretion profile has been mostly shown to be caused in endothelial cells by cancer cell‐derived hypoxia‐inducible factors (HIFs) (Carmeliet, 2000; Carmeliet and Jain, 2000). As in the case of CAFs and TAMs, proteomic technologies could be a promising and valuable tool to study the interactions between cancer and endothelial cells. In the co‐culture model of murine lung cancer along with stromal cells, including murine endothelial cells, as established by Zhong et al., a wide multitude of cytokines were increasingly expressed in the co‐cultures compared to the monocultures, when quantitated with SILAC. This analysis pointed out that endothelial cells are essential and able to stimulate in vitro and probably in vivo the production of various soluble factors that assist in tumor development and progression. In addition, the importance of studying tumor angiogenesis with tools of secretome analysis should not be underestimated; this neoplasia‐driven process has been considered as a target for chemotherapies in the past and present (Grothey and Galanis, 2009; Ivy et al., 2009), making it quite clear that the elucidation of key participants of angiogenesis will definitely support future research in cancer therapeutics and management.
4.4. The adipocytic secretome in breast cancers
Other than the reported stromal cells, certain tissue‐specific tumor‐host cell interactions have also been investigated. For instance, in the breast, where fat tissue is abundant, the tumor cell–adipocyte interactions have been documented to play pivotal roles in cancer development and progression. Although adipose tissue has been considered metabolically inactive, and has been assigned a role as energy storage depot, there is now recent evidence demonstrating that this tissue is an active endocrine organ that produces hormones, growth factors, adipokines and other molecules that not only affect physiological cellular responses but also contribute to paracrine and autocrine signalling networks, especially in tumor microenvironments where hormonal dependence mediates cancer progression (Kim et al., 2009, 2008, 2009, 2009, 2009). An effort to dissect the molecular circuitry of epithelial‐adipocyte stromal cell interactions was performed by Celis et al., where the secretome of fat interstitial fluid from breast cancer patients was analyzed with mass spectrometry. Their analysis enabled the identification of numerous (a) proinflammatory cytokines (IL‐6, IL‐8, TNF‐α, TGF‐β) that are known to mediate inflammatory responses within tumors, (b) growth factors (IGF‐1, macrophage stimulating growth factor) that are known to enhance cell proliferation, (c) angiogenic factors (VEGF, angiopoietin‐2, granulocyte CSF), (d) tissue inhibitors of metalloproteinases (TIMPs) that participate in extracellular matrix remodelling (Celis et al., 2005). This diversity of secreted factors suggests that these molecules may directly participate in a mutual growth with the adjacent breast cancer cells, invading the stroma and also keep specific communications with other cancer‐associated stromal cells. In addition, the fact that several cytokines and growth factors that have not been previously reported to be secreted by fat cells were identified in this study, points out that the depth of mass spectrometry‐based proteomic mining and the high‐throughput nature of the secretome analysis are capable of delineating novel signalling networks in complex cancer microenvironments.
5. Conclusions and future perspectives
In the next few years, important developments in cancer biomarker discovery as well as in the identification of putative therapeutic targets for cancer are anticipated. Secretome analysis can facilitate applications with clinical value and provide insight in tumorigenesis at the molecular level. Nevertheless, specific challenges remain. However, the emergence of technologically advanced mass spectrometers opens the path for identification of very low abundance proteins, even in complex biological samples, which will certainly bring major discoveries in near term (5 years).
Certain sub‐fields of secretome analysis have been briefly described in this review due to their emerging significance in the proteomics community. For example, proteolysis, a biological process that adds additional layers of complexity to the cellular proteomes and secretomes, is now widely explored with the assistance of powerful proteomic technologies that enable the quantification and identification of protease activity in tumors, as well as the delineation of the substrate repertoire of poorly characterized extracellular proteases.
Perhaps, of astounding interest is the emerging roles of exosomes in malignant conditions; it has been extensively shown and documented that tumor‐derived exosomes identified either in cancer cell lines or in relevant biological fluids, such as ascites and serum, could serve the purposes of valuable diagnostic biomarkers of the disease or even provide insights in mechanisms of neoplastic development and progression (Simpson et al., 2008). In this review, we have focused to the contribution of proteomics technologies to exosome purification and characterization. Over the last years, as we have witnessed an enormous understanding of the molecular composition of exosomes, a notion that is also shown by the publication of proteome exosome datasets from various cell types and biological fluids (Mathivanan and Simpson, 2009). All these observations provide an exciting platform of opportunities for proteomic investigations on tumor‐derived exosomes and increasing efforts are expected towards this direction in the near future.
In this review, we proposed the term “heterotypic cancer secretome”, to better define and describe the cancer secretome; the heterotypic nature of the cancer secretome is not a new idea, but has been used to denote the importance of the associated stromal cells in the modulation and regulation of the tumor microenvironment. It is evident that in vitro approaches, with the use of various co‐culture systems, represent the basis for investigating the tumor‐host cell interactions. These in vitro co‐culture experiments are easy to perform, and, at the same time, the depth of the mass spectrometric analysis allows for the generation of large protein datasets, originating from complex microenvironments consisting of many cell types. We have already been witnessing an increasing interest and number of publications, attempting to elucidate mechanisms of tumor‐host cell interactions with the assistance of proteomic technologies and this trend will likely continue.
To briefly conclude, cancer secretome analysis is currently facing certain challenges, but the future is bright. Many applications of secretome analysis are expected to be integrated within the oncoproteomics arena and hopefully provide a handful of clinically relevant tools for patient management.
Acknowledgements
The authors have no financial involvement in any of the technologies, products or companies mentioned in this publication. There is no conflict of interest to be reported. The authors wish to thank Shalini Makawita, for helpful suggestions.
Karagiannis George S., Pavlou Maria P., Diamandis Eleftherios P., (2010), Cancer secretomics reveal pathophysiological pathways in cancer molecular oncology, Molecular Oncology, 4, doi: 10.1016/j.molonc.2010.09.001.
References
- Aebersold, R. , Mann, M. , 2003. Mass spectrometry-based proteomics. Nature. 422, (6928) 198–207. [DOI] [PubMed] [Google Scholar]
- Bendtsen, J.D. , Jensen, L.J. , 2004. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des Sel. 17, (4) 349–356. [DOI] [PubMed] [Google Scholar]
- Bendtsen, J.D. , Nielsen, H. , 2004. Improved prediction of signal peptides: signalP 3.0. J. Mol. Biol. 340, (4) 783–795. [DOI] [PubMed] [Google Scholar]
- Bertout, J.A. , Patel, S.A. , 2008. The impact of O2 availability on human cancer. Nat. Rev. Cancer. 8, (12) 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialkowska, K. , Zaffran, Y. , 2003. 14-3-3 zeta mediates integrin-induced activation of Cdc42 and Rac. Platelet glycoprotein Ib-IX regulates integrin-induced signaling by sequestering 14-3-3 zeta. J. Biol. Chem. 278, (35) 33342–33350. [DOI] [PubMed] [Google Scholar]
- Birkenfeld, J. , Betz, H. , 2003. Identification of cofilin and LIM-domain-containing protein kinase 1 as novel interaction partners of 14-3-3 zeta. Biochem. J. 369, (Pt 1) 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum, G. , von Degenfeld, G. , 2007. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nat. Chem. Biol. 3, (10) 668–677. [DOI] [PubMed] [Google Scholar]
- Boraldi, F. , Annovi, G. , 2007. Hypoxia influences the cellular cross-talk of human dermal fibroblasts. A proteomic approach. Biochim. Biophys. Acta. 1774, (11) 1402–1413. [DOI] [PubMed] [Google Scholar]
- Borgono, C.A. , Diamandis, E.P. , 2004. The emerging roles of human tissue kallikreins in cancer. Nat. Rev. Cancer. 4, (11) 876–890. [DOI] [PubMed] [Google Scholar]
- Cancemi, P. , Albanese, N.N. , 2009. Multiple changes induced by fibroblasts on breast cancer cells. Connect. Tissue Res. 51, (2) 88–104. [DOI] [PubMed] [Google Scholar]
- Cao, J. , Hu, Y. , 2009. Nanozeolite-driven approach for enrichment of secretory proteins in human hepatocellular carcinoma cells. Proteomics. 9, (21) 4881–4888. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P. , 2000. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6, (4) 389–395. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P. , Jain, R.K. , 2000. Angiogenesis in cancer and other diseases. Nature. 407, (6801) 249–257. [DOI] [PubMed] [Google Scholar]
- Celis, A. , Rasmussen, H.H. , 1999. Short-term culturing of low-grade superficial bladder transitional cell carcinomas leads to changes in the expression levels of several proteins involved in key cellular activities. Electrophoresis. 20, (2) 355–361. [DOI] [PubMed] [Google Scholar]
- Celis, J.E. , Moreira, J.M. , 2005. Identification of extracellular and intracellular signaling components of the mammary adipose tissue and its interstitial fluid in high risk breast cancer patients: toward dissecting the molecular circuitry of epithelial-adipocyte stromal cell interactions. Mol. Cell Proteomics. 4, (4) 492–522. [DOI] [PubMed] [Google Scholar]
- Chang, Y.H. , Wu, C.C. , 2009. Cell secretome analysis using hollow fiber culture system leads to the discovery of CLIC1 protein as a novel plasma marker for nasopharyngeal carcinoma. J. Proteome Res. 8, (12) 5465–5474. [DOI] [PubMed] [Google Scholar]
- Chung, S.D. , Yu, H.J. , 2009. Re: Ching-Chia Li, Tu-Hao Chang, Wen-Jeng Wu, et al. Significant predictive factors for prognosis of primary upper urinary tract cancer after radical nephroureterectomy in taiwanese patients. Eur. Urol. 2008, (54) 1127–1135. Eur Urol 55(4): e69-70; author reply e71 [DOI] [PubMed] [Google Scholar]
- Ciborowski, P. , Kadiu, I. , 2007. Investigating the human immunodeficiency virus type 1-infected monocyte-derived macrophage secretome. Virology. 363, (1) 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colzani, M. , Waridel, P. , 2009. Metabolic labeling and protein linearization technology allow the study of proteins secreted by cultured cells in serum-containing media. J. Proteome Res. 8, (10) 4779–4788. [DOI] [PubMed] [Google Scholar]
- Conn, E.M. , Madsen, M.A. , 2008. Cell surface proteomics identifies molecules functionally linked to tumor cell intravasation. J. Biol. Chem. 283, (39) 26518–26527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, S. , 2003. Reappraisal of serum starvation, the restriction point, G0, and G1 phase arrest points. Faseb J. 17, (3) 333–340. [DOI] [PubMed] [Google Scholar]
- Coussens, L.M. , Werb, Z. , 2002. Inflammation and cancer. Nature. 420, (6917) 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wever, O. , Nguyen, Q.D. , 2004. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. Faseb J. 18, (9) 1016–1018. [DOI] [PubMed] [Google Scholar]
- Dean, R.A. , Overall, C.M. , 2007. Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol. Cell Proteomics. 6, (4) 611–623. [DOI] [PubMed] [Google Scholar]
- Delany, A.M. , Canalis, E. , 1998. Dual regulation of stromelysin-3 by fibroblast growth factor-2 in murine osteoblasts. J. Biol. Chem. 273, (26) 16595–16600. [DOI] [PubMed] [Google Scholar]
- Denny, P.W. , Gokool, S. , 2000. Acylation-dependent protein export in Leishmania. J. Biol. Chem. 275, (15) 11017–11025. [DOI] [PubMed] [Google Scholar]
- Doucet, A. , Butler, G.S. , 2008. Metadegradomics: toward in vivo quantitative degradomics of proteolytic post-translational modifications of the cancer proteome. Mol. Cell Proteomics. 7, (10) 1925–1951. [DOI] [PubMed] [Google Scholar]
- Eck, S.M. , Cote, A.L. , 2009. CXCR4 and matrix metalloproteinase-1 are elevated in breast carcinoma-associated fibroblasts and in normal mammary fibroblasts exposed to factors secreted by breast cancer cells. Mol. Cancer Res. 7, (7) 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emami, N. , Diamandis, E.P. , 2008. Utility of kallikrein-related peptidases (KLKs) as cancer biomarkers. Clin. Chem. 54, (10) 1600–1607. [DOI] [PubMed] [Google Scholar]
- Everley, P.A. , Zetter, B.R. , 2005. Proteomics in tumor progression and metastasis. Ann. N. Y Acad. Sci. 1059, 1–10. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. , Gerber, H.P. , 2003. The biology of VEGF and its receptors. Nat. Med. 9, (6) 669–676. [DOI] [PubMed] [Google Scholar]
- Finley, D.S. , Calvert, V.S. , 2009. Periprostatic adipose tissue as a modulator of prostate cancer aggressiveness. J. Urol. 182, (4) 1621–1627. [DOI] [PubMed] [Google Scholar]
- Geho, D.H. , Bandle, R.W. , 2005. Physiological mechanisms of tumor-cell invasion and migration. Physiology (Bethesda). 20, 194–200. [DOI] [PubMed] [Google Scholar]
- Gromov, P. , Gromova, I. , 2010. Up-regulated proteins in the fluid bathing the tumour cell microenvironment as potential serological markers for early detection of cancer of the breast. Mol. Oncol. 4, (1) 65–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothey, A. , Galanis, E. , 2009. Targeting angiogenesis: progress with anti-VEGF treatment with large molecules. Nat. Rev. Clin. Oncol. 6, (9) 507–518. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2000. The hallmarks of cancer. Cell. 100, (1) 57–70. [DOI] [PubMed] [Google Scholar]
- Hasan, N.M. , Adams, G.E. , 1999. Effect of serum starvation on expression and phosphorylation of PKC-alpha and p53 in V79 cells: implications for cell death. Int. J. Cancer. 80, (3) 400–405. [DOI] [PubMed] [Google Scholar]
- Hegmans, J.P. , Bard, M.P. , 2004. Proteomic analysis of exosomes secreted by human mesothelioma cells. Am. J. Pathol. 164, (5) 1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, J.J. , Moreno, M.J. , 2009. Identification of secreted proteins regulated by cAMP in glioblastoma cells using glycopeptide capture and label-free quantification. Proteomics. 9, (3) 535–549. [DOI] [PubMed] [Google Scholar]
- Hood, J.L. , Pan, H. , 2009. Paracrine induction of endothelium by tumor exosomes. Lab. Invest. 89, (11) 1317–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannetti, A. , Pacifico, F. , 2008. The neutrophil gelatinase-associated lipocalin (NGAL), a NF-kappaB-regulated gene, is a survival factor for thyroid neoplastic cells. Proc. Natl. Acad. Sci. U S A. 105, (37) 14058–14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy, S.P. , Wick, J.Y. , 2009. An overview of small-molecule inhibitors of VEGFR signaling. Nat. Rev. Clin. Oncol. 6, (10) 569–579. [DOI] [PubMed] [Google Scholar]
- Jain, K.K. , 2008. Innovations, challenges and future prospects of oncoproteomics. Mol. Oncol. 2, (2) 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, H. , Erfani, N. , 2008. Difference gel electrophoresis analysis of Ras-transformed fibroblast cell-derived exosomes. Electrophoresis. 29, (12) 2660–2671. [DOI] [PubMed] [Google Scholar]
- Joyce, J.A. , Pollard, J.W. , 2009. Microenvironmental regulation of metastasis. Nat. Rev. Cancer. 9, (4) 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, T. , Castellana, D. , 2009. CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia. 11, (10) 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri, R. , Zeisberg, M. , 2006. Fibroblasts in cancer. Nat. Rev. Cancer. 6, (5) 392–401. [DOI] [PubMed] [Google Scholar]
- Kim, J.H. , Kim, K.Y. , 2008. Adipocyte culture medium stimulates production of macrophage inhibitory cytokine 1 in MDA-MB-231 cells. Cancer Lett. 261, (2) 253–262. [DOI] [PubMed] [Google Scholar]
- Kim, K.Y. , Baek, A. , 2009. Adipocyte culture medium stimulates invasiveness of MDA-MB-231 cell via CCL20 production. Oncol. Rep. 22, (6) 1497–1504. [DOI] [PubMed] [Google Scholar]
- Klee, E.W. , Ellis, L.B. , 2005. Evaluating eukaryotic secreted protein prediction. BMC Bioinform. 6, 256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, R. , Deavers, M. , 2009. 14-3-3 zeta protein secreted by tumor associated monocytes/macrophages from ascites of epithelial ovarian cancer patients. Cancer Immunol. Immunother. 58, (2) 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga, K. , Matsumoto, K. , 2005. Purification, characterization and biological significance of tumor-derived exosomes. Anticancer Res. 25, (6A) 3703–3707. [PubMed] [Google Scholar]
- Koperek, O. , Scheuba, C. , 2007. Molecular characterization of the desmoplastic tumor stroma in medullary thyroid carcinoma. Int. J. Oncol. 31, (1) 59–67. [PubMed] [Google Scholar]
- Kulasingam, V. , Diamandis, E.P. , 2008. Tissue culture-based breast cancer biomarker discovery platform. Int. J. Cancer. 123, (9) 2007–2012. [DOI] [PubMed] [Google Scholar]
- Kunz-Schughart, L.A. , Knuechel, R. , 2002. Tumor-associated fibroblasts (part I): active stromal participants in tumor development and progression?. Histol. Histopathol. 17, (2) 599–621. [DOI] [PubMed] [Google Scholar]
- Levin, V.A. , Panchabhai, S.C. , 2010. Different changes in protein and phosphoprotein levels result from serum starvation of high-grade glioma and adenocarcinoma cell lines. J. Proteome Res. 9, (1) 179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, M.P. , Lygoe, K.A. , 2004. Tumour-derived TGF-beta1 modulates myofibroblast differentiation and promotes HGF/SF-dependent invasion of squamous carcinoma cells. Br. J. Cancer. 90, (4) 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M.X. , Xiao, Z.Q. , 2009. Quantitative proteomic analysis of differential proteins in the stroma of nasopharyngeal carcinoma and normal nasopharyngeal epithelial tissue. J. Cell Biochem. 106, (4) 570–579. [DOI] [PubMed] [Google Scholar]
- Liang, X. , Huuskonen, J. , 2009. Identification and quantification of proteins differentially secreted by a pair of normal and malignant breast-cancer cell lines. Proteomics. 9, (1) 182–193. [DOI] [PubMed] [Google Scholar]
- Liang, X. , Zhao, J. , 2006. Quantification of membrane and membrane-bound proteins in normal and malignant breast cancer cells isolated from the same patient with primary breast carcinoma. J. Proteome Res. 5, (10) 2632–2641. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Patricelli, M.P. , 1999. Activity-based protein profiling: the serine hydrolases. Proc. Natl. Acad. Sci. U.S.A. 96, (26) 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marconi, C. , Bianchini, F. , 2008. Tumoral and macrophage uPAR and MMP-9 contribute to the invasiveness of B16 murine melanoma cells. Clin. Exp. Metastasis. 25, (3) 225–231. [DOI] [PubMed] [Google Scholar]
- Mareel, M. , Leroy, A. , 2003. Clinical, cellular, and molecular aspects of cancer invasion. Physiol. Rev. 83, (2) 337–376. [DOI] [PubMed] [Google Scholar]
- Mathias, R.A. , Chen, Y.S. , 2010. Extracellular remodelling during oncogenic Ras-induced epithelial-mesenchymal transition facilitates MDCK cell migration. J. Proteome Res. 9, (2) 1007–1019. [DOI] [PubMed] [Google Scholar]
- Mathias, R.A. , Wang, B. , 2009. Secretome-based proteomic profiling of Ras-transformed MDCK cells reveals extracellular modulators of epithelial-mesenchymal transition. J. Proteome Res. 8, (6) 2827–2837. [DOI] [PubMed] [Google Scholar]
- Mathivanan, S. , Ahmed, M. , 2008. Human Proteinpedia enables sharing of human protein data. Nat. Biotechnol. 26, (2) 164–167. [DOI] [PubMed] [Google Scholar]
- Mathivanan, S. , Lim, J.W. , 2009. Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Mol. Cell Proteomics. 9, (2) 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathivanan, S. , Simpson, R.J. , 2009. ExoCarta: a compendium of exosomal proteins and RNA. Proteomics. 9, (21) 4997–5000. [DOI] [PubMed] [Google Scholar]
- Mears, R. , Craven, R.A. , 2004. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics. 4, (12) 4019–4031. [DOI] [PubMed] [Google Scholar]
- Mellman, I. , Warren, G. , 2000. The road taken: past and future foundations of membrane traffic. Cell. 100, (1) 99–112. [DOI] [PubMed] [Google Scholar]
- Mignatti, P. , Morimoto, T. , 1992. Basic fibroblast growth factor, a protein devoid of secretory signal sequence, is released by cells via a pathway independent of the endoplasmic reticulum-Golgi complex. J. Cell Physiol. 151, (1) 81–93. [DOI] [PubMed] [Google Scholar]
- Moller, S. , Croning, M.D. , 2001. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 17, (7) 646–653. [DOI] [PubMed] [Google Scholar]
- Mueller, L. , Goumas, F.A. , 2007. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am. J. Pathol. 171, (5) 1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, M.M. , Fusenig, N.E. , 2004. Friends or foes – bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer. 4, (11) 839–849. [DOI] [PubMed] [Google Scholar]
- Nickel, W. , 2003. The mystery of nonclassical protein secretion. A current view on cargo proteins and potential export routes. Eur. J. Biochem. 270, (10) 2109–2119. [DOI] [PubMed] [Google Scholar]
- Ochieng, J. , Pratap, S. , 2009. Anchorage-independent growth of breast carcinoma cells is mediated by serum exosomes. Exp. Cell Res. 315, (11) 1875–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo, A. , Tomioka, Y. , 2001. Cancer-associated myofibroblasts possess various factors to promote endometrial tumor progression. Clin. Cancer Res. 7, (10) 3097–3105. [PubMed] [Google Scholar]
- Ornstein, D.K. , Gillespie, J.W. , 2000. Proteomic analysis of laser capture microdissected human prostate cancer and in vitro prostate cell lines. Electrophoresis. 21, (11) 2235–2242. [DOI] [PubMed] [Google Scholar]
- Overall, C.M. , Kleifeld, O. , 2006. Tumour microenvironment – opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer. 6, (3) 227–239. [DOI] [PubMed] [Google Scholar]
- Pages, F. , Galon, J. , 2010. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 29, (8) 1093–1102. [DOI] [PubMed] [Google Scholar]
- Park, J.E. , Tan, H.S. , 2010. Hypoxia modulates tumor microenvironment to enhance angiogenic and metastastic potential by secretion of proteins and exosomes. Mol Cell Proteomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. , Cheon, S. , 2007. The dual effects of interleukin-18 in tumor progression. Cell Mol. Immunol. 4, (5) 329–335. [PubMed] [Google Scholar]
- Paschos, K.A. , Canovas, D. , 2009. The engagement of selectins and their ligands in colorectal cancer liver metastases. J. Cell Mol. Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschos, K.A. , Canovas, D. , 2009. The role of cell adhesion molecules in the progression of colorectal cancer and the development of liver metastasis. Cell Signal. 21, (5) 665–674. [DOI] [PubMed] [Google Scholar]
- Paulitschke, V. , Kunstfeld, R. , 2009. Entering a new era of rational biomarker discovery for early detection of melanoma metastases: secretome analysis of associated stroma cells. J. Proteome Res. 8, (5) 2501–2510. [DOI] [PubMed] [Google Scholar]
- Pavlides, S. , Whitaker-Menezes, D. , 2009. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 8, (23) 3984–4001. [DOI] [PubMed] [Google Scholar]
- Pinto, A.F. , Ma, L. , 2007. Use of SILAC for exploring sheddase and matrix degradation of fibroblasts in culture by the PIII SVMP atrolysin A: identification of two novel substrates with functional relevance. Arch. Biochem. Biophys. 465, (1) 11–15. [DOI] [PubMed] [Google Scholar]
- Pugh, C.W. , Ratcliffe, P.J. , 2003. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9, (6) 677–684. [DOI] [PubMed] [Google Scholar]
- Qu, J.L. , Qu, X.J. , 2009. The role of cbl family of ubiquitin ligases in gastric cancer exosome-induced apoptosis of Jurkat T cells. Acta Oncol. 48, (8) 1173–1180. [DOI] [PubMed] [Google Scholar]
- Qu, J.L. , Qu, X.J. , 2009. Gastric cancer exosomes promote tumour cell proliferation through PI3K/Akt and MAPK/ERK activation. Dig. Liver Dis. 41, (12) 875–880. [DOI] [PubMed] [Google Scholar]
- Rhee, S.Y. , Wood, V. , 2008. Use and misuse of the gene ontology annotations. Nat. Rev. Genet. 9, (7) 509–515. [DOI] [PubMed] [Google Scholar]
- Rho, J.H. , Roehrl, M.H. , 2009. Tissue proteomics reveals differential and compartment-specific expression of the homologs transgelin and transgelin-2 in lung adenocarcinoma and its stroma. J. Proteome Res. 8, (12) 5610–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondepierre, F. , Bouchon, B. , 2009. B16 melanoma secretomes and in vitro invasiveness: syntenin as an invasion modulator. Melanoma Res. 20, (2) 77–84. [DOI] [PubMed] [Google Scholar]
- Rubartelli, A. , Cozzolino, F. , 1990. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. Embo J. 9, (5) 1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad, S. , Gottlieb, D.J. , 2002. Cancer cell-associated fibronectin induces release of matrix metalloproteinase-2 from normal fibroblasts. Cancer Res. 62, (1) 283–289. [PubMed] [Google Scholar]
- Saghatelian, A. , Jessani, N. , 2004. Activity-based probes for the proteomic profiling of metalloproteases. Proc. Natl. Acad. Sci. U.S.A. 101, (27) 10000–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidinger, H. , Hermetter, A. , 2006. Activity-based proteomics: enzymatic activity profiling in complex proteomes. Amino Acids. 30, (4) 333–350. [DOI] [PubMed] [Google Scholar]
- Schnabele, K. , Roser, S. , 2009. Effects of adipocyte-secreted factors on cell cycle progression in HT29 cells. Eur. J. Nutr. 48, (3) 154–161. [DOI] [PubMed] [Google Scholar]
- Schoppmann, S.F. , Birner, P. , 2002. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am. J. Pathol. 161, (3) 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, H.J. , Stubbs, R. , 2009. Characterization of de novo synthesized proteins released from human colorectal tumour explants. Electrophoresis. 30, (14) 2442–2453. [DOI] [PubMed] [Google Scholar]
- Shin, J.S. , Hong, S.W. , 2008. Serum starvation induces G1 arrest through suppression of Skp2-CDK2 and CDK4 in SK-OV-3 cells. Int. J. Oncol. 32, (2) 435–439. [PubMed] [Google Scholar]
- Shoemaker, R.H. , 2006. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer. 6, (10) 813–823. [DOI] [PubMed] [Google Scholar]
- Sierra, J.R. , Corso, S. , 2008. Tumor angiogenesis and progression are enhanced by Sema4D produced by tumor-associated macrophages. J. Exp. Med. 205, (7) 1673–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, R.J. , Jensen, S.S. , 2008. Proteomic profiling of exosomes: current perspectives. Proteomics. 8, (19) 4083–4099. [DOI] [PubMed] [Google Scholar]
- Singer, C.F. , Kronsteiner, N. , 2002. MMP-2 and MMP-9 expression in breast cancer-derived human fibroblasts is differentially regulated by stromal-epithelial interactions. Breast Cancer Res. Treat. 72, (1) 69–77. [DOI] [PubMed] [Google Scholar]
- Slany, A. , Haudek, V.J. , 2010. Cell characterization by proteome profiling applied to primary hepatocytes and hepatocyte cell lines Hep-G2 and Hep-3B. J. Proteome Res. 9, (1) 6–21. [DOI] [PubMed] [Google Scholar]
- Srisomsap, C. , Sawangareetrakul, P. , 2004. Proteomic analysis of cholangiocarcinoma cell line. Proteomics. 4, (4) 1135–1144. [DOI] [PubMed] [Google Scholar]
- Tam, E.M. , Morrison, C.J. , 2004. Membrane protease proteomics: isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc. Natl. Acad. Sci. U.S.A. 101, (18) 6917–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery, J.P. , 2003. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 15, (6) 740–746. [DOI] [PubMed] [Google Scholar]
- Thiery, J.P. , Sleeman, J.P. , 2006. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, (2) 131–142. [DOI] [PubMed] [Google Scholar]
- Tjalsma, H. , Bolhuis, A. , 2000. Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol. Mol. Biol. Rev. 64, (3) 515–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudel, C. , Faure-Desire, V. , 2000. Translocation of FGF2 to the cell surface without release into conditioned media. J. Cell Physiol. 185, (2) 260–268. [DOI] [PubMed] [Google Scholar]
- Tsukinoki, K. , Yasuda, M. , 2004. Hepatocyte growth factor and c-Met immunoreactivity are associated with metastasis in high grade salivary gland carcinoma. Oncol. Rep. 12, (5) 1017–1021. [PubMed] [Google Scholar]
- Vajkoczy, P. , Knyazev, P. , 2006. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc. Natl. Acad. Sci. U.S.A. 103, (15) 5799–5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Hoeven, P.C. , Van Der Wal, J.C. , 2000. Protein kinase C activation by acidic proteins including 14-3-3. Biochem. J. 347, (Pt 3) 781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, P. , Gilmore, R. , 1984. Protein translocation across the endoplasmic reticulum. Cell. 38, (1) 5–8. [DOI] [PubMed] [Google Scholar]
- Weng, L.P. , Wu, C.C. , 2008. Secretome-based identification of Mac-2 binding protein as a potential oral cancer marker involved in cell growth and motility. J. Proteome Res. 7, (9) 3765–3775. [DOI] [PubMed] [Google Scholar]
- Xiao, Z. , Blonder, J. , 2009. Proteomic analysis of extracellular matrix and vesicles. J. Proteomics. 72, (1) 34–45. [DOI] [PubMed] [Google Scholar]
- Xing, F. , Saidou, J. , 2010. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 15, 166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, H. , Lu, B. , 2010. Identification of serum biomarkers for colorectal cancer metastasis using a differential secretome approach. J. Proteome Res. 9, (1) 545–555. [DOI] [PubMed] [Google Scholar]
- Yilmaz, M. , Christofori, G. , 2009. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 28, (1–2) 15–33. [DOI] [PubMed] [Google Scholar]
- Zander, L. , Bemark, M. , 2008. Identification of genes deregulated during serum-free medium adaptation of a Burkitt's lymphoma cell line. Cell Prolif. 41, (1) 136–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg, M. , Neilson, E.G. , 2009. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 119, (6) 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, L. , Roybal, J. , 2008. Identification of secreted proteins that mediate cell-cell interactions in an in vitro model of the lung cancer microenvironment. Cancer Res. 68, (17) 7237–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]