

Abstract
Activating mutations in BRAF, a constituent of the map kinase pathway, were first discovered as being most prevalent in melanoma in 2002. Only recently have potent and selective, orally available inhibitors of BRAF emerged for clinical testing and demonstrated clear evidence of tumor regression in the majority of patients whose tumors harbor a BRAF mutation. While these early observations suggest that the BRAF targeted therapy will become part of the standard treatment paradigm for patients with advanced melanoma, it is also clear that a majority of these responses are incomplete and temporary. Therefore, the focus of the melanoma field has shifted to understanding the limits of the first generation of selective BRAF inhibitors with regard to safety and efficacy, the context of somatic genetic changes that accompany BRAF, and the combination regimens that target distinct elements of melanoma pathophysiology.
Keywords: BRAF inhibitors, Targeted therapies, Cancer, Oncogenic BRAF signaling
1. Introduction
BRAF is a key protein kinase component of the RAS‐RAF pathway. This critical intracellular signaling pathway relays extracellular signals to the nucleus in order to regulate the gene expression (Figure 1). The extracellular signals may be growth factors or hormones present in the extracellular milieu, which bind to and activate cell surface or intracellular receptors, respectively. The activated receptor then, in turn, activates downstream components of the signaling pathway, propagating the signal to the nucleus where nuclear transcription factors regulate target gene transcription. By regulating the expression of target genes, the cell can respond to the extracellular environment in a variety of ways, including proliferation and survival via prevention of the cell's innate cell death mechanism (McCubrey et al., 2007; Sebolt‐Leopold and Herrera, 2004).
Figure 1.
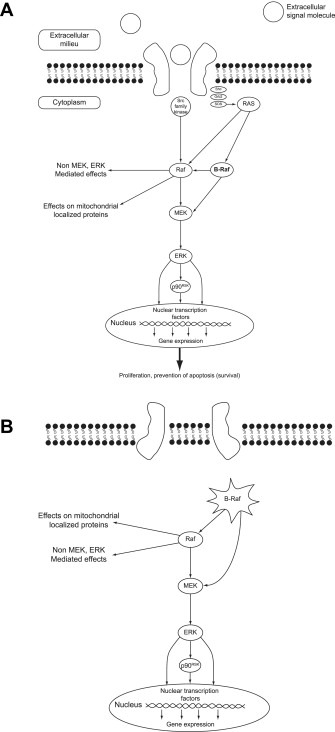
The RAF‐MEK‐ERK signaling pathway (panel A) and activation via constitutive BRAF activation (panel B) (McCubrey et al., 2007).
Around 8% of all solid human tumors are thought to harbor mutated BRAF and over 30 mutations in the BRAF gene have been associated with human cancers (Wan et al., 2004). The most commonly identified mutation in the BRAF gene occurs in the region that encodes the kinase domain of the protein at position V600 and results in constitutive activation of the kinase. The mutated BRAF kinase activates downstream components of the pathway in the absence of an upstream (external) signal, when cessation of proliferation and/or cell death may be appropriate or required. The result of this deregulated downstream signaling is an alteration in gene expression leading to unregulated cell proliferation and survival, factors that contribute to oncogenesis (Hoeflich et al., 2009; McCubrey et al., 2007; Wan et al., 2004; Zhang and Guan, 2000). Oncogenic BRAF signaling is implicated in approximately 50% of melanomas, 30–70% of thyroid cancers, 30% of serous low‐grade ovarian cancers, and 10% of colorectal cancers (CRCs) (Davies et al., 2002; Fransen et al., 2004; Garnett and Marais, 2004; Goydos et al., 2005; Libra et al., 2005; McCubrey et al., 2007). The pervasive nature of oncogenic BRAF signaling across human cancers makes this an important area of focus for the development of anticancer agents specifically targeted to the aberrant signaling generated by the mutant BRAF kinases.
RG7204 (also referred to as PLX4032 and RO5185426) (Figure. 2), is a potent inhibitor of the V600E mutation‐containing BRAF kinase, and has shown promising preclinical and early clinical efficacy against mutant BRAF cell lines and tumors (Bollag et al., 2010; Joseph et al., 2010; Sala et al., 2008; Tsai et al., 2008). This agent is currently in clinical development for the treatment of a range of human cancers and here we review the preclinical studies, pharmacokinetics, clinical toxicity, early clinical efficacy and possible mechanisms of resistance and toxicity. GSK2118436 is another inhibitor of activated BRAF, with a similar preclinical and clinical profile, but has been in clinical development for less time and therefore less data are available.
Figure 2.
Molecular structure of RG7204/PLX4032 (Bollag et al., 2010). Reprinted by permission from Macmillan Publishers Ltd: Nature (Bollag et. al.), copyright (2010).
2. Preclinical studies
In vitro biochemical assays have shown that RG7204/PLX4032 exhibits selectivity against a broad range of kinases. In a panel of over 200 kinases, RG7204/PLX4032 showed a similar potency for BRAFV600E (31 nM) and CRAF (48 nM), and selectivity with respect to other kinases including wild‐type BRAF (100 nM). The vast majority of kinases were only minimally affected, with IC50 values of >10 μM, thought to be irrelevant in clinically achievable drug concentrations. However, several kinases (CRAF, SRMS, ACK1, MAP4K5 and FGR) were inhibited at <100 nM concentrations and could be relevant contributors to efficacy or toxicity. The in vitro selectivity of RG7204/PLX4032 translate into remarkable cellular selectivity in a series of experiments designed to evaluate the effect of RG7204/PLX4032 on RAF–MEK–ERK pathway inhibition and proliferation suppression in a panel of cancer cell lines (Yang et al., 2010). Cell lines tested for inhibition of MEK and ERK phosphorylation included the melanoma cell lines expressing BRAFV600E, BRAF V600D, BRAF V600R or BRAFWT. RG7204/PLX4032 inhibits both phosphorylation of MEK and ERK, and cellular proliferation in all BRAFV600E‐expressing melanoma cell lines tested, including Colo829 and LOX. RG7204/PLX4032 also exhibited potent inhibitory effects on MEK and ERK phosphorylation and cellular proliferation in melanoma cell lines which expressed other mutations at the V600 position, such as BRAF V600D, BRAF V600R and BRAF V600K (Yang et al., 2010; Halaban et al., 2010).
RG7204/PLX4032 lacked antiproliferative activity in cell lines expressing wild‐type BRAF proteins, including those from melanomas and other tumor types such as lung, gastric, breast, pancreatic, and skin tumors. Activity was reported in one additional breast cancer cell line (MDA‐MB‐435) which expressed BRAFV600E and wild‐type RAS (Yang et al., 2010). Suppression of ERK and MEK phosphorylation by RG7204/PLX4032 correlates with the inhibition of cellular proliferation in melanoma cells harboring mutations at the V600 position. Thus, RG7204/PLX4032 displays a high degree of selectivity against BRAFV600E kinase in mechanistic and antiproliferative cellular assays (Lee et al., 2010). The studies described above show that RG7204/PLX4032 potently inhibits MEK phosphorylation and activation, which consequently inhibits ERK phosphorylation and ultimately cell proliferation in tumor cells expressing the mutant BRAF gene.
3. Pharmacokinetics
In a LOX BRAFV600E‐mutant melanoma‐xenograft model exposure‐dependent tumor responses have been reported. This was determined by comparison of the percentage of pMEK/MEK and pERK/ERK inhibition in RG7204/PLX4032‐treated vs. vehicle‐treated tumor samples. The highest plasma concentration of RG7204/PLX4032 corresponded to the highest mean percentage inhibition of MEK and ERK phosphorylation (Yang et al., 2010).
Phase I pharmacokinetic analyses in humans have shown that exposure increased with dose (Flaherty et al., 2010). A total of 87 patients (49 of whom had melanoma and a further 32 who had BRAFV600E metastatic melanoma) were recruited to a Phase I study and received doses of up to 1120 mg twice daily. Exposure was dose‐proportional through the range 240 mg twice daily to 960 mg twice daily (Figure 3A). Patients were exposed to relatively constant daily drug levels at steady state that were between 6 and 9 times the mean level on Day 1 at the 960 mg twice daily dose (Figure 3B). The mean half‐life of RG7204/PLX4032 was ∼50 h and the area under the plasma concentration–time curve (AUC) for the 960 mg twice daily dose was 1741 ± 639 μM/h (Flaherty et al., 2010). The 960 mg twice daily dose has been selected for Phase II evaluation based on its acceptable toxicity profile and the steady state drug concentration that remained in the therapeutic range.
Figure 3.
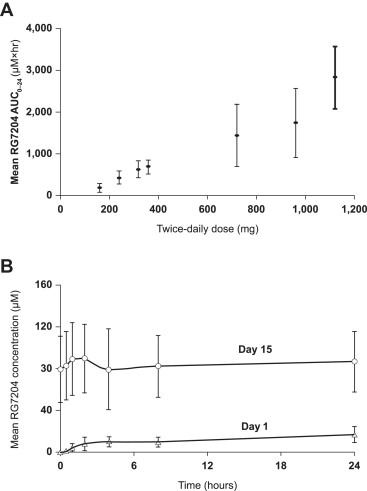
Pharmacokinetic profile of RG7204/PLX4032 in humans. Dose‐dependent exposure (panel A) and relatively constant daily exposure at steady state (panel B) (Flaherty et al., 2010).
Phase I pharmacodynamic analyses showed that tumor levels of phosphorylated ERK, cyclin D1 and Ki‐67 were markedly reduced at Day 15 compared with baseline (n = 7 patients treated with RG7204/PLX4032 960 mg twice daily), indicating that RG7204/PLX4032 inhibited the MAPK pathway, resulting in decreased cell proliferation (Flaherty et al., 2010). Furthermore, a smaller subset of patients underwent serial tumor biopsies before and after 15 days on therapy. These patients were treated at lower doses that resulted in only one quarter of the steady state drug exposure achieved with 960 mg twice daily. While evidence of suppression of ERK phosphorylation was demonstrated, the level of inhibition was far lower compared to the patients treated with higher doses/drug exposure. Therefore, the dose–response relationship between MAP kinase pathway inhibition and tumor regression observed in preclinical studies was recapitulated in patients.
4. Toxicity profile
RG7204/PLX4032 was well tolerated in the in vivo preclinical studies. No treatment‐related mortality or clinical signs of toxicity were reported at any time (Yang et al., 2010). These data supported the potentially high therapeutic index for this agent in subsequent clinical trials.
The Phase I trial reported by Flaherty et al. (2010) consisted of a dose escalation phase followed by an extension phase at the maximum dose that could be administered without frequent, severe adverse effects. The most frequent adverse events (AEs) at the recommended Phase II dose of 960 mg twice daily were arthralgia, rash, nausea, photosensitivity, fatigue, pruritus and palmar‐plantar dysesthesia. Most events were mild to moderate in intensity. Cutaneous squamous cell carcinoma occurred at a rate of 31% (10/32 patients) during the extension phase of the study during which all patients received a 960 mg twice daily starting dose. The median time to occurrence of these carcinomas was 8 weeks; the majority was resected and none led to discontinuation of treatment. These tumors were not limited to the sun exposed areas where typical cutaneous squamous cell carcinomas were frequently diagnosed, nor were they usually associated with a clinically detectable pre‐existing lesion such as an actinic keratosis or verruca. A subsequent clinical and histological evaluation of these therapy‐associated cutaneous neoplasms has shown that these are generally well differentiated, often showing features of keratoacanthoma, with a low likelihood of invasive or metastatic potential (Lacouture et al., 2010). However, at least one case of squamous cell carcinoma with perineural invasion occurred during treatment with RG7204/PLX4032 in a patient with no previous history of non‐melanoma skin cancer. This tumor was completely resected with Mohs micrographic surgery.
Other cutaneous AEs included the report of rash in 40.2% of patients of the extension cohort (Lacouture et al., 2010). This rash was usually self limited or responded to systemic or topical steroids and included a typical morbilliform drug eruption as well as a diffuse eruption of keratosis pilaris‐like erythematous papules. A mild nodular form of cutaneous vasculitis was observed in several patients and was more persistent than other cutaneous eruptions, but was not progressive. Photosensitivity was observed in 20% of patients in the extension cohort (Lacouture et al., 2010). Depending on the level of the patient's sun exposure, in some cases this led to extensive sun burns with bullae. Based on this AE profile in the skin, it is recommended that patients undergoing treatment with selective BRAF inhibitors be counseled to perform regular self examination of their skin and report any new or changing lesions to their physician. Additionally, patients starting therapy should take precautions to avoid prolonged sun exposure, wear sun protective clothing, and sun screen in order to avoid significant burning. It has not been determined if there is a relationship between photosensitivity and therapy‐associated squamous cell carcinoma in this patient population.
Paradoxical activation of ERK by RAF inhibitors has been reported by several groups. Three recent reports have explored the potential mechanism(s) for this activation by showing that selective BRAF inhibitors, such as PLX4720 (an analog of RG7204/PLX4032), 885‐A and GDC‐0879 (selective BRAF inhibitors of different chemical series), stimulate MEK–ERK signaling via CRAF activation in the presence of an upstream activator (e.g. activated RTK, RAS mutation) in melanoma and other cell lines with only wild‐type BRAF (Hatzivassiliou et al., 2010; Heidorn et al., 2010; Poulikakos et al., 2010). These studies support a model in which BRAF‐specific inhibitors induce RAS‐GTP‐dependent CRAF activation via the formation of BRAF–CRAF heterodimers or BRAF and/or CRAF homodimers, followed by recruitment of RAF complexes to the plasma membrane, triggering activation of the MEK–ERK pathway. In the context of the BRAF/CRAF heterodimer, a selective BRAF inhibitor will inhibit the kinase activity of BRAF which is otherwise involved in the negative regulation of CRAF activity. An alternative explanation is that BRAF kinase activity of one molecule in a BRAF/CRAF, dimer is inhibited while the other remains unbound by drug and therefore hyperactivated by the loss of the activity of one BRAF molecule. Neither of these mechanisms appears to be relevant in the context of an activating BRAF mutation. It is worthy of noting that HRAS mutations are commonly found in cutaneous squamous cell carcinomas and keratoacanthomas (Corominas et al., 1989). The pre‐existence of keratinocytes harboring HRAS mutations might provide the perfect context for a growth stimulatory effect of a selective BRAF inhibitor to produce the squamous cell carcinomas with keratoacanthoma features that have been observed in the clinical trials with RG7204/PLX4032.
This emphasizes the importance of selecting patients with BRAFV600E mutations for treatment with RG7024/PLX4032. The Cobas® 4800 BRAFV600 mutation assay is a diagnostic test designed to detect the BRAFV600E mutation, the primary BRAF mutation in melanoma. It is being codeveloped by Roche Molecular Systems, Inc. and Plexxikon Inc. as a clinically validated companion diagnostic test to identify patients who are candidates for treatment with RG7204. This test is currently being used to identify patients for clinical trials evaluating RG7204 treatment. Additional assays are being developed to ensure sensitive and reliable detection of activating BRAF mutations prior to the initiation of the selective BRAF targeted therapy.
5. Efficacy in clinical studies
5.1. Phase I
The Phase I study reported by Flaherty et al. (2010) consisted of a dose escalation portion followed by an extension phase during which patients received the recommended Phase II dose of 960 mg twice daily until disease progression. The two extension cohorts consisted of 32 patients with melanoma, and 21 patients with colorectal cancer (Kopetz et al., 2010). Among the 32 patients with melanoma carrying a BRAFV600E mutation who took part in the extension phase, a total of 26 patients (81%) responded to therapy with two patients achieving a complete response (Figure 4) (Flaherty et al., 2010). A subset of these patients was evaluated with FDG‐PET scan and responses were observed in all 19 examined, indicating that FDG‐PET is a potentially useful marker for early biological response (McArthur et al., 2010). Among patients with metastatic disease, responses were recorded at all metastatic sites and there was an improvement in symptoms for those with symptomatic disease and a reduction in the requirement for narcotic pain relief in three patients within one to two weeks. Longer‐term evaluation of patients on this extension arm is ongoing; and has revealed a confirmed response rate of 59%, and a progression free survival (PFS) of 7.61 months at the most recent analysis. In the colorectal cancer extension cohort, 19 of the 21 patients were evaluable for efficacy at the time of latest reporting (Kopetz et al., 2010). All patients received RG7204/PLX4032 960 mg twice daily. One patient achieved a partial response and four patients achieved minor responses (≥10% shrinkage, but less than the 30% required to meet the threshold for partial response). The median PFS was 3.7 months with two patients still on study (Kopetz et al., 2010). Three patients with thyroid cancer carrying BRAFV600E mutation took part in the Phase I study. One had a partial response and PFS was 11, 12 and 13 months (Flaherty et al., 2010).
Figure 4.
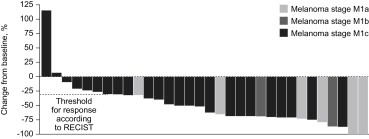
Best overall response among 32 patients with melanoma treated with RG7204/PLX4032 960 mg twice daily (Flaherty et al., 2010).
5.2. Phase II
A Phase II (BRIM 2) study of RG7204/PLX4032 in patients with metastatic melanoma is currently ongoing (Sosman et al., 2010). Interim data were presented at the Society for Melanoma Research Congress in Sydney, Australia in November 2010. One hundred and thirty‐two patients with metastatic melanoma harboring a V600E mutation and whose disease had progressed after receiving at least one other standard therapy were enrolled. In this trial, all radiographic evaluations were reviewed by the central panel of radiologists.52% of patients achieved partial (49%) or complete (3%) responses. An additional 30% had disease stabilization, with many having minor degrees of tumor regression. The median PFS was 6.2 months and median duration of response was 6.8 months, although follow‐up time for this cohort was quite short. The toxicity profile observed in the larger study recapitulated the Phase I trial.
A number of additional studies are currently ongoing including additional Phase I studies to further characterize the pharmacokinetics, optimal dosing regimen, and possible drug–drug interactions for RG7204/PLX4032 (NCT01164891, NCT01107418, NCT01001299). A Phase III study (BRIM3; NCT01006980) is also underway to compare the efficacy of RG7204/PLX4032 with that of dacarbazine in patients with previously untreated metastatic melanoma harboring a V600E BRAF mutation. Enrollment for this study (target of 680 patients) is expected to be completed by December 2010 with primary outcome data (overall survival) expected in 2011/2012.
6. Mechanisms of resistance
The mechanisms by which a subpopulation of tumor cells survive BRAF inhibition upon initial treatment and subsequently emerge as radiographically and clinically evident growing tumors following a months‐long response have not yet been elucidated. However, the framework for understanding them has been divided into primary/acute and secondary/acquired resistance. While the first wave of results from studies in which resistance studies purely in vitro or analyzing the small numbers of tumor samples that have been acquired at the time of disease progression are awaited, hypotheses can be formulated based on available molecular data in BRAF mutant tumors and the precedents set by other oncogene targeted therapies in other cancers.
6.1. Tumor cell heterogeneity
One hypothesis to explain the survival of large or small subpopulations of melanoma cells in patients treated with selective BRAF inhibitors is the presence of BRAF wild‐type cells within one or more metastatic lesions which otherwise contain predominantly BRAF mutant tumor cells. However, several groups investigated this possibility by analyzing multiple surgically resected or biopsied tumors from the same patient. The nearly uniform conclusion of these analyses is that BRAF mutation is homogeneously present in all tumor cells within a single metastatic nodule and across the spectrum of multiple metastatic sites (Gorden et al., 2003; Omholt et al., 2003; Sensi et al., 2006) while such heterogeneity may be observed in premalignant melanocytic proliferations (Lin et al., 2009).If such heterogeneity existed, even for a tiny subpopulation of cells, one would expect that an analysis of BRAF mutation in tumors from progressing patients would reveal that the dominant population at that time was BRAF wild‐type.
Another dimension of which heterogeneity may manifest in BRAF mutant tumors is the percentage of cells that are going through the cell cycle during the course of BRAF inhibitor therapy. Laboratory investigation suggests that such a non‐cycling fraction may exist and render such tumor cells immune to the effects of a BRAF inhibitor (Jaksch et al., 2008; Roesch et al., 2010). However, it has not yet been possible to characterize such a subpopulation in a tumor specimen taken directly from a patient. It is known that within a BRAF mutant melanoma tumor, there is heterogeneity regarding MEK and ERK phosphorylation, suggesting that other factors can regulate pathway activity beyond activated BRAF itself (Houben et al., 2008; Venesio et al., 2008).
6.2. Concomitant mutations
For almost as long as BRAF mutations have been known in melanoma, there has been evidence that other somatic genetic changes commonly occur within the same tumors. This is clearly the case for PTEN mutation or deletion, p16 inactivation by mutation, deletion, or promoter hypermethylation, AKT amplification, cyclin D amplification, and CDK4 amplification or mutation (Stahl et al., 2004; Tsao et al., 2004; Curtin et al., 2005; Gray‐Schopfer et al., 2006; Muthusamy et al., 2006; Smalley et al., 2008). The exact percentage of human melanoma tumors that harbors each of these genetic alterations in addition to BRAF mutation is not currently known, but is being cataloged in clinical trial populations currently. In preclinical system it appears that PTEN loss, AKT activation, cyclin D/CDK4 activation confer resistance to BRAF inhibitors in cell lines that harbor an activating BRAF mutation (Smalley et al., 2006, 2008).
6.3. Drug delivery
In diverse cancer models systems, including melanoma, heterogeneity and tumor, blood supply and nutrient delivery have been documented. This has been related to effective delivery of cytotoxic chemotherapies in other tumors. And, the administration of anti‐angiogenic therapy with a result reduction in regions of tumor hypoxia has been associated with improved delivery of chemotherapy (Willett et al., 2004). This is thought to be the basis by which anti‐angiogenic therapies enhanced the antitumor activity of chemotherapy in colon and lung cancer (Hurwitz et al., 2004; Sandler et al., 2006). Whether limited delivery of BRAF inhibitors into hypoxic compartment within melanoma tumors might account for low local drug concentration and lack of efficacy remains unknown.
6.4. Compensatory signaling and resistance mutations
Chronic myelogenous leukemia, gastrointestinal stromal tumor, and non‐small cell lung cancer, oncogene directed therapy targeting abl kinase, mutated c‐KIT, or mutated epidermal growth factor receptor creates a selective pressure for the emergence of mutations in the target enzymes that resistance inhibitory effects of these therapies (Branford et al., 2002; Antonescu et al., 2005; Bean et al., 2007). If such a mechanism underlies acquired resistance to BRAF in melanoma, simply sequencing BRAF in tumors biopsied at the time of disease progression should uncover them. The acquisition of additional genetic alterations that restore MAP kinase pathway signaling even in the presence of oncogenic BRAF and a BRAF inhibitor is another possibility that requires exploration. In some cases of EGFR mutant lung cancer treated with EGFR kinase inhibitors, there is evidence that amplification of c‐MET is capable of restoring downstream signal transduction even while EGFR is still being inhibited (Bean et al., 2007). The number of signaling molecules and pathways that ultimately activate the MAP kinase pathway is large, therefore the investigation into such a mechanism of acquired resistance must be broad‐based.
7. Combination regimens
As in the cases where oncogene targeted therapy has proven successful in other cancers, the discovery of newly acquired genetic alterations that mediate resistance following initial response to BRAF targeted therapy would logically lead to sequential treatment strategies. As much attention has been paid to this line of investigation, combination strategies are being investigated preclinically and clinically to counter some of the known or anticipated mechanism of primary/acute resistance.
It has been known since the Phase I trial of RG7204/PLX4032 that significant, but incomplete, inhibition of the MAP kinase pathway is achieved at the maximum tolerated doses (Flaherty et al., 2010). And, preclinical evidence suggests that even a small amount of residual MAP kinase pathway activity is sufficient to mediate cell survival (Paraiso et al., 2010). Thus, one strategy that is supported by in vitro data is to combine an allosteric MEK inhibitor with a selective BRAF inhibitor, in the hope of driving down MAP kinase pathway signaling to even lower levels. It is possible that such a combination will not be tolerable in terms of on‐target toxicity to normal tissues, given that selective BRAF inhibitors cause upregulation of the MAP kinase pathway in cells with wild‐type BRAF, while MEK inhibitors would be expected to counter that (Hatzivassiliou et al., 2010; Heidorn et al., 2010; Poulikakos et al., 2010). Such a combination is currently being investigated in the phase 1/2 trial combining GSK2118436 and GSK1120212 (NCT01072175).
A similarly high priority strategy for building upon single agent BRAF targeted therapy is to take advantage of the knowledge that PTEN deletion, AKT amplification, and p16/cyclin D/CDK4 aberrations are commonly found in association with BRAF mutation. As there are pharmacologic inhibitors of both CDK4 and the PI3‐kinase pathway, which are dysregulated by PTEN deletion and AKT amplification, combination regimens with these agents should be feasible in the near future in appropriately genetically defined subpopulations of melanoma patients harboring BRAF mutations and the relevant alterations in these other pathways.
The emergence of a new generation of clinically effective molecularly targeted agents that reverse anergy in T‐cells by blocking inhibitory receptors on their surface has provided another class of novel therapies to consider combining with selective BRAF inhibitors. Ipilimumab, a CTLA4 blocking antibody, has demonstrated a survival advantage in a Phase III trial for the first time in history of clinical investigation in metastatic melanoma (Hodi et al., 2010). It has been hypothesized that therapies which cause cell death acutely named prime the immune system for a secondary antitumor response and there is preclinical evidence to support this (Schumacher et al., 2006; Begley and Ribas, 2008). Furthermore, there is recent evidence that selective BRAF inhibitors cause regulation of melanocyte lineage antigens on the surface of BRAF mutant melanoma cells allowing for better T‐cell recognition (Boni et al., 2010). And, there appears to be no inhibitory effect on T‐cell function with such selective BRAF inhibitors, presumably because T‐cells rely on CRAF as the isoform that mediates MAP kinase pathway activity at that level. With several lines of evidence supporting a potentially positive interaction between BRAF targeted therapy and immunotherapy, there is increasing enthusiasm in the melanoma field to initiate investigations with the newer generation of immunologic agents as well as cytokine‐based immunotherapy.
Lastly, the combination of anti‐angiogenic therapy with BRAF targeted therapy is of interest due to the success of this approach in other cancers where cytotoxic chemotherapy was utilized as the tumor directed backbone. The difficulty in knowing how to proceed with such a strategy in melanoma is that individually effective anti‐angiogenic therapy has not yet been clinically developed. There is some evidence that the VEGF ligand targeted antibody, bevacizumab, can modestly augment the activity of largely inactive cytotoxic chemotherapies in melanoma (O'Day et al., 2009). However, there is also evidence that melanoma angiogenesis is driven by other factors that are at least as important as VEGF in the setting. Basic FGF is one such factor that has been implicated in melanoma pathophysiology, and is notable that the most highly active single agent anti‐angiogenic agent evaluated to date is an orally available inhibitor of both VEGF and FGF receptors (Fruehauf et al., 2008).
8. Conclusions
The discovery of BRAF mutations in melanoma and the emergence of highly selective inhibitors that have produced evidence of unprecedented clinical benefit have energized the melanoma field. At the same time, the clinical limitations of this therapy are already evident and attention must be focused on understanding the other molecular mediators of melanoma cell survival that maintain a subpopulation of tumor cells in the face of BRAF targeted therapy. Similarly, it is critical to understand what prior changes occur in the tumor that permit growth following theresponse to BRAF inhibitors. These two lines of investigation will provide the most rational basis for assembling combinations for sequential treatment strategies to make the next quantum leap in outcome for this patient population.
Puzanov Igor, Burnett Patrick and Flaherty Keith T., (2011), Biological challenges of BRAF inhibitor therapy, Molecular Oncology, 5, doi: 10.1016/j.molonc.2011.01.005.
References
- Antonescu, C.R. , Besmer, P. , Guo, T. , Arkun, K. , Hom, G. , Koryotowski, B. , Leversha, M.A. , Jeffrey, P.D. , Desantis, D. , Singer, S. , Brennan, M.F. , Maki, R.G. , DeMatteo, R.P. , 2005. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin. Cancer Res.. 11, 4182–4190. [DOI] [PubMed] [Google Scholar]
- Bean, J. , Brennan, C. , Shih, J.Y. , Riely, G. , Viale, A. , Wang, L. , Chitale, D. , Motoi, N. , Szoke, J. , Broderick, S. , Balak, M. , Chang, W.C. , Yu, C.J. , Gazdar, A. , Pass, H. , Rusch, V. , Gerald, W. , Huang, S.F. , Yang, P.C. , Miller, V. , Ladanyi, M. , Yang, C.H. , Pao, W. , 2007. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. U.S.A.. 104, 20932–20937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley, J. , Ribas, A. , 2008. Targeted therapies to improve tumor immunotherapy. Clin. Cancer Res.. 14, 4385–4391. [DOI] [PubMed] [Google Scholar]
- Bollag, G. , Hirth, P. , Tsai, J. , Zhang, J. , Ibrahim, P.N. , Cho, H. , 2010. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 467, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni, A. , Cogdill, A.P. , Dang, P. , Udayakumar, D. , Njauw, C.N. , Sloss, C.M. , Ferrone, C.R. , Flaherty, K.T. , Lawrence, D.P. , Fisher, D.E. , Tsao, H. , Wargo, J.A. , 2010. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res.. 70, 5213–5219. [DOI] [PubMed] [Google Scholar]
- Branford, S. , Rudzki, Z. , Walsh, S. , Grigg, A. , Arthur, C. , Taylor, K. , Herrmann, R. , Lynch, K.P. , Hughes, T.P. , 2002. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 99, 3472–3475. [DOI] [PubMed] [Google Scholar]
- Corominas, M. , Kamino, H. , Leon, J. , Pellicer, A. , 1989. Oncogene activation in human benign tumors of the skin (keratoacanthomas): is HRAS involved in differentiation as well as proliferation?. Proc. Natl. Acad. Sci. U.S.A.. 86, 6372–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin, J.A. , Fridlyand, J. , Kageshita, T. , Patel, H.N. , Busam, K.J. , Kutzner, H. , Cho, K.H. , Aiba, S. , Brocker, E.B. , LeBoit, P.E. , Pinkel, D. , Bastian, B.C. , 2005. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med.. 353, 2135–2147. [DOI] [PubMed] [Google Scholar]
- Davies, H. , Bignell, G.R. , Cox, C. , Stephens, P. , Edkins, S. , Clegg, S. , 2002. Mutations of the BRAF gene in human cancer. Nature. 417, 949–954. [DOI] [PubMed] [Google Scholar]
- Flaherty, K.T. , Puzanov, I. , Kim, K.B. , Ribas, A. , McArthur, G.A. , Sosman, J.A. , 2010. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med.. 363, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen, K. , Klintenas, M. , Osterstrom, A. , Dimberg, J. , Monstein, H.J. , Soderkvist, P. , 2004. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis. 25, 527–533. [DOI] [PubMed] [Google Scholar]
- Fruehauf, J.P. , Lutzky, J. , McDermott, D.F. , Brown, C.K. , Pithavala, Y.K. , Bycott, P.W. , Shalinsky, D. , Liau, K.F. , Niethammer, A. , Rixe, O. , 2008. Axitinib (AG-013736) in patients with metastatic melanoma. J. Clin. Oncol.. 26, (Suppl) (Abstract No. 9006) [Google Scholar]
- Garnett, M.J. , Marais, R. , 2004. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 6, 313–319. [DOI] [PubMed] [Google Scholar]
- Gorden, A. , Osman, I. , Gai, W. , He, D. , Huang, W. , Davidson, A. , Houghton, A.N. , Busam, K. , Polsky, D. , 2003. Analysis of BRAF and N-RAS mutations in metastatic melanoma tissues. Cancer Res.. 63, 3955–3957. [PubMed] [Google Scholar]
- Goydos, J.S. , Mann, B. , Kim, H.J. , Gabriel, E.M. , Alsina, J. , Germino, F.J. , 2005. Detection of B-RAF and N-RAS mutations in human melanoma. J. Am. Coll. Surg.. 200, 362–370. [DOI] [PubMed] [Google Scholar]
- Gray-Schopfer, V.C. , Cheong, S.C. , Chong, H. , Chow, J. , Moss, T. , Abdel-Malek, Z.A. , Marais, R. , Wynford-Thomas, D. , Bennett, D.C. , 2006. Cellular senescence in naevi and immortalisation in melanoma: a role for p16?. Br. J. Cancer. 95, 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaban, R. , Zhang, W. , Bacchiocchi, A. , Cheng, E. , Parisi, F. , Ariyan, S. , 2010. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment Cell Melanoma Res.. 23, 190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou, G. , Song, K. , Yen, I. , Brandhuber, B.J. , Anderson, D.J. , Alvarado, R. , 2010. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 464, 431–435. [DOI] [PubMed] [Google Scholar]
- Heidorn, S.J. , Milagre, C. , Whittaker, S. , Nourry, A. , Niculescu-Duvas, I. , Dhomen, N. , 2010. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 140, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi, F.S. , O'Day, S.J. , McDermott, D.F. , Weber, R.W. , Sosman, J.A. , Haanen, J.B. , Gonzalez, R. , Robert, C. , Schadendorf, D. , Hassel, J.C. , Akerley, W. , van den Eertwegh, A.J. , Lutzky, J. , Lorigan, P. , Vaubel, J.M. , Linette, G.P. , Hogg, D. , Ottensmeier, C.H. , Lebbe, C. , Peschel, C. , Quirt, I. , Clark, J.I. , Wolchok, J.D. , Weber, J.S. , Tian, J. , Yellin, M.J. , Nichol, G.M. , Hoos, A. , Urba, W.J. , 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med.. 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich, K.P. , Herter, S. , Tien, J. , Wong, L. , Berry, L. , Chan, J. , 2009. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res.. 69, 3042–3051. [DOI] [PubMed] [Google Scholar]
- Houben, R. , Vetter-Kauczok, C.S. , Ortmann, S. , Rapp, U.R. , Broecker, E.B. , Becker, J.C. , 2008. Phospho-ERK staining is a poor indicator of the mutational status of BRAF and NRAS in human melanoma. J. Invest. Dermatol.. 128, 2003–2012. [DOI] [PubMed] [Google Scholar]
- Hurwitz, H. , Fehrenbacher, L. , Novotny, W. , Cartwright, T. , Hainsworth, J. , Heim, W. , Berlin, J. , Baron, A. , Griffing, S. , Holmgren, E. , Ferrara, N. , Fyfe, G. , Rogers, B. , Ross, R. , Kabbinavar, F. , 2004. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med.. 350, 2335–2342. [DOI] [PubMed] [Google Scholar]
- Jaksch, M. , Munera, J. , Bajpai, R. , Terskikh, A. , Oshima, R.G. , 2008. Cell cycle-dependent variation of a CD133 epitope in human embryonic stem cell, colon cancer, and melanoma cell lines. Cancer Res.. 68, 7882–7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph, E.W. , Pratilas, C.A. , Poulikakos, P.I. , Tadi, M. , Wang, W. , Taylor, B.S. , 2010. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc. Natl. Acad. Sci. U.S.A.. 107, 14903–14908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopetz, S. , Desai, J. , Chan, E. , Hecht, J.R. , O'Dwyer, P.J. , Lee, R.J. , 2010. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J. Clin. Oncol.. 28, (Suppl. 15) (Abstract No. 3534) [Google Scholar]
- Lacouture, M.E. , McArthur, G.A. , Chapman, P.B. , Ribas, K. , Flaherty, K.T. , Lee, R.J. , 2010. PLX4032 (RG7204), a selective mutant RAF inhibitor: clinical and histologic characteristics of therapy-associated cutaneous neoplasms in a Phase I trial. J. Clin. Oncol.. 28, (15 Suppl.) (Abstract No. 8592) [Google Scholar]
- Lee, J.T. , Li, L. , Brafford, P.A. , van den, E.M. , Halloran, M.B. , Sproesser, K. , 2010. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res.. 23, 820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libra, M. , Malaponte, G. , Navolanic, P.M. , Gangemi, P. , Bevelacqua, V. , Proietti, L. , 2005. Analysis of BRAF mutation in primary and metastatic melanoma. Cell Cycle. 4, 1382–1384. [DOI] [PubMed] [Google Scholar]
- Lin, J. , Takata, M. , Murata, H. , Goto, Y. , Kido, K. , Ferrone, S. , Saida, T. , 2009. Polyclonality of BRAF mutations in acquired melanocytic nevi. J. Natl. Cancer Inst.. 101, 1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur, G.A. , Puzanov, I. , Ribas, A. , Chapman, P.B. , Kim, K.B. , Sosman, J.A. , 2010. Early FDG-PET responses to PLX4032 in BRAF-mutant advanced melanoma. J. Clin. Oncol.. 28, (Suppl. 15) [Abstract No. 8529] [Google Scholar]
- McCubrey, J.A. , Steelman, L.S. , Chappell, W.H. , Abrams, S.L. , Wong, E.W. , Chang, F. , 2007. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta. 1773, 1263–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthusamy, V. , Hobbs, C. , Nogueira, C. , Cordon-Cardo, C. , McKee, P.H. , Chin, L. , Bosenberg, M.W. , 2006. Amplification of CDK4 and MDM2 in malignant melanoma. Genes Chromosomes Cancer. 45, 447–454. [DOI] [PubMed] [Google Scholar]
- O'Day, S.J. , Kim, K.B. , Sosman, J.A. , 2009. BEAM: a randomized Phase II study evaluating the activity of bevacizumab in combination with carboplatin plus paclitaxel in patients with previously untreated advanced melanoma. Eur. J. Cancer Suppl.. 7, (3) (Abstract No. 23LBA) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omholt, K. , Platz, A. , Kanter, L. , Ringborg, U. , Hanson, J. , 2003. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin. Cancer. Res.. 9, 6483–6488. [PubMed] [Google Scholar]
- Paraiso, K.H. , Fedorenko, I.V. , Cantini, L.P. , Munko, A.C. , Hall, M. , Sondak, V.K. , Messina, J.L. , Flaherty, K.T. , Smalley, K.S. , 2010. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer. 102, 1724–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos, P.I. , Zhang, C. , Bollag, G. , Shokat, K.M. , Rosen, N. , 2010. RAF inhibitors transactivate RAF dimers and ERK signaling in cells with wild-type BRAF. Nature. 464, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch, A. , Fukunaga-Kalabis, M. , Schmidt, E.C. , Zabierowski, S.E. , Brafford, P.A. , Vultur, A. , Basu, D. , Gimotty, P. , Vogt, T. , Herlyn, M. , 2010. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 141, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala, E. , Mologni, L. , Truffa, S. , Gaetano, C. , Bollag, G.E. , Gambacorti-Passerini, C. , 2008. BRAF silencing by short hairpin RNA or chemical blockade by PLX4032 leads to different responses in melanoma and thyroid carcinoma cells. Mol. Cancer Res.. 6, 751–759. [DOI] [PubMed] [Google Scholar]
- Sandler, A. , Gray, R. , Perry, M.C. , Brahmer, J. , Schiller, J.H. , Dowlati, A. , Lilenbaum, R. , Johnson, D.H. , 2006. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med.. 355, 2542–2550. [DOI] [PubMed] [Google Scholar]
- Schumacher, L.Y. , Vo, D.D. , Garban, H.J. , Comin-Anduix, B. , Owens, S.K. , Dissette, V.B. , Glaspy, J.A. , McBride, W.H. , Bonavida, B. , Economou, J.S. , Ribas, A. , 2006. Immunosensitization of tumor cells to dendritic cell-activated immune responses with the proteasome inhibitor bortezomib (PS-341, Velcade). J. Immunol.. 176, 4757–4765. [DOI] [PubMed] [Google Scholar]
- Sebolt-Leopold, J.S. , Herrera, R. , 2004. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer. 4, 937–947. [DOI] [PubMed] [Google Scholar]
- Sensi, M. , Nicolini, G. , Petti, C. , Bersani, I. , Lozupone, F. , Molla, A. , Vegetti, C. , Nonaka, D. , Mortarini, R. , Parmiani, G. , Fais, S. , Anichini, A. , 2006. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene. 25, 3357–3364. [DOI] [PubMed] [Google Scholar]
- Smalley, K.S. , Haass, N.K. , Brafford, P.A. , Lioni, M. , Flaherty, K.T. , Herlyn, M. , 2006. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol. Cancer Ther.. 5, 1136–1144. [DOI] [PubMed] [Google Scholar]
- Smalley, K.S. , Lioni, M. , Dalla Palma, M. , Xiao, M. , Desai, B. , Egyhazi, S. , Hansson, J. , Wu, H. , King, A.J. , Van Belle, P. , Elder, D.E. , Flaherty, K.T. , Herlyn, M. , Nathanson, K.L. , 2008. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAFV600E-mutated melanomas. Mol. Cancer Ther.. 7, 2876–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosman, J., Kim, K., Schuchter, L., Gonzales, R., Pavlick, A., Weber, J., McArthur, G., Hutson, T., Lawrence, D., Moschos, S., Flaherty, K., Hersey, P., Kefford, R., Chmielowski, B., Amaravadi, R., Puzanov, I., Li, J., Bhattacharya, S., Nolop, K., Lee, R., Joe, A., Ribas, A. An open-label, multicenter Phase II study of continuous oral dosing of RG7204 (PLX4032) in previously treated patients with BRAFV600E mutation-positive metastatic melanoma. Society of Melanoma Research 2010; 7th International Melanoma Congress & 4th Meeting of Interdisciplinary Melanoma/Skin Cancer Centers; 4–7 November 2010; Sydney, Australia.
- Stahl, J.M. , Sharma, A. , Cheung, M. , Zimmerman, M. , Cheng, J.Q. , Bosenberg, M.W. , Kester, M. , Sandirasegarane, L. , Robertson, G.P. , 2004. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res.. 64, 7002–7010. [DOI] [PubMed] [Google Scholar]
- Tsai, J. , Lee, J.T. , Wang, W. , Zhang, J. , Cho, H. , Mamo, S. , 2008. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. U.S.A.. 105, 3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao, H. , Goel, V. , Wu, H. , Yang, G. , Haluska, F.G. , 2004 Feb. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J. Invest. Dermatol.. 122, (2) 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venesio, T. , Chiorino, G. , Balsamo, A. , Zaccagna, A. , Petti, C. , Scatolini, M. , Pisacane, A. , Sarotto, I. , Picciotto, F. , Risio, M. , 2008. In melanocytic lesions the fraction of BRAFV600E alleles is associated with sun exposure but unrelated to ERK phosphorylation. Mod. Pathol.. 21, 716–726. [DOI] [PubMed] [Google Scholar]
- Wan, P.T. , Garnett, M.J. , Roe, S.M. , Lee, S. , Niculescu-Duvaz, D. , Good, V.M. , 2004. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 116, 855–867. [DOI] [PubMed] [Google Scholar]
- Willett, C.G. , Boucher, Y. , di Tomaso, E. , Duda, D.G. , Munn, L.L. , Tong, R.T. , Chung, D.C. , Sahani, D.V. , Kalva, S.P. , Kozin, S.V. , Mino, M. , Cohen, K.S. , Scadden, D.T. , Hartford, A.C. , Fischman, A.J. , Clark, J.W. , Ryan, D.P. , Zhu, A.X. , Blaszkowsky, L.S. , Chen, H.X. , Shellito, P.C. , Lauwers, G.Y. , Jain, R.K. , 2004. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat. Med.. 10, 145–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Higgins, B. , Kolinsky, K. , Packman, K. , Go, Z. , Iyer, R. , 2010. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res.. 70, 5518–5527. [DOI] [PubMed] [Google Scholar]
- Zhang, B.H. , Guan, K.L. , 2000. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J.. 19, 5429–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]