

Abstract
For a panel of cancer related proteins, the aim was to shed light on which molecular level the expression of each protein was mainly regulated in breast tumors, and to investigate whether differences in regulation were reflected in different molecular subtypes. DNA, mRNA and protein lysates from 251 breast tumor specimens were analyzed using appropriate microarray technologies. Data from all three levels were available for 52 proteins selected for their known involvement in cancer, primarily through the PI3K/Akt pathway. For every protein, in cis Spearman rank correlations between the three molecular levels were calculated across all samples and within each intrinsic gene expression subtype, enabling 63 comparisons altogether due to multiple gene probes matching to single proteins. Subtype‐specific relationships between the three molecular levels were studied by calculating the variance of subtype‐specific correlation and differences between overall and average subtype‐specific correlation. The findings were validated in an external dataset comprising 703 breast tumor specimens. The proteins were sorted into four groups based on the calculated rank correlation values between the three molecular levels. Group A consisted of eight proteins with significant correlation between DNA copy number levels and mRNA expression, and between mRNA expression and protein expression (Bonferroni adjusted p < 0.05). Group B consisted of 14 proteins with significant correlation between mRNA expression and protein expression. Group C consisted of 15 proteins with significant correlation between copy number levels and mRNA expression. For the remaining 25 proteins (group D), no significant correlations was observed. Stratification of tumors according to intrinsic subtype enabled identification of positive correlations between copy number levels, mRNA and protein expression that were undetectable when considering the entire sample set. Protein pairings that either demonstrated high variance in correlation values between subtypes, or between subtypes and the total dataset were studied in particular. The protein expression of cleaved caspase 7 was most highly expressed, and correlated highest to CASP7 gene expression within the basal‐like subtype, accompanied by the lowest amounts of hsa‐miR‐29c. Luminal A‐like subtype demonstrated highest amounts of hsa‐miR‐29c (a miRNA with a putative target sequence in CASP7 mRNA), low expression of cleaved caspase 7 and low correlation to CASP7 gene expression. Such pattern might be an indication of hsa‐miR‐29c miRNA functioning as a repressor of translation of CASP7 within the luminal‐A subtype. Across the entire cohort no correlation was found between CCNB1 copy number and gene expression. However, within most gene intrinsic subtypes, mRNA and protein expression of cyclin B1 was found positively correlated to copy number data, suggesting that copy number can affect the overall expression of this protein. Aberrations of cyclin B1 copy number also identified patients with reduced overall survival within each subtype. Based on correlation between the three molecular levels, genes and their products could be sorted into four groups for which the expression was likely to be regulated at different molecular levels. Further stratification suggested subtype‐specific regulation that was not evident across the entire sample set.
Keywords: Breast cancer, Gene expression, DNA copy number, Protein expression, Molecular subtypes, Correlation, RPPA, Cyclin B1, CASP7
Highlights
Correlation analyses indicate regulatory relationships between genes and proteins.
Stratification by molecular subtype disclosed relationships between molecular levels.
Cyclin B1 copy number aberrations show subtype‐dependent prognostic relevance.
Elevated CASP7 protein in basal‐like tumors is accompanied by low levels of hsa‐mir‐29c.
Abbreviations
- RPPA
reverse phase protein array
- DBCG
Danish Breast Cancer Group
- RT
radiotherapy
- CMF
cyclophosphamide methotrexate fluorouracil
- LRR
locoregional recurrence
- DM
distant metastasis
- CBC
contralateral breast cancer
- TCGA
The Cancer Genome Atlas
- HE
Hematoxylin and Eosin
- BCA
bicinchoninic acid
- PCF
piecewise constant fit
- aCGH
array Comparative Genomic Hybridization
- FE
Feature Extraction
- KS
Kolmogorov–Smirnov
- VSC
variance of subtype-specific correlation
1. Introduction
Breast cancer is a heterogeneous disease from a molecular as well as a clinical point of view. Features at the DNA, mRNA and protein level (in the following referred to as “molecular levels”) have been utilized to identify clinically relevant subgroups of breast cancers. Protein expression of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2/ERBB2) are routinely analyzed by immunohistochemical staining methods and utilized in prognostic evaluation and for treatment decision (Fisher et al., 1989, 1980; Varley et al., 1987). ER and HER2 are important in regulation of proliferation of cancer cells, and therapies targeting these receptors have improved the prognosis for patients with tumors expressing these proteins (Geyer et al., 2006; Murphy and Fornier, 2010; Early Breast Cancer Trialists' Collaborative Group, 1998). Elevated HER2 protein expression is highly correlated to both elevated ERBB2 gene expression and amplification of the ERBB2 chromosomal region 17q11.2–q12 (Pauletti et al., 1996). Protein expression of ER is highly correlated to the expression of the ESR1 gene, however copy number gain of ESR1 is rarely observed (Horlings et al., 2008). Evidently, the major regulatory mechanisms driving the expression of these two receptors are different, as the low frequency of ESR1 aberrations at the chromosomal level cannot explain the broad span of gene and protein expression (ESR1 and ER, respectively). Regulatory mechanisms involving transcription factors, miRNA, linc‐RNA, DNA methylation, and other translational and post‐translational modifications as well as protein stability all play roles in regulating mRNA and protein abundance (deConinck et al., 1995; Jaenisch and Bird, 2003; Wilusz et al., 2009; Zhao and Jensen, 2009; Stingele et al., 2012). Phosphorylation and de‐phosphorylation of specific protein epitopes regulate protein activity and how proteins communicate in signal transduction pathways such as the PI3K/Akt pathway (Supplementary Figure 1). The Akt proteins phosphorylate serine and threonine residues on a number of proteins, and thus regulate cellular processes such as protein synthesis, proliferation, apoptosis, synaptic signaling, and homeostasis and glucose metabolism. In cellular quiescence, the PI3K/Akt pathway remains dormant, but during carcinogenesis, activation and dysregulation of this pathway commonly occur. The molecular mechanisms involved in dysregulation of this pathway are important for understanding the biology of cancer and for discovering potential new drug targets (Castaneda et al., 2010). We therefore wanted to study the influence of copy number levels and mRNA expression on protein levels of selected proteins, and whether the relationship between these molecular levels differed between the previously identified molecular intrinsic subtypes of breast cancer (Sorlie et al., 2001). In this study, 52 proteins known to be involved in the PI3K/Akt pathway and other cancer related processes were selected. We compared DNA copy number, mRNA expression and protein expression (assessed by aCGH, whole genome gene expression microarrays and RPPA respectively) across intersections of a total of 251 breast tumors (Supplementary Figure 2). Correlation analyses were utilized to sort 63 possible pairings of the 52 unique proteins into four groups on the basis of the inferred molecular level of regulation.
2. Materials and methods
2.1. Patient material
A total of 251 fresh frozen tumor specimens were available for patients in the “DBCG82 b & c” trials diagnosed between October 1982 and March 1990. Patients in these studies were defined as “high‐risk”; positive lymph nodes, and/or tumor invasion to surrounding skin or pectoral fascia, and/or tumor size larger than five cm. The patients were randomized to receive radiation therapy (RT) or no radiation therapy after mastectomy. All patients received adjuvant therapy. Premenopausal women were given the “DBCG b”‐protocol involving CMF (cyclophosphamide, methotrexate, fluorouracil; eight cycles if they got RT or 9 cycles if no RT) (Kyndi et al., 2009). Postmenopausal women were given the “DBCG c”‐protocol involving 30 mg Tamoxifen daily for 1 year. The patients were followed up at regular intervals the first 10 years or until the first recurrence, death or new primary cancer. Recorded endpoints were locoregional recurrence (LRR), distant metastases (DM), contralateral breast cancer (CBC) and death (Nielsen et al., 2006). In the survival analyses of CCNB1 copy number, the original dataset was expanded by additional four patient cohorts named “MDG” (Haakensen et al., 2010),”Uppsala” (Muggerud et al., 2010), “ULL”(Langerod et al., 2007; Russnes et al., 2010), and “MicMa” (Naume et al., 2007; Wiedswang et al., 2003). Validation of the correlations was performed utilizing copy number, mRNA expression and protein expression data for 703 invasive breast carcinomas in The Cancer Genome Atlas data portal (TCGA; http://tcga‐data.nci.nih.gov/tcga/ (2012a)).
2.1.1. Sample handling
All tumor specimens were cut in three while placed on dry ice. Individual sections from the center piece were used for total RNA extraction, DNA and protein extraction. Sections were prepared from the two flanking pieces and used for histopathological analyses including Hematoxylin and Eosin (HE) staining. In total, 251 tumor biopsies were utilized to generate DNA copy number, mRNA expression and/or protein expression data, the intersection of samples is shown in Supplementary Figure 2.
2.2. Microarray experiments
2.2.1. Reverse phase protein array
Protein expression was analyzed using reverse phase protein array (RPPA) developed for analyzing the expression of single proteins across multiple samples simultaneously (Tibes et al., 2006). In contrast to forward phase array format, the reverse phase array immobilizes the samples to be analyzed (to a nitrocellulose‐coated slide), and each array is incubated with an antibody specific for a protein of interest (Charboneau et al., 2002; Espina et al., 2003). The antibodies utilized in this study were primarily targeting proteins involved in PI3K/Akt pathway or otherwise cancer related. The RPPA data were load control‐adjusted, log2‐transformed and protein centered. RPPA data was available for 210 of the 251 tumors. The RPPA dataset has been previously published and described (Hennessy et al., 2010).
2.2.2. aCGH – array Comparative Genomic Hybridization
DNA copy number was assessed using Comparative Genomic Hybridization 244K Agilent Microarrays (Agilent Technologies, Santa Clara, CA). The array contains ∼236,000 oligonucleotide probes (60‐mers) spanning coding and non‐coding genomic regions. The median spacing in coding and non‐coding regions is 7.4 kb and 16.5 kb respectively (Kirchhoff et al., 2009). Experimental handling of the data has been described previously (Baumbusch et al., 2008; Russnes et al., 2010). aCGH data were available for 196 out of 251 patients.
2.2.3. Gene expression analyses
To measure gene (mRNA) expression, we utilized the Human Genome Survey Microarray version 2.0 (Applied Biosystem). The whole genome array contains 32,878 probes (60‐mers) covering 29,098 transcripts. Quality was assessed by the QC‐parameters of the internal controls, and 20 arrays were removed due to reduced quality or low present call, returning a total of 194 unique samples with good quality gene expression data. No filtering of probes was applied in order to gain data for all genes coding for the selected proteins in the study. Data were log2‐transformed, quantile normalized and median gene centered. Gene expression data were available for 194 out of 251 patients. The mRNA expression data are available from GEO (GSE24117), and experimental details of this dataset have been described previously (Myhre et al., 2010).
2.2.4. miRNA expression analyses
miRNA expression was measured by one‐color microarray “Human miRNA Microarray Kit (V2) (Agilent Technologies) according to protocol supplied by manufacturer (miRNA Microarray System v1.5). This miRNA array is based on miRBase release 10.1 and contains 723 human and 76 human viral miRNAs. The array contains 15,744 features (60‐mers) including 953 control probes, thus each miRNA is in average replicated ∼18 times on the array. In brief, total RNA was isolated with TRIzol (Invitrogen), and 100 ng RNA was used as input for labeling and hybridization to the array. Scanning was performed on Agilent Scanner G2565A. miRNA data were processed using Feature Extraction (FE) version 11, log2‐transformed and quantile normalized in Genespring v.10.0 (Agilent Technologies). Quality was assessed by the QC parameters in FE. miRNAs with a present call in less than 20% of samples were removed resulting in 408 miRNAs considered to be expressed in this set of human breast tumor.
2.3. Matching of proteins and gene‐probes
To perform cis‐correlation (correlation between DNA, mRNA and protein for the same genomic region) analyzes across the three molecular levels, the aCGH‐ and mRNA probes were matched to the corresponding 52 proteins from the RPPA platform. For proteins, where expression data were available for both total protein and phosphorylated epitopes, data from each antibody were matched to the same gene expression probe (e.g. ER, ERp118, ERp167 were all matched to ESR1 gene probe). In cases where one protein was composed of several subunits (multiple compartment protein) encoded for by more than one mRNA transcript, expression of that total protein was matched to each individual gene probe (e.g. AMPK protein consists of gene products from seven different genes PRKAA1, PRKAA2, PRKAB1, PRKAB2, PRKAG1, PRKAG2, and PRKAG3). In cases where multiple gene probes were available for the same gene (i.e. one gene product), each gene probe was matched to the corresponding protein (total protein and phosphorylated epitopes). E.g. IGF1R‐probe id “186013” and “159991” were both matched to IGFR1 and IGFR1p. Finally, the gene expression probes were matched to all aCGH probes that spanned the genomic region of the gene. For each sample the probes representing the genomic region of a single gene showed identical copy number values. Hence, the signal from one single probe was utilized as representative for that genomic region. In instances where the annotated genomic region for a specific gene differed between the gene expression and aCGH platforms, the aCGH probe showing the best correlation value was selected. The complete matching details are listed in Supplementary Table 1.
The multiple protein epitopes (including phosphorylated epitopes), aCGH probes and gene probes were arranged in 135 possible cis‐combinations for 52 unique proteins. In order to reduce redundancy in the comparison, one DNA and gene‐probe for each unique gene/protein representative for each molecular level (DNA, mRNA and protein) was selected, favoring probes that returned the highest in cis‐correlation for each pairing. At the protein level antibodies targeting the total protein were selected for most pairings except for HER2 and Stat6, due to technical issues for these two total‐protein directed antibodies. Hence, for HER2 and Stat6, the phosphorylated epitopes HER2‐p1248 and Stat6‐p641 were included instead of total protein. HER2‐p1248 represents the activated form of the protein, which thus enabled us to study the functional form of HER2. For the proteins caspase 7 and PARP, RPPA data was only available for antibodies targeting the cleaved (and active) version of the proteins. For proteins consisting of multiple gene products, all genes were included. The comparisons of correlations between phosphorylated epitopes‐ and total protein and copy number and gene expression are available as Supplementary material (Supplementary Table 2). Copy number logR‐values, mRNA expression and protein expression for the pairings are available in supplementary data files 1, 2 and 3.
2.4. Gene expression subtyping
Gene expression based subtyping of the tumor samples was performed based on the intrinsic gene list of Sorlie et al. (2003). From the original intrinsic gene list containing 561 gene probes, we identified 374 gene probes on our gene expression platform by matching entrez gene id and gene symbol. Gene expression data for these genes were median‐centered per probe across 194 samples. Pearson correlation between each tumor specimen and each of the 5 subtype centroids was estimated using the 374 gene probes. Each sample was annotated with the subtype of the respective centroid to which the sample correlated highest. A minimum Pearson correlation threshold >0.15 was applied. Samples achieving a maximum correlation below 0.15 were annotated “unclassified”. The following classification was obtained Luminal A (n = 63), Luminal B (n = 25), HER2‐enriched (n = 27), Basal‐like (n = 29) and Normal‐like (n = 32), unclassified (n = 18).
2.5. Inter‐platform correlation between genes and proteins
A Kolmogorov–Smirnov (KS) test was utilized to test for a normal distribution in each of the three datasets. A KS‐test calculates maximum distance between the cumulative frequency curve and the best‐fit normal curve of the data, and determines the significance of this distance. If the KS‐test returns a low (significant) p‐value (p < 0.05) the normal distribution hypothesis is rejected. If the KS test returns a high p‐value the normality assumption is not falsified. In SPSS a KS test for normality was done for each genomic DNA probe, mRNA expression probe and antibody (protein expression). The KS tests returned highly significant p‐values for the copy number data, and mostly non‐significant p‐values for the mRNA‐ and protein expression data. Due to the non‐normally distribution of aCGH data, Spearman rank correlation was selected as correlation estimator in all comparisons (Gry et al., 2009; Spearman, 2010). For each pair of gene–gene or gene–protein the Spearman rank cis‐correlation was estimated across the three platforms using SPSS 17. After Bonferroni adjustment Spearman correlation values >0.3 reflected statistically significant positive correlations (p‐value < 0.05). Correlation values were categorized as “high” >0.5, “medium” between 0.5 and 0.3, “low” between 0.3 and 0.15, “none” between 0.15 and −0.15 and “negative” <−0.15. Correlations between copy number and mRNA expression were estimated for the selected genes, using 154 breast tumor samples. Correlations between mRNA expression and protein expression were found using 164 breast tumor samples, and correlations between copy number and protein expression for the 62 pairings (52 unique proteins) of gene/proteins were found using 161 breast tumors (Supplementary Figure 2).
2.5.1. Correlation within five gene expression subtypes
Correlation analyses between the three molecular levels of expression were performed within each of the five molecular subtypes. The in cis‐correlations between copy number and mRNA expression were calculated for a total of 140 samples divided into 5 subtypes. The in cis‐correlations between mRNA expression and protein expression were calculated for a total of 151 samples divided into 5 subtypes. The in cis‐correlation between copy number and protein expression were calculated for a total of 122 samples divided into 5 subtypes (Table 1).
Table 1.
Number of samples in the five gene‐expression subtypes utilized in subtype specific correlation estimates across three molecular levels.
| Comparison | Luminal A | Luminal B | HER2‐enr. | Basal | Normal | Total |
|---|---|---|---|---|---|---|
| CN‐GX | 52 | 20 | 20 | 25 | 23 | 140 |
| GX‐PX | 55 | 22 | 21 | 25 | 29 | 151 |
| CN‐PX | 44 | 19 | 16 | 22 | 21 | 122 |
In order to assess subtype‐specific correlations the variance of the subtype‐specific correlations was calculated (the variance of these correlations were called VSC). A high VSC pointed to candidate genes and proteins with potential regulatory differences between subtypes.
In order to compare overall correlation to subtype‐specific correlations, a variable “Z” was defined. Let “A i” be overall Spearman correlation coefficient “A” of protein “i”, and “B i” be the weighted mean (weighted by number of samples in each subtype) of the five subtype‐specific correlations “B” of protein “i”. Given the difference A i − B i = Z , a positive Z indicates lower subtype‐specific correlations, while a negative Z indicates higher subtype‐specific correlations compared to the overall correlation for the given protein.
2.6. miRNA target prediction
For the two gene (proteins) PECAM1 (CD31), and CASP7, all miRNAs predicted to have “conserved” targets in the base‐sequences of these two genes were identified using Targetscan [www.targetscan.org] (2012b). After filtering the miRNA data by present call in at least 20% of the samples, five miRNAs with predicted target in CASP7, and two miRNAs with predicted target in PECAM1 were identified. Analysis of variance (ANOVA) was utilized to test for significant differences in expression of proteins and miRNAs (cleaved caspase 7 and CD31 and targeted miRNAs) between the five expression subtypes.
2.7. Cyclin B1 in relation to outcome
For each molecular level the samples were divided in three groups (GAIN/HIGH, NONE/MEDIUM and LOSS/LOW) according to their copy number and expression levels of cyclin B1. At the copy number level an extended dataset of 506 cancer patients with 15 years overall survival was utilized to increase the numbers of gains and loss in this region. Samples with a copy number value (LogR) >0.20 were assigned to the GAIN‐group and <−0.20 were assigned to the LOSS group, and the remaining to the NONE‐group. For mRNA and protein expression the samples were assigned to the groups using the 25%–75% percentile borders of expression. Using 15 years overall survival as outcome, Kaplan–Meier plots and log rank tests were used for assessing significance of overall survival for CN, GX and PX expression of CCNB1/cyclin B1.
2.8. Validation
An external validation set was utilized to validate the correlation values observed in the original dataset. Data on copy number‐, mRNA expression‐ and RPPA data were available for 39 out of the 52 protein pairings for a total of 703 invasive breast carcinomas in The Cancer Genome Atlas database (TCGA (2012a)). With a minimum overlap of 388 samples (Supplementary Figure 3), Spearman rank correlations were calculated for the three comparisons and the 39 protein pairings (Supplementary Table 9). In order to test whether the pattern of correlation was similar, the correlation values from the validation set were correlated to the correlation values of the original set across these 39 protein pairings both overall and within each subtype (PAM50). Data were available for luminal A (n = 220), luminal B (n = 127), ERBB2‐like (n = 55), basal‐like (n = 93) and normal‐like (n = 8). Spearman correlation was calculated across all 503 samples, and within each subtype (except for the normal‐like subtype that had too few samples).
3. Results
3.1. Classification of proteins based on correlation between molecular levels
In‐cis correlations were calculated between all three molecular levels (DNA copy number, mRNA and protein expression) for the 52 selected proteins (63 pairings to mRNA probes). Based on the pattern of the correlation values, the proteins were categorized into four groups A–D (Figure 1a). Figure 2 exemplifies scatterplots between the three levels for one protein from each of the four groups.
Figure 1.
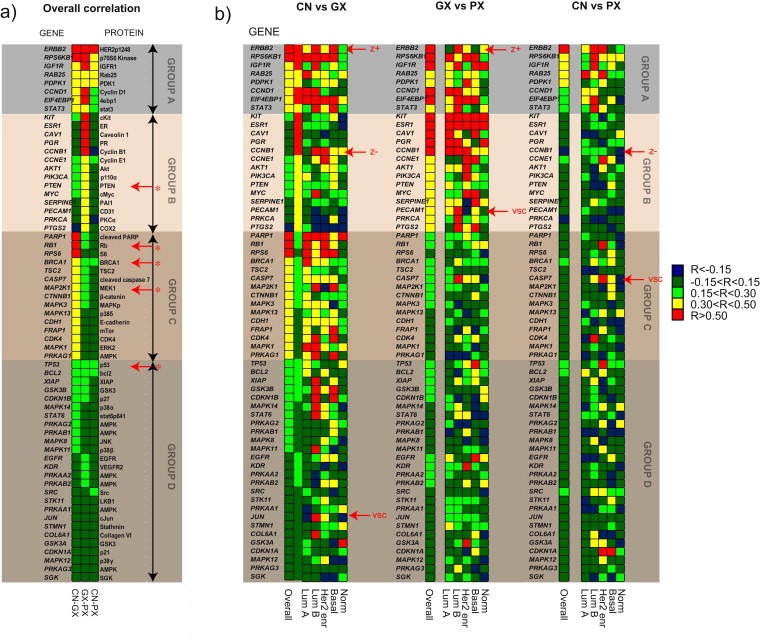
Multilevel correlation. a) Overall correlation between copy number and mRNA expression (CN‐GX), mRNA expression and protein expression (GX‐PX) and copy number and protein expression (CN‐PX). Gene and proteins are divided into 4 groups: “A” protein expression influenced by copy number, “B” protein expression significantly influenced by mRNA expression but not copy number, “C” mRNA expression significantly influenced by copy number, but not correlated to protein. “D” gene/proteins show no significant correlation between any molecular levels. Red arrows with “*” denote proteins demonstrating a significant correlation to copy number after reducing the effect of non‐aberrant samples. b) Subtype specific correlations for all three comparisons. Proteins with highest VSC (Variance in subtype‐specific correlation) and |Z‐scores| (absolute difference in average subtype‐specific correlation and overall correlation) are indicated with red arrows.
Figure 2.
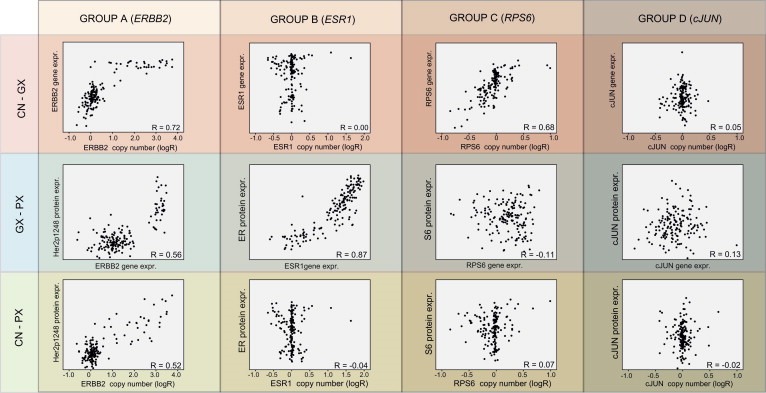
Group A–D scatterplot examples. The four groups of correlation (A–D) are exemplified with scatterplots for one gene/protein pairing within each group and for all three comparisons. Group A is exemplified by ERBB2 that demonstrates how copy number correlates to protein expression via high correlation to mRNA. Group B is exemplified by ESR1 for which mRNA expression correlates highly with protein expression but not with copy number. Group C is exemplified by RPS6 that demonstrates a high correlation between copy number and mRNA expression, but not with protein expression. Group D is exemplified by cJUN that demonstrates no evidence of correlation between any molecular levels of expression.
3.1.1. Group A: protein‐expression influenced by copy number levels (CN‐PX)
Group A consisted of eight pairs for which copy number was significantly correlated to mRNA expression, and mRNA expression was significantly correlated to protein expression (Spearman's rank correlation >0.3) (Figure 1a, Figure 2). Copy number level was significantly correlated to protein expression for five of the proteins (HER2p1248, p70S6 Kinase, IGFR1, Rab25, and PDK1) and correlated positively (albeit not significantly) for the remaining three (4ebp1, Cyclin D1 and stat3). The positive correlation across the three molecular levels indicated that copy number level variations made an impact on expression of these proteins. High copy number gain or loss (for this purpose defined as |logR| > 0.4) in corresponding genomic regions, were observed in more than 15% of the samples for 6 of the 8 proteins. For the remaining two genes, IGF1R and STAT3, high copy number gain or loss was 5%, and 7% respectively. Variance in copy number values was on average four‐fold higher in group A compared to group B, C and D (Supplementary Table 3) supporting copy number as a major driver of expression of the group A proteins.
3.1.2. Group B: protein‐expression influenced by mRNA expression levels only (GX‐PX)
Group B consisted of 14 protein pairings for which the only significant correlation observed was between mRNA expression and protein expression (Spearman's rank correlation >0.3) (Figure 1a, Figure 2). These proteins were cKit, ER, Caveolin 1, PR, cyclin B1, Cyclin E1, Akt, p110α, cMYC, PTEN, PAI1, CD31, PKCα, and COX2. In contrast to group A, only 5 out of 14 proteins in group B showed |logR| > 0.4 in corresponding genomic regions in more than 15% of the samples. Thus, the lower variance in copy number levels reduced the likelihood for copy number aberrations of these genes to be major drivers of mRNA and protein expression. As observed for PTEN, Rb, BRCA1, MEK1 and p53 (see Section 3.1.5).
3.1.3. Group C: copy number levels influence mRNA expression but not protein expression (CN‐GX)
Group C consisted of 15 pairings for which the only significant correlation was between copy number and mRNA expression (Spearman's rank correlation > 0.3) (Figure 1a, Figure 2). In this group, we find Rb, cleaved PARP, S6, TSC2, β‐catenin, MAPKp, p38δ, BRCA1, MEK1, E‐cadherin, cleaved caspase 7, mTor, CDK4, ERK2 and AMPK (for AMPK significant correlation was found for one out of 7 pairings to genes encoding subunits of this protein). Strikingly, only 4 out of 15 genes in group C showed |logR| > 0.4 in more than 15% of the samples, less than what was observed for group B which demonstrated no positive correlation to copy number. Hence, low copy number variance was not the sole explanation for low correlation between copy number and mRNA expression in group B.
3.1.4. Group D: protein expression with no significant correlation to mRNA expression and copy number levels
Group D consisted of the remaining 25 pairings that showed no significant correlation between any of the molecular levels (Figure 1a, Figure 2). This group included p53, bcl2, AMPK (6 genes encoding 6 out of 7 subunits of this protein‐complex), p27, GSK3 (two genes encoding two subunits), XIAP, Stat6p641, LKB1, cJun, VEGFR2, Src, p21, Collagen VI, Stathmin, EGFR, JNK, p38, and SGK. Only 2 out of 25 genomic regions in group D showed |logR| > 0.4 in more than 15% of the samples. This low number of copy number aberrations was a probable explanation for the lack of copy number driven mRNA and/or protein expression in this group.
3.1.5. Correlation among samples with most extreme copy number values
Low observed correlation between copy number and mRNA/protein expression may be caused either by an actual lack of association or insufficient variability in copy number levels in the respective genomic regions. To minimize the dilutive effect of non‐aberrant cases, samples populating the top and bottom 10 percentiles of copy number values were selected for each protein pairing (data not shown). The correlations obtained from these subsets of analyses increased, and despite the reduced power due to reduced sample‐size, five additional proteins for which copy number levels correlated significantly to protein expression were identified: Rb (RB1), p53 (TP53), BRCA1 (BRCA1), MEK1 (MAP2K1), PTEN (PTEN) (Highlighted in Figure 1a). In addition, an increase in copy number to mRNA expression correlation was observed for Akt, p110α, cMyc, EGFR, CD31 and PAI1 (data not shown). This demonstrates that lack of copy number variation can result in lowered correlation.
3.2. Within‐subtype correlation
To explore subtype‐specific regulatory relationships, correlations were calculated within each intrinsic molecular subtype separately (Sorlie et al., 2001). Next, the variance of these within‐subtype correlations (henceforth referred to as variance of subtype‐specific correlations, or VSC) was calculated for each protein for each comparison. A high VSC points to protein pairings that demonstrate large differences in correlation between the molecular levels. These differences could be a result of variable levels of heterogeneity in expression values at one or more molecular levels between the subtypes, or be an indication of differences in molecular regulatory relationships between subtypes. CASP7 and PECAM1 were selected based on high VSC as potential candidates for subtype‐specific miRNA regulation. (Figure 1, Figure 3, Supplementary Tables 4–6, see Section 3.2.1). Validation of subtype‐specific correlation was performed using an external breast tumor data set from the Cancer Genome Atlas (TCGA) (http://tcga‐data.nci.nih.gov/) (2012a) (see Materials and Methods). Positive and significant correlations were observed for most subtypes between the within subtype correlation values of the original dataset and the validation set (Supplementary Table 7).
Figure 3.
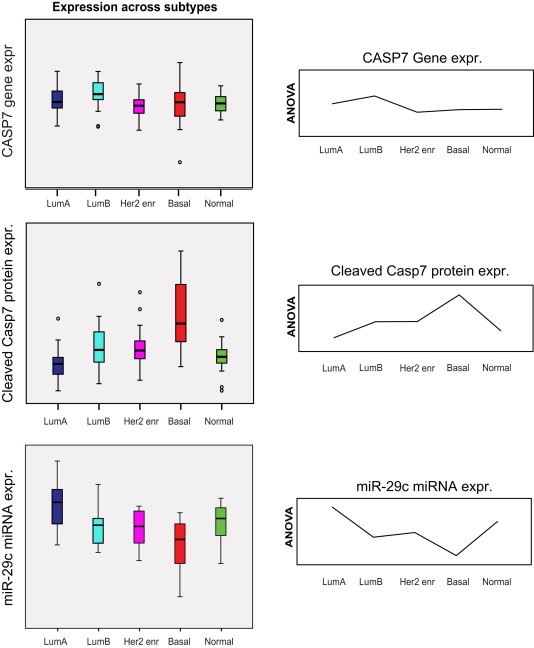
CASP7‐, cleaved caspase 7‐ and hsa‐mir29c‐expression across subtypes. Box‐plot of expression of CASP7 mRNA in the top panel demonstrates no significant difference in expression levels between subtypes (ANOVA p‐value = 0.061). A highly significant difference in protein expression across subtypes is observed for cleaved caspase 7 (ANOVA p‐value < 0.001), and a significant but opposite pattern of expression is observed for the miRNA hsa‐mir‐29c (p < 0.001), predicted to target CASP7 mRNA. The mean expression curves from the ANOVA analyses are included for visualization of the mirror like expression patterns across subtypes for cleaved caspase 7 and the hsa‐mir‐29c.
3.2.1. Differences in cleaved caspase 7 expression across subtypes may be due to miRNA repressed translation
Cleaved caspase 7 protein demonstrated a high VSC in comparisons of both copy number and mRNA expression correlated to protein expression (Supplementary Table 6). The subtype‐specific correlations ranged from −0.27 (Normal‐like) to 0.65 (HER2 enriched) (Supplementary Table 5, Figure 1b). In order to test whether these correlation differences could be assigned to differences in CASP7 expression between subtypes, multigroup comparisons were performed at the three molecular levels. Although, a Kruskal–Wallis test identified a significant difference in CASP7 copy number‐levels between the HER2‐enriched and normal‐like subtypes (p = 0.04), ANOVA analyses of mRNA and protein expression identified no significant differences in mRNA expression levels of CASP7 between any of the subtypes (most significant p‐value = 0.07 between luminal B and Her2‐enriched). At the protein level the basal‐like subtype demonstrated significantly higher level of cleaved caspase 7 protein expression compared to all other subtypes (p < 0.001) (Figure 3). This was surprising considering that the antibody selectively reacts with cleaved caspase 7 suggesting that total protein level contributes at least in part to cleavage. Since mRNA expression of CASP7 could not explain the differences in protein expression, expression of six miRNAs predicted (www.targetscan.org) to target CASP7 mRNA was explored (supplementary data file 4). The hypothesis was that reduced correlation to protein expression could be caused by a corresponding repression of translation due to increased expression of miRNAs targeting CASP7. Three miRNAs predicted to target CASP7 mRNA were found significantly differentially expressed between subtypes; hsa‐miR‐29a, hsa‐miR‐29c and hsa‐miR‐29c*. Strikingly, the expression pattern of hsa‐miR‐29c (and hsa‐miR‐29c*) showed a mirrored‐like image to expression of cleaved caspase 7 protein across the five subtypes (Figure 3). Thus, the higher level of cleaved caspase 7 expression observed in basal‐like tumors could be an effect of less repression of translation by hsa‐miR‐29c.
3.2.2. Within‐subtype correlation versus between‐subtype correlation
For each of the three comparisons of correlation and for each protein a parameter Z was calculated. Z describes the difference between the “overall correlation” and the “average of the five subtype‐specific correlation values”, weighted by the number of samples in each subtype. Hence, Z quantifies the discrepancy between across‐subtype correlation and average within‐subtype correlation. A large positive Z signified the presence of low correlations within subtypes, combined with substantial inter‐subtype differences that resulted in high overall correlation. A large negative Z on the other hand, indicates the presence of high correlations within subtypes, masked by inter‐subtype differences that resulted in low overall correlation (Supplementary Tables 4–6). HER2p1248 (ERBB2) obtained the largest positive Z score in the copy number to mRNA expression and mRNA expression to protein expression comparisons, and the second largest score (after PDK1) in the copy number to protein expression comparison. The lower subtype‐specific correlation values can be explained by a more homogenous expression of ERBB2 in most subtypes, except for luminal B, as shown in Supplementary Figure 4.
3.2.3. Subtype stratification identifies copy number as a minor driver of cyclin B1 expression
The largest negative Z was found for cyclin B1 (CCNB1) in both the copy number to mRNA expression and the copy number to protein expression comparison. A negative Z means that the overall correlation (across the entire dataset) is lower than the mean correlation value across the five subtypes for this pairing. Since supervised subtyping in principle reduces the within‐group variance and thus penalizes conditions for obtaining a positive correlation, a negative Z is a rare phenomenon that can disclose hidden regulatory relationship being masked in the overall dataset. The overall correlation between CCNB1 copy number and mRNA expression was −0.07, while the weighted mean subtype correlation was 0.33 (Z = −0.40) (Supplementary Table 4, Figure 1b). Thus, although no overall correlation between copy number and mRNA expression of CCNB1 was observed, a positive copy number to mRNA expression correlation across tumors within each of the five subtypes was found (significant p < 0.05 in four subtypes, not significant in luminal A (Figure 4a)). Box‐plots demonstrated a similar pattern for mRNA and protein levels, and distinct from what was observed for copy number of CCNB1 between subtypes (Figure 4b). The subtype‐dependent correlations between copy number and gene expression of CCNB1 were validated in an external breast tumor dataset (TCGA, http://tcga‐data.nci.nih.gov/). As observed in the original dataset, no evidence for significant correlation between CCNB1 copy number and gene expression was found across a sample set of 481 breast tumors (R = 0.01), however significant positive correlations ranging from R = 0.22 to R = 0.47 were observed within each subtype (classified by PAM50, Supplementary Table 8). The normal‐like class was omitted due to few samples (for details see materials and methods).
Figure 4.
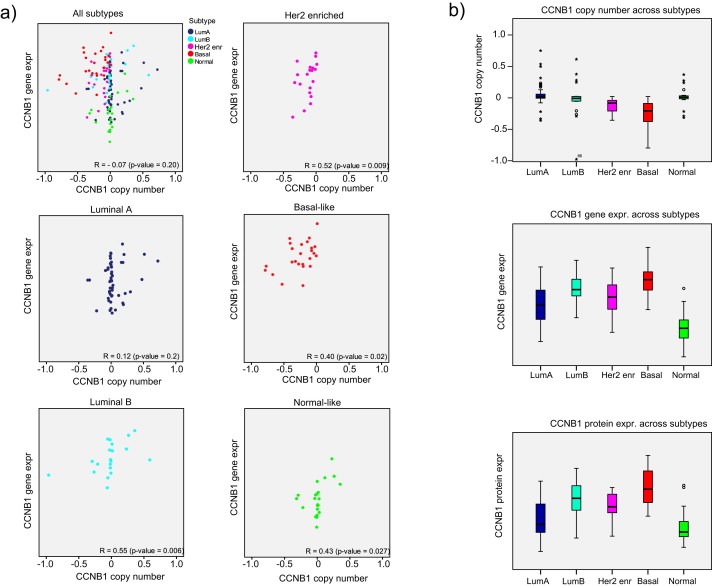
CCNB1 copy number and mRNA expression overall and within subtypes a) Correlation plots between CN (x‐axis) and GX (y‐axis) for all samples and the five individual subtypes on the same scale. Stratification according to molecular subtypes reveals significant and positive CN‐GX‐correlation in 4 out of 5 subtypes. b) Box plots showing copy number of CCNB1 across subtypes (top panel), mean mRNA expression of CCNB1 across subtypes (middle panel), and mean protein expression of cyclin B1 across subtypes (bottom panel).
3.2.4. Copy number, mRNA and protein expression of cyclin B1 and overall survival
Increased levels of cyclin B1 protein have been reported as a marker for poor prognosis in breast cancer (Aaltonen et al., 2009; Agarwal et al., 2009; Nimeus‐Malmstrom et al., 2010). In accordance with these studies, we observed that both elevated mRNA and protein expression were associated with significantly worse 15 years overall survival (Figure 5a–c). Since the number of samples with CCNB1 copy number aberrations was low (Figure 4a), this effect on overall survival was further assessed in an extended tumor set including copy number data (aCGH) from 506 breast tumors (Supplementary datafile 5). Since, mRNA and protein expression data were not available for this extended sample set, subtypes could not be constructed for all samples. Hence, a stratification according to ER‐status was performed in order to obtain groups with sufficient events (Table 2, Figure 5d–f). Copy number loss of CCNB1 (LOSS‐group) was associated with reduced overall survival (p‐value < 0.001). Also, copy number gain of CCNB1 (GAIN‐group) demonstrated borderline significant reduced overall survival compared to NONE‐group (p‐value = 0.068).
Figure 5.
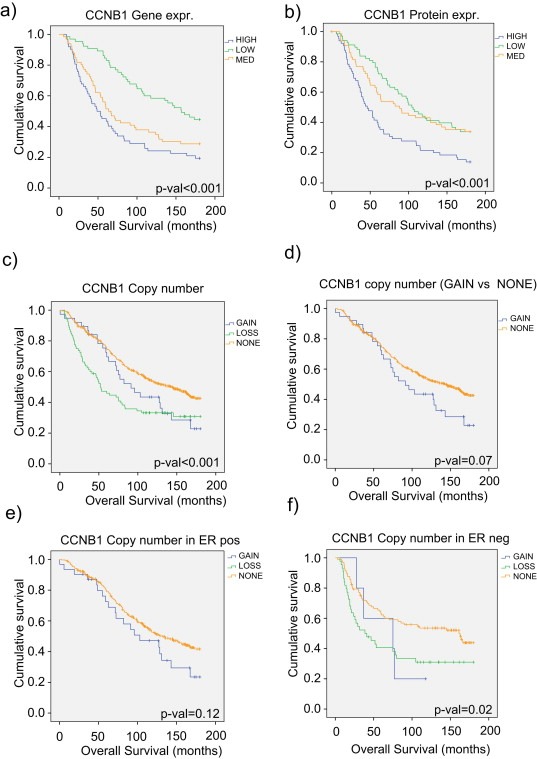
Breast cancer survival in different subgroups of patients stratified by CCNB1. Kaplan–Meier curves (15 years overall survival) for patients classified into groups according to a) mRNA expression and b) protein expression of cyclin B1 (HIGH = blue (25% of samples with highest expression values), MEDIUM = orange, LOW = green (25% with lowest expression values). c) Copy number levels of the CCNB1 genomic region in the extended 506 sample set (GAIN = blue (logR > 0.2), NONE = orange, LOSS = green (logR < −0.2)). d) Copy number levels of CCNB1, GAIN vs NONE groups only, in the 506 sample set, demonstrating borderline significant reduced 15 years survival for the gain group. e) Copy number levels in ER positive patients (no copy number loss of CCNB1 was observed within this group), and f) copy number levels within ER negative patients.
Table 2.
Number of samples with gain, loss or none‐aberrations of CCNB1 within subtypes, ER positive, and ER negative tumors of the 506‐tumor dataset.
| Luminal A | Luminal B | HER2 enr | Basal‐like | Normal‐like | ER + | ER − | |
|---|---|---|---|---|---|---|---|
| None | 123 | 31 | 43 | 19 | 35 | 273 | 97 |
| Gain | 15 | 4 | 1 | 0 | 6 | 31 | 5 |
| Loss | 4 | 6 | 10 | 31 | 4 | 34 | 44 |
As observed earlier, cyclin B1 measured at all three molecular levels differed between breast cancer subtypes. Basal‐like subtype (represented in here with the ER‐negative group) was a confounder with copy number loss in the CCNB1 region, and luminal subtypes (associated with ER‐positivity) were confounding with CCNB1 copy number gain. Within the ER‐positive tumors, the CCNB1 GAIN‐group demonstrated worse overall survival although not significant (p‐value = 0.112) compared to NONE‐group (no tumors in the ER‐positive group showed loss of CCNB1) (Figure 5d–f). Reduced overall survival observed in the LOSS‐group was most likely related to the adverse prognosis of ER‐negative and/or basal‐like tumors. While the reduced overall survival in the GAIN‐group could be a result of copy number driven elevated cyclin B1 expression.
4. Discussion
Protein levels are influenced by regulation of transcription, translation and protein stability. Estimations of how much of the variance of protein abundance can be explained by mRNA abundance varies (Lu et al., 2007; Stranger et al., 2007). Our results from the panel of 52 proteins indicate that mRNA expression correlates significantly to protein expression for about ∼35% of the proteins (in cis). However, although statistically significant these correlation values are in range between 0.3 and 0.9, and mRNA expression data cannot account for all protein expression data. The impact copy number levels have on mRNA expression depends on the extent of chromosomal aberrations in the genomic region in question, and it has been proposed that copy number variation capture ∼18% of variation in gene expression across diseases (Stranger et al., 2007). Breast cancer is a heterogeneous disease and copy number aberrations vary within and between breast tumors, and between breast cancer subtypes (Russnes et al., 2010; Hyman et al., 2002; Pollack et al., 2002). Therefore the number of genes and proteins having their expression directly influenced by copy number aberrations is likely to vary between subtypes. Overall, we observed that copy number had a significant impact on expression of 35% of the genes (genome‐wide), and on about 12–15% of the selected proteins in this study. However, the study was based on a set of proteins, selected for their proposed roles in breast cancer, and not for the purpose of representing the proteome as a whole. Several proteins were also measured by using antibodies directed toward their functionally active configuration (eg. HER2p1248, Cleaved caspase 7, Cleaved PARP). Extrapolating the percentages of significantly correlated pairings onto a whole genome or proteome setting is therefore not valid.
4.1. Correlation as a basis for proposing regulation of protein expression
Although correlation does not imply causation, the biological established relationship between DNA copy number levels, mRNA and protein expression makes the assumption of causation less bold. For copy number levels of a genomic region to be identified as a possible driver of increased or decreased mRNA and protein expression the region must be affected by aberrations. The majority of the genomic regions corresponding to the proteins in group A have been reported to be commonly affected by copy number aberrations in breast cancer (Barbareschi et al., 1997; Cheng et al., 2004; Hui et al., 1996; Maurer et al., 2009; Reis‐Filho et al., 2006; Rojo et al., 2007; Roy et al., 2010). Hence, based on the positive correlation observed, expression of the eight proteins in group A was proposed to be under significant copy number influence (in cis).
The 14 proteins in group B demonstrated medium (R > 0.3) or high (R > 0.5) mRNA expression to protein expression correlation but low or no correlation of either to copy number. Hence, expression of the proteins in group B was likely to be under direct influence of mRNA expression levels (in cis) but not copy number. The mechanisms regulating expression of these proteins are likely to be others than copy number aberrations (e.g. transcription factor activity, methylation, mutations).
The mRNA expression of proteins in group C was influenced by copy number levels, but no evidence was found for correlation between mRNA and protein expression. For some proteins this lack of correlation might be due to post‐translational cleavage. For instance caspase 7 is activated through its cleavage by caspase 3 and calpain‐1 (Gafni et al., 2009), and PARP1 is cleaved by caspases in order to prevent energy depletion and necrosis, and to promote apoptosis (D'Amours et al., 2001). In this study only antibodies targeting the cleaved version of these two proteins were utilized and correlation can be disrupted if the extent of protein cleavage is independent of levels of mRNA expression.
Low correlation between all levels, as observed for the remaining proteins (group D), could be an effect of multiple factors. A high proportion of non‐aberrant tumors may dilute potential direct relationships between copy number and mRNA expression. Indeed, a general increase in copy number to mRNA expression correlations was observed when only including tumor samples with copy number aberrations in the analysis.
Degradation of both mRNA and protein would make an impact on the mRNA expression to protein expression correlation (in both group C and D). In addition, two of the proteins in group (C) and D consist of two or more subunits, and each subunit is encoded by different genes (two genes for GSK3 and seven for AMPK). Proteins encoded by a single gene have higher probability of achieving positive correlation, simply due to a more linear relationship. Noise can also be introduced by an unspecific antibody. This was observed for EGFR, as the antibody targeting EGFR used in this study also detected high levels of HER2 protein. Hence, nonspecific detection of HER2 may have disrupted mRNA expression to protein expression correlation of EGFR, rendering unreliable data for this protein. Also, the genetic libraries are under constant review, and although the human genome sequence is known, probe annotation is often modified and erroneous matching of probes to genes and proteins may occur. Gene probes on the microarray might also detect alternative splice variants which can contribute noise into the comparisons (Shi et al., 2006). Improvement and optimization of antibodies and technologies have likely led to the higher correlations observed for the validation set. However, the correlation values obtained from comparing the two datasets were highly significant, giving support to the proposed regulatory relationships.
4.2. Variance, heterogeneity and possible impact on correlation
High correlation requires systematic heterogeneity in the data, and variance is a measure of heterogeneity. Indeed, the variance of copy number values was on average four‐fold higher for group A proteins compared to those in group B, C and D (Supplementary Table 3). Similarly, proteins in group B showed the highest variance of both mRNA, and protein expression values. Since different proteins and genes can differ in their range of expression (signal distribution), the level of variance is not directly comparable between different proteins. However, the variance of expression values (copy number, mRNA and protein expression) for the same protein was comparable between subtypes. When low correlation was observed in subtypes showing equal or higher variance as a subtype demonstrating high correlation, the resulting high VSC indicates subtype‐specific differences in regulatory mechanisms (e.g CASP7).
4.3. Tissue heterogeneity
Tissue heterogeneity within each tumor can potentially bring noise to the data. Microdissection was not performed, and different cell types could potentially contribute differently into the amounts of DNA, mRNA and protein. The correlations were a study of tumors as heterogeneous “organ” and not an examination of tumor epithelial cells specifically. However, a previous gene expression study of the DBCG 82 b & c cohort, took pathologic information regarding amount of tumor epithelial‐, stromal‐ and adipocytic cells into account, and concluded that variance in mRNA expression related to metastatic disease was derived from tumor epithelial cells (Myhre et al., 2010). Although the DNA, mRNA and protein lysates were prepared from adjacent pieces of the same tumor, they were not derived from the exact same cell fraction. Retrospectively, it would have been preferable to prepare isolates of DNA, mRNA and protein from one piece of the tumor and divided it into three fractions for downstream isolations. The issue of intratumoral heterogeneity has previously been studied in a technical assessment of RPPA data in comparison with gene expression and the levels of intratumor protein levels were found to be much less variable than intertumoral levels. Hence, despite challenges with intratumoral heterogeneity, RPPA provided accurate and reproducible analysis of protein expression (Hennessy et al., 2010).
4.4. miRNA regulation of cleaved caspase 7 expression
High VSC was observed for both copy number level to protein expression and mRNA expression to protein expression for cleaved caspase 7. Except for the normal‐like subtype the variance in expression values were similar between subtypes that demonstrated high (Luminal B, HER2‐enriched and basal) and low (luminal A) correlation values. Hence, differences in variance could not solely explain differences in correlation. Surprisingly cleaved caspase 7 correlated with mRNA levels suggesting that total caspase 7 contributes to the frequency of cleaved caspase 7 potentially due to increase substrate levels. Caspase 7 is cleaved by Calpain 1 (CAPN1) and caspase 3 (CASP3) but the basal‐like subtype did not demonstrate higher expression levels of these two genes that potentially could explain the higher levels of cleaved caspase 7 protein. However, at the miRNA level the basal‐like subtype demonstrated lowest expression of hsa‐miR‐29c which is predicted to target CASP7. Thus, the increased protein level in basal‐like tumors could be an effect of less translational repression, rather than increased level of CASP7 mRNA expression or excessive cleavage activity by CAPN1 or CASP3. The higher expression of these miRNAs in basal‐like subtype was further supported by a recent publication of miRNA expression from 101 early‐stage breast carcinomas where both hsa‐miR‐29c and hsa‐mir‐29c* were found to be among the top miRNAs that discriminated basal‐like from luminal A subtypes (Enerly et al., 2011). Elevated expression of hsa‐miR‐29c has been reported to be associated with favorable prognosis and decreased proliferation, migration, invasion and colony formation in mesothelioma cell lines (Pass et al., 2010). However, miRNAs exhibit a somewhat promiscuous nature in terms of which mRNA they can target, target prediction methods are uncertain, and their proposed “fine‐tuning” regulatory functions can both repress translation as well as destabilize mRNA. Therefore the biological signal‐to‐noise ratio in such data is expected to be low (Baek et al., 2008; Bartel, 2009; Guo et al., 2010), and functional studies are necessary to investigate the specific effect of these miRNAs.
4.5. Correlation within mRNA expression subtypes compared to overall correlation
Subdivision of samples into molecular gene‐expression subtypes was expected to influence correlation since variance in the overall group was divided into more homogeneous groups with expectedly less within‐group variance. Thus, subtyping penalized correlation as the variance in copy number and/or mRNA and protein expression values was reduced. In addition the sample sizes were smaller, making an impact on statistical power, and since more tests were performed more false positives would appear. A large positive Z (high overall correlation across all samples and low mean subtype‐specific correlation) was observed for ERBB2/HER2p1248 correlations, and was a consequence of dividing heterogeneous data into homogenous subgroups (such as the HER2‐enriched group). However, negative Z (low overall correlation and a high mean subtype‐specific correlation) as observed for CCNB1 copy number to mRNA correlation cannot be explained by the same mechanism. Due to the before mentioned sample size reduction and additional test issue, it was important to be able to validate the positive cyclin B1 correlation between copy number and mRNA expression within each subtype in the TCGA dataset. Stratification according to subtypes in both the original and the validation sample set demonstrated that copy number levels of the CCNB1 genomic region indeed did influence the total expression level of CCNB1. However, the majority of expression differences of CCNB1 observed between subtypes seemed to be driven by regulatory features other than copy number. This further emphasizes that analyses of larger heterogeneous datasets might mask associations that become evident when including subtype‐specific information.
4.6. Cyclin B1 and clinical outcome
In a study of three cyclins (cyclin B1, D1 and E1) across molecular subtypes, cyclin B1 was found over‐expressed in basal‐like breast tumors, both at the gene and protein level (Agarwal et al., 2009). Cyclin B1 is known to have important regulatory functions in the G2‐M checkpoint, and other studies have reported cyclin B1 as a marker for neoplasticity and aggressiveness in breast cancer (Aaltonen et al., 2009; Androic et al., 2008; Megha et al., 1999). In this study copy number loss of CCNB1 was primarily observed in the basal‐like subtype (Supplementary Figure 5). Hence, “basal‐likeness” was a confounder of CCNB1 copy number loss and the poor prognosis observed in the LOSS‐group was likely a confounding effect (Agarwal et al., 2009). Gain of CCNB1 was observed solely in the luminal‐like and the normal‐like groups (mostly ER‐positive tumors) (Table 2) and showed slightly worse prognosis compared to the non‐aberration group (p‐value = 0.12) of patients with ER‐positive tumors (Figure 5e). The reduced survival in this group may be due to the resulting elevated CCNB1 expression within this group (Figure 5b–c).
5. Conclusion
Correlation studies involving multiple molecular levels can propose regulatory relationships between DNA copy number, mRNA and protein expression. Based on correlation patterns across such comparisons, we were able to sort genes and proteins into different groups with proposed different regulatory mechanisms. Further stratification into gene expression subtypes was instrumental in discovering hidden associations across these levels. The variance in correlation between subtypes was utilized to identify possible subtype‐specific regulatory differences. High expression of cleaved caspase 7 protein was associated with low expression of hsa‐miR‐29c. Loss of CCNB1 copy number was associated with, but was not a probable driver of the poor overall survival observed for basal‐like tumors. Copy number gain of CCNB1 identified patients with increased cyclin B1 protein (and gene) expression and worse prognosis within the ER‐positive (luminal‐like) group.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SM carried out expression microarray experiments, performed statistical analyses and wrote the article. OCL performed statistical analyses and wrote the article. BH carried out RPPA experiments, study design, and critically read the article. MRA carried out miRNA experiments, wrote the article. MSC carried out RPPA experiments, analyzed RPPA data and critically read the article. JA was involved in sample handling (DBCG 82 b&c), clinical data, study design and critically read the article. JO was responsible for sample handling (DBCG 82 b&c), clinical data, study design, and critically read the article. Trine Tramm was involved in sample handling (DBCG 82 b&c), pathology and clinical data, and critically read the article. GM provided RPPA analyses of test (DBCG 82 b&c) and validation set (TCGA), study design, and wrote the article. TS was responsible for study design, supervised analyses and wrote the article. ALBD conceived the study, supervised analyses and wrote the article.
Supporting information
The following are the supplementary data related to this article:
Supplementary data file 1 DBCG 82 b & c data files: Segmented aCGH data (PCF‐values) for probes that each represents a genomic region that match to a corresponding gene‐probe in the 135 possible protein pairings. The pairings are numbered from 1 to 135 and the entity number in each data file refers to the same pairing.
Supplementary data file 2 DBCG 82 b & c data files: Gene expression data for probes that each represents one gene‐probe in the 135 possible protein pairings.
Supplementary data file 3 DBCG 82 b & c data files: Protein expression‐data for antibodies directed against total protein or phosphoprotein in the 135 possible protein pairings.
Supplementary data file 4 DBCG82 data file: miRNA expression data for six miRNAs that are predicted to target the CASP7 gene. Sample IDs and subtype information are included.
Supplementary data file 5 Extended data set containing 506 breast tumors with overall survival data (time in months), Status 1 = dead, 0 = Alive. LogR <> |0.2| defined as loss/gain.
Supplementary Table 1 Protein‐to‐gene matching table describing chromosomal position and gene name. First column shows the order of pairings in Figure 1. Second column corresponds to the pairing‐number in the three supplementary data files. Third column denotes the selected protein pairings used in the comparisons (marked “u”). Fourth column includes a comment annotation to highlight the phosphorylated epitopes. The remaining columns include gene/protein information as indicated.
Supplementary Table 2 Complete overall correlation table including all possible 135 protein pairings, spearman rank correlation and p‐value for the CN‐GX, PX‐GX and CN‐PX comparisons.
Supplementary Table 3 Table and histograms of variance in copy number and gene – and protein expression values within group A, B, C and D.
Supplementary Tables 4–6 Complete tables of all overall and within subtype correlations for CN‐GX (4).
Supplementary Tables 4–6 Complete tables of all overall and within subtype correlations for GX‐PX (5).
Supplementary Tables 4–6 Complete tables of all overall and within subtype correlations for CN‐PX (6).
Supplementary Table 7 Spearman rank correlation values between correlation values in the original dataset and the validation set, per subtype. The positive correlations validate the correlations found between the three combinations of CN, GX and PX for the selected pairings in the original dataset.
Supplementary Table 8 Validation set with PAM50‐subtyping; number of samples in each subtype and the correlation values of CN‐GX for CCNB1 (overall and within each subtype).
Supplementary Table 9 Spearman correlation values for 39 available protein pairings in the TCGA validation set. For each available protein pairing the correlation values from the validation set are found in the leftmost column and the correlation values from the original dataset are found in the rightmost column. The comparisons are sorted accordingly (“copy number vs gene expression”, “gene expression vs protein expression” and “copy number vs protein expression”).
Supplementary Figure 1 PI3K/Akt‐pathway. Illustration of the PI3K/Akt‐pathway. Proteins with available data on all three levels of measurements in this study are outlined in red. Green arrows indicate direct stimulatory modification and red lines indicate direct inhibitory modification between proteins.
Supplementary Figure 2 Sample sizes in the DBCG 82 b & c dataset (original). Venn‐diagram showing number of tumor samples with available copy number, mRNA expression and protein expression data in the DBCG 82 b & c dataset (original dataset).
Supplementary Figure 3 Sample sizes in the TCGA dataset (validation set). Venn‐diagram showing number of tumor samples with available copy number, mRNA expression and protein expression data in the TCGA dataset (validation set).
Supplementary Figure 4 a–d: Subtype‐specific correlation of the HER2 gene p1248 protein pairing. Scatter plots of three levels of comparisons, overall and subtype‐stratified, of the ERBB2/Her2p1248 gene‐protein pairing. a) Copy number plotted against mRNA expression. Correlation values and corresponding p‐values are given in each panel.
Supplementary Figure 4 a–d: Subtype‐specific correlation of the HER2 gene p1248 protein pairing. Scatter plots of three levels of comparisons, overall and subtype‐stratified, of the ERBB2/Her2p1248 gene‐protein pairing. b) mRNA expression plotted against protein expression. Correlation values and corresponding p‐values are given in each panel.
Supplementary Figure 4 a–d: Subtype‐specific correlation of the HER2 gene p1248 protein pairing. Scatter plots of three levels of comparisons, overall and subtype‐stratified, of the ERBB2/Her2p1248 gene‐protein pairing. c) copy number plotted against protein expression across all samples and for each of the five subtypes, separately. Correlation values and corresponding p‐values are given in each panel.
Supplementary Figure 4 a–d: Subtype‐specific correlation of the HER2 gene p1248 protein pairing. Scatter plots of three levels of comparisons, overall and subtype‐stratified, of the ERBB2/Her2p1248 gene‐protein pairing. Correlation values and corresponding p‐values are given in each panel. d) Box plots of copy number and gene and protein expression values illustrating the difference in heterogeneity (variance) between the subtypes at each molecular level.
Supplementary Figure 5 Copy number plot for CCNB1 across intrinsic gene expression subtypes. The overall frequency plot of copy number levels (gain = green, loss = red) across the whole sample set (upper panel) and frequency plots for each subtype (lower five panels). CCNB1 copy number aberrations differ between subtypes; loss of CCNB1 is mostly observed in the basal‐like subtype, while gain is mostly observed in the luminal‐like subtypes.
Acknowledgments
We gratefully acknowledge the TCGA Program for the availability of breast tumor data and for support in the analyses. This work was supported by grants from the Norwegian Cancer Society (To ALBD: 138296 – PR‐2008‐0108, To TS: 0332) and The Norwegian Research Council (To ALBD: 175240/S10 and 193387/V50, To TS: 163027)
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.02.018.
Myhre Simen, Lingjærde Ole-Christian, Hennessy Bryan T., Aure Miriam R., Carey Mark S., Alsner Jan, Tramm Trine, Overgaard Jens, Mills Gordon B., Børresen-Dale Anne-Lise, Sørlie Therese, (2013), Influence of DNA copy number and mRNA levels on the expression of breast cancer related proteins, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.02.018.
Contributor Information
Simen Myhre, Email: simen@amh.no.
Ole-Christian Lingjærde, Email: ole@ifi.uio.no.
Bryan T. Hennessy, Email: bryanhennessy74@gmail.com
Miriam R. Aure, Email: mirrag@rr-research.no
Mark S. Carey, Email: mark.carey@vch.ca
Jan Alsner, Email: jan@oncology.dk.
Trine Tramm, Email: tramm@oncology.dk.
Jens Overgaard, Email: jens@oncology.dk.
Gordon B. Mills, Email: gmills@mdanderson.org
Anne-Lise Børresen-Dale, Email: a.l.borresen-dale@medisin.uio.no.
Therese Sørlie, Email: Therese.Sorlie@rr-research.no, Email: tsorlie@rr-research.no.
References
- Aaltonen, K. , Amini, R.M. , Heikkila, P. , Aittomaki, K. , Tamminen, A. , Nevanlinna, H. , Blomqvist, C. , 2009. High cyclin B1 expression is associated with poor survival in breast cancer. Br. J. Cancer 100, 1055–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal, R. , Gonzalez-Angulo, A.M. , Myhre, S. , Carey, M. , Lee, J.S. , Overgaard, J. , 2009. Integrative analysis of cyclin protein levels identifies cyclin b1 as a classifier and predictor of outcomes in breast cancer. Clin. Cancer Res. 15, 3654–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androic, I. , Kramer, A. , Yan, R. , Rodel, F. , Gatje, R. , Kaufmann, M. , 2008. Targeting cyclin B1 inhibits proliferation and sensitizes breast cancer cells to taxol. BMC Cancer 8, 391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek, D. , Villen, J. , Shin, C. , Camargo, F.D. , Gygi, S.P. , Bartel, D.P. , 2008. The impact of microRNAs on protein output. Nature 455, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbareschi, M. , Pelosio, P. , Caffo, O. , Buttitta, F. , Pellegrini, S. , Barbazza, R. , 1997. Cyclin-D1-gene amplification and expression in breast carcinoma: relation with clinicopathologic characteristics and with retinoblastoma gene product, p53 and p21WAF1 immunohistochemical expression. Int. J. Cancer 74, 171–174. [DOI] [PubMed] [Google Scholar]
- Bartel, D.P. , 2009. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbusch, L.O. , Aaroe, J. , Johansen, F.E. , Hicks, J. , Sun, H. , Bruhn, L. , 2008. Comparison of the Agilent, ROMA/NimbleGen and Illumina platforms for classification of copy number alterations in human breast tumors. BMC Genomics 9, 379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda, C.A. , Cortes-Funes, H. , Gomez, H.L. , Ciruelos, E.M. , 2010. The phosphatidyl inositol 3-kinase/AKT signaling pathway in breast cancer. Cancer Metastasis Rev. 29, 751–759. [DOI] [PubMed] [Google Scholar]
- Charboneau, L. , Tory, H. , Chen, T. , Winters, M. , Petricoin, E.F. , Liotta, L.A. , Paweletz, C.P. , 2002. Utility of reverse phase protein arrays: applications to signalling pathways and human body arrays. Brief. Funct. Genom. Proteom. 1, 305–315. [DOI] [PubMed] [Google Scholar]
- Cheng, K.W. , Lahad, J.P. , Kuo, W.L. , Lapuk, A. , Yamada, K. , Auersperg, N. , 2004. The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat. Med. 10, 1251–1256. [DOI] [PubMed] [Google Scholar]
- D'Amours, D. , Sallmann, F.R. , Dixit, V.M. , Poirier, G.G. , 2001. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: implications for apoptosis. J. Cell. Sci. 114, 3771–3778. [DOI] [PubMed] [Google Scholar]
- deConinck, E.C. , McPherson, L.A. , Weigel, R.J. , 1995. Transcriptional regulation of estrogen receptor in breast carcinomas. Mol. Cell. Biol. 15, 2191–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early Breast Cancer Trialists' Collaborative Group 1998. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 351, 1451–1467. [PubMed] [Google Scholar]
- Enerly, E. , Steinfeld, I. , Kleivi, K. , Leivonen, S.K. , Aure, M.R. , Russnes, H.G. , 2011. miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS One 6, e16915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espina, V. , Mehta, A.I. , Winters, M.E. , Calvert, V. , Wulfkuhle, J. , Petricoin, E.F. , Liotta, L.A. , 2003. Protein microarrays: molecular profiling technologies for clinical specimens. Proteomics 3, 2091–2100. [DOI] [PubMed] [Google Scholar]
- Fisher, E.R. , Redmond, C.K. , Liu, H. , Rockette, H. , Fisher, B. , 1980. Correlation of estrogen receptor and pathologic characteristics of invasive breast cancer. Cancer 45, 349–353. [DOI] [PubMed] [Google Scholar]
- Fisher, B. , Costantino, J. , Redmond, C. , Poisson, R. , Bowman, D. , Couture, J. , 1989. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor-positive tumors. N. Engl. J. Med. 320, 479–484. [DOI] [PubMed] [Google Scholar]
- Gafni, J. , Cong, X. , Chen, S.F. , Gibson, B.W. , Ellerby, L.M. , 2009. Calpain-1 cleaves and activates caspase-7. J. Biol. Chem. 284, 25441–25449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer, C.E. , Forster, J. , Lindquist, D. , Chan, S. , Romieu, C.G. , Pienkowski, T. , 2006. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 355, 2733–2743. [DOI] [PubMed] [Google Scholar]
- Gry, M. , Rimini, R. , Stromberg, S. , Asplund, A. , Ponten, F. , Uhlen, M. , Nilsson, P. , 2009. Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genomics 10, 365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, H. , Ingolia, N.T. , Weissman, J.S. , Bartel, D.P. , 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haakensen, V.D. , Biong, M. , Lingjaerde, O.C. , Holmen, M.M. , Frantzen, J.O. , Chen, Y. , 2010. Expression levels of uridine 5'-diphospho-glucuronosyltransferase genes in breast tissue from healthy women are associated with mammographic density. Breast Cancer Res. 12, R65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy, B.T. , Lu, Y. , Gonzalez-Angulo, A.M. , Carey, M.S. , Myhre, S. , Ju, Z. , 2010. A technical assessment of the utility of reverse phase protein arrays for the study of the functional proteome in non-microdissected human breast cancers. Clin. Proteomics 6, 129–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlings, H.M. , Bergamaschi, A. , Nordgard, S.H. , Kim, Y.H. , Han, W. , Noh, D.Y. , 2008. ESR1 gene amplification in breast cancer: a common phenomenon?. Nat. Genet. 40, 807–808. [DOI] [PubMed] [Google Scholar]
- Hui, R. , Cornish, A.L. , McClelland, R.A. , Robertson, J.F. , Blamey, R.W. , Musgrove, E.A. , 1996. Cyclin D1 and estrogen receptor messenger RNA levels are positively correlated in primary breast cancer. Clin. Cancer Res. 2, 923–928. [PubMed] [Google Scholar]
- Hyman, E. , Kauraniemi, P. , Hautaniemi, S. , Wolf, M. , Mousses, S. , Rozenblum, E. , 2002. Impact of DNA amplification on gene expression patterns in breast cancer. Cancer Res. 62, 6240–6245. [PubMed] [Google Scholar]
- Jaenisch, R. , Bird, A. , 2003. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33, (Suppl.) 245–254. [DOI] [PubMed] [Google Scholar]
- Kirchhoff, M. , Bisgaard, A.M. , Stoeva, R. , Dimitrov, B. , Gillessen-Kaesbach, G. , Fryns, J.P. , 2009. Phenotype and 244k array-CGH characterization of chromosome 13q deletions: an update of the phenotypic map of 13q21.1-qter. Am. J. Med. Genet. A 149A, 894–905. [DOI] [PubMed] [Google Scholar]
- Kyndi, M. , Overgaard, M. , Nielsen, H.M. , Sorensen, F.B. , Knudsen, H. , Overgaard, J. , 2009. High local recurrence risk is not associated with large survival reduction after postmastectomy radiotherapy in high-risk breast cancer: a subgroup analysis of DBCG 82 b&c. Radiother. Oncol. 90, 74–79. [DOI] [PubMed] [Google Scholar]
- Langerod, A. , Zhao, H. , Borgan, O. , Nesland, J.M. , Bukholm, I.R. , Ikdahl, T. , 2007. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 9, R30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, P. , Vogel, C. , Wang, R. , Yao, X. , Marcotte, E.M. , 2007. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 25, 117–124. [DOI] [PubMed] [Google Scholar]
- Maurer, M. , Su, T. , Saal, L.H. , Koujak, S. , Hopkins, B.D. , Barkley, C.R. , 2009. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 69, 6299–6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megha, T. , Lazzi, S. , Ferrari, F. , Vatti, R. , Howard, C.M. , Cevenini, G. , 1999. Expression of the G2-M checkpoint regulators cyclin B1 and P34CDC2 in breast cancer: a correlation with cellular kinetics. Anticancer Res. 19, 163–169. [PubMed] [Google Scholar]
- Muggerud, A.A. , Hallett, M. , Johnsen, H. , Kleivi, K. , Zhou, W. , Tahmasebpoor, S. , 2010. Molecular diversity in ductal carcinoma in situ (DCIS) and early invasive breast cancer. Mol. Oncol. 4, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, C.G. , Fornier, M. , 2010. HER2-positive breast cancer: beyond trastuzumab. Oncol. (Williston Park) 24, 410–415. [PubMed] [Google Scholar]
- Myhre, S. , Mohammed, H. , Tramm, T. , Alsner, J. , Finak, G. , Park, M. , 2010. In silico ascription of gene expression differences to tumor and stromal cells in a model to study impact on breast cancer outcome. PLoS One 5, e14002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naume, B. , Zhao, X. , Synnestvedt, M. , Borgen, E. , Russnes, H.G. , Lingjaerde, O.C. , 2007. Presence of bone marrow micrometastasis is associated with different recurrence risk within molecular subtypes of breast cancer. Mol. Oncol. 1, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, H.M. , Overgaard, M. , Grau, C. , Jensen, A.R. , Overgaard, J. , 2006. Study of failure pattern among high-risk breast cancer patients with or without postmastectomy radiotherapy in addition to adjuvant systemic therapy: long-term results from the Danish Breast Cancer Cooperative Group DBCG 82 b and c randomized studies. J. Clin. Oncol. 24, 2268–2275. [DOI] [PubMed] [Google Scholar]
- Nimeus-Malmstrom, E. , Koliadi, A. , Ahlin, C. , Holmqvist, M. , Holmberg, L. , Amini, R.M. , 2010. Cyclin B1 is a prognostic proliferation marker with a high reproducibility in a population-based lymph node negative breast cancer cohort. Int. J. Cancer 127, 961–967. [DOI] [PubMed] [Google Scholar]
- Pass, H.I. , Goparaju, C. , Ivanov, S. , Donington, J. , Carbone, M. , Hoshen, M. , 2010. hsa-miR-29c* is linked to the prognosis of malignant pleural mesothelioma. Cancer Res. 70, 1916–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauletti, G. , Godolphin, W. , Press, M.F. , Slamon, D.J. , 1996. Detection and quantitation of HER-2/neu gene amplification in human breast cancer archival material using fluorescence in situ hybridization. Oncogene 13, 63–72. [PubMed] [Google Scholar]
- Pollack, J.R. , Sorlie, T. , Perou, C.M. , Rees, C.A. , Jeffrey, S.S. , Lonning, P.E. , 2002. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc. Natl. Acad. Sci. U. S. A. 99, 12963–12968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis-Filho, J.S. , Savage, K. , Lambros, M.B. , James, M. , Steele, D. , Jones, R.L. , Dowsett, M. , 2006. Cyclin D1 protein overexpression and CCND1 amplification in breast carcinomas: an immunohistochemical and chromogenic in situ hybridisation analysis. Mod. Pathol. 19, 999–1009. [DOI] [PubMed] [Google Scholar]
- Rojo, F. , Najera, L. , Lirola, J. , Jimenez, J. , Guzman, M. , Sabadell, M.D. , 2007. 4E-binding protein 1, a cell signaling hallmark in breast cancer that correlates with pathologic grade and prognosis. Clin. Cancer Res. 13, 81–89. [DOI] [PubMed] [Google Scholar]
- Roy, P.G. , Pratt, N. , Purdie, C.A. , Baker, L. , Ashfield, A. , Quinlan, P. , Thompson, A.M. , 2010. High CCND1 amplification identifies a group of poor prognosis women with estrogen receptor positive breast cancer. Int. J. Cancer 127, 355–360. [DOI] [PubMed] [Google Scholar]
- Russnes, H.G. , Vollan, H.K. , Lingjaerde, O.C. , Krasnitz, A. , Lundin, P. , Naume, B. , 2010. Genomic architecture characterizes tumor progression paths and fate in breast cancer patients. Sci. Transl. Med. 2, 38ra47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, L. , Reid, L.H. , Jones, W.D. , Shippy, R. , Warrington, J.A. , Baker, S.C. , 2006. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 24, 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie, T. , Perou, C.M. , Tibshirani, R. , Aas, T. , Geisler, S. , Johnsen, H. , 2001. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. U. S. A. 98, 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie, T. , Tibshirani, R. , Parker, J. , Hastie, T. , Marron, J.S. , Nobel, A. , 2003. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. U. S. A. 100, 8418–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spearman, C. , 2010. The proof and measurement of association between two things. Int. J. Epidemiol. 39, 1137–1150. [DOI] [PubMed] [Google Scholar]
- Stingele, S. , Stoehr, G. , Peplowska, K. , Cox, J. , Mann, M. , Storchova, Z. , 2012. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 8, 608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger, B.E. , Forrest, M.S. , Dunning, M. , Ingle, C.E. , Beazley, C. , Thorne, N. , 2007. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes, R. , Qiu, Y. , Lu, Y. , Hennessy, B. , Andreeff, M. , Mills, G.B. , Kornblau, S.M. , 2006. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Ther. 5, 2512–2521. [DOI] [PubMed] [Google Scholar]
- http://tcga-data.nci.nih.gov/tcga/. 2012a.
- www.targetscan.org. 2012b.
- Varley, J.M. , Swallow, J.E. , Brammar, W.J. , Whittaker, J.L. , Walker, R.A. , 1987. Alterations to either c-erbB-2(neu) or c-myc proto-oncogenes in breast carcinomas correlate with poor short-term prognosis. Oncogene 1, 423–430. [PubMed] [Google Scholar]
- Wiedswang, G. , Borgen, E. , Karesen, R. , Kvalheim, G. , Nesland, J.M. , Qvist, H. , 2003. Detection of isolated tumor cells in bone marrow is an independent prognostic factor in breast cancer. J. Clin. Oncol. 21, 3469–3478. [DOI] [PubMed] [Google Scholar]
- Wilusz, J.E. , Sunwoo, H. , Spector, D.L. , 2009. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 23, 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Jensen, O.N. , 2009. Modification-specific proteomics: strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 9, 4632–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data file 1 DBCG 82 b & c data files: Segmented aCGH data (PCF‐values) for probes that each represents a genomic region that match to a corresponding gene‐probe in the 135 possible protein pairings. The pairings are numbered from 1 to 135 and the entity number in each data file refers to the same pairing.
Supplementary data file 2 DBCG 82 b & c data files: Gene expression data for probes that each represents one gene‐probe in the 135 possible protein pairings.
Supplementary data file 3 DBCG 82 b & c data files: Protein expression‐data for antibodies directed against total protein or phosphoprotein in the 135 possible protein pairings.
Supplementary data file 4 DBCG82 data file: miRNA expression data for six miRNAs that are predicted to target the CASP7 gene. Sample IDs and subtype information are included.
Supplementary data file 5 Extended data set containing 506 breast tumors with overall survival data (time in months), Status 1 = dead, 0 = Alive. LogR <> |0.2| defined as loss/gain.
Supplementary Table 1 Protein‐to‐gene matching table describing chromosomal position and gene name. First column shows the order of pairings in Figure 1. Second column corresponds to the pairing‐number in the three supplementary data files. Third column denotes the selected protein pairings used in the comparisons (marked “u”). Fourth column includes a comment annotation to highlight the phosphorylated epitopes. The remaining columns include gene/protein information as indicated.
Supplementary Table 2 Complete overall correlation table including all possible 135 protein pairings, spearman rank correlation and p‐value for the CN‐GX, PX‐GX and CN‐PX comparisons.
Supplementary Table 3 Table and histograms of variance in copy number and gene – and protein expression values within group A, B, C and D.
Supplementary Tables 4–6 Complete tables of all overall and within subtype correlations for CN‐GX (4).
Supplementary Tables 4–6 Complete tables of all overall and within subtype correlations for GX‐PX (5).
Supplementary Tables 4–6 Complete tables of all overall and within subtype correlations for CN‐PX (6).
Supplementary Table 7 Spearman rank correlation values between correlation values in the original dataset and the validation set, per subtype. The positive correlations validate the correlations found between the three combinations of CN, GX and PX for the selected pairings in the original dataset.
Supplementary Table 8 Validation set with PAM50‐subtyping; number of samples in each subtype and the correlation values of CN‐GX for CCNB1 (overall and within each subtype).
Supplementary Table 9 Spearman correlation values for 39 available protein pairings in the TCGA validation set. For each available protein pairing the correlation values from the validation set are found in the leftmost column and the correlation values from the original dataset are found in the rightmost column. The comparisons are sorted accordingly (“copy number vs gene expression”, “gene expression vs protein expression” and “copy number vs protein expression”).
Supplementary Figure 1 PI3K/Akt‐pathway. Illustration of the PI3K/Akt‐pathway. Proteins with available data on all three levels of measurements in this study are outlined in red. Green arrows indicate direct stimulatory modification and red lines indicate direct inhibitory modification between proteins.
Supplementary Figure 2 Sample sizes in the DBCG 82 b & c dataset (original). Venn‐diagram showing number of tumor samples with available copy number, mRNA expression and protein expression data in the DBCG 82 b & c dataset (original dataset).
Supplementary Figure 3 Sample sizes in the TCGA dataset (validation set). Venn‐diagram showing number of tumor samples with available copy number, mRNA expression and protein expression data in the TCGA dataset (validation set).
Supplementary Figure 4 a–d: Subtype‐specific correlation of the HER2 gene p1248 protein pairing. Scatter plots of three levels of comparisons, overall and subtype‐stratified, of the ERBB2/Her2p1248 gene‐protein pairing. a) Copy number plotted against mRNA expression. Correlation values and corresponding p‐values are given in each panel.
Supplementary Figure 4 a–d: Subtype‐specific correlation of the HER2 gene p1248 protein pairing. Scatter plots of three levels of comparisons, overall and subtype‐stratified, of the ERBB2/Her2p1248 gene‐protein pairing. b) mRNA expression plotted against protein expression. Correlation values and corresponding p‐values are given in each panel.
Supplementary Figure 4 a–d: Subtype‐specific correlation of the HER2 gene p1248 protein pairing. Scatter plots of three levels of comparisons, overall and subtype‐stratified, of the ERBB2/Her2p1248 gene‐protein pairing. c) copy number plotted against protein expression across all samples and for each of the five subtypes, separately. Correlation values and corresponding p‐values are given in each panel.
Supplementary Figure 4 a–d: Subtype‐specific correlation of the HER2 gene p1248 protein pairing. Scatter plots of three levels of comparisons, overall and subtype‐stratified, of the ERBB2/Her2p1248 gene‐protein pairing. Correlation values and corresponding p‐values are given in each panel. d) Box plots of copy number and gene and protein expression values illustrating the difference in heterogeneity (variance) between the subtypes at each molecular level.
Supplementary Figure 5 Copy number plot for CCNB1 across intrinsic gene expression subtypes. The overall frequency plot of copy number levels (gain = green, loss = red) across the whole sample set (upper panel) and frequency plots for each subtype (lower five panels). CCNB1 copy number aberrations differ between subtypes; loss of CCNB1 is mostly observed in the basal‐like subtype, while gain is mostly observed in the luminal‐like subtypes.