

Abstract
Uveal melanoma (UM) is the most common primary tumor of the eye in adults. There is no standard adjuvant treatment to prevent metastasis and no effective therapy in the metastatic setting. We have established a unique panel of 7 UM cell lines from either patient's tumors or patient‐derived tumor xenografts (PDXs). This panel recapitulates the molecular landscape of the disease in terms of genetic alterations and mutations. All the cell lines display GNAQ or GNA11 activating mutations, and importantly four of them display BAP1 (BRCA1 associated protein‐1) deficiency, a hallmark of aggressive disease. The mTOR pathway was shown to be activated in most of the cell lines independent of AKT signaling. mTOR inhibitor Everolimus reduced the viability of UM cell lines and significantly delayed tumor growth in 4 PDXs. Our data suggest that mTOR inhibition with Everolimus, possibly in combination with other agents, may be considered as a therapeutic option for the management of uveal melanoma.
Keywords: Uveal melanoma, BAP1, Everolimus, mTOR, Cell lines, Patients‐derived tumor xenografts
Highlights
A panel of UM cell lines including BAP1‐deficient cells has been established.
mTOR pathway is a potential therapeutic target in UM.
Everolimus displays therapeutical efficacy in vivo.
1. Introduction
Uveal melanoma (UM) is the most frequent and aggressive ocular primary tumor in adults with approximately 5 new cases per million per year in the United States and in Europe (Mallone et al., 2012; Singh et al., 2011). Even if local control rate with photon radiotherapy exceeds 90% at 10 years (Dunavoelgyi et al., 2011) enucleation remains the treatment of choice for large tumors (Singh and Topham, 2003; Singh et al., 2011). Up to 50% of patients develop metastasis, which occur only via hematogenous spread because of the absence of lymphatic drainage of the eye and are rarely detected at the time of initial diagnosis (2–4% of the patients)(Harbour and Chen, 2013). In 90% of cases, metastatic spread involves the liver usually leading to death within a few months despite medical treatment (Gragoudas et al., 1991). Currently, no effective adjuvant therapy is available to prevent metastases, neither is there any effective treatment once metastases have developed.
Genome‐wide genetic analysis (Trolet et al., 2009) and expression profiling (Onken et al., 2004) divide UM in two subgroups according to the risk of metastatic spreading. UM at high risk for metastasis are associated with monosomy of chromosome 3, loss of 6q and gain of 8q (Trolet et al., 2009). Although occurring in the same cell lineage, uveal and skin melanomas represent different diseases: we have recently demonstrated that uveal melanomas display a remarkably low mutation burden with ∼2000 predicted somatic single nucleotide variants per tumor and low levels of aneuploidy. Moreover, no ultraviolet radiation DNA‐damage signature has been found in UM (Furney et al., 2013) and BRAF or NRAS mutations commonly found in cutaneous melanoma are not observed in UM (Cohen et al., 2003; Cruz et al., 2003; Edmunds et al., 2003; Kiliç et al., 2004; Rimoldi et al., 2003; Weber et al., 2003). Mutually exclusive mutations in the GNAQ/11 genes activating the MAP kinase pathway have been described in the majority of UM (Van Raamsdonk et al., 2008, 2010). Although GNAQ/11 mutational status is not correlated with disease‐free survival, these mutations are considered oncogenic drivers and consequently potential good targets for therapeutic intervention. Inactivating mutations of the tumor suppressor BAP1 occur in ∼85% of aggressive tumors and are associated with metastatic disease (Harbour et al., 2010). Recently, exome and whole genome sequencing of uveal melanomas identified recurrent mutations in SF3B1 (Furney et al., 2013; Harbour et al., 2013; Martin et al., 2013), which encodes a component of the spliceosome, and in the translation initiation factor EIF1AX (Martin et al., 2013). SF3B1 and EIF1AX mutations are inversely correlated with chromosome 3 monosomy and associated with good prognosis (Furney et al., 2013; Harbour et al., 2013; Martin et al., 2013).
The currently available UM cell lines do not completely reflect the genetic alterations recurrently found in UM (Griewank et al., 2012). Some cell lines display BRAF mutations, which are not found in UM samples and to our knowledge no UM cell line harboring BAP1 mutations, which represent a hallmark of aggressive UM, have been described so far. The first goal of our study was to develop cellular models of UM representing the genetic landscape (genetic alterations and mutations) of this disease, to provide a good model for assessing the efficacy of new drugs and drug combinations. Next we looked at the activation status of PI3K/mTOR signaling pathway and assessed the effect of Everolimus on cell viability. Last, to provide in vivo data, we examined the effect of mTOR inhibition using several previously described patient‐derived UM xenografts (Némati et al., 2010).
2. Material and methods
2.1. Tumor samples
Eighty‐seven tumor samples were obtained either from patients (60 from primary tumors and 13 from metastasis) or from 14 patient‐derived xenografts (PDXs), which were established as described (Némati et al., 2010). All patients had previously given their informed consent for experimental research on residual tumor tissue available after histopathologic and cytogenetic analyses.
2.2. Establishment of uveal melanoma cell lines
Fresh or DMSO frozen tumor samples obtained from pathologists were mechanically fragmented, passed in a 40 μM Nylon filter and resuspended in RPMI 1640 (Gibco, France), supplemented with 20% (vol/vol) fetal bovine serum (FBS, Invitrogen, France), 100 U/ml penicillin and 100 μg/ml streptomycin (P/S, Invitrogen, France). Once cell lines showed unlimited proliferation and were cultured for more than 40 passages, they were considered established. Optic microscopy images were taken with a Leica DM IL microscope and a Nikon DS‐L1 camera.
2.3. Cell culture
92.1 (De Waard‐Siebinga et al., 1995), Mel202 (Ksander et al., 1991), were purchased from The European Searchable Tumour Line Database (Tubingen University, Germany). OMM1, OMM2.5 (Luyten et al., 1996; Chen et al., 1997) were kindly provided by P.A. Van Der Velden (Leiden University, The Netherlands). Cell lines were cultured in RPMI‐1640 supplemented with 20% (MM28, MM33, MP46, MP41, MP65, and MM66) or 10% (Mel202, OMM1, and OMM2.5) FBS (Life Technologies), Penicillin 100 U/ml – Streptomycin 100 μg/ml (Life Technologies). All cell lines were tested for Mycoplasma and proved to be Mycoplasma free. Cell lines were maintained in a humidified atmosphere (5% CO2) at 37 °C. All cell lines were genotyped: Short Tandem repeat Polymorphism (STR) profiles of 92.1, Mel202, OMM1, OMM2.5 matched at 100% those presented in reference (Griewank et al., 2012).
2.4. Chemicals
mTOR inhibitor Everolimus/Rad001, MEK inhibitor GSK1120212, and AKT inhibitor KRX‐0401 were supplied by Euromedex (France) and dissolved in DMSO (Rad001,GSK1120212) or ethanol (KRX0401) at 10 mM and stored at −20 °C.
2.5. Cell viability assays
We determined cell viability using a colorimetric assay based on 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5 diphenyltetrazolium bromide (MTT; M‐2128, Sigma) as explained previously (Marty et al., 2008). Cells were seeded at appropriate concentration in 96‐well plates at day 0 (MM28:3500 cells/well; MP38:8000 cells/well; MP41:1500; MP46:6000 cells/well; MP65:8000 cells/well; MM66:6000 cells/well; 92.1; Mel202:4000 cells/well; OMM1:1500 cells/well; OMM2.5:3500 cells/well); drug was added to the medium at day 2 and cell viability tested by MTT assay at day 7. Results are expressed as relative percentages of metabolically active cells compared with untreated controls. Drug sensitivity curves were calculated using GraphPad Prism 4.
2.6. Genomic analysis
The DNA was extracted from cell pellets using a standard phenol/chloroform procedure. The total RNA was isolated from cell pellets using a miRNeasy mini kit (Qiagen, Courtaboeuf, France).cDNA synthesis was performed with MuLV Reverse Transcriptase in accordance with the manufacturers' instructions (Invitrogen, Cergy‐Pontoise, France) and quality verified on an Agilent 2100 bioanalyzer. For Sanger sequencing, gDNA was amplified by PCR and the products were sequenced using dye‐terminator chemistry as previously described (16). Primer sequences for BAP1, GNAQ, GNQ11, SF3B1 and EIF1AX are available upon request. Sequences were visualized using Sequencher software. To perform Loss of heterozygosity and copy number analysis and to detect other abnormalities, genetic analyses of the cell lines were done using Affymetrix Genome‐Wide SNP Arrays 6.0. or Cytoscan HD (Affymetrix, High Wycombe, UK). DNA was used to perform Affymetrix Human mapping SNP 6.0 assay as described in (Tuefferd et al., 2008) or Cytoscan assay according to the manufacturer's protocol at the Institut Curie microarray core facility. Genetic profiles were compared to the profiles of the corresponding tumors and PDXs by Chromosome Analysis Suite (Affymetrix). To perform Short Tandem repeat Polymorphism (STR) analysis GenePrint 10 system kit (Promega, France) was used according to manufacturer's instructions.
2.7. Cytopathologic analysis
Cells were fixed in a 4% formalin solution and embedded in paraffin. 4 μm sections were cut from the embedded blocks, and then dewaxed for immunostaining. Heat‐induced epitope retrieval was performed at 97° for 20 min in EDTA buffer pH 9.0 (Dako S2367). Mouse antihuman BAP1 antibody (monoclonal mouse anti BAP1 (C4) Santa Cruz Biotechnology, Inc, Santa Cruz, CA) was applied for 1 h at a concentration of 1:200. For antibody revelation polymer HRP (DAKO Envision, Denmark) was used followed by application of di‐aminobenzidin (DAB) for 5 min. The immunostaining was performed on a Dako Autostainer Platform. A brown coloration of nuclear localization of strong intensity was observed in the presence of the protein. Cell nuclei were counterstained with Herris' Hematoxylin. Epithelial cells of normal breast glands were used as positive control for BAP1.
2.8. Western blotting
Tissue lysates were loaded onto gels, transferred to nitrocellulose and revealed as described (Marty et al., 2008). Quantification was performed using a LAS‐3000 Luminescent Image analyzer and Image Gauge software (Fuji, FSVT, Courbevoie, France). Beta‐Actin was used for normalization between samples and detected using anti‐beta‐actin primary antibodies at the dilution of 1:5000 (Sigma–Aldrich, Saint Quentin Fallavier, France). AKT, phospho‐AKT (S473), phospho‐AKT (T308), S6, phospho‐S6 (Ser 235/236) (Cell Signaling Technology, Ozyme, Saint Quentin en Yveline, France) and BAP1 (C4) (Santa Cruz Biotechnologies) antibodies were used at 1:1000 dilution.
2.9. In vivo antitumor efficacy of an mTOR inhibitor
Female SCID mice were grafted with a tumor fragment of 15 mm3. Mice bearing tumors with a volume of 40–200 mm3 were individually identified and randomly assigned to the control or treatment groups (6–10 animals per group). Number of mice used were respectively: for PDXs MP34: 8 mice for the control group, 8 for the treatment group; for PDXs MP41: 10 mice for controls and 9 for the treatment group; for PDX MP55: 10 mice for the control group and 8 mice for the treatment group; for PDX MP46: 8 mice for the control group and 6 for the treatment group. Mice were weighed twice a week. Tumor volumes were calculated by measuring two perpendicular diameters with calipers. Xenografted mice were sacrificed at the end of treatment or when their tumor reached a volume of 2000 mm3. Each tumor volume (V) was calculated according to the following formula: V = a × b 2/2, where a and b are the largest and smallest perpendicular tumor diameters. Relative tumor volumes (RTV) were calculated with the following formula: RTV = (V x/V 1), where V x is the tumor volume on day x and V 1 is the tumor volume on the first day of treatment. Growth curves were obtained by plotting the mean values of RTV on the Y axis against time (X axis, expressed as days of treatment). Antitumor activity was evaluated according to tumor growth inhibition (TGI), calculated with the following formula: percent TGI = 100 − (RTVt/RTVc × 100), where RTVt is the median RTV for a treatment group and RTVc is the median RTV for its control group at the end of the therapy. mTOR inhibitor (Everolimus) was reconstituted in PEG300/HPBCD/Glucose 5% (10/10/80), and administered PO at a dose of 2 mg/kg 3 times a week, for 4–6 weeks. In all in vivo experiments, mice of the control groups received 0.2 ml of the drug‐formulating vehicle with the same schedule as the treated animals. The experimental protocol and animal housing were in accordance with institutional guidelines as put forth by the French Ethical Committee (Agreement C75‐05 – 18, France), and the ethics committee of the Institut Curie that approved this project.
2.10. Expression of tumor‐specific antigens
Expression of tumor‐specific antigens was assessed by reverse transcription‐PCR on RNA extracted from cellular culture as described (Némati et al., 2010).
2.11. Assessment of synergy in drug combination experiments
Synergy computed as excess over Bliss (Straussman et al., 2012) was assessed by calculation, for each combination of doses tested, of its fractional inhibition value (1 – fraction of viable cells compared to controls) and by successive subtraction of the fractional inhibition value calculated according to the Bliss independence model. Therefore Excess over Bliss = c − (a + b − 2*a*b) where a is the fractional inhibition obtained with an x concentration of drug A, b is the fractional inhibition obtained with an y concentration of drug B and c is the fractional inhibition obtained with x concentration of drug A combined with y concentration of drug B. Synergy calculated as Combination Index was obtained using Chu and Talalay median‐effect equation (Chou, 2006) with the software Compusyn ComboSyn, Inc., Paramus, NJ. USA, 2005 (Chou, 2010).
2.12. Statistical methods
For in vitro experiments 95% Confidence Intervals on 3 independent replicates were calculated to assess statistical significance for synergic effects of drug combinations. For in vivo experiments the statistical significance of the difference between calculated RTVs for treatments versus control groups was calculated by the two‐tailed Student's t test.
3. Results
3.1. Establishment of UM cell lines
We have established 7 UM cell lines: 2 of them, MP38 and MP65, were obtained directly from human primary tumors (success rate of 3%), 3 cell lines derived from PDX models (Némati et al., 2010) of liver (MM28 and MM66) or skin (MM33) metastasis, while MP41 and MP46 derived from PDX models of primary tumors (See Table 1). MP38 and MP65 display a fusiform morphology, MP41 shows a predominant epithelioid appearance while MP46, MM28, MM33 and MM66 have a mixed morphology (see Figure 1). All the cell lines are adherent with MM66 having a minor component growing in suspension. Estimated doubling times (shown in Table 1) ranged between 40 and 120 h.
Table 1.
Characteristics of UM cell lines and Xenografts used in this study.
| Model | Origin | Morphology | Doubling time | Status of chromosomes 1; 3;6; 8 and 16 | LOH of chromosome 3 | BAP1 mutations | BAP1 protein expression | GNAQ mutations | GNA11 mutations | SF3B1 mutations | EIF1AX mutations |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MP38CL | Primary Tumor | S | 80 h | L3q; G8; L16q | Yesa | c.68‐9_72del | No | c.626 a > T | _ | _ | _ |
| MP41CL | PDX established from Primary Tumor | M | 41 h | L1p; G1q; L3; G6P; L6q; L8p; G8q; L16 | Yesb | _ | Yes | _ | c.626 a > A/T | _ | _ |
| MP46CL | PDX established from Primary Tumor | M | 110 h | G1q; G6p; L6q; L8p; G8q; L16q | Yes | _ | No | c.626 a > T | _ | _ | _ |
| MP65CL | Primary Tumor | S | 120 h | G1q; G6p; G8 | Yes | c.1717del | No | _ | c.626A > T | _ | _ |
| MM28CL | PDX established from Liver Metastasis | M | 109 h | L1p; G1q; L3q; G6p; L6q; L8p; G8q; L16 | Yesa | c.1881C > A | No | _ | c.626A > T | _ | _ |
| MM33CL | PDX established from Skin Metastasis | S | 91 h | G1; G6p; L6q; G8; G16 | No | Yes | c.626 a > C | _ | _ | c.22G/A | |
| MM66CL | PDX established from Liver Metastasis | M | 80 h | G1q; L6q; G8 | No | _ | Yes | _ | c.626A > T | _ | _ |
| 92.1CL | Primary tumor | M | 38 h | der (X) t (X; 6)(q28; p11),+8d | ND | ND | Yes | c.626 a > Tc | _ | – | c.17G/A |
| Mel202CL | Primary tumor | M | 43 h | ND | ND | ND | Yes | c.629 G > Ac | _ | c.1793c > T | _ |
| OMM1CL | SubcutisMetastasis | M | 34 h | der(1)t (1; 3)(p31; p13),+3[50%], add (8)p11),add (16)(p12)e | ND | ND | Yes | _ | 626A > Tc | _ | _ |
| 0MM2.5CL | Liver Metastasis | M | 50 h | ND | ND | ND | Yes | c.626 a > Cc | _ | _ | _ |
| MP34X | Primary tumor | E | 7 d | L1p; L6q | Yesa | _ | Yes | _ | c.626A > T | c.1793c > T | _ |
| MP41X | Primary tumor | E | 15 d | L1p; G1q.L6q; L8p; G8q; G16p; L16q | No | _ | Yes | _ | 626 a > A/T | _ | _ |
| MP55X | Primary tumor | E | 8 d | L3; G6p; Lq; G8p; G8q; | Yes | c.516C > G | No | _ | c.626A > T | _ | _ |
| MP46X | Primary tumor | M | 11 d | G1q; L3; G6p; L8p; G8q; L16q | Yes | _ | No | c.626 a > T | _ | _ | _ |
Model: CL, cell line; X, Xenograft.
Morphology: S, spindle cell; M, mixed; E, Epithelioid.
Doubling time. h: hours; d: days.
ND: not determined.
3 As determined by Western Blot and Immunocytochemistry.
Uniparental disomy of 3q.
Uniparental disomy of chromosome 3.
92.1 and Mel202 were tested for GNAQ 626A > C, GNAQ 626A > T, GNA11 626A > T; the other data on GNA mutations were issued from (Griewank et al., 2012).
(De Waard‐Siebinga et al., 1995).
(Luyten et al., 1996).
Figure 1.
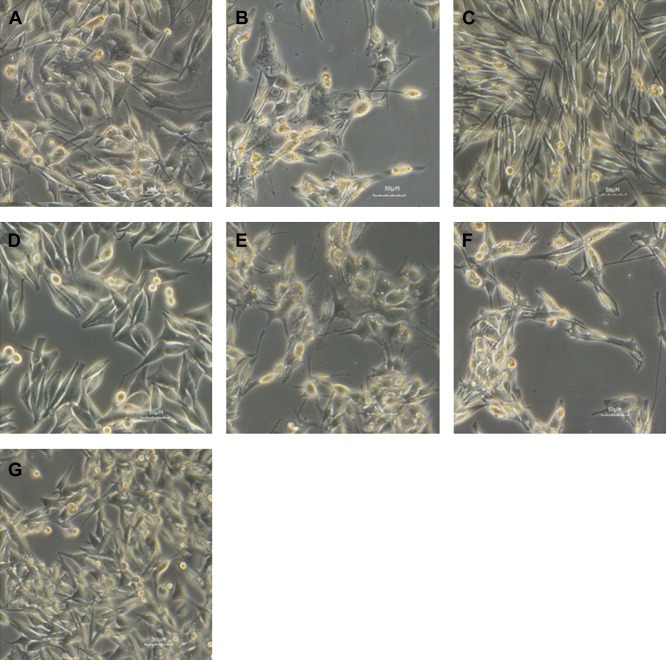
Morphological analysis of established uveal melanoma cell lines. Light microscopy image of UM cell lines showing predominant epithelioid (MP41) spindle (MP38; MP65) or mixed morphology (MM28; MM33; MP46; MM66). MM28 (A), MM33 (B), MP38 (C), MP41 (D), MP46 (E), MP65 (F), MM66 (G).
3.2. Characterization of UM cell lines
Copy number and SNP profiles were generated for each cell line and compared to the profiles obtained from the tumors of origin (patients or PDXs). DNA arrays profiles are represented in Supplementary Figure 1. Genotype analysis by Affymetrix mapping SNPs arrays confirmed the overall conservation of chromosome alterations between cell lines and corresponding tumor specimens, in particular for chromosomes 1, 3, 6, 8 and 16 whose status are known to have an impact on classification and prognosis of the disease (Couturier and Saule, 2012; Harbour, 2012). Six cell lines display loss or LOH of 1p or gain of 1q; five cell lines display chromosome 3 monosomy or isodisomy. Five cell lines show gain of 6p and loss or LOH of 6q and one shows loss of 6q only. A gain of 8q was observed in six cell lines except for MP38, with three showing also 8p loss. Loss of 16q was found in four cell lines.
As shown in Table 1 all cell lines harbor mutually exclusive mutations in either GNAQ or GNA11 as occurred in the corresponding tumor of origin: GNAQ c.626A > C; p.Gln209Pro in MM33 and GNAQ c.626A > T p.Gln209Pro in MP46 and MP38, while MP41, MP65, MM28 and MM66 bear GNA11 mutations (GNA11 c.626 a > T; p.Gln209Leu). MP38, MP65, and MM28 display loss of function mutations of the BAP1 gene associated with LOH of chromosome 3 as follows: MP38 harbors a deletion of 14 bp (c.68‐9_72del) leading eventually to the loss of a splice site. MP65 displays a frame‐shift deletion of 1 pb (c.1717del; p.Leu573TrpfsX3) and MM28 harbors a BAP1 point mutation (c.1881C > A; p.Y627). Western blot showed expression of BAP1 in MP41, MM33 and MM66 cell lines and absence of the protein in the 3 BAP1 mutated cells and in MP46 (Figure 2). The expression of BAP1 was also checked by immunocytochemistry (data not shown) confirming nuclear localization of BAP1 in MP41 M33 and MM66 lines, and absence of nuclear staining in the remaining cell lines. A strong BAP1 nuclear staining was observed as well in a series of previously described UM cell lines including 92.1, Mel202, OMM1, and OMM2.5 (Griewank et al., 2012).
Figure 2.
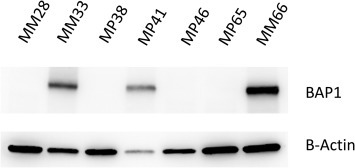
Western blot analysis of BAP1 protein expression in UM cell lines. Immunostaining on cell lines MM33, MP41 and MM66 reveals presence of the protein BAP1 while MP28, MP38, MP46 and MP65 show loss of BAP1 protein expression.
All the cell lines established in this study as well as cell lines received from other laboratories were tested for known SF3B1 mutations. Only Mel202 proved to be mutated for SF3B1 (c.1793c > T; p.Arg625Gly). EIF1AX gene were also tested at exons 1 and 2 and proved mutated in cell lines MM33 (c.22G/A; p.Gly8Arg) and 92.1 (c.17G/A; p.Gly6Asp). Short Tandem repeat Polymorphism (STR) genotyping was performed and results are reported in Supplementary Table 1.
The expression of 12 tumor‐specific antigens (i.e., MAGE1, MAGE2, MAGE3, MAGE4, MAGE6, MAGE10, MAGE‐C2, LAGE1, LAGE2, NA17, tyrosinase, and Melan‐A) was assessed on cell lines; data are shown in Supplementary Table 2. All the cell lines except MM33 showed a strong expression of Tyrosinase, NA‐17 or both. Expression of MAGE and LAGE antigens was found to be negative or very low in our cell lines except MP46 which exhibits a 20% and 100% expression of MAGE2 and MAGE3 respectively). This expression pattern corresponds to what has been already described for the original models and patients (Némati et al., 2010).
3.3. Activation of mTOR pathway and effect of Everolimus on UM cell lines
UM cells have been reported to display activation of the PKC, MEK‐ERK and PI3K/mTOR pathways (Abdel‐Rahman et al., 2006, 2012, 2011, 2010, 2005). Clinical trials with PKC and MEK inhibitors are in progress. The MEK inhibitor Selumetinib has been shown to increase progression free survival compared to standard of care, but failed to demonstrate a statistically significant increase in overall survival (Carvajal et al., 2013). No clinical data concerning the use of PI3K/mTOR inhibitors in UM have been reported so far. Some in vitro studies have addressed the effect of these inhibitors using UM cell lines but in a BAP1‐proficient context and sometimes with cell lines displaying activating B‐RAF mutations (Babchia et al., 2010; Ho et al., 2012; Khalili et al., 2012). We therefore decided to assess the activation status of PI3K/mTOR pathway on our panel of cell lines, which recapitulate the genetic features of the disease.
First, we tested the activation of the pathway on 2 BAP1 mutated (MP38 and MP65) and 2 BAP1 wild‐type cell lines (MP41 and MM66). BT20, a cell line displaying a PI3KCA mutation conferring a constitutive activity to the kinase, was used as control for the activation of PI3K/mTOR pathway. Analysis of the phosphorylation of mTOR downstream target S6 ribosomal protein (El‐Hashemite et al., 2003) showed an activation of mTOR pathway comparable to that of BT20, with evidence of phosphorylation of the protein also after 24 h of serum starvation in 3 out of 4 uveal melanoma cell lines (Figure 3). Phospho‐AKT was barely detectable on western blot, and the ratio between phospho AKT and total AKT was found dramatically low as compared to BT20 (Figure 3). This suggests that mTOR activation of UM cell lines is not dependent of AKT phosphorylation. In agreement with this hypothesis, the AKT inhibitor Perifosine did not significantly alter cell proliferation of UM cell lines (supplementary Figure 2). Viability of 10 UM cell lines (MM28, MP38, MP41, MP46, MP56 and MM66, 92.1, Mel202, OMM1 and OMM2.5) was significant affected by Everolimus at relative low doses even if a full inhibition of cellular viability was not reached (Figure 4A). The slopes of curves obtained with Everolimus suggest a cytostatic rather than cytotoxic effect. As depicted in Figure 4B, a dramatic reduction in S6 phosphorylation could be observed in 6 different UM cell lines treated with Everolimus at 1 nM. The most sensitive cell lines in terms of cellular viability (MM66, OMM1 and OMM2.5) display the higher reduction in S6 phosphorylation, whereas MP65 and MP41 are the more resistant to Everolimus in terms of both cell viability and S6 phosphorylation. However a statistically significant correlation between the effect of Everolimus on S6 phosphorylation and cellular viability in the different cell lines could not be demonstrated. Altogether our data demonstrate that UM cell lines display mTOR signaling activation and that Everolimus significantly affects cell proliferation at doses at which it inhibits mTOR downstream signaling.
Figure 3.
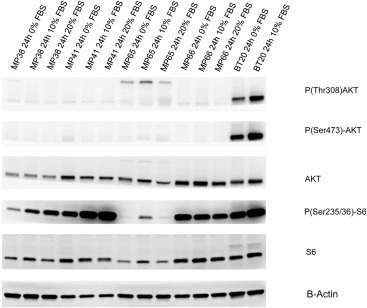
Analysis of mTOR and AKT signaling pathway in UM cell. UM cell lines were cultured for 24 h at different serum concentrations. P(Ser473)‐AKT, P(Thr308)‐AKT, AKT, P(Ser235/236)‐S6, S6 and B‐Actin were evaluated on cellular lysates by Western blot analysis.
Figure 4.
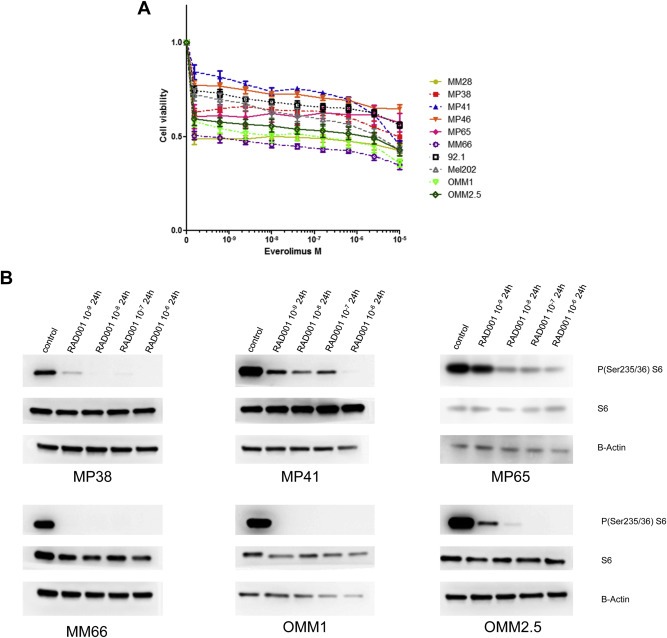
Sensitivity of a representative panel of uveal melanoma cell lines to mTOR inhibitor Everolimus and effect of Everolimus on UM cell lines viability. A. UM cell lines were treated for 24 h with different concentrations of Everolimus and P(Ser235/236)‐S6, S6 and B‐Actin assessed by Western blot analysis. B. MM28 (GNAQ 11 mutated, BAP1 deficient) MP38 (GNAQ mutated, BAP1 deficient), MP41 (GNA11 mutated), MP46 (GNAQ mutated, BAP1 deficient) MP65 (GNA11 mutated, BAP1 deficient), MM66 (GNA11 mutated), 92.1 (GNAQ mutated, EIF1AX mutated), Mel202 (GNAQ mutated, SF3B1 mutated), OMM1 (GNA11 mutated), OMM2.5 (GNAQ mutated) were seeded at adequate concentration and incubated with the drugs for 5 days. Cell viability was quantified with the MTT assay. Results are expressed as the mean of at least 3 separate experiments. Error bars represent standard errors of the mean.
3.4. Everolimus effects in vivo
We then tested the effect of Everolimus in vivo using our UM PDX panel previously characterized (Laurent et al., 2013; Némati et al., 2010) that represents the genetic landscape of UM as described above. Four models were tested for this purpose: MP34, MP41, MP55, and MP46. Of note, we did not succeed in establishing cell lines from MP34 and MP55 PDXs. MP34 displays a mutation in GNAQ and MP41, MP55, and MP46 harbor GNA11 mutations. Two of them (MP46 and MP55) do not express BAP1 protein as assessed by immunohistochemistry (Laurent et al., 2013). MP34 harbors an SF3B1 mutation. Mice were treated with Everolimus per os at 2 mg/kg 3 times per week for 4–6 weeks. As depicted in Figure 5, treatment with the mTOR inhibitor resulted in a significant tumor growth delay in the models MP41, MP55 and MP34, with a Tumor Growth Inhibition (TGI) of 57%, 51% and 47% respectively, and a moderate effect in MP46 with a TGI of 38%. Taken together, our results show that Everolimus significantly reduced tumor growth of uveal melanoma in vivo.
Figure 5.
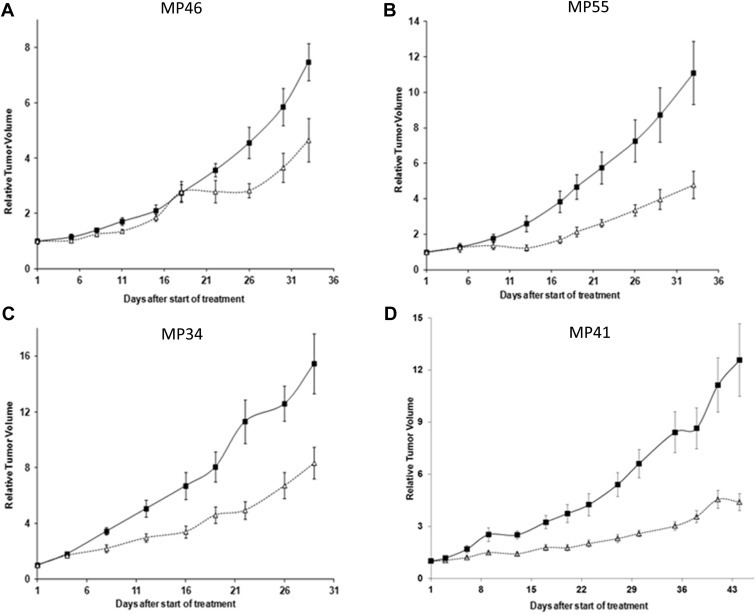
Effects of mTOR inhibitor Everolimus in the growth of four UM PDXs in vivo. Growth curves of four human uveal melanoma xenografts: MP46 (A), MP55 (B), MP34(C), and MP41(D), treated with Everolimus (Δ) per os at 2 mg/kg 3 times a week, or receiving vehicle (■) with the same schedule as the treated animals for 4 (MP46, MP55, MP34) to 6 (MP41) weeks. Tumor volume and RTV were calculated as described in Materials and Methods. Growth curves were obtained by plotting mean RTV against time. Bars, SD. For the treated groups n = 6–8 mice; for the control groups n = 8–10 mice. P values calculated at the end of the treatment were <0.05 for the four models.
3.5. Effect of combined MEK inhibitor and Everolimus on UM cell proliferation
Given that tumor regression was not achieved with Everolimus alone and since mTOR inhibitors have been reported to have a rather cytostatic than cytotoxic effect (Weigelt et al., 2011), combinatorial approaches need to be addressed to implement efficient therapeutic schedules. MAPK inhibitors clearly represent good candidates to be tested in combination with Everolimus in UM given that GNAQ/11 activating mutations result in MAPK upregulated activity and this gene is mutated in >85% of UM patients. Our data argue that the MEK inhibitor Trametinib displays the lowest IC50 among a panel of compounds tested on UM cell lines (data not shown). Moreover, recent data testing MEK inhibitors in uveal melanoma metastatic patients were promising (Carvajal et al., 2013). We, therefore, tested whether the MEK inhibitor GSK1120212 (Trametinib) on the already described panel of 10 UM cell lines could enhance the in vitro efficacy of Everolimus. Figure 6 shows the effect of single drug and of the combination on the 10 different cell lines. Analysis of synergism was performed according to two different models: Bliss independence (Keith et al., 2005) and combination Index described by Chu (2006). Although both analyses gave roughly the same results, the first method was more reproducible in our hands and therefore only the data generated with it are shown in Supplementary Figure 3. A significant fraction of UM cell lines exhibited moderate synergy between Everolimus and Trametinib supporting the development of combinatorial approaches with agents targeting MEK and mTOR pathways in UM patients. This needs to be addressed in preclinical in vivo models. Under our in vitro experimental conditions, the combination of Everolimus and Trametinib did not result in induction of apoptosis (examining cleaved PARP by Western blot) in UM cell lines with the exception of 92.1 cells in which Everolimus was shown to increase the apoptosis induced by Trametinib (data not shown). Further investigation is necessary to better understand the molecular mechanisms resulting in the observed synergy of these two compounds.
Figure 6.
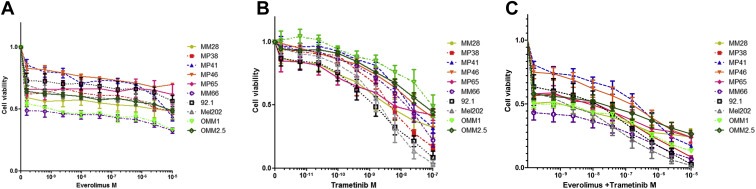
Effect of the combination of MEK inhibitor Trametinib and mTOR inhibitor Everolimus on the viability of a panel of 10 UM cell lines. Cell lines were treated at the indicated doses of inhibitors for 5 days and cell viability was determined by MTT as described in Material and Methods. Drug concentration is expressed as Molarity; Drug concentration in (C) is expressed as sum of the concentration of each drug. A and B: single drug curves for Everolimus and Trametinib, C: combination. Drug concentrations for the combination had been selected maintaining a constant ratio between the two drugs in order to facilitate synergy evaluation.
4. Discussion
Efficient management of UM patients requires a better understanding of the genetic and molecular abnormalities implicated in the development and progression of this disease. With the emergence of an armamentarium of targeted drugs, in vitro and in vivo preclinical models for testing new drugs and drug combinations is mandatory to rationally set up clinical trials. We have recently described a panel of patient‐derived UM PDXs, which recapitulates the genetic features of primary human UMs and exhibit genetic stability over the course of their in vivo maintenance (Laurent et al., 2013; Némati et al., 2010). Although this panel represents a powerful preclinical tool for both pharmacologic and biological analyses, it is useful for functional studies to have access to a panel of well‐characterized tumor cell lines. Unfortunately, obtaining UM cell lines from patients is not easy and the cell lines reported to be of uveal origin do not always display the genetic alterations described in UM. For example, some UM cell lines described in the literature have activating mutations in BRAF (Calipel et al., 2003; Griewank et al., 2012) despite the absence of these mutations in UM tissues. Moreover no UM cell line harboring BAP1 mutations, a hallmark of metastasizing UM, has been reported. In this paper, we have established and characterized 7 new human UM cell lines. Five of them were obtained from PDXs models and the other two directly from human primary tumors. This suggests that the success in establishing UM cell lines could be significantly improved by previously engrafting the UM samples in immunodeficient mice as already reported for colorectal tumors (Dangles‐Marie et al., 2007). We are continuing to develop UM cell lines from our entire collection of PDX and aim to expand our cell lines panel in the future. The UM cell lines described here match the genotype of the tumors of origin. All of them harbor mutually exclusive activating mutation in either GNAQ or GNA11. In addition, we have established 4 unprecedented BAP1‐deficient UM cell lines. we could not demonstrate any BAP1 mutation in the BAP1 deficient model MP46, which display a LOH with isodisomy of chromosome 3. For all the cell lines established, the absence of nuclear BAP1 correlated with LOH of chromosome 3. The 7 cell lines were found to be wild type for SF3B1 while one was found mutated in the EIF1AX gene. Together, this describes the genetic landscape of our UM cell lines.
We show that Everolimus significantly affects the cell growth of our UM cell line panel and other UM cell lines previously described. It has been reported that Everolimus very slightly affects cell proliferation of two UM cell lines (92.1 and Mel270) at doses at which it entirely inhibit mTOR downstream signaling (Babchia et al., 2010). Interestingly, the cell lines displaying the highest sensitivity to Everolimus in terms of cell viability exhibited a more pronounced reduction in the phosphorylation S6 ribosomal protein, a target of mTOR. We also show that mTOR signaling is activated in the absence of significant AKT upregulation. The activation of mTOR can be a consequence of MAPK activation resulting from GNAQ/11 activating mutations present in >85% of UM. In a recent study the PI3K inhibitor GSK2126458 showed a reduced efficacy on GNAQ or GNA11 mutated UM cell lines compared to wild type uveal melanoma cells (Khalili et al., 2012). In the same study RPPA analysis showed a reduced phosphorylation of AKT in GNAQ mutated cells compared to GNAQ wild type, thus supporting our findings. In contrast, basal P‐4EBP and basal P‐S6 were higher in the GNAQ mutated cell lines, suggesting a key role of the pathway downstream of mTOR in GNAQ mutant cells. This is supported by the observation that in our cellular models phosphorylation of AKT was very weak in comparison with a cell line (BT20) displaying a constitutive active PI3K/AKT pathway. On the contrary, phosphorylation of S6 in our cellular models and in BT20 cell line was similar. Interestingly MP41 and MM66 showed significant phosphorylation of S6 even after 24 h serum starvation at the same levels of the controls, suggesting a constitutive activation of the pathway.
Inhibiting PI3K axis alone or in combination with mTOR inhibition has been proposed as a therapeutic strategy for UM (Babchia et al., 2010). This study showed that PI3K inhibition by LY294002 is more effective than mTOR inhibition by Everolimus, but these differences were significant only in a GNAQ/11 wild‐type context.
Few studies have addressed the effect of PI3K/mTOR pathway in vivo. Results were non‐conclusive or conducted with cell lines not perfectly representing the genetic landscape of UM (Ho et al., 2012). Here we show that the mTOR inhibitor Everolimus significantly delayed tumor growth in 4 different UM PDX models. The in vivo effect of Everolimus is not dependent on BAP1 status. However, since this conclusion is based on four PDX models, it is possible that this finding is due to small sampling size. Our in vitro data also suggest that genetic differences and, specifically, BAP1 mutations does not influence the response to Everolimus.
Although cell lines established from UM metastases were at least as sensitive to Everolimus as cell lines established from primary tumors, it is important to note that the four UM PDX models used in this work were established from primary tumors and not metastatic lesions. In the absence of a comprehensive study using metastatic tissue in UM, caution is required in making conclusions about potential effects of Everolimus on metastatic UM patients.
Given that treatment with Everolimus did not result in tumor regression, combination strategies need to be addressed in vitro and in vivo. Our data supports the cytostatic effect of Everolimus alone, which would benefit from combination with MEK inhibitors or low doses of dual mTOR/PI3K inhibitors as others have argued (Mitsiades et al., 2011; Nyfeler et al., 2012).
Everolimus has indications in oncology and a clinical phase 2 trial is currently ongoing at Sloan‐Kettering cancer center with the aim of assessing its efficacy in combination with a somatostatin receptor inhibitor Pasireotide on patients with metastatic UM (clinicaltrial.gov identifier NCT01252251). Our preliminary data indicates a synergy of Everolimus and the MEK inhibitor Trametinib. It would be of future interest to evaluate the synergy displayed by other combinations of currently available inhibitors of PI3K/mTOR and MEK‐ERK pathways across a heterogeneous panel of UM cell lines and then to assess their efficacy in vivo. We believe our approach using in vitro and in vivo models will help orient future innovative clinical trials in uveal melanoma patients.
5. Conclusions
We have established 7 UM cell lines from either patient surgical specimens or patient‐derived xenografts (PDXs). This panel of cell lines has been fully characterized in terms of genetic alterations and recurrent mutations and recapitulates together with our previously described panel of PDXs (Laurent et al., 2013; Némati et al., 2010) the diversity of the UM genetic landscape. Moreover we have demonstrated in our UM cellular models the activation of mTOR pathway in the absence of significant AKT phosphorylation. Treatment with the mTOR inhibitor Everolimus resulted in the reduction of cell viability of all the studied UM cell lines and significantly delayed in vivo tumor growth of 4 independent UM PDXs. Although efficient therapeutic combinations need to be carefully evaluated, our data suggest that Everolimus could be considered as a therapeutic option for managing UM.
Grant support
This project is supported by French national cancer Institute (INCa).
Disclosure of potential conflicts of interest
No potential conflicts of interest were, disclosed.
Database linking
MM28, MM33, MP38, MP41, MP46, MP65, MM66 cell lines Affimetrix SNP Arrays 6.0 and Cytoscan HD Arrays are available at http://microarrays.curie.fr/publications/recherche_translationnelle/uveal_melanoma/.
Supporting information
The following are the supplementary data related to this article:
Supplementary Figure S1. DNA microarray analysis of the 7 Uveal Melanoma cell lines. Allele ratio (top) copy number values (middle), and Log2ratio (bottom) for MM28 (A), MM33 (B), MP38 (C), MP41 (D), MP46 (E), MP65 (F), MM66 (G); red line: LOH.
Supplementary Figure S2. Sensitivity of a representative panel of uveal melanoma cell lines to AKT inhibitor Perifosine. MM28 (GNAQ 11 mutated, BAP1 deficient) MP38 (GNAQ mutated, BAP1 deficient), MP41 (GNA11 mutated), MP46 (GNAQ mutated, BAP1 deficient) MP65 (GNA11 mutated, BAP1 deficient), MM66 (GNA11 mutated), 92.1 (GNAQ mutated, EIF1AX mutated), Mel202 (GNAQ mutated, SF3B1 mutated), OMM1 (GNA11 mutated), OMM2.5 (GNAQ mutated) were seeded at adequate concentration and left in contact with the drugs for 5 days. Cell viability was quantified with MTT assay. Results are expressed as mean of at least 3 separate experiments. Error bars represent standard errors of the mean.
Supplementary Figure S3. Synergy by excess over Bliss (A) and corresponding normalized cell viability fractions (B) for the combination of Everolimus and Trametinib on a panel of 10 UM cell lines. Fractional inhibition calculated for Trametinib and Everolimus used as single agent and in combination were used to assess synergy as explained in paragraph 2.11. Results from 3 independent experiments were used to calculate 95% confidence intervals. A shows synergy values as mean of three separate experiments, values that resulted significantly >0 with a p value <0.05 are colored in red (synergy), values that resulted significantly < 0 with a p value <0.05 are colored in blue (antagonism), values that produced synergy values not significantly different from 0 are colored in white (antagonism). B. express the corresponding percentage of fractional inhibition for each condition tested.
Supplementary data
Supplementary data
Acknowledgments
The authors wish to thank Delphine Lequin, Julie Gayet, Sandrine Arrufat and Jordan Madic for technical help, Marika Pla, Isabelle Grandjean and colleagues for animal facilities support, Virginie Maire and the team of Thierry Dubois for useful technical advices, Olivier Lantz for helpful discussions, and Ronald Lebofsky for carefully reading the manuscript.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.06.004.
Amirouchene-Angelozzi Nabil, Nemati Fariba, Gentien David, Nicolas André, Dumont Amaury, Carita Guillaume, Camonis Jacques, Desjardins Laurence, Cassoux Nathalie, Piperno-Neumann Sophie, Mariani Pascale, Sastre Xavier, Decaudin Didier, Roman-Roman Sergio, (2014), Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.06.004.
Contributor Information
Nabil Amirouchene-Angelozzi, Email: Nabil.Amirouchene-Angelozzi@curie.fr.
Fariba Nemati, Email: Fariba.Nemati@curie.fr.
David Gentien, Email: David.Gentien@curie.fr.
André Nicolas, Email: Andre.Nicolas@curie.fr.
Amaury Dumont, Email: Amaury.Dumont@curie.fr.
Guillaume Carita, Email: Guillaume.Carita@curie.fr.
Jacques Camonis, Email: Jacques.Camonis@curie.fr.
Laurence Desjardins, Email: Laurance.Desjardins@curie.net.
Nathalie Cassoux, Email: Nathalie.Cassoux@curie.net.
Sophie Piperno-Neumann, Email: Sophie.Piperno-Neumann@curie.net.
Pascale Mariani, Email: Pascale.Mariani@curie.net.
Xavier Sastre, Email: Xavier.Sastre@curie.net.
Didier Decaudin, Email: Didier.Decaudin@curie.net.
Sergio Roman-Roman, Email: Sergio.Roman-Roman@curie.fr.
References
- Abdel-Rahman, M.H. , Yang, Y. , Zhou, X.-P. , Craig, E.L. , Davidorf, F.H. , Eng, C. , 2006. High frequency of submicroscopic hemizygous deletion is a major mechanism of loss of expression of PTEN in uveal melanoma. JCO. 24, 288–295. [DOI] [PubMed] [Google Scholar]
- Babchia, N. , Calipel, A. , Mouriaux, F. , Faussat, A.-M. , Mascarelli, F. , 2010. The PI3K/Akt and mTOR/P70S6K signaling pathways in human uveal melanoma cells: interaction with B-Raf/ERK. Invest. Ophthalmol. Vis. Sci. 51, 421–429. [DOI] [PubMed] [Google Scholar]
- Calipel, A. , Lefevre, G. , Pouponnot, C. , Mouriaux, F. , Eychène, A. , Mascarelli, F. , 2003. Mutation of B-Raf in human choroidal melanoma cells mediates cell proliferation and transformation through the MEK/ERK pathway. J. Biol. Chem. 278, 42409–42418. [DOI] [PubMed] [Google Scholar]
- Carvajal, R.D. , Sosman, J.A. , Quevedo, F. , Milhem, M.M. , Joshua, A.M. , Kudchadkar, R.R. , Linette, G.P. , Gajewski, T. , Lutzky, J. , Lawson, D.H. , Lao, C.D. , Flynn, P.J. , Albertini, M.R. , Sato, T. , Paucar, D. , Panageas, K.S. , Dickson, M.A. , Wolchok, J.D. , Chapman, P.B. , Schwartz, D.K. , 2013. Phase II study of selumetinib (sel) versus temozolomide (TMZ) in gnaq/Gna11 (Gq/11) mutant (mut) uveal melanoma (UM). J. Clin. Oncol. 31, (ASCO annual Meeting Abstracts) [Google Scholar]
- Chen, P.W. , Murray, T.G. , Uno, T. , Salgaller, M.L. , Reddy, R. , Ksander, B.R. , 1997. Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. Clin. Exp. Metastasis. 15, 509–518. [DOI] [PubMed] [Google Scholar]
- Chou, T.-C. , 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58, 621–681. [DOI] [PubMed] [Google Scholar]
- Chou, T.-C. , 2010. Drug combination studies and their synergy quantification using the Chou–Talalay method. Cancer Res. 70, 440–446. [DOI] [PubMed] [Google Scholar]
- Cohen, Y. , Goldenberg-Cohen, N. , Parrella, P. , Chowers, I. , Merbs, S.L. , Pe'er, J. , Sidransky, D. , 2003. Lack of BRAF mutation in primary uveal melanoma. IOVS. 44, 2876–2878. [DOI] [PubMed] [Google Scholar]
- Couturier, J. , Saule, S. , 2012. Genetic determinants of uveal melanoma. Dev. Ophthalmol. 49, 150–165. [DOI] [PubMed] [Google Scholar]
- Cruz, F. , Rubin, B.P. , Wilson, D. , Town, A. , Schroeder, A. , Haley, A. , Bainbridge, T. , Heinrich, M.C. , Corless, C.L. , 2003. Absence of BRAF and NRAS mutations in uveal melanoma. Cancer Res. 63, 5761–5766. [PubMed] [Google Scholar]
- Dangles-Marie, V. , Pocard, M. , Richon, S. , Weiswald, L.-B. , Assayag, F. , Saulnier, P. , Judde, J.-G. , Janneau, J.-L. , Auger, N. , Validire, P. , Dutrillaux, B. , Praz, F. , Bellet, D. , Poupon, M.-F. , 2007. Establishment of human colon cancer cell lines from fresh tumors versus xenografts: comparison of success rate and cell line features. Cancer Res. 67, 398–407. [DOI] [PubMed] [Google Scholar]
- De Waard-Siebinga, I. , Blom, D.J. , Griffioen, M. , Schrier, P.I. , Hoogendoorn, E. , Beverstock, G. , Danen, E.H. , Jager, M.J. , 1995. Establishment and characterization of an uveal-melanoma cell line. Int. J. Cancer. 62, 155–161. [DOI] [PubMed] [Google Scholar]
- Dunavoelgyi, R. , Dieckmann, K. , Gleiss, A. , Sacu, S. , Kircher, K. , Georgopoulos, M. , Georg, D. , Zehetmayer, M. , Poetter, R. , 2011. Local tumor control, visual acuity, and survival after hypofractionated stereotactic photon radiotherapy of choroidal melanoma in 212 patients treated between 1997 and 2007. Int. J. Radiat. Oncol. Biol. Phys. 81, 199–205. [DOI] [PubMed] [Google Scholar]
- Edmunds, S.C. , Cree, I.A. , Dí Nícolantonío, F. , Hungerford, J.L. , Hurren, J.S. , Kelsell, D.P. , 2003. Absence of BRAF gene mutations in uveal melanomas in contrast to cutaneous melanomas. Br. J. Cancer. 88, 1403–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hashemite, N. , Zhang, H. , Henske, E.P. , Kwiatkowski, D.J. , 2003. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. The Lancet. 361, 1348–1349. [DOI] [PubMed] [Google Scholar]
- Furney, S.J. , Pedersen, M. , Gentien, D. , Dumont, A.G. , Rapinat, A. , Desjardins, L. , Turajlic, S. , Piperno-Neumann, S. , De la Grange, P. , Roman-Roman, S. , Stern, M.-H. , Marais, R. , 2013. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 3, 1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gragoudas, E.S. , Egan, K.M. , Seddon, J.M. , Glynn, R.J. , Walsh, S.M. , Finn, S.M. , Munzenrider, J.E. , Spar, M.D. , 1991. Survival of patients with metastases from uveal melanoma. Ophthalmology. 98, 383–390. [DOI] [PubMed] [Google Scholar]
- Griewank, K.G. , Yu, X. , Khalili, J. , Sozen, M.M. , Stempke-Hale, K. , Bernatchez, C. , Wardell, S. , Bastian, B.C. , Woodman, S.E. , 2012. Genetic and molecular characterization of uveal melanoma cell lines. Pigment Cell Melanoma Res. 25, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour, J.W. , 2012. The genetics of uveal melanoma: an emerging framework for targeted therapy. Pigment Cell Melanoma Res. 25, 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour, J.W. , Chen, R. , 2013. The DecisionDx-UM gene expression profile test provides risk stratification and individualized patient care in uveal melanoma. PLoS Curr. 5, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour, J.W. , Onken, M.D. , Roberson, E.D.O. , Duan, S. , Cao, L. , Worley, L.A. , Council, M.L. , Matatall, K.A. , Helms, C. , Bowcock, A.M. , 2010. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 330, 1410–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour, J.W. , Roberson, E.D.O. , Anbunathan, H. , Onken, M.D. , Worley, L.A. , Bowcock, A.M. , 2013. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 45, 133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, A.L. , Musi, E. , Ambrosini, G. , Nair, J.S. , Deraje Vasudeva, S. , De Stanchina, E. , Schwartz, G.K. , 2012. Impact of combined mTOR and MEK inhibition in uveal melanoma is driven by tumor genotype. PLoS One. 7, e40439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith, C.T. , Borisy, A.A. , Stockwell, B.R. , 2005. Multicomponent therapeutics for networked systems. Nat. Rev. Drug Discov. 4, 71–78. [DOI] [PubMed] [Google Scholar]
- Khalili, J.S. , Yu, X. , Wang, J. , Hayes, B.C. , Davies, M.A. , Lizee, G. , Esmaeli, B. , Woodman, S.E. , 2012. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin. Cancer Res. 18, 4345–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiliç, E. , Brüggenwirth, H.T. , Verbiest, M.M.P.J. , Zwarthoff, E.C. , Mooy, N.M. , Luyten, G.P.M. , De Klein, A. , 2004. The RAS-BRAF kinase pathway is not involved in uveal melanoma. Melanoma Res. 14, 203–205. [DOI] [PubMed] [Google Scholar]
- Ksander, B.R. , Rubsamen, P.E. , Olsen, K.R. , Cousins, S.W. , Streilein, J.W. , 1991. Studies of tumor-infiltrating lymphocytes from a human choroidal melanoma. Invest. Ophthalmol. Vis. Sci. 32, 3198–3208. [PubMed] [Google Scholar]
- Laurent, C. , Gentien, D. , Piperno-Neumann, S. , Némati, F. , Nicolas, A. , Tesson, B. , Desjardins, L. , Mariani, P. , Rapinat, A. , Sastre-Garau, X. , Couturier, J. , Hupé, P. , De Koning, L. , Dubois, T. , Roman-Roman, S. , Stern, M.-H. , Barillot, E. , Harbour, J.W. , Saule, S. , Decaudin, D. , 2013. Patient-derived xenografts recapitulate molecular features of human uveal melanomas. Mol. Oncol. 7, 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyten, G.P. , Naus, N.C. , Mooy, C.M. , Hagemeijer, A. , Kan-Mitchell, J. , Van Drunen, E. , Vuzevski, V. , De Jong, P.T. , Luider, T.M. , 1996. Establishment and characterization of primary and metastatic uveal melanoma cell lines. Int. J. Cancer. 66, 380–387. [DOI] [PubMed] [Google Scholar]
- Mallone, S. , De Vries, E. , Guzzo, M. , Midena, E. , Verne, J. , Coebergh, J.W. , Marcos-Gragera, R. , Ardanaz, E. , Martinez, R. , Chirlaque, M.D. , Navarro, C. , Virgili, G. , 2012. Descriptive epidemiology of malignant mucosal and uveal melanomas and adnexal skin carcinomas in Europe. Eur. J. Cancer. 48, 1167–1175. [DOI] [PubMed] [Google Scholar]
- Martin, M. , Maßhöfer, L. , Temming, P. , Rahmann, S. , Metz, C. , Bornfeld, N. , Van de Nes, J. , Klein-Hitpass, L. , Hinnebusch, A.G. , Horsthemke, B. , Lohmann, D.R. , Zeschnigk, M. , 2013. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 45, 933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty, B. , Maire, V. , Gravier, E. , Rigaill, G. , Vincent-Salomon, A. , Kappler, M. , Lebigot, I. , Djelti, F. , Tourdès, A. , Gestraud, P. , Hupé, P. , Barillot, E. , Cruzalegui, F. , Tucker, G.C. , Stern, M.-H. , Thiery, J.-P. , Hickman, J.A. , Dubois, T. , 2008. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 10, R101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades, N. , Chew, S.A. , He, B. , Riechardt, A.I. , Karadedou, T. , Kotoula, V. , Poulaki, V. , 2011. Genotype-dependent sensitivity of uveal melanoma cell lines to inhibition of b-Raf, MEK, and Akt kinases: rationale for personalized therapy. Invest. Ophthalmol. Vis. Sci. 52, 7248–7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Némati, F. , Sastre-Garau, X. , Laurent, C. , Couturier, J. , Mariani, P. , Desjardins, L. , Piperno-Neumann, S. , Lantz, O. , Asselain, B. , Plancher, C. , Robert, D. , Péguillet, I. , Donnadieu, M.-H. , Dahmani, A. , Bessard, M.-A. , Gentien, D. , Reyes, C. , Saule, S. , Barillot, E. , Roman-Roman, S. , Decaudin, D. , 2010. Establishment and characterization of a panel of human uveal melanoma xenografts derived from primary and/or metastatic tumors. Clin. Cancer Res. 16, 2352–2362. [DOI] [PubMed] [Google Scholar]
- Nyfeler, B. , Chen, Y. , Li, X. , Pinzon-Ortiz, M. , Wang, Z. , Reddy, A. , Pradhan, E. , Das, R. , Lehár, J. , Schlegel, R. , Finan, P.M. , Cao, Z.A. , Murphy, L.O. , Huang, A. , 2012. RAD001 enhances the potency of BEZ235 to inhibit mTOR signaling and tumor growth. PLoS One. 7, e48548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken, M.D. , Worley, L.A. , Ehlers, J.P. , Harbour, J.W. , 2004. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 64, 7205–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pópulo, H. , Soares, P. , Faustino, A. , Rocha, A.S. , Silva, P. , Azevedo, F. , Lopes, J.M. , 2011. mTOR pathway activation in cutaneous melanoma is associated with poorer prognosis characteristics. Pigment Cell Melanoma Res. 24, 254–257. [DOI] [PubMed] [Google Scholar]
- Pópulo, H. , Soares, P. , Rocha, A.S. , Silva, P. , Lopes, J.M. , 2010. Evaluation of the mTOR pathway in ocular (uvea and conjunctiva) melanoma. Melanoma Res. 20, 107–117. [DOI] [PubMed] [Google Scholar]
- Rimoldi, D. , Salvi, S. , Liénard, D. , Lejeune, F.J. , Speiser, D. , Zografos, L. , Cerottini, J.-C. , 2003. Lack of BRAF mutations in uveal melanoma. Cancer Res. 63, 5712–5715. [PubMed] [Google Scholar]
- Saraiva, V.S. , Caissie, A.L. , Segal, L. , Edelstein, C. , Burnier, M.N. , 2005. Immunohistochemical expression of phospho-Akt in uveal melanoma. Melanoma Res. 15, 245–250. [DOI] [PubMed] [Google Scholar]
- Singh, A.D. , Topham, A. , 2003. Survival rates with uveal melanoma in the United States: 1973–1997. Ophthalmology. 110, 962–965. [DOI] [PubMed] [Google Scholar]
- Singh, A.D. , Turell, M.E. , Topham, A.K. , 2011. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 118, 1881–1885. [DOI] [PubMed] [Google Scholar]
- Straussman, R. , Morikawa, T. , Shee, K. , Barzily-Rokni, M. , Qian, Z.R. , Du, J. , Davis, A. , Mongare, M.M. , Gould, J. , Frederick, D.T. , Cooper, Z.A. , Chapman, P.B. , Solit, D.B. , Ribas, A. , Lo, R.S. , Flaherty, K.T. , Ogino, S. , Wargo, J.A. , Golub, T.R. , 2012. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 487, 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trolet, J. , Hupé, P. , Huon, I. , Lebigot, I. , Decraene, C. , Delattre, O. , Sastre-Garau, X. , Saule, S. , Thiéry, J.-P. , Plancher, C. , Asselain, B. , Desjardins, L. , Mariani, P. , Piperno-Neumann, S. , Barillot, E. , Couturier, J. , 2009. Genomic profiling and identification of high-risk uveal melanoma by array CGH analysis of primary tumors and liver metastases. Invest. Ophthalmol. Vis. Sci. 50, 2572–2580. [DOI] [PubMed] [Google Scholar]
- Tuefferd, M. , De Bondt, A. , Van Den Wyngaert, I. , Talloen, W. , Verbeke, T. , Carvalho, B. , Clevert, D.-A. , Alifano, M. , Raghavan, N. , Amaratunga, D. , Göhlmann, H. , Broët, P. , Camilleri-Broët, S. , 2008. Genome-wide copy number alterations detection in fresh frozen and matched FFPE samples using SNP 6.0 arrays. Genes Chrom. Cancer. 47, 957–964. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk, C.D. , Bezrookove, V. , Green, G. , Bauer, J. , Gaugler, L. , O'Brien, J.M. , Simpson, E.M. , Barsh, G.S. , Bastian, B.C. , 2008. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 457, 599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk, C.D. , Griewank, K.G. , Crosby, M.B. , Garrido, M.C. , Vemula, S. , Wiesner, T. , Obenauf, A.C. , Wackernagel, W. , Green, G. , Bouvier, N. , Sozen, M.M. , Baimukanova, G. , Roy, R. , Heguy, A. , Dolgalev, I. , Khanin, R. , Busam, K. , Speicher, M.R. , O'Brien, J. , Bastian, B.C. , 2010. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 363, 2191–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, A. , Hengge, U.R. , Urbanik, D. , Markwart, A. , Mirmohammadsaegh, A. , Reichel, M.B. , Wittekind, C. , Wiedemann, P. , Tannapfel, A. , 2003. Absence of mutations of the BRAF gene and constitutive activation of extracellular-regulated kinase in malignant melanomas of the uvea. Lab. Invest. 83, 1771–1776. [DOI] [PubMed] [Google Scholar]
- Weigelt, B. , Warne, P.H. , Downward, J. , 2011. PIK3CA mutation, but not PTEN loss of function, determines the sensitivity of breast cancer cells to mTOR inhibitory drugs. Oncogene. 30, 3222–3233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary Figure S1. DNA microarray analysis of the 7 Uveal Melanoma cell lines. Allele ratio (top) copy number values (middle), and Log2ratio (bottom) for MM28 (A), MM33 (B), MP38 (C), MP41 (D), MP46 (E), MP65 (F), MM66 (G); red line: LOH.
Supplementary Figure S2. Sensitivity of a representative panel of uveal melanoma cell lines to AKT inhibitor Perifosine. MM28 (GNAQ 11 mutated, BAP1 deficient) MP38 (GNAQ mutated, BAP1 deficient), MP41 (GNA11 mutated), MP46 (GNAQ mutated, BAP1 deficient) MP65 (GNA11 mutated, BAP1 deficient), MM66 (GNA11 mutated), 92.1 (GNAQ mutated, EIF1AX mutated), Mel202 (GNAQ mutated, SF3B1 mutated), OMM1 (GNA11 mutated), OMM2.5 (GNAQ mutated) were seeded at adequate concentration and left in contact with the drugs for 5 days. Cell viability was quantified with MTT assay. Results are expressed as mean of at least 3 separate experiments. Error bars represent standard errors of the mean.
Supplementary Figure S3. Synergy by excess over Bliss (A) and corresponding normalized cell viability fractions (B) for the combination of Everolimus and Trametinib on a panel of 10 UM cell lines. Fractional inhibition calculated for Trametinib and Everolimus used as single agent and in combination were used to assess synergy as explained in paragraph 2.11. Results from 3 independent experiments were used to calculate 95% confidence intervals. A shows synergy values as mean of three separate experiments, values that resulted significantly >0 with a p value <0.05 are colored in red (synergy), values that resulted significantly < 0 with a p value <0.05 are colored in blue (antagonism), values that produced synergy values not significantly different from 0 are colored in white (antagonism). B. express the corresponding percentage of fractional inhibition for each condition tested.
Supplementary data
Supplementary data