

Abstract
Bone metastasis represents one of the most deleterious clinical consequences arising in the context of many solid tumors. Severe osteolysis results from tumor cell colonization of the bone compartment, a process which entails reciprocal exchange of soluble signals between tumor cells and their osseous microenvironment. Recent evidence indicates that tumor‐intrinsic miRNAs are pleiotropic regulators of gene expression. But they are also frequently released in exosome‐like vesicles (ELV). Yet the functional relevance of the transference of tumor‐derived ELV and their miRNA cargo to the extracellular milieu during osseous colonization is unknown.
Comparative transcriptomic profiling using an in vivo murine model of bone metastasis identified a repressed miRNA signature associated with high prometastatic activity. Forced expression of single miRNAs identified miR‐192 that markedly appeased osseous metastasis in vivo, as shown by X‐ray, bioluminescence imaging and microCT scans. Histological examination of metastatic lesions revealed impaired tumor‐induced angiogenesis in vivo, an effect that was associated in vitro with decreased hallmarks of angiogenesis. Isolation and characterization of ELV by flow cytometry, Western blot analysis, transmission electron microscopy and nanoparticle tracking analysis revealed the ELV cargo enrichment in miR‐192. Consistent with these findings, fluorescent labeled miR‐192‐enriched‐ELV showed the in vitro transfer and release of miR‐192 in target endothelial cells and abrogation of the angiogenic program by repression of proangiogenic IL‐8, ICAM and CXCL1. Moreover, in vivo infusion of fluorescent labeled ELV efficiently targeted cells of the osseous compartment. Furthermore, treatment with miR‐192 enriched ELV in a model of in vivo bone metastasis pre‐conditioned osseous milieu and impaired tumor‐induced angiogenesis, thereby reducing the metastatic burden and tumor colonization.
Changes in the miRNA‐cargo content within ELV represent a novel mechanism heavily influencing bone metastatic colonization, which is most likely relevant in other target organs. Mechanistic mimicry of this phenomenon by synthetic nanoparticles could eventually emerge as a novel therapeutic approach.
Keywords: Lung cancer, Metastasis, Cell communication, Exosome, HUVEC, Angiogenesis, Adenocarcinoma
Highlights
miRNAs are released by tumor cells in exosome‐like vesicles (ELV) to the local milieu.
ELV are incorporated into endothelial cells where their miRNA‐cargo is released.
Transferred cargo modulates their angiogenic program and bone metastatic colonization.
This novel mechanism could be relevant in other steps of the metastatic cascade.
Abbreviations
- ADC
Adenocarcinoma
- CM
Conditioned Medium
- BLI
Bioluminescence imaging
- ELV
Exosome-like vesicles
- HUVEC
Human umbilical vein endothelial cells
- SCDC
Single-cell derived colonies
- HMS
Highly metastatic subpopulations
- MV
Microvesicles
- TRAP
Tartrate-resistant acid phosphatase
- MMP
Metalloproteinase
- i.c.: intracardiac
i.t.: intratibial
1. Introduction
Bone constitutes a preferred site of metastasis for many solid tumors (Mundy, 2002), including those of prostate, breast, kidney, thyroid and lung (Weilbaecher et al., 2011). But as opposed to other neoplasms, lung cancer is diagnosed at advanced stages when metastases are already established. As many as 30–40% of patients with non‐small cell lung cancer (NSCLC) develop bone metastases characterized by osteolytic lesions associated with significant reduction of quality of life and dismal prognosis (Coleman, 1997).
During the multistep course of osseous metastasis development, survival in the circulation, engagement in the target organ, and other functions allow for the efficient colonization of the skeletal compartment (Fidler, 2003; Nguyen and Massague, 2007). This later step frequently entails the formation of tumor‐induced osteolysis and the progressive infiltration of malignant cells in the bone marrow compartment, where a variety of mechanisms contribute to bone colonization.
Along this process, intercellular signals mediated by soluble factors released by the osseous stroma contribute as chemoattractants for receptors expressed in tumor cells (Muller et al., 2001) and facilitate metastatic homing. Subsequently, survival and engagement in the bone microenvironment occur by the activation of critical pathways triggered by complex intercellular and/or cell–matrix interactions. Activation of collagen receptor DDR1 (Valencia et al., 2012) or other intracellular pathways such as Src in tumor cells (Zhang et al., 2009) mediate survival required for effective engagement to bone and also confer resistance to apoptosis in the osseous microenvironment. Moreover, signals via PDGFR/VEGFR axis were highly involved in tumor‐bone marrow stroma interactions in bone metastases, and the exclusive blockade of this pathway in the stromal compartment severely impaired ossesous colonization (Catena et al., 2011), since ligands acting through this pathway allow efficient tumor engraftment (Coenegrachts et al., 2010). This pathway also elicited osteoclast activity leading to tumor‐induced osteolysis a hallmark of bone colonization (Guise et al., 1996; Yin et al., 1999) and allowed for proteolytic degradation of the ECM achieved by the complex secretion of MMP and other proteases. Similarly to other metastatic sites, full metastatic expansion proceeds by tumor‐induced angiogenesis a process also regulated by signals released upon host–tumor interactions at the tumor endothelial/stromal bone interphase (Vicent et al., 2008). Yet, the complexity of soluble signals that mediate this process that entails overt growth of bone metastases is far from being understood.
miRNAs, small non‐coding RNAs involved in the regulation of gene expression, play an important role during tumorogenesis (Croce and Calin, 2005; Antonyak et al., 2011; Kumar et al., 2007). Previous studies have also suggested their involvement as regulators of metastasis (Ma et al., 2007; Tavazoie et al., 2008). A single miRNA, inducing a tumor‐intrinsic regulation of a multigenic network, elicited endothelial recruitment leading to metastatic initiation and colonization (Png et al., 2012). MiRNAs are also frequently released to the extracellular milieu into microvesicles and exosomes (Skog et al., 2008) that are detectable in body fluids (Yu et al., 2010). Besides miRNAs, exosomes (30–100 nm of size) (Simons and Raposo, 2009) also contain a discrete set of membrane receptors, such as CD63, intracellular proteins, cytoskeletal components and nucleic acids (Montecalvo et al., 2012) and are constitutively released in response to activation or stress. They act as unique vehicles for the transport of their cargo to neighboring cells or distant organs. This transfer constitutes an emerging paradigm of cellular communication and reprogramming in physiological and pathological processes (Valadi et al., 2007).
Here, in the context of metastatic expansion, we describe a previously unappreciated mechanism in which tumor cells, by decreasing anti‐angiogenic miRNAs and their release and transfer through exosome‐like vesicles, perturb the surrounding endothelial compartment during tumor‐induced angiogenesis, leading to overt osseous colonization. Changes in exosome cargo by re‐expression of a single anti‐angiogenic miRNA dramatically repressed the tumor‐induced angiogenic program leading to a blatant reduction of bone metastatic lesions. Thus, our findings underscore the pleiotropic functions orchestrated by a single miR‐192 and identify novel mediators of relevance in key steps of the metastatic cascade. Our work further emphasizes the process of metastasis‐induced angiogenesis in bone as critically involved in metastatic colonization.
2. Materials and methods
2.1. Cell lines and reagents
Lung cancer cell lines were obtained from the ATCC (Manassas, VA) and were grown in RPMI 1640 with l‐glutamine (Invitrogen, Barcelona, Spain) supplemented with 10% fetal bovine serum (Invitrogen), 1% penicillin and streptomycin (Invitrogen). A549 cells and derived subpopulations were authenticated by sequencing of two distinctive mutations in KRAS and STK4. HUVECs were seeded onto gelatin‐coated culture dishes and cultured in EGM‐2 medium (Lonza) supplemented with 5% fetal bovine serum. ST‐2 murine stromal cells were grown using Dulbecco's modified Eagle's medium with ultraglutamine (Cambrex). VEGFα was obtained from RELIATech GmbH, and bFGF, luciferin was from Promega. Retroviral MirVec for the overexpression of each miRNA were obtained from Source Bioscience (Nottingham, UK), and sequenced for assessment of the integrity of the insert. The following antibodies were used: mouse anti‐Alix, rabbit anti‐Calnexin and rabbit anti‐TSG101 (all from Cell Signaling Technology, Inc., Danvers, MA).
2.2. Coculture in Boyden chamber
HUVEC cells were seeded in a 6‐well plates at density of 1.25 × 105 per well using EGM‐2 medium with exosome‐depleted serum. After 1–2 h, 6.2 × 104 tumor cells were seeded in the upper chamber in exosome‐free RPMI. After 48–72 h incubation, HUVEC cells were recovered to perform subsequent experiments.
2.3. BrdU proliferation assay
HUVEC per well were seeded (3000 cell/well) into 96‐well plate coated with gelatin 0.2% and allowed to adhere for 16 h. Between 6 and 10 replicates per condition were seeded. The medium was washed with PBS and replaced with EBM‐2 medium supplemented with bFGF (10 ng/ml) and VEGF‐α (50 ng/ml). Sixteen hours later, BrdU was added and the culture was incubated for 8 h. Incorporated BrdU was measured with ELISA (Roche Applied Sciences) according to the manufacturer's recommendations.
2.4. Scratch wound migration assay
HUVEC were seeded (5 × 104 cells/well) into a 0.2% gelatin precoated 48‐well plate in EGM‐2 medium and incubated for 24 h. Using a tip, a “scratch” was performed in the monolayer (at time 0). Cells were then washed with PBS and incubated with EBM‐2 medium containing 10 ng/ml recombinant bFGF and 50 ng/ml recombinant VEGFα for 7 h. The distance between the two sides of the wound was measured with a graduated ocular lens coupled with an Olympus CKX41 microscope (Olympus) and subtracted from the distance at time 0.
2.5. Capillary network formation
The ability of HUVECs that have been treated with tumoral CM to form capillary networks was evaluated in a Matrigel™ angiogenesis assay. Briefly, 8750 pretreated HUVEC cells were plated in a 96‐well plate precoated with 35 μl Matrigel per well (BD Biosciences). Cells were incubated in EGM‐2 medium for 16 h (Lonza). Fifteen randomly chosen fields per condition were counted. Quantitative analysis of network structure was performed with ImageJ® software (http://rsbweb.nih.gov/ij/) by counting the number of intersections in the network.
2.6. Exosome like‐vesicles (ELV) isolation
ELV were isolated from cell culture supernatants of A549, mock M1 and miR‐192 M1 overexpressing cells. To remove cells, conditioned media were centrifuged for 20 min at 2000 × g at 4 °C. To remove the remaining debris, another centrifugation step was performed for 40 min at 8200 × g. To remove all particles >200 nm, supernatants were filtered through low‐protein binding 0.22 μm pore filters. To concentrate the exosomes, supernatants were ultracentifuged at 110 000 × g for 2 h at 4 °C. Then, pelleted exosomes were washed with PBS and ultracentrifuged again at 110 000 × g for 2 h at 4 °C. The precipitates were resuspended in 50 μl PBS and stored at −80 °C. The protein quantification was performed using Pierce BCA Protein Assay Kit (Thermo). Similar protocol was used to deplete exosomes from fetal bovine serum employed in all subsequent cellular experiments.
2.7. Dynamic light scattering (DLS)
DLS and zeta potential determinations were performed with a Zetasizer nanoseries instrument (Malvern Nano‐Zetasizer, λ = 532 nm laser wavelength). The ELV size data refers to the scattering intensity distribution (z‐average).
2.8. Nanoparticle tracking analysis (NTA)
NTA measurements were performed with a NanoSight LM20 (NanoSight, Amesbury, United Kingdom), equipped with a sample chamber with a 640‐nm laser and a Viton fluoroelastomer O‐ring. The samples were diluted at ∼2–5 ng/μL and injected in the sample chamber with sterile syringes (BD Discardit II, New Jersey, USA) until the liquid reached the tip of the nozzle. All measurements were performed at room temperature.
2.9. Transmission electron microscopy
Sample was mounted on a carbon film mesh (CF300‐Cu, EMS, Hatfield, PA) with 2% Kellenberger uranyl acetate. Samples were visualized on TEM (Philips CM 120 Biotwin) and images were captured on a digital camera (Olympus SIS).
2.10. Exosome‐like vesicle (ELV) staining
Isolated ELV were labeled with PKH67 Green Fluorescent Cell Linker Kit for General Cell Membrane Labelling (Sigma–Aldrich) according to the manufacturer's recommendations. For immunofluorescence, cells were seeded in gelatin precoated glass. After overnight culture, cells were incubated with ELV for 24 h. After fixation cells were incubated for 30 min in INMUNO, washed several times with PBS and mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA).
2.11. CD63 staining
Briefly, 5 μg of ELV were mixed with 5 μl of 4 μm‐latex‐beads (Invitrogen) and the mixture was incubated 30 min at 4 °C. After resuspension in 400 μl PBS, 5% BSA was added and incubated for 30 min. The ELV conjugated with the latex beads were pelleted by centrifugation 100 × g for 10 min at 4 °C. The pellet was resuspended in 50 μl PBS and 5% BSA. Incubation with primary antibody 1/50 was performed 1 h at 4 °C (CD63 anti‐human BD Biosciences) followed by several PBS washes and centrifuged at 1000 × g 10 min at 4 °C. Subsequently, incubation with secondary biotinylated antibody 1/200 was performed for 30 min at 4 °C (Biotin‐conjugated‐ IgG2 goat anti‐mouse, Dako) and a final incubation with streptavidin‐phycoeritrin × 28 (Jackson Inmuno Research), at 1/50 incubated for 30 min was performed in the dark. After centrifugation the sample was resuspended in 300 μl of PBS before FACS scan analysis. Non‐incubated ELV with the primary antibody were used as controls.
2.12. Target screening
We used three publicly available search engines for target prediction to obtain the putative targets: TargetScan (Release 2.1), http://genes.mit.edu/targetscan and miRanda.
2.13. Statistical analysis
Statistical analysis was performed using SPSS 15.0. To study differences in proliferation rates, invasion and migration assays, differences in metastatic area, number of osteoclasts, SCC number, adhesion to cell monolayers or precoated wells, and MMP activity, data were analyzed by parametric test (Anova) or non‐parametric homologue Kruskal–Wallis test depending on data distribution. Multiple comparisons were studied with Dunnet's test or U Mann–Whitney adjusted by Bonferroni test. Values were expressed as means ± SEM and statistical significance was defined as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
Other methods including intracardiac and intratibial injection (i.t.), radiographic, microcomputed tomographic (μCT) analysis and Bioluminescence Imaging (BLI), and histological analysis, subcutaneous tumor growth, have been described elsewhere (Yin et al., 1999). A detailed version of “Material and Methods” is available in the supplementary section.
3. Results
3.1. Identification of miR‐192
We took advantage of highly metastatic subpopulations (HMS) with marked propensity to form osseous metastases derived from a parental human adenocarcinoma A549 cell line, as described previously (Vicent et al., 2013, 2013, 2008). To identify miRNAs with robust role in metastasis, we established a dual screening strategy by transcriptomic selection of miRNAs, and subsequent functional test of their contribution to in vitro invasiveness and to the in vivo prometastatic activity.
We utilized human microarrays to identify miRNAs differentially expressed in highly metastatic subpopulations (HMS), M1, M3 and M4, compared to the parental cell line. Most of the differentially expressed miRNAs were downregulated in HMS, with the exception of miR‐21 and miR‐101 (Figure 1A). We confirmed these results using real‐time PCR (Figure 1B). These two miRNAs, together with miR‐34a and miR‐335, have been previously reported as dysregulated in tumor development and metastasis (Liu et al., 2011). To identify miRNAs that exhibit functional relevance in metastasis, we performed an invasion assay using the HMS M1 transduced with a retrovirus for overexpression of single miRNAs or empty vector (mock) (Figure 1C). The invasiveness of cells overexpressing miR‐192, miR‐215, and miR‐138 was dramatically decreased suggesting that these miRNAs were potentially involved as repressors of the regulatory network associated with metastasis (Figure 1D). These data indicate that miR‐192, miR‐215, and miR‐138 modulate invasiveness, a function relevant to metastatic activity.
Figure 1.
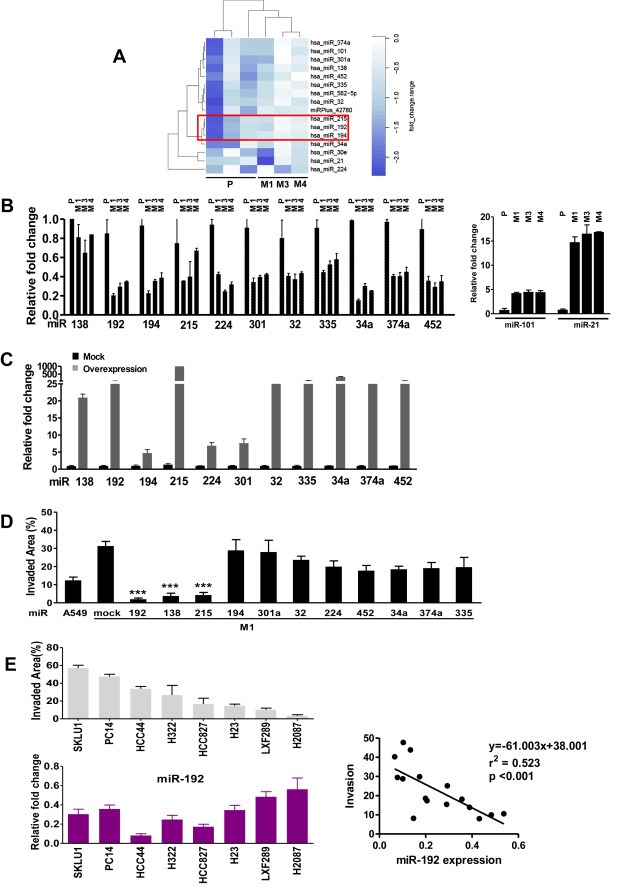
Identification of metastatic associated‐miR signature. A. Unsupervised clustering of HMS (M1, M3 and M4) and parental A549 cells (P). Dark blue denotes strong repression, whereas white denotes “no change”. B. Validation of all single differentially expressed miRNAs in the HMS (M1, M3 and M4) and A549 by qPCR. C. Relative expression of different miRNA in M1 highly‐metastatic‐subpopulation retrovirally transduced with a single miRNA as compared to mock transduced M1 cells. D. Invasive assay with collagen type I in Boyden chambers of M1 cells overexpressing each single miRNA compared to mock transfected M1 cells. A number of 2 × 105 cells was seeded with >95% viability for each cell line. E. Top: Invasion assay in a panel of human ADC cell lines. Bottom: Relative expression levels of miR‐192 in the panel of ADC cell lines. Right: A robust correlation was shown between invasiveness and miR‐192 expression levels. *p < 0.05, **p < 0.01, **p < 0.001.
To confirm the relevance of this observation, we utilized a panel of human lung adenocarcinoma cell lines and investigated the correlations between the expression levels of these three miRNAs and invasive ability. There was a highly significant inverse correlation (R = 0.523, p < 0.001) between miR‐192 levels and invasiveness (Figure 1D and E). In contrast, levels of miR‐215 and miR‐138 were not significantly correlated (Sup Fig. S1A, B). Taken together, these data suggest that miR‐192 potentially participates in the cellular invasiveness of lung adenocarcinoma. Finally, because proteolytic degradation is an important cellular function involved in metastatic invasion, MMP activity was determined using a fluorogenic assay. As expected, MMP activity was affected in cell lines overexpressing miRNAs. MMP‐3/10 activity was downregulated in CM of miR‐192 cells. A global MMP inhibitor GM6001 was used as a positive control of inhibition (Sup. Fig. S2).
3.2. miR‐192 confers antimetastatic activity in vivo
Given the limitations related to tumor–host interactions of in vitro assays, we tested the association between miR‐192, miR‐215, and miR‐138 and the pro‐metastatic activity of lung cancer cells in vivo. After intracardiac (i.c.) inoculation of cells overexpressing single miRNAs, mice (n = 9 per group) inoculated with miR‐192 tumor cells demonstrated a significant decrease in tumor burden from day 14 to day 21, as evaluated by bioluminescence imaging (BLI) (Figure 2A). In comparison to controls (mock), miR‐192‐tumor cells inoculated mice exhibited a reduction in bone osteolytic lesions at the level of parental cells according to X‐ray imaging and microcomputed tomography (μCT) analysis (Figure 2B and C). As a consequence of striking differences between bone metastatic lesions induced by mock and miR‐192 overexpressing cells, angiogenesis of incipient lesions observed in miR‐192 inoculated mice could not be compared with overt mock‐induced metastases (data not shown). However, the number of TRAP+ multinucleated cells at the tumor–bone interface at day 21 was dramatically reduced in miR‐192 animals (Sup Figure 3A). Taken together, these data indicate that increased miR‐192 levels decrease the pro‐metastatic activity of lung cancer cells in the bone compartment. Interestingly, no differences in metastases were detected in mice inoculated with miR‐215 and miR‐138 tumor cells as compared to controls (mock) (Sup Fig S3B,C). Cell growth kinetics in vitro was unchanged (Sup Fig S3D). Similarly, the cell growth kinetics of miR‐192 tumor cells did not exhibit differences in vitro or in vivo (Sup Fig S4A,B). Cell cycle components including TP53, p21, p‐Rb, CDK6, cyclin D1 and CNEE were also unaffected (Sup Fig S4C). Taken together, these data indicate that miR‐192 overexpression suppresses the pro‐metastatic activity of lung cancer cells by diminishing tumor‐induced osteolysis.
Figure 2.
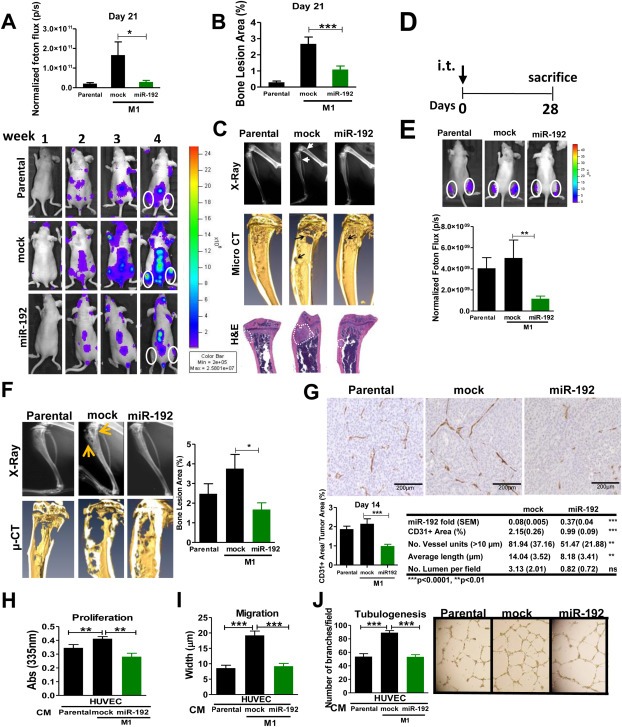
Effect of miR‐192 in bone metastasis and colonization in vivo. A. Cells overexpressing miR‐192 levels, vector‐transduced (mock), and parental (A549) cells were inoculated into the left cardiac ventricle of athymic nude mice. Top: Quantification of photon flux at day 21 post‐inoculation and Bottom: representative BLI. B. Quantification of osteolytic bone area of X‐ray imaging at day 21 post‐inoculation. C. Representative images of X‐ray (top), micro‐CT scans (middle), and H&E sections (bottom) showing the dramatic decrease of bone metastasis burden in animals inoculated with miR‐192 cells. Arrowhead indicates the location of osteolytic lesions. Metastatic area is depicted by a punctate line. D. Experimental regimen of bone colonization assay after intratibial injection of miR‐192 cells. E. Top: BLI quantification. Bottom: Representative photon flux images in the metaphyses of tumor‐bearing mice. F. Left: Bones were analyzed by X‐ray and μCT scans. Right: Quantification of osteolytic lesions in miR‐192 overexpressed cells of injected animals demonstrated a decreased tumor burden in the metaphyses. G. Immunohistochemical analysis of CD31+ cells in tumors. Top: M1 overexpressing miR‐192 cells exhibited a significant decrease in tumoral vessels. Representative images. Scale bar = 200 μm. Bottom left: Quantification of CD31+ area. Bottom right: Expression levels of miR‐192 were assessed by RT‐qPCR after microdissection of paraffin sections derived from mice i.c. inoculated with mock and miR‐192 overexpressing cells. Angiogenic parameters in metastatic bone lesions were assessed by image analysis. H. Cell proliferation of HUVEC cells induced after 72 h co‐culture with parental A549 cells, mock transduced, and miR‐192 overexpressing cells. I. Cell migration assay (scratch assay) of HUVEC cells after 72 h co‐culture with parental A549 cells, mock transduced, and miR‐192 overexpressing cells. J. Left: Tubulogenesis assay of HUVEC cells after 72 h co‐culture with parental A549 cells, mock transduced and miR‐192 overexpressing cells. Right: Representative images.
3.3. miR‐192 impairs tumor‐induced angiogenesis in bone metastasis
To further explore whether miR‐192 endows tumor cells with additional functions that could be relevant in the process of osseous colonization, we used an intratibial (i.t.) injection model (Figure 2D). As opposed to the i.c. injection approach and because the high number of cells injected, this model obviates the contribution of tumor‐induced osteoclastogenesis, since the accelerated tumor growth prevents the mobilization and fusion of mononuclear precursor cells. Rather, this model relies on the tumor cell growth highly‐dependent on host mediated effects, MMP activities and tumor‐induced angiogenesis (unpublished observations). BLI of the tibias at day 14 post‐injection, showed marked differences between the groups (Figure 2E). Moreover, image analysis demonstrated a decrease in tumor size in miR‐192‐injected mice compared to controls (Figure 2F). Because, we have previously shown that metastasis‐induced angiogenesis process is required for osseous colonization (Catena et al., 2011), we analyzed angiogenesis by CD31+ staining. Interestingly, the number of intra‐tumoral vessels detected in miR‐192 injected animals was dramatically decreased compared to mice injected with mock‐transduced cells (Figure 2G). Digital image analysis and quantification of vessel structure demonstrated a severely reduced average number of discrete vessel units and length in miR‐192 overexpressing tumors, whereas the number of lumens did not reach statistical significance (Figure 2G). Assessment of miR‐192 levels in vivo after microdissection of tumor cells from formalin fixed paraffin embedded tissue sections revealed that tumor cells still express high levels of miR‐192 in vivo as compared to mock cells (Figure 2G). Notably, no differences were found in cell proliferation marker Ki‐67 and in cleaved caspase‐3 in osseous tumor sections at day 28 post‐injection (Sup Fig. S5).
Next, we investigated the effects of tumor cell secreted factors on the functions required for tumor‐induced angiogenesis such as endothelial proliferation, migration and tubulogenesis. We co‐cultured tumor cells with HUVEC cells in Boyden chambers. In accordance with previous findings, co‐incubation of metastatic mock M1 cells slightly increased HUVEC proliferation (Figure 2H). Notably, no effect on apoptosis was observed in all three conditions tested (Sup. Fig. S6A). Similarly, migration activity was assessed by the scratch assay in HUVEC cells. Enhanced endothelial cell migration was observed when metastatic mock M1 cells were used in the co‐culture, an effect that was appeased using miR‐192 overexpressing cells in the co‐culture (Figure 2I). More importantly, in a tubulogenesis assay, increased vessel connectivity in HUVEC cells, a hallmark of angiogenesis, was detected when mock cells were co‐cultured (Figure 2J). In contrast, co‐culture with miR‐192 cells negated this effect on tubule connectivity (Figure 2J), preventing the pro‐angiogenic effects induced by the CM derived from mock cells. Taken together, these data suggest that CM enriched with miR‐192 repressed the strong colonization of the highly metastatic subpopulation in the bone compartment an effect highly associated with the tumor‐induced angiogenic activity.
3.4. Extracellular release of miR‐192 into exosome‐like vesicles (ELV)
Because miRNAs modulate angiogenesis and are frequently released to the extracellular milieu in microvesicles and exosomes, we sought to investigate the potential effects of secreted miR‐192 in regulating angiogenesis in metastatic colonization. As with our analysis of miRNA levels, we detected low levels of miR‐192 in the conditioned‐medium (CM) derived from mock cells (mock‐transduced M1 cells) in relation to CM derived from parental A549 cells. As expected, levels of miR‐192 were restored in miR‐192 cells (M1 cells overexpressing miR‐192) (Sup. Figure 6B). We isolated microvesicles (MV) from parental, mock, and miR‐192 overexpressing cells; these MV were detected by the presence of CD63+, TSG101 and ALIX in all extracts, general hallmarks found in exosomes, whereas the cytoplasmic protein calnexin was used as a negative control (Figure 3A). Moreover, transmission electron microscopy analysis revealed small vesicles heterogeneous in size in the range of ∼40–150 nm for all three cell lines analyzed (Figure 3B). Fused MV were not detected in any of the extracts analyzed by this technique. Thus, the MV isolated contained a high abundance of exosome‐like vesicles (ELV).
Figure 3.
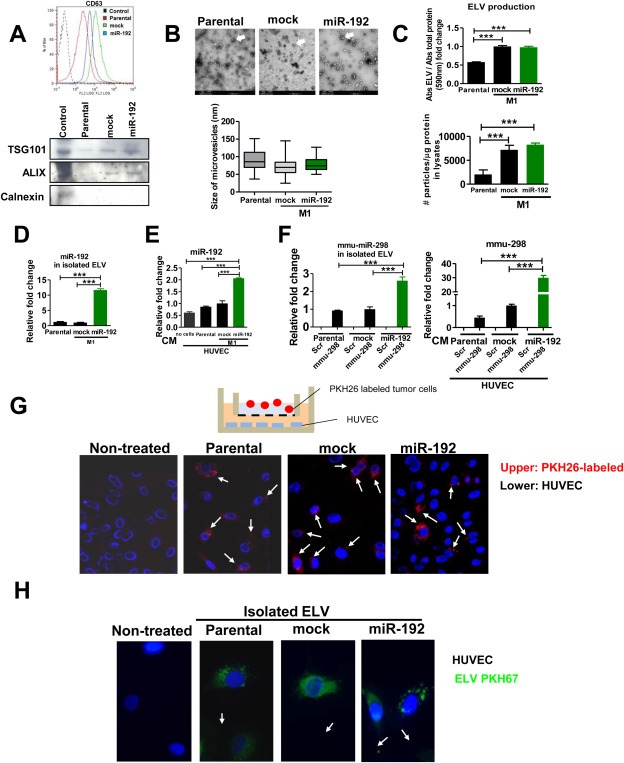
Characterization of tumor‐derived ELV. A. Top: FACS analysis demonstrating the presence of CD63+ vesicles in ELV in ultracentrifugated CM from parental (A549), mock (M1, mock‐transduced), and miR‐192‐overexpressing cells (M1, transduced with miR‐192). Bottom: western blot analysis of ELV proteins such as TSG101 and Alix, as compared to Calnexin, a cytoplasmic protein not present in exosomes, in ELV isolated from parental, mock and miR‐192‐overexpressing cells. B. Top: Transmission electron microscopy images of microvesicles derived from their respective cells, with a range ∼40–150 nm, compatible with exosomal like vesicles (ELV). Bar = 500 nm. Bottom: Quantification in random fields was performed by image analysis. C. Top: ELV production levels were determined by measuring the amount of protein in the ELVs isolated from supernatants normalized with the total amount of protein in cell lysates. A greater amount of ELV protein from mock and miR‐192 cells were detected compared to the parental cell line A549. Bottom: The number of particles assessed by Nanosight technology was normalized with the total amount of protein of cell lysates from which they derived. Of note, a greater number of particles was released from mock and miR‐192 cells than from parental cells. D. Relative fold content of miR‐192 levels in isolated ELVs assessed by qPCR. E. Quantification of miR‐192 measured in HUVEC cells co‐cultured in Boyden chambers with parental, mock and miR‐192 overexpressing cells. We observed high levels of miR‐192 in HUVEC cells co‐cultured with tumor miR‐192 cells as compared to HUVEC cultured alone. F. Transitory overexpression with a murine miRNA, mmu‐miR‐298 or scramble (scr), was performed in parental, mock, and miR‐192 tumor cells. Left: Quantification by qPCR of mmu‐miR‐298 levels in isolated ELVs released by parental, mock and miR‐192 cells previously transfected with scramble (scr) or mmu‐miR‐298. Right: Quantification by qPCR of mmu‐miR‐298 levels in HUVEC cells co‐cultured in Boyden chambers with parental, mock and miR‐192 tumor cells previously transfected with scramble (scr) or mmu‐miR‐298. G. Tumor cells (parental, mock, and miR‐192) were fluorescently‐labeled with PKH26 and cocultured in Boyden chambers with HUVEC as in the figure. Unlabeled cells were used a negative control and nuclei were counterstained with DAPI. Positive red fluorescence was detected in HUVEC cells in the three conditions. H. Isolated ELVs from indicated cells were fluorescently labeled with PKH67 and incubated with HUVEC cells. Nuclei were counterstained with DAPI and exhibited the presence of incorporated ELVs in the cytoplasm of HUVEC cells (in green).
Consistent with previous findings showing increased amount of ELV released by aggressive cells (Ginestra et al., 1998), the total protein levels of the ELV increased in M1‐derived metastasic cell lines (mock and miR‐192 overexpressing cells) as compared to parental cells. However, no differences were detected between M1‐derived metastatic mock and miR‐192 cells (Figure 3C). Similarly the assessment of the number of particles normalized to the protein content of cell lysates revealed an increased number of particles in metastatic M1 cells (mock and miR‐192 overexpressing cells), as compared to the parental cell line (Figure 3C). These data indicate that metastatic M1‐derived cell lines associate with an increased amount of ELV release as compared to the parental cells.
3.5. miR‐192 enriched ELV can be transferred to endothelial cells
We analyzed the levels of miR‐192 in isolated ELV derived from each cell line. Interestingly, miR‐192 levels detected in isolated ELV were similar to levels previously detected in CM for all three cell lines (Figure 3D), whereas the protein secretion of ELV was similar between mock and miR‐192 overexpressing cells (Sup Figure 6C). To investigate the potential role of released ELV fusing with other cells in intercellular communication, we co‐cultured tumor cells with HUVEC in Boyden chambers. Endothelial lysates revealed the presence of endogenous miR‐192 in HUVEC cells, whereas the presence of miR‐192 was slightly higher in HUVEC cells co‐cultured with parental A549 and M1 metastatic mock cells (Figure 3E). Interestingly, a 4‐fold increase in the level of miR‐192 in HUVEC cells was detected when the cells were co‐cultured with miR‐192 overexpressing cells (Figure 3E). Similar results were obtained with another human endothelial cell line HBMEC (Sup Figure 6D). These data strongly suggest that miR‐192 released from miR‐192‐overexpressing cells could be transferred to endothelial cells. However, it could be speculated that miR‐192 could be secreted by HUVEC cells upon ELV incubation. To disprove this possibility, we transfected parental, mock and miR‐192 cells with murine miR‐298 (mmiR‐298), a miR not present in human cells, or with a pre‐miR‐control (Scr). After ELV isolation, quantification of total levels of mmiR‐298 demonstrated the presence of mmiR‐298 in transfected cells. Interestingly, mmiR‐298 levels were higher in miR‐192 cells as compared to mock and parental cells transduced with mmiR‐298 (Figure 3F). After co‐culturing these cells on Boyden chambers together with HUVEC we detected mmiR‐298 in HUVEC cells by qPCR, suggesting that mmiR‐298 was released from the tumoral cells and crossed the cell membrane of HUVEC cells (Figure 3F). We observed an increase in the release of mmiR‐298 in miR‐192 overexpressing cells as compared to mock, most likely due to a spurious cross‐species effect of murine mmiR‐298 in human cells. Indeed, fluorescence was detected in the cytoplasmic compartment of HUVEC cells after incubation of cells with fluorescently‐labeled tumor cells in Boyden chamber assays, suggesting tumor release and transfer of ELV to endothelial cells (Figure 3G). Similar results were obtained when ELV previously isolated from all three cell lines were fluorescently labeled and incubated into HUVEC cells (Figure 3H). Taken together, these findings unambiguously indicate that miRNAs can be released within ELV by tumor cells into the CM and can fuse releasing their cargo into the cytosol of neighboring cells.
3.6. miR‐192 enriched ELV attenuates the angiogenic program in vitro
Next, we assessed whether ELV isolated from each cell line could affect in vitro endothelial connectivity, a hallmark of angiogenesis. As expected, HUVEC incubation with ELV derived from miR‐192 overexpressing cells led to increased miR‐192 levels in HUVEC cells as compared to HUVEC incubated with ELV derived from mock cells (Figure 4A). Interestingly, HUVEC co‐cultured with miR‐192–derived ELV impaired vessel connectivity as compared to mock‐derived suggesting that miR‐192‐ELV decreased the tubulogenic activity induced by mock cells (Figure 4A). To further prove that this effect was mediated by miR‐192 transfer to HUVEC cells, we transfected HUVEC with pre‐miR‐192 or mock oligonucleotides. Interestingly, miR‐192‐transfected HUVEC showed a decreased migration and tubulogenic activities in vitro (Figure 4B), two key cell functions associated with angiogenesis. In order to identify target genes of this anti‐angiogenic program, we used an integrative transcriptomic approach in HUVEC cells overexpressing miR‐192 (Figure 4C). Significantly repressed genes are listed in Sup. Figure 7. Analysis of hexamer‐seed sequence in 3′UTR identified 17 putative targets. Target prediction tools identified 7 out of the 17 as potential direct targets. Based on biological criteria, we selected relevant genes related to angiogenesis. Among these factors, only ICAM‐1 and PTPRJ could potentially represent direct targets, whereas, IL‐8, CXCL‐1 and LAMB1, could be considered indirect targets (Sup. Table 1).
Figure 4.
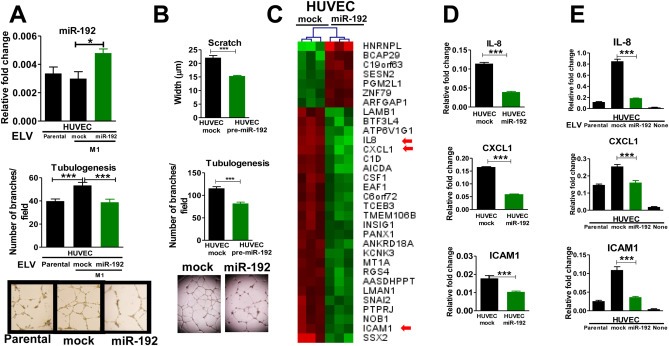
In vitro effects of ELV transfer in endothelial cells. A. Top: Quantification of miR‐192 assessed by qPCR in HUVEC previously incubated with 2 μg of ELVs from parental, mock, and miR‐192 cells for 72 h. Bottom: Tubulogenesis assay of HUVEC cells after 72 h of treatment with ELV isolated from parental, mock, and miR‐192 cells and representative images. B. Scratch and tubulogenesis assay of HUVEC cells after transfection with a pre‐miR‐192 or mock empty vector. C. Hierarchical cluster after integrative transcriptomic analysis of HUVEC cells overexpressing miR‐192 and mock transfected. Top list of the most significantly overexpressed (red) and repressed (green) genes are represented. Several angiogenesis‐related genes were found to be altered (red arrows). D. qRT‐PCR analysis confirmed the alteration of ICAM1, CXCL1, and IL‐8 in HUVEC cells transfected with miR‐192 or mock. E. qRT‐PCR analysis of HUVEC cells after 72 h incubation with 2 μg ELV isolated from parental, mock, and miR‐192 tumor cells.
Previous results were validated by qPCR analysis (Figure 4D). Moreover, incubation of HUVEC cells with ELV derived from cells (parental, mock and miR‐192) induced a significant downregulation of ICAM‐1, CXCL‐1, and IL‐8 expression levels in HUVEC cells (Figure 4E). Similar results were obtained for the murine cell line PY4.1 and another human endothelial cell line HBMEC (Sup Figure 8). Of note, endogenous extremely low levels of miR‐192 in the murine PY4.1 cell line and HBMEC prevented the use of an antagomir strategy. These data suggest that transfer of ELV carrying miR‐192 to endothelial cells modulates crucial angiogenic functions by repressing either directly or indirectly key target pro‐angiogenic genes.
Because previous findings (Sup Figure 3A) indicated that tumor‐released factors induced different osteoclastogenic activities in vivo, we tested the ability of tumor cells to induce osteoclastogenesis in vitro. CM derived from miR‐192 overexpressing cells induced a decreased TRAP+ formation in vitro (Sup Figure 9A). A similar experiment was performed using isolated ELV from mock and miR‐192 cells. Interestingly, ELV did not have any effect in the osteoclastogenesis assay in vitro (Sup. Figure 9). Thus, these findings indicate that tumor‐secreted factors independent of ELV released modulated by miR‐192 had an impact in tumor‐induced osteolysis. Concomitantly, these results strongly suggest that ELV could exert a distinct and relevant effect in vivo during bone colonization by modulating tumor‐induced angiogenesis.
To explore whether these findings could have any relevance in vivo, we subcutaneously injected cells (A549, M1mock and M1miR‐192) into nude mice. After tumor growth, analysis by RT‐qPCR showed a decrease in murine ICAM1 expression levels in bulk tumors derived from miR‐192 cells as compared to that of mock cells. No changes were found in CXCL1 and LAMB1 levels (Sup.Figure 10). Of note, IL‐8 does not have a homologue gene in mouse. These findings indicate that forced expression of miR‐192 modulates target genes in tumor cells and suggest that tumor cells could modulate the expression levels of other tumor‐associated murine cells.
3.7. ELV is transferred to the bone marrow after systemic administration
To demonstrate the role of ELV transfer in vivo, we first assessed whether isolated ELVs could reach the bone marrow compartment after systemic delivery.
To this end, ELV previously isolated from M1 cells were fluorescently‐labeled with PKH26 and were subsequently administered in mice. Control mice were injected with vehicle non‐containing ELV (Figure 5A). After sacrifice, cells were isolated from bone marrow co‐labeled with fluorescent anti‐CD31, anti‐CD45 and anti‐F4/80 antibodies and analyzed by flow cytometry. As shown in Fig.5B, fluorescent ELV were transferred to CD31+, CD45+ and F4/80+ cells in the bone marrow as compared to control animals. These findings indicate that systemically injected ELV are able to fuse in vivo with a variety of target cells of the bone marrow. Interestingly, the transfer varies in different cell types.
Figure 5.
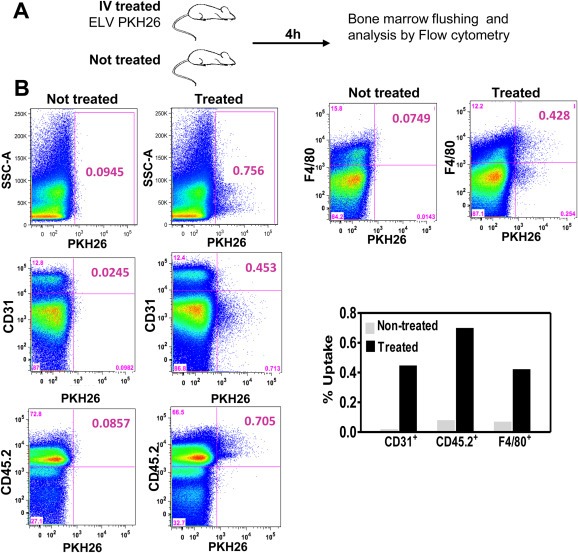
In vivo transfer of ELV to cells in the bone marrrow compartment. A. Outline of the experimental setting. After PKH26‐labeling, fluorescently‐labeled ELV were injected and 4 h later, bone marrow flushing was performed and cells were analyzed by flow cytometry. Untreated mice were used as control. B. Bivariate displays of flow cytometry analysis showing an increase labeling of CD31+, CD45+ and F4/80+ cell populations in mice treated with PKH26–labeled ELV (right column) as compared to untreated mice (left column).
3.8. miR‐192 enriched ELV appeases osseous colonization in vivo
To explore the role of ELV transfer in the osseous colonization in vivo, we treated by intravenous injection 3 groups of mice (n = 4 per group) with isolated ELV derived from A549, M1mock and M1miR‐192 cells at a dosage of 5 μg/mouse/day starting one day before intratibial injection of identical highly‐metastatic cells M1 to all the groups in both hind limbs per animal, according to the regimen presented in Figure 6A. As an additional control, a group of mice was injected with PBS (vehicle). At day 21 postinjection, bioluminescence imaging revealed a dramatic decrease tumor burden in mice treated with miR‐192‐derived ELV as compared to the rest of the groups, although these differences did not reach significant statistical differences (Figure 6A). More remarkably, X‐rays image analysis, revealed 6‐fold increase in osteolytic lesions in mice treated with ELV derived from M1 cells than those treated with the ones derived from A549. Moreover, M1miR‐192‐derived ELV decreased the overt lesions induced in mice treated with M1mock derived ELV (Figure 6B). Furthermore, tumors derived from mice treated with ELV from M1miR‐192 overexpressing cells showed a complete absence of CD31+ immunostaining (Figure 6C).
Figure 6.
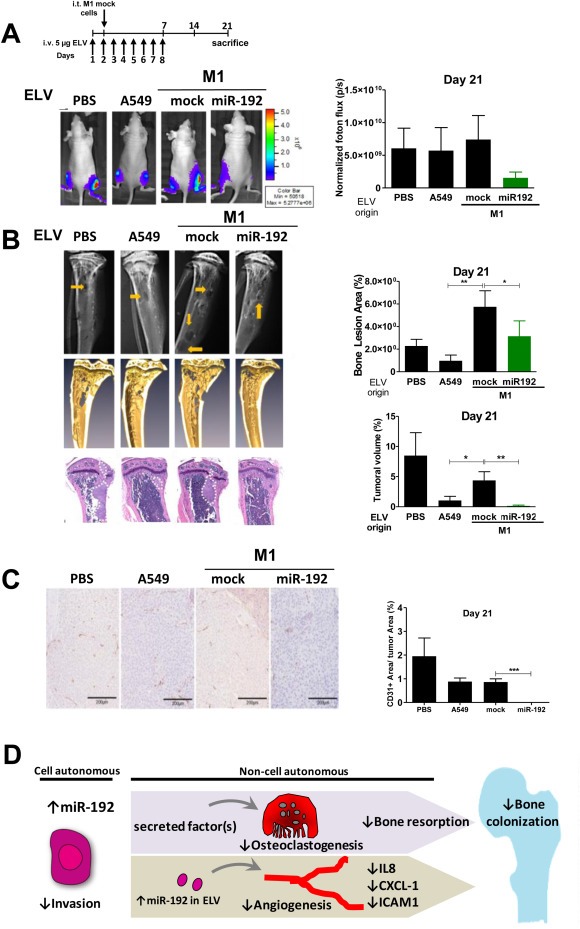
In vivo effects of ELV treatment in metastatic activity. A. Top: Experimental regimen preconditioning the animals with isolated ELVs from each of the indicated cell lines. The same M1 cells were i.c. inoculated in all groups one day after the initiation of the treatment. Bottom: Representative images of BLI (left) and quantification (right). B. Left: Representative X‐rays, microCT scans, and H&E staining from representative bones. Right: Quantification of osteolytic lesions in X‐rays (Top) and tumor volume in histological sections (bottom). C. Left: Representative images of CD31+ staining. Right: quantification. D. Model of the multimodal mechanisms elicited by miR‐192. These mechanisms include tumor cell intrinsic and non‐cell autonomous effects acting on host cells of the bone microenvironment.
Taken together, these data suggest that ELV derived from miR‐192 overexpressing cells precondition the bone compartment creating a conducive microenvironment that deranges osteolytic lesions and bone colonization presumably by decreasing tumor‐induced angiogenesis in vivo (Figure 6D).
Finally, we assessed miR192 kinetics during bone metastasis progression in the serum of animals i.c. inoculated with M1 metastatic cells according to the regimen outlined in Sup Figure 11A. Serum levels of miR192 decreased in serum over time and were inversely correlated with the increased tumor burden and osteolytic lesions (Sup Figure 11 B, C). These data suggest that miR192 levels inversely associate the progression of bone metastasis.
4. Discussion
This study unveils the relevance of a novel mechanism of a short‐range intercellular communication system between tumor cells and the host microenvironment leading to metastatic bone colonization. Metastases were appeased by tumor transfer of miR‐192‐enriched ELV to the endothelial compartment of the osseous milieu. These findings were remarkable during bone colonization, at which point increased levels of miR‐192‐transferred in the microenvironment suppressed in a paracrine manner, key factors in endothelial precursor cells such as IL8 and CXCL1, thus preventing effective metastatic angiogenesis and resulting in impaired colonization. Indeed, IL8 promotes the expression of VEGF, and recruits inflammatory cells that also contribute to the release of proangiogenic factors (Martin et al., 2009). Similarly, CXCL1 participates in the recruitment of inflammatory and endothelial precursors for angiogenesis (Hristov et al., 2007). Our findings establish that endothelial functional impairment was achieved by manipulating the cargo content of a single miRNA in tumor cells and its subsequent release and transfer in ELV to the extracellular milieu where they can fuse to endothelial cells. This transference to endothelial cells led to the repression of the angiogenic program since endotehlial incubation with ELV demonstrated a functional modulation of tubulogenesis in vitro, a hallmark of angiogenesis. Moreover, in vivo treatment with ELV isolated from cells displaying different metastatic degree, led to a cargo transfer towards cells of the bone marrow microenvironment and a striking modulation of the prometastatic activity, in mice injected with identical metastatic cells. These findings indicate a prominent role of ELV in preconditioning the metastatic site in vivo. This finding further underscores the major importance of cancer–endothelial interactions in metastatic initiation and colonization (Catena et al., 2011; Png et al., 2012).
This miRNA‐ELV transfer mechanism might also be germane in a more general context of metastatic colonization in other organs. More importantly, it might potentially be relevant from early steps of tumorigenesis mediating colonization at the primary site through the multistep process of metastasis (Park et al., 2010). Indeed, melanoma‐derived exosomes from highly metastatic cells increased the metastatic behavior of primary tumors by 'pre‐conditioning' bone marrow progenitors through the receptor tyrosine kinase MET, and inducing a pro‐vasculogenic phenotype in bone marrow progenitors (Peinado et al., 2012). Thus, this mechanism of long‐range intercellular signals adds to previously described mechanism of chemoattractants release by the osseous stroma facilitating metastatic homing (Muller et al., 2001), and hence might emerge as a novel paradigm of cell‐to‐cell communication involved in this “seed and soil” cross‐talk for the establishment of metastasis (Grange et al., 2011) (Hood et al., 2011). Thus, elucidation of crucial components secreted by the tumor via ELV, at early stages of tumorogenesis that could potentially influence metastatic initiation or development, are of great therapeutic interest, and further research concerning miRNA delivery to target organs is warranted (Krutzfeldt et al., 2005).
The observation that serum levels of miR‐192 decreased along osseous metastatic progression after i.c. inoculation, suggests that a sub‐population of M1 cells, which express low levels of miR‐192, could survive and colonize the bone compartment contributing to the decrease in miR‐192 levels detected in serum. In addition, serum detection of increasing levels of tumor‐derived miR‐326 during bone metastatic progression, is consistent with the ELV release into the circulation, and the serum enrichment of some circulating miRNAs (Valencia et al., 2013).
Although miR‐192 was found to be abundant in ELV, we cannot dismiss the possibility that other molecules co‐assembled in these vesicles, such as different miRNAs, mRNA, lipids, or protein receptors co‐regulated by miR‐192 overexpression, cooperatively contribute to the observed effects. The observation, that cells with forced co‐expression of mmiR‐298 in miR‐192 background also secrete more mmiR‐298 in their ELV, supports this contention. This increased transcriptional activity observed in the forced miR‐192 background could be explained by the direct or indirect regulation of proteins of the miRNA processing machinery or spurious cross‐species effects of murine mmiR‐298 in human cells. Furthermore, although in theory other non‐exosomal secreted factors released by tumor cells could participate in the antiangiogenic effects, our in vitro tubulogenic assay using CM or isolated ELV yielded similar results. Complementary to this view, these components, especially protein receptors assembled in ELV, could also play a significant role in mediating target specificity and functional effects in the metastatic organ, as they might selectively direct their fusion with specific acceptor cells. At present, the molecules assembled in the ELV cargo are highly complex, and further studies are needed to allow for a deeper understanding. Future experiments will address this important issue.
Concomitant to this mechanism, miR‐192 also contributed to a decrease of tumor‐induced osteolysis, probably by diminishing osteoclastogenic factors. Interestingly, this effect was independent of ELV transfer. Indeed, CCL2 (or MCP‐1) a potent osteoclastogenic chemokine was dramatically downregulated in miR‐192 tumor cells (data not shown). Of note, CCL2 a chemokine that also recruits myeloid cells, an event closely associated with cancer progression and metastasis (Zhang et al., 2010), was involved in tumor‐induced osteolytic lesions in a model of bone metastasis (Cai et al., 2009).
In addition, miR‐192 also acted in a tumor cell‐autonomous manner by suppressing gene expression in tumor cells, modulating invasiveness and metalloprotease activities, two functions required for effective tumor cell infiltration in the osseous milieu. The significant downregulation of miR‐192 and miR‐215 were particularly interesting findings because they are located, together with miR‐194, in two related microRNA clusters. Both clusters have the same seed sequence and are frequently expressed together upon physiological or pathological stimuli, such as p53 (Pichiorri et al., 2010). Despite sharing the same seed sequence, only miR‐192 and miR‐215 induced similar invasive activity in vitro but not in vivo, which suggests that other cell‐specific factors were required, along with other miRNA target genes, to modulate cellular functions.
Finally, miR‐192 potentially elicits different responses in a cell‐specific and context‐specific manner. For instance, no effects on cell growth kinetics were found in several cell types such as HeLa or HEK293 cells upon miR‐192 overexpression (Feng et al., 2011). In contrast, miR‐192 decreases cell proliferation and contributes to cell apoptosis in lung cancer cells such as A549 cells (Feng et al., 2011; Braun et al., 2008). The arrest in the cell cycle upon miR‐192 overexpression is influenced by multiple factors including TP53 status, and the levels of MDM2 (Pichiorri et al., 2010). This discrepancy with our findings could be related to the in vivo selection of our HMS cells, since they could display different levels of MDM2 or other factors as compared to parental A549 cells. Variations in the methodological approach could help explain differences in the expression levels achieved, and possible toxic effects cannot be underestimated either. Our main conclusions however, are not affected by these differences.
5. Conclusions
In summary, as depicted in Figure 6D, our results demonstrated that miR‐192 elicits pleiotropic functions that cooperatively appease osseous metastasis. This multimodal mechanism orchestrated by a single miRNA, includes repression of intrinsic tumor cell functions, non‐cell‐autonomously regulating invasiveness, tumor‐induced osteoclastogenesis, and interfering with metastatic angiogenesis via EVL transfer. Because the multigenic regulatory network inducing such a repertoire of cellular functions was triggered by a single miRNA, targeting one or more miRNAs could represent a potentially beneficial strategy to block the metastatic process.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Acknowledgments
The authors thank Inés López and Marisol González for their outstanding technical assistance. We are also grateful to L. Guembe and E. Remírez from the Core Histology Facility and J. Mateo. This work was supported by “UTE project FIMA” agreement, the Cancer Research Thematic Network of the Health Institute Carlos III (RTICC RD12/0036/0066), European Regional Development Fund (ERDF) “Una manera de hacer Europa”, SAF‐2009‐11280, SAF2012‐40056, the Government of Navarra (09/2009) awards to F.L.. K.V. and D.L‐R were funded by the FIMA and FPU. K.V. was the recipient of a Young Investigator Award at the ASBMR 2012. I.A. was funded by the Basque Government. F.L. is an investigator from the I3 Program.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.01.012.
Valencia Karmele, Luis-Ravelo Diego, Bovy Nicolas, Antón Iker, Martínez-Canarias Susana, Zandueta Carolina, Ormazábal Cristina, Struman Ingrid, Tabruyn Sébastien, Rebmann Vera, De Las Rivas J., Guruceaga Elisabet, Bandrés Eva and Lecanda Fernando, (2014), miRNA cargo within exosome‐like vesicle transfer influences metastatic bone colonization, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.01.012.
References
- Antonyak, M.A. , Li, B. , Boroughs, L.K. , Johnson, J.L. , Druso, J.E. , 2011. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. U. S. A.. 108, 4852–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, C.J. , Zhang, X. , Savelyeva, I. , Wolff, S. , Moll, U.M. , 2008. p53-Responsive micrornas 192 and 215 are capable of inducing cell cycle arrest. Cancer Res.. 68, 10094–10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Z. , Chen, Q. , Chen, J. , Lu, Y. , Xiao, G. , 2009. Monocyte chemotactic protein 1 promotes lung cancer-induced bone resorptive lesions in vivo. Neoplasia. 11, 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catena, R. , Luis-Ravelo, D. , Anton, I. , Zandueta, C. , Salazar-Colocho, P. , 2011. PDGFR signaling blockade in marrow stroma impairs lung Cancer bone metastasis. Cancer Res.. 71, 164–174. [DOI] [PubMed] [Google Scholar]
- Coenegrachts, L. , Maes, C. , Torrekens, S. , Van Looveren, R. , Mazzone, M. , 2010. Anti-placental growth factor reduces bone metastasis by blocking tumor cell engraftment and osteoclast differentiation. Cancer Res.. 70, 6537–6547. [DOI] [PubMed] [Google Scholar]
- Coleman, R.E. , 1997. Skeletal complications of malignancy. Cancer. 80, 1588–1594. [DOI] [PubMed] [Google Scholar]
- Croce, C.M. , Calin, G.A. , 2005. miRNAs, cancer, and stem cell division. Cell. 122, 6–7. [DOI] [PubMed] [Google Scholar]
- Feng, S. , Cong, S. , Zhang, X. , Bao, X. , Wang, W. , 2011. MicroRNA-192 targeting retinoblastoma 1 inhibits cell proliferation and induces cell apoptosis in lung cancer cells. Nucleic Acids Res.. 39, 6669–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler, I.J. , 2003. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat. Rev. Cancer. 3, 453–458. [DOI] [PubMed] [Google Scholar]
- Ginestra, A. , La Placa, M.D. , Saladino, F. , Cassara, D. , Nagase, H. , 1998. The amount and proteolytic content of vesicles shed by human cancer cell lines correlates with their in vitro invasiveness. Anticancer Res.. 18, 3433–3437. [PubMed] [Google Scholar]
- Grange, C. , Tapparo, M. , Collino, F. , Vitillo, L. , Damasco, C. , 2011. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res.. 71, 5346–5356. [DOI] [PubMed] [Google Scholar]
- Guise, T.A. , Yin, J.J. , Taylor, S.D. , Kumagai, Y. , Dallas, M. , 1996. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Invest.. 98, 1544–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood, J.L. , San, R.S. , Wickline, S.A. , 2011. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res.. 71, 3792–3801. [DOI] [PubMed] [Google Scholar]
- Hristov, M. , Zernecke, A. , Bidzhekov, K. , Liehn, E.A. , Shagdarsuren, E. , 2007. Importance of CXC chemokine receptor 2 in the homing of human peripheral blood endothelial progenitor cells to sites of arterial injury. Circ. Res.. 100, 590–597. [DOI] [PubMed] [Google Scholar]
- Krutzfeldt, J. , Rajewsky, N. , Braich, R. , Rajeev, K.G. , Tuschl, T. , 2005. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 438, 685–689. [DOI] [PubMed] [Google Scholar]
- Kumar, M.S. , Lu, J. , Mercer, K.L. , Golub, T.R. , Jacks, T. , 2007. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet.. 39, 673–677. [DOI] [PubMed] [Google Scholar]
- Liu, C. , Kelnar, K. , Liu, B. , Chen, X. , Calhoun-Davis, T. , 2011. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med.. 17, 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis-Ravelo, D. , Anton, I. , Zandueta, C. , Valencia, K. , Ormazabal, C. , 2013. A gene signature of bone metastatic colonization sensitizes for tumor-induced osteolysis and predicts survival in lung cancer. Oncogene. in press. [DOI] [PubMed] [Google Scholar]
- Luis-Ravelo, D. , Anton, I. , Zandueta, C. , Valencia, K. , Pajares, M.J. , 2013. RHOB influences lung adenocarcinoma metastasis and resistance in a host-sensitive manner. Mol. Oncol.. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L. , Teruya-Feldstein, J. , Weinberg, R.A. , 2007. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 449, 682–688. [DOI] [PubMed] [Google Scholar]
- Martin, D. , Galisteo, R. , Gutkind, J.S. , 2009. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J. Biol. Chem.. 284, 6038–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecalvo, A. , Larregina, A.T. , Shufesky, W.J. , Stolz, D.B. , Sullivan, M.L. , 2012. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 119, 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, A. , Homey, B. , Soto, H. , Ge, N. , Catron, D. , 2001. Involvement of chemokine receptors in breast cancer metastasis. Nature. 410, 50–56. [DOI] [PubMed] [Google Scholar]
- Mundy, G.R. , 2002. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat. Rev. Cancer. 2, 584–593. [DOI] [PubMed] [Google Scholar]
- Nguyen, D.X. , Massague, J. , 2007. Genetic determinants of cancer metastasis. Nat. Rev. Genet.. 8, 341–352. [DOI] [PubMed] [Google Scholar]
- Park, J.E. , Tan, H.S. , Datta, A. , Lai, R.C. , Zhang, H. , 2010. Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol. Cell Proteomics. 9, 1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado, H. , Aleckovic, M. , Lavotshkin, S. , Matei, I. , Costa-Silva, B. , 2012. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med.. 18, (6) 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichiorri, F. , Suh, S.S. , Rocci, A. , De Luca, L. , Taccioli, C. , 2010. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 18, 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Png, K.J. , Halberg, N. , Yoshida, M. , Tavazoie, S.F. , 2012. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature. 481, 190–194. [DOI] [PubMed] [Google Scholar]
- Simons, M. , Raposo, G. , 2009. Exosomes–vesicular carriers for intercellular communication. Curr. Opin. Cell Biol.. 21, 575–581. [DOI] [PubMed] [Google Scholar]
- Skog, J. , Wurdinger, T. , van Rijn, S. , Meijer, D.H. , Gainche, L. , 2008. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol.. 10, 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie, S.F. , Alarcon, C. , Oskarsson, T. , Padua, D. , Wang, Q. , 2008. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 451, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadi, H. , Ekstrom, K. , Bossios, A. , Sjostrand, M. , Lee, J.J. , 2007. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol.. 9, 654–659. [DOI] [PubMed] [Google Scholar]
- Valencia, K. , Ormazabal, C. , Zandueta, C. , Luis-Ravelo, D. , Anton, I. , 2012. Inhibition of collagen receptor Discoidin Domain Receptor-1 (DDR1) reduces cell survival, homing, and colonization in lung Cancer bone metastasis. Clin. Cancer Res.. 18, 969–980. [DOI] [PubMed] [Google Scholar]
- Valencia, K. , Martin-Fernandez, M. , Zandueta, C. , Ormazabal, C. , Martinez-Canarias, S. , 2013. miR-326 associates with biochemical markers of bone turnover in lung cancer bone metastasis. Bone. 52, 532–539. [DOI] [PubMed] [Google Scholar]
- Vicent, S. , Luis-Ravelo, D. , Anton, I. , Garcia-Tunon, I. , Borras-Cuesta, F. , 2008. A novel lung cancer signature mediates metastatic bone colonization by a dual mechanism. Cancer Res.. 68, 2275–2285. [DOI] [PubMed] [Google Scholar]
- Weilbaecher, K.N. , Guise, T.A. , McCauley, L.K. , 2011. Cancer to bone: a fatal attraction. Nat. Rev. Cancer. 11, 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, J.J. , Selander, K. , Chirgwin, J.M. , Dallas, M. , Grubbs, B.G. , 1999. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Invest.. 103, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L. , Todd, N.W. , Xing, L. , Xie, Y. , Zhang, H. , 2010. Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int. J. Cancer. 127, 2870–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.H. , Wang, Q. , Gerald, W. , Hudis, C.A. , Norton, L. , 2009. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 16, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Lu, Y. , Pienta, K.J. , 2010. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J. Natl. Cancer Inst.. 102, 522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data