

Abstract
Cancer is a multifactorial and heterogeneous disease. The corresponding complexity appears at multiple levels: from the molecular and the cellular constitution to the macroscopic phenotype, and at the diagnostic and therapeutic management stages. The overall complexity can be approximated to a certain extent, e.g. characterized by a set of quantitative phenotypic observables recorded in time‐space resolved dimensions by using multimodal imaging approaches. The transition from measures to data can be made effective through various computational inference methods, including networks, which are inherently capable of mapping variables and data to node‐ and/or edge‐valued topological properties, dynamic modularity configurations, and functional motifs. We illustrate how networks can integrate imaging data to explain cancer complexity, and assess potential pre‐clinical and clinical impact.
Keywords: Cancer hallmarks, Molecular imaging, Networks, Integrative inference
Highlights
Computational Multiplexing Imaging merges imaging and networks.
Networks show signatures of tumor heterogeneity and phenotypic profiles observed in‐vivo.
A profile ensemble establishes a tumor fingerprint, and this constitutes a novel type of marker.
Personalized treatment is embedded in a systems medicine approach.
1. Cancer as heterogeneous complex disease
Heterogeneity is an intrinsic feature of essentially any tumor and manifests itself at different levels (Burrell et al., 2013). First, heterogeneity is expressed at clinical level (Bedard et al., 2013), due to disease evolution and therapeutic responses varying among patients with the same kind of tumor (Marusyk and Polyak, 2010). Second, heterogeneity characterizes the anatomical and physiological properties of tumors (Meacham and Morrison, 2013). Notably, while heterogeneous features drive tumor histological classification, unfortunately they do not allow a common treatment option to be defined. A third level of heterogeneity refers to molecular constitution and processes in view of the significant amount of aberrant signaling events in any particular tumor. Finally, these phenotypic characteristics are the results of genetic aberrations occurring in a variable dynamic environment, and causing tissue to develop abnormally.
The main problem concerning heterogeneity is how to account for it, from a biomedical point of view. It is crucial to understand how to effectively develop ideally targeted therapeutic strategies for a condition that is intrinsically heterogeneous and potentially unstable over time. Also, it is necessary to identify specific models to treat the complexity which is transferred over the data. The set of observations should be characterized quantitatively and concisely. With multimodal imaging, for instance, a spatially resolved distribution of structural, functional and/or molecular parameters linked to phenotypes can be derived, leading to the mapping of biological heterogeneity of neoplastic tissue onto data not necessarily cross‐correlated. In this context, we are referring to observational phenotypic complexity, which embeds a hierarchy of other complexities (genetic, molecular, physiological, microstructural). Defining this hierarchy is beyond the scopes of this work, whose objective is instead to describe cancer‐related complexity following a novel integrative approach.
1.1. Hallmarks
In order to rationalize the complexities of neoplastic disease, Hanahan and Weinberg (2000) have defined six phenotypic hallmarks of cancer, which correspond to six biological abilities acquired during tumor development. They include: 1) Sustained proliferative signaling, 2) Evasion of effects of growth suppressor, 3) Resistance to cell death program, 4) Acquisition of replicative immortality 5) Development of a vascular network (angiogenesis), 6) Invasion of adjacent healthy tissue formation of distant metastases.
In a more recent publication (Hanahan and Weinberg, 2011), four new complementary features were added to the list: 7) Genome instability, 8) Inflammation, 9) Reprogramming of energy metabolism and 10) Evasion of immune surveillance. The hallmarks indicate to a large extent the multifaceted complexity of cancer disease. The latter finds a rationale in the basic remark that wide‐spectrum cellular perturbations are needed for cancer to take place, and they translate into highly specialized functions of biological processes and pathways that interact in an open system through a rich web of communication exchanges between the cancer cells and their microenvironment.
Figure 1 reports all these hallmarks and some of their main references. By looking at them, and considering the continuing advances in the field, two questions emerge: Is the Hallmarks' list sufficient to cover the complexities of neoplastic disease? Can we specialize the study of hallmarks based on sub‐classifications like sources of data, approach of investigation, types of interrelationships, etc.? In an attempt to provide possible answers to these two questions, we set a context for description and integration of the listed hallmarks, making primarily use of concepts belonging to molecular imaging and biological networks.
Figure 1.
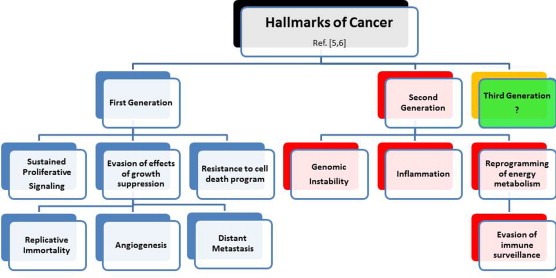
Hallmarks of cancer. Hallmarks are listed, with corresponding examples of references. They are divided according to generation: first six and then four hallmarks have appeared in the literature, inspiring worldwide research work. Conceiving the third generation is left to the future, knowing that new synergies and integrations can potentially derive from imaging and networks.
1.2. Imaging
A starting point is to establish under which type of “lens” the hallmarks can be analyzed. In such regards, molecular imaging (MI) offers a variety of such lenses aimed to visualize molecular and cellular events in living subjects, either animals or human beings. Naturally enough, MI accounts for different images modalities designed to study individual cells behavior, for instance via intravital microscopy (IM). Instead, the study of the formation of macroscopic tumor regions is made possible by Computer Tomography (CT). Among these two extremes, other techniques such as Positron Emission Tomography (PET), Magnetic Resonance Imaging (MRI) and Optical Imaging (OI) allow the study of molecular‐ and cellular‐related processes underlying tumor physiological behavior and anatomical architecture.
In particular, PET detects pairs of gamma‐rays produced by the annihilation of positron emitted by a radionuclide, which is bound to an active molecule that targets the specific biologic process to be evaluated. In the context of cancer hallmarks, PET is used to detect the formation of metastasis (Tateishi et al., 2008), to study hypoxia levels (Chitneni et al., 2011) and glucose metabolism (Zhu et al., 2011), to quantify apoptotic cell population (Blankenberg, 2008) and to stage several types of tumors. MRI differs from PET since it does not use any radioactive compound, but instead measures the nuclear magnetic properties of nuclei or molecules that are considered markers of physiological processes. MRI allows monitoring of angiogenesis processes (Barrett et al., 2007), tumor metabolism (Zhang et al., 2010), acidosis (Kleijn et al., 2011), inflammation (McCann et al., 2011) and invasion of surrounding healthy tissue by tumor cells (Wu et al., 2013). Also, OI measures the light emitted by an optical reporter, which can be administered to the subject, or genetically engineering into protein or DNA. Such reporter is composed of a fluorescent or bioluminescent dye, and an active molecule. Optical reporter probes that target pH, hypoxia and proteases are widely used in the characterization of tumor microenvironment (Wenzl and Wilkens, 2011). Compared to PET or MRI, OI is more sensitive (order of femto‐molar) to detect low concentration of target molecules in tissue, but does not offer an equivalent spatial resolution. Moreover, due to the limited penetration of light in tissue, OI is confined to small animals.
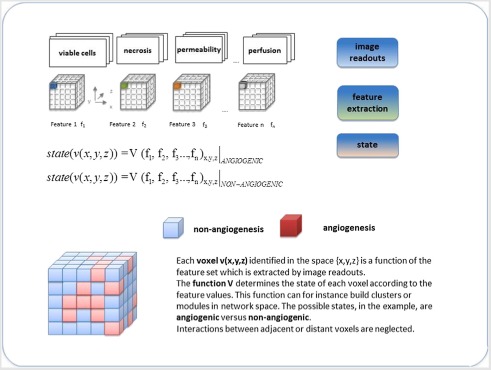
Notably, multiple combinations of different MI techniques can be used, in principle, to study the evolution of cancer hallmarks in‐vivo in and in a non‐invasive fashion. This integrated approach is definitely attractive since ensures measurements of a large number of phenotypic variables of tumor tissue from which inference on gene expression, signaling pathway activity and tumor microenvironment can be conducted. Table 1 below introduces a list of features characterizing tumor tissues in terms of morphology, status of the inflammatory process, physiology and metabolism. The list we report is not complete, being only a sample from the possible feature space which could be considered. We thus focus on the listed examples assuming as the available knowledge base all the image readouts obtained by the described MI techniques. Box 1 is then built to show the use of the features to obtain for each image voxel a quantitative assessment, which can be then translated into corresponding terms.
Table 1.
List of features characterizing tumor tissues (left), and corresponding MI modality through which feature measurements are obtained (middle). References are reported for each example.
| Features | MI modality | Reference |
|---|---|---|
| Morphology: | ||
| shape | MRI (T1, T2, ρ), CT | (Young, 2007) |
| solid vs liquid regions | MRI (T1, T2, ρ), CT | (Young, 2007) |
| necrotic regions | MRI (T1, T2, ρ), CT | (Berry et al., 2008) |
| viable regions | MRI (T1, T2, ρ) | (Berry et al., 2008) |
| alteration in cellularity | MRI – ADC | (Drevelegas, 2011; Haacke, 1999) |
| apoptosis | PET (124I‐annexin V) | (Blankenberg, 2008) |
| Inflammatory status | ||
| edema formation | MRI (T1, T2, FLAIR), CT | (Young, 2007) |
| infiltration of immune cells | MRI (cell tracking SPIO) | (Bulte, 2009; Ahrens and Bulte, 2013) |
| Physiology | ||
| angiogenesis | MRI (DCE) | (Barrett et al., 2007) |
| vascular architecture | CT (angiography), MRI (angiography) | (Matsumotoa et al., 2007; Hartung et al., 2011) |
| vessel density | MRI (VSI) | (Troprès et al., 2011) |
| hemodynamic response | MRI (DSC) | (Kim et al., 2013) |
| vascular permeability | MRI (DCE) | (Kickingereder et al., 2014) |
| tumor oxygenation | PET (18F‐MISO), MRI (BOLD, 17O) | (Hendrickson et al., 2011; Nilesh and Quarles, 2011) |
| acidosis | MRI, MRS | (Raghunand, 2006) |
| Tumor metabolism | ||
| glucose consumption | 18FDG PET | (Chen, 2007) |
| metabolites concentration | MRS | (Kurhanewicz et al., 2011) |
Symbols: MRI = Magnetic Resonance Imaging (T1 = T1‐weighted, T2 = T2‐weighted, ρ = proton density, ADC = Apparent Diffusion Coefficient, FLAIR = Fluid Attenuated Inversion Recovery, SPIO = Super Paramagnetic Iron Oxide, VSI = Vessel Size Index, DSC = Dynamic Susceptibility Enhancement, DCE = Dynamic Contrast Enhancement), MRS = Magnetic Resonance Spectroscopy, PET = Positron Emission Tomography (18F‐FDG = 18F‐Fluordesoxyglucose, 18F‐MISO = 18F‐fluoromisonidazole), CT = Computer Tomography.
Box 1 Computational approach: from imaging readouts to networks.
Panel A shows the image readout – voxel – feature chain for two states, angiogenic and non. Based on the feature values, the state of the tumor processes can be determined. If a voxel presents high values for perfusion, permeability and vascular size index (VSI) features, it is reasonable to assume that it is angiogenic. On the contrary, low values of these features, together with high levels of necrosis, identify a non‐angiogenic voxel.
Panel B shows the bridge between images and networks starting from voxels and nodes, and correspondingly from clusters and modules.
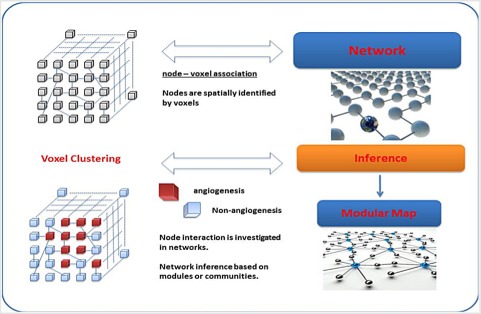
1.3. Integrated approach to cancer
An emerging need is therefore integrating information from multi‐evidence sources, and this step requires harmonization of multiple heterogeneous findings. In particular, as imaging measurements lie at the interface of experimental and technological developments, the obtained information brings a variable potential, depending on the changes occurring in both fields. MI tools support therapy decisions as they address the need of responding effectively to key cancer mechanisms such as multidrug resistance. However, due to the fact that a data multitude on vascularity, hypoxia, signaling and metabolic activity etc. (Paulmurugan et al., 2013) is at play, a thorough analysis requires a new generation of computational instruments. MI is especially valuable for the goal of identifying markers of angiogenesis and advancing early diagnosis of cancer, but also for optimizing the response to anti‐angiogenic treatment and combining drug therapies. Therefore, personalized solutions urgently require support from such techniques for improving the identification of responders.
Consider the tumor vascular network: an important goal is to measure abnormalities observed at a molecular level, and indicate the presence of cancer phenotypes. Such phenotypes can be useful to define the associated marker potential, depending on specific features (molecular processes triggering vessel formation, architecture, functions such as perfusion, leakage, etc.). Clearly enough, the accurate measurement of a few specific tumor features is not sufficient to characterize the wide spectrum of tumor phenotypes. Studies performed in the past decades (for instance, Goh et al., 2007) have concluded that cancer is a network disease, with many players acting in combination at different space and temporal scales, and with different kinds of feedback mechanisms as part of a complex interactive framework. The development of a new neoplastic mass, in this perspective, cannot only be viewed as a genetic disease but actually as a network of both genome‐based and transcriptome‐based abnormalities, co‐operating to the progression of tumor growth.
As we aim to elucidate networks in the context of cancer, we anticipate that three aspects are worth consideration when choosing network inference tools:
The data diversity, reflected into similarity/dissimilarity aspects at network scale, can reveal dependence/independence data structure;
The inference approach to be derived. If data integration is a component of the analysis, the methodology is conditioned on such factor, and networks offer high flexibility in such regards;
The data heterogeneity, which calls for suitable transformations and normalizations, and leads to a preference towards computational frames that harmonize such complexity.
2. Complex networks in tumor environment
A remarkable property of networks is that they adapt to a variety of contexts, and widely differentiate their impact on the basis of only two structural components, nodes and edges. However, biological data can reflect measurements of highly correlated entities, and these correlations are often not analytically known or statistically predictable. This limitation explains in part the success that clustering and network modularity techniques have over more traditional quantitative approaches requiring hypotheses or assumptions to build ad hoc models. In particular, inferring through networks on the basis of connectivity patterns allows for the identification of forms of data dependencies at multiple scales, a concept summarized by the scale‐free property of networks (Barabási and Albert, 1999).
Central to the cancer field are features such as progression, angiogenesis, immune response, interaction with microenvironment and mechanisms leading to the development of drug resistance. Rather than conceiving networks as structures designed to reproduce in silico pathophysiological processes, we consider them as inference tools that elucidate their complex connectivity patterns and their cross‐linked pathways. Including MI information obtained from a multitude of observed data types represents an injection of prediction power in the system. As each data type brings some characteristic features, a rich classification of these features accounting for the specificity of each imaging technique/tool is obtained by stacking them together as attributes of the network nodes. The advantages of such strategy are:
Multiscale information (molecular and biological variety) at node level;
Multiple evidences (technical/technological variety) at network scale;
Direct data‐driven mathematical treatment;
Fast generalization to new types of features;
Reproducibility to other imaging contexts.
Networks usually represent static associations (edges) between biological variables (nodes). If the associative relationships have a regulatory nature, inference can be conducted by using ad hoc causal methods. Unfortunately, spatiotemporal dynamics are lacking in network maps, and typically require volume‐averaged parameters at specific time points. By addressing an ensemble view of network (Marras et al., 2010) (see Box 2), inference can be conducted at the systems' level, providing results which are less sensitive to possible outlying influences and non‐equilibrium effects.
Box 2 Networks as integrative models: theory.
A network generating process can be conceived as a stochastic mechanism responsible for the construction of gene regulatory, protein–protein, metabolic, inflammatory and other specialized networks. In general, a partition of network space as N[Θ] = [N[Θ](O), N[Θ](L)] can consist of observed (O) and latent (L) components, according to the incidence of unknown parameters Θ.
This is equivalent to considering a subset of variables which are function of Θ and are separated in this parameter space according to V[Θ] = [O1, L2], with [Θ = (Θ1,Θ2)]. An example is provided by a network configuration in which the links between the nodes are only partially measured, typical case for PIN for instance, and thus only a few links can be drawn. The rest of missing links can only be predicted. Thus, the network partition N[Θ] creates a dichotomy between observed and latent variables, which in this case are links differentiated according to the possibility of quantifying them.
Mixed combinations of such components yield a network N0 which is a noisy approximation to an optimal (according to defined criteria) one, say N*. Due to the power of random networks, the mixtures allow to approximate Poisson or Gaussian, multiplied by a hidden variable distribution. Notably, an ensemble view of random network dynamics considers the average connectivity as a random variable of unknown distribution.
Note that the observed degree distribution of a network, say a Poisson distribution, is: P(k) = ∫ П(λ) p(k|λ)dλ, for (0,∞) integral range. This simply represents a marginalization of the degree distribution based on the conditional and the hidden variable distributions. Depending on the latter distribution, a wide class of complex networks can be recovered. For example, a scale‐free network approximation is associated with power law distributions. The inference problem of interest is to recover the unknown entity, i.e. the latent distribution which drives the fluctuations. An inverse problem is therefore presented, i.e. recovering П(λ) as a function of the observed degree distribution. This degree distribution in random networks can be associated to a Gaussian, g(k), which would lead to efficient computation of П(μ,σ).
Multiscale analysis would also be computable by a wavelet‐like transform, i.e. Ψ[(k–μ)/σ], which can also be associated to a Gaussian. Other parametric families may fit into the networks, and also non‐parametric families (such as principal and independent components, factor analyzers, kernels, splines, etc.) can be exploited in their approximation power. Finally, variational modeling and Dirichlet Processes (infinite‐order mixtures) extends the flexibility with which networks can be stochastically characterized. Notably, a stochastic model can be defined by a specified mixture of densities, allowing for unsupervised learning and probabilistic clustering (modularization) in networks (see for further details Marras et al., 2010).
Networks naturally address the concept of integrative inference (Bakal et al., 2007), as they adapt the nodes to a multitude of data types, define the edges linking the nodes as either interactive or regulative (causal) connecting mechanisms underlying the phenotypic parameters, and enable the computational predictions. In principle, systems‐based network inference can model features representing the hallmarks of oncogenic signaling networks (see for instance Huang and Fraenkel, 2009; Bandyopadhyay et al., 2010; Morris et al., 2010; Vinayagam et al., 2011) or cell–cell communication (see Goke et al., 2013). We believe that network inference approaches inspiring network medicine (Zanzoni et al., 2009; Barabasi et al., 2011), can be further specialized to allow characterization of cancer hallmarks through their many interfaces with clinical data, but also genetics, omics, and imaging modalities.
How functional sub‐networks can apply to cancer is illustrated in some recent work (Wu et al., 2010; Nibbe et al., 2010; Wen et al., 2013). Sub‐networks are network partitions that can be obtained in various ways. Each partition can generate a module or a hierarchy of sub‐modules, depending on the resolution with which the partitioning occurs. Figure 2 is centered on modularity, from which a series of properties can be derived (see also Kirouac and Onsum, 2013). Modularity stands at the very nexus of network inference, acting in two directions. First, it generates those properties, in particular robustness, which allow for an adaptive response to the environment. Second, it mitigates the complexity associated with heterogeneity by dissecting the system into components, while limiting the negative influences of system's redundancies.
Figure 2.
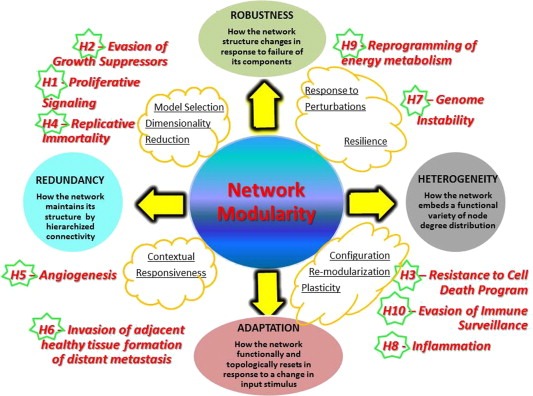
Network modularity and properties. Modularity is key, and properties are indicated and defined (circles). The yellow clouds include action guidelines enabled by network inference. Robustness brings better stability of modular configurations, inducing resilience. Biotype heterogeneity can be handled by configuration plasticity and ability to re‐modularize, leading to better adaptation to change of conditions in a contextual way. Possible redundancy can be tackled by dimensionality reduction and model selection. An adjustment of the basic network structure presents the advantage of optimizing the fit of individual traits (personalized therapy approaches). Cancer hallmarks (H1… H10) are mapped and positioned in correspondence with network properties that most suitably refer to them.
2.1. Importance of modularity
Modular configurations offer the advantage of being highly adaptable structures to pursue the examination the distribution in space and in time of cellular components. Assuming that a panel of imaging measurements is available, the temporal monitoring focused on modules would allow inference on cancer progression and therapy response. Also, a spatial control over the modules can elucidate both methodological and cancer‐specific aspects, the former referred to the integration of evidences from different MI techniques, and the latter related to angiogenesis, proliferation, etc., whose changes would likely determine differential configurations useful to discriminate across the marker potential of modules.
Modular cancer maps can link molecular pathways to phenotypes at variable resolution (hierarchically, sub‐modularly), and sort apparently disparate mutations into conserved sets of oncogenic modules by a variety of similarity metrics underlying the network topology. In order to explain the configurations of modules that can be generated under the influence of cancer, synergistic dysregulation dynamics should be measured in terms of both connectivity (characterizing the modules) and activity through expression levels (associated to cancer phenotypes such as sub‐types, functional modules, splicing motifs etc.). In particular, network topology identifies through hub nodes (degree‐based, measuring node connectivity) and central nodes (betweenness‐based, measuring node load and importance) possible cancer hotspots. Correspondingly, the warning signals associated to them may allow monitoring of cancer evolution. Warning signals emerge from dynamic patterns or motifs which are observable at network or at modular scale, and their utility is alerting that the system is likely undergoing a change of state having an impact on its global configuration. Examples are provided by warning signals related to events such as switching between disease states or measurement of therapy effects, whose influences can be monitored at systems level.
Modeling tumor processes is quite different from modeling the same processes in healthy tissue. The simultaneous presence of different states of the same cells and the apparently chaotic alteration of cellular behavior make the formulation of a deterministic mathematical model very hard at a cellular level. This hurdle increases when considering the physiological behavior of tumor tissues. Nevertheless, the possibility to measure different features at different scales suggests a data‐driven model for the system as a whole. The complexity of the model, together with its reliability, increases with the number of features that we can consider and measure.
When a single entity (say, a gene or a protein) can be used under certain conditions to predict the state of the whole module to which it belongs, the corresponding changes induce effects subject to better control at both intra‐ and inter‐modular scales.
2.2. Attractor states
Network modules do not act as separate entities, but as cross‐linked ones. Consequently, a certain redundancy from overlapping pathway activity can be observed when signals converge to restricted network regions in which sets of interconnected proteins function as state attractors (Kauffman, 1969; Huang et al., 2010; Creixell et al., 2012).
In general, biological networks should lead to the construction of models that predict the change of configuration following perturbation events. Depending on the latter, we might expect that some significant differential aspects emerge from networks, and they might involve hubs, modules, etc. Differential network analysis should identify the causes of a change of configuration and the propagation of the effects ultimately affecting the phenotypes of interest, with reference to cancer hallmarks. The issue of localizing regions in the network identified as target of any possible intervention is even more important (Pe'er and Hacohen, 2011), but also hard to obtain because network models reflect static rather than dynamic context relationships, thus simplifying the representation of their internal state fluctuations due to a variety of signals (stimuli, drugs, etc.) and their time series measurements. The example provided by these authors involves mTOR as a therapeutic target in view of its critical role for cell growth, and the complications in terms of increased cell proliferation arising from crosstalks between the joint activation of Akt and ERK pathways induced by mTOR inhibition.
Establishing through networks a tumor fingerprint is thus possible even if complicated to model due to the need of considering network re‐configuration or re‐modulation, and changes in attractor or stable equilibrium states. While advances in network medicine are expected toward modeling both observed and latent cancer systems dynamics, network states can be associated to points of an N‐dimensional state space, and network modules would represent sets or communities of states in communication with each other, either transiently or persistently. An advantage offered by networks is that the modularized dynamics might be entropically measured and topologically‐characterized. Attractors as stable points in the expression state space, represent points to which the system would return after small perturbations. However, this idea might be simplistic, as it assumes a smooth expression landscape, occasionally subject to abrupt events.
This simply hypothetical condition might not account for the impacts of a variety of possible perturbations (external cues or internal microenvironment), including the treatment at systems scale of phenotypic response patterns, for instance induced by drug resistance (Mar and Quackenbush, 2009) or by mutations that perturb signaling dynamics. In both cases, non‐stationarity in the system disrupts cancer network attractors. An important effect is noticed when the elimination of a specific pathway by an intervention may be counteracted by the up‐regulation of interconnected pathways. This is a pattern observed for kinases, described as frequent pathway switching after inhibitory treatments. Accordingly, these dynamic should drive state transitions within the network and through mechanisms of adaptation based on complex feedback controls that modulate cascades of events. Compensatory effects also occur at the pathway level within an integrative (genetic, environmental, omics) context. For instance, responsiveness to drug treatments presents the critical aspect of sensitivity and resistance mechanisms. A common inference strategy is to compare signaling states across tumor phenotypes following perturbation experiments targeted to key modules and/or component nodes/edges (i.e. edgetic perturbations, see del Sol et al. (2010); Zhong et al., 2009).
Network states reflect specific conditions, for instance normal versus disease at a coarse level, but also tissue‐specific physiological variability or disease stages at finer levels. Such states are characterized by the measurement of ad hoc parameters, which become then relevant to the output assessment and interpretation. Networks represent a methodological tool for conducting inference, indeed one of the several tools which are available. For example, Janes et al. (2005) have proposed a systems approach centered on data‐driven models for the analysis of large‐scale experimental signaling network measurements, and specifically designed to reduce the redundancy and complexity of the corresponding data sets.
In particular, distinct apoptotic outputs were identified and measured by flow cytometry at multiple times after stimulation. The output measurements were useful to build an apoptotic signature covering early‐to‐late apoptotic responses measured across cytokine input combinations. Both time‐related and dose‐dependent features were uncovered from such complex measurement system. Such signatures were clearly dependent on the initial state conditions, in addition to the input stimulus, and two aspects concerned: (a) The possibility of controlling separately the signature components can simplify the task of linking signatures to intracellular network dynamics; (b) Mapping a variety of molecular signals to systems responses allows for the identification of variables which are critical in predicting apoptosis within the target network.
Defining the salient features of such critical variables is a very important task in every application context. In the work of the above mentioned authors, such goal was achieved by organizing a panel of time‐dependent signaling metrics used as dimensions to evaluate the projected stimuli. Similarly, a systems approach based on networks – the one we propose – can merge quantitative imaging experiments with data‐driven analysis aimed to identify at molecular scale a basis set of diagnostic and/or therapeutic features explaining the signaling dynamics of cancer‐modified biological processes and pathways.
2.3. Endogenous and exogenous network dynamics
The complexity of signaling networks involves convoluted dynamics originated by level and activation states of proteins exerting effects on multiple key pathways at different time points. These dynamics occur in part in response to exposure to drugs, and in part refer to other measurements, such as expression profiles that enrich in additional pathways, and phenotypic responses that refer to key processes (cell cycle phases, apoptosis, etc.). The organization of signaling data in a mathematical framework is a complex problem. Data‐driven and exploratory tools may be preferred to models based on distributional or probabilistic assumptions. In Lee et al. (2012) networks design and inference are discussed, with the aim of controlling data connectivity and dynamic patterns underlying a combinatorial drug context.
Inference of angiogenesis, apoptosis, therapeutic targeting can be conducted through multiple types of experimental imaging which allow to monitor patients responsiveness to therapeutic drugs, thus assessing their efficacy and toxicity. The advantage of using networks in support to such observations is that they can integrate such features with other cancer‐intrinsic features measured at various biological levels, and translate the whole information into a topological representation. From the changes observed in the network configurations, one can infer the dynamics of complex signaling pathways and elucidate the effects of their inhibition with regard to key mechanisms such as drug resistance. In general, the network topology change induced by cancer appears to some extent controlled, thus preventing global alterations while remaining significant compared to the variation typical of a random network (Serra‐Musach et al., 2012). This described is a sort of localized impact observed with networks that may reflect the property of robustness in relation with cancer onset and progression, and also response to therapy. Even after the inhibition of cellular targets by some drugs, the outcome of treatment varies adaptively with cancer cells response and with the dysfunctionality level present in pathways. Depending on the tissues, resistance to the same targeted agent in cancer with identical oncogenic mutations can develop.
A classic example of chemotherapy resistance is provided by inhibition of EGFR linked to possible unresponsiveness, as with KRAS‐mutant colorectal cancer or the mechanism linking HER3 to MET in lung cancer, indicating a so‐called oncogenic bypass, i.e. the fact that a primary drug target remains unaffected by inhibition while an alternative kinase can instead be activated (Holohan et al., 2013). Instead, for BRAF in melanoma other types of resistance mechanisms have been identified, in particular acute adaptive responses through activation of alternative BRAF isoforms. However, BRAF inhibitors are less effective in colorectal cancer than in melanoma due to the activation of EGFR‐AKT signaling responsible for resistance.
3. Integration of two worlds: imaging and networks
The potential of joining spatially resolved features (e.g. derived from MI) and network descriptions to account for tumor heterogeneity remains largely unexplored. The simultaneous acquisition of different MI modalities offers the possibility to consider together both spatial and temporal aspects of physiological processes related to tumor development. These aspects plays a crucial role in the understating of complex and multiscale mechanisms such as angiogenesis or metastasis formation that are driven by a network of processes at a different spatial and temporal scale (Qutub et al., 2009).
For example, angiogenesis (Figure 3) at a molecular level is regulated by a balance between pro‐ and anti‐angiogenic factors, microenvironment conditions and hypoxia level: an alteration of this equilibrium drives the recruitment of endothelial cells, which in turn start in cascade the capillary formation, vessel maturation (veins and arteries) and building of vascular network. At this stage the vascular network fully integrates tumor and hosting tissue and determines the exchange of blood, oxygen and nutrients, which has effects at molecular level. Through MI it is possible to study all these events at their specific spatial scale and it is also possible to follow their dynamic evolution. By taking into account these two aspects, a comprehensive physiological model of angiogenic process can be built and an added value achieved.
Figure 3.
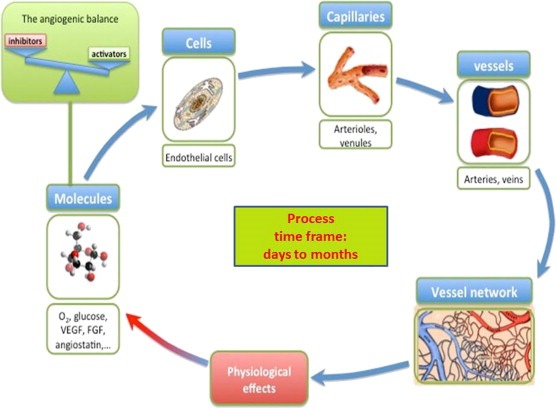
Sketch of angiogenic process through its specific components. At a molecular level, angiogenesis can be modeled as a balance between pro‐angiogenic and anti‐angiogenic factors, respectively promoting and inhibiting the formation of the new vessels. Once the process has started, the endothelial cells are recruited and the formation of the smaller capillaries (5–20 μm) takes place. The next step is the formation of bigger vessels as arterioles and venules, (20–200 μm), and further arteries and veins 200–500 μm. This vascular system grows like a network and the connection between the different parts will determine the perfusion of the tissues. A better network architecture leads to a better blood delivery. Afterward, the link between tumor vascular network and the vascular system of the surrounding tissue will be tightly established and tumor can be considered an organ which interacts with other organs but in abnormal conditions. Newly formed vessels are now able to produce important physiological effects as draining blood from the vascular system, changing the local level of pH and hypoxia, mechanically compressing the host tissue, introducing tumor cells in the bloodstream or in the lymphatic system, and invading surrounding tissues.
The hybrid approach combining observational inference with data‐driven analysis represents a strategy to characterize proliferative diseases. In particular, the complexity of phenotypic expression represented within the network topology implies that a change in the latter should reflect a variation in phenotypic expression, which then translates into physiological, anatomical and molecular changes of tumor tissue. Network topologies are informative for diagnosis, especially for differentiating benign from malignant tumor, for assessing the efficacy of tumor treatment, and for predicting therapy outcome.
Box 3 illustrates the concept of multiplexing, re‐adapted to merge the two objects of our investigation. The combination between imaging and networks is framed within an approach named Computational Multiplexing Imaging (COMI). In particular, a non‐invasive readout such as imaging is ideal to study adaptive processes, and naturally integrates with both experimental and computational multiplexing components, including networks.
Box 3 Multiplexing.
Originally developed in the experimental context, and then transferred into the computational domain, the idea of multiplexing is in principle as simple to define as extremely hard to implement, implying that a new generation of tools would be needed.
Computational Multiplexing Imaging (COMI) is a very desirable research direction.
COMI is a natural derivation of the seminal idea recently inspired by Welch et al. (2011), who discussed of Experimental and Computational Multiplexing by focusing on the reconstruction of complex multi‐component pathways, and involving concurrent activity measurements obtained from spatiotemporal dynamics. Another contribution has just appeared about pre‐clinical cancer imaging applied to therapeutics and drug discovery (Conway et al., 2014).
COMI would thus emerge in two principal ways:
Integrating the observational power available from imaging, thus associating with the experimental evidence;
Expanding the system's prediction power by combining information in a unifying computational frame.
Network for instance organize the information flowing across connectivity patterns. The integration in the network context implies that a specific computational treatment should be adapted to its structure. The variables and parameters measured quantify an abundance of observables (related to cell morphology, morphodynamic pattern analysis, etc.) that refer to molecular activities. Integrating such wealth of data necessarily requires that networks produce inference, also causative and not just correlative, multiresoluted, modular and robust to perturbations rather than globally optimized and model‐dependent.
COMI's impact is expected in diagnostics, prognostics, and by considering therapeutics, also in theranostics.
3.1. Bottlenecks in cancer modeling
The (ten) hallmarks may be related to potential barriers that the neoplastic mass has to overcome in order to survive and proliferate. Despite the knowledge that has been acquired in the last decades on tumor biology, substantially improving our understanding of the molecular and cellular processes involved, mathematical modeling of these processes remains a challenge. The reason for the complexity is in the variety of such processes, and in both the amount and the nature of their interdependent component variables that need to be considered.
Angiogenic complexity
Among the tumor hallmarks, two of them are of particular relevance from a clinical perspective, being critical determinants of patient prognosis: angiogenesis, and activation of invasion and metastasis formation. In particular, angiogenesis refer to the establishment of a vascular network, and is prerequisite for the tumor to outgrow its limits dictated by compound diffusion. Without its vascular system, the tumor mass would be confined to a size of a millimeter in diameter or even less, with no clinical evidence. Angiogenesis therefore constitutes and essential switch in tumorigenesis and, due to this peculiarity, an ideal target for therapy. In this context, modeling the angiogenic processes would be fostering our intrinsic understanding and potentially supporting the development of anti‐angiogenic therapy.
Angiogenesis involves remodeling of the blood vasculature according to a network with a mix of functions characterized by distinct cellular features. Some of the latter are induced by cancer and not specifically referred to its primary site. The changes that such features cause are relevant not just to the disease progression because of an increased vessel subtype heterogeneity, but also for the prognosis and the response to treatment (Farnsworth et al., 2014). The concept of anti‐angiogenic drugs aims at re‐normalizing the cancer vasculature to allow efficacy of the drug due to an easier cell access. A commonly associated effect is drug resistance because alternative signaling routes or other mechanisms such as hypoxia may facilitate revascularization.
A network‐centered assessment of angiogenesis may require that node aggregates are considered at a spectrum of localized scales to measure the influence exerted by the state at which each cell is referred. However, encompassing structural diversity comes with the price of weakening the accuracy of estimating global properties. This is basically due to the presence of under‐sampled networks. Typically, models hold for the entire network, but the data may consist of just fractions of it, i.e. a sampled sub‐network. The problem is that estimates for the whole network parameters are obtained by applying the model to the sub‐network, assuming the model is consistent under sampling. A general way to reduce the typical complexity of highly dimensional networks enriching hierarchies of relations, is to employ ensemble models that average out possible errors and avoid the singularity of very specific results.
Despite three decades of efforts in this direction, a comprehensive model is not yet available. Beyond the difficulty related to the huge number of players, angiogenesis events span multiple length and time scales. Molecular processes such as the expression of angiogenic growth factors triggered by hypoxia once the tumor has outgrown the distance of oxygen diffusion occur in a confined space of micrometers. These mediators then diffuse through tissue, prompting sprouts in the direction of the concentration gradient nearby host vessels. The formation of a capillary network including feeding and draining vessels extends over a range from millimeter to centimeter. Then, characteristic times range from micro‐to milli‐seconds for molecular events, while a time frame from weeks to months is required for the formation of vascular structures.
Metastatic complexity
Death caused by cancer is not due to primary tumor formation, which usually is surgically removed, but rather to the dissemination of cancer cells into adjacent tissue (recurrence) or in other organs (metastases). Hence, tumor invasion and metastasis formation are determinants of patient survival. Activation of these processes require the acquisition of specific phenotypes that make cells able to detach from the tumor mass, migrate and survive in a different tissue. The interaction of cancer cells with the surrounding microenvironment therefore constitutes a critical node in a tumor network (see for instance Johansson et al., 2014).
Studies of tumor microenvironment (Ungefroren et al., 2011) have revealed some specific mechanism involved in tissue infiltration and metastasis formation, such as epithelial‐to‐mesenchymal transition and degradation of the extracellular matrix. Together, they are responsible for the change of cell morphology enabling the detachment from the primary tumor mass and the migration through the extracellular space for invasion of the surrounding tissue. If such detached cells manage to reach lymphatic or blood vessels, these circulating tumor cells may form tumor colonies (metastases) in other parts of the body.
The possibility of determining features that characterize tumor microenvironment in a 3D fashion (Cox and Erler, 2012) is crucial for understanding the infiltration capacity of the tumor cells into healthy tissue. The alteration of cellular pH and oxygenation in a specific direction, for example, will promote the infiltration in that direction instead of another one. Moreover, the increase of mechanical pressure of surrounding tissue in a region will decrease the possibility for the cells to proliferate or migrate there. Therefore, being able to determine a group of 3D features that drive proliferation allows identifying the preferential direction of tumor evolution. Microenvironment plays a role in tumor development, influencing in a significant way the response to therapy. High level of oxygenation, for example, promotes the formation of free radicals that enhance the effect of radiotherapy treatment (Wenzl and Wilkens, 2011).
In the last decade, multiscale models aimed to describe tumor invasion (Anderson, 2007), cell migration (Zaman, 2006; Rangarajan and Zaman, 2008), and metastases formation (Zaman, 2007), have been proposed. Nevertheless, a unifying model which accounts for every aspect is not available. For both angiogenesis and metastases formation, the main problem for a model is to cover multiple temporal and spatial scales. Complex networks offer an integrative multiscale modeling framework (Capobianco et al., 2011; Ryan et al., 2013; Capobianco, 2013).
3.2. Network profiling toward tumor fingerprint
Recently, it has been demonstrated (Curtis et al., 2012) that what is commonly classified as breast cancer, may include ten different pathological conditions at genetic level. This example suggests that a single biomarker may unlikely target tumor features for all types of breast cancer, even considering the expression of possible common traits. Instead, a set of biomarkers or a marker panel targeting different phenotypic characteristics should be considered in order to significantly enhance the diagnostic accuracy with complementary information (Erler and Linding, 2010). This has been demonstrated ex‐vivo in tumor samples using a set of five different labeled antibodies enabling the differentiation of malignant from non‐malignant lesions (Weissleder et al., 2010).
Networks offer a unique opportunity to characterize tumor phenotypes (Pe'er and Hacohen, 2011). In particular, modularity allows the implementation of marker panel strategies to face combinatorial treatments. In order to describe the complexity of heterogeneous proliferative mechanisms, multi‐marker signatures rather than single marker ones build a more predictive fingerprint, owing to the connectivity of the multiple readouts at play. Network models can de facto be specialized in cancer for detecting modules induced by markers (Dao et al., 2011; Ben‐Hamo and Efroni, 2013) with the result of complementing gene selection approaches based on expression thresholds and pathway enrichment.
Network profiles can therefore be built from the obtained modular configurations, based on specific hallmarks or phenotypes that are observed in‐vivo. In turn, an ensemble of such profiles establishes a sort of unique fingerprint by which physiological processes can be simultaneously encoded, for instance gene expressions, pathway landscapes, morphological abnormalities, and molecular mechanisms occurring at different spatial and temporal scales. Such fingerprint is obviously strictly correlated with the stage of tumor evolution, which in turn, is defined by the hallmarks. Even if different regions of the tumor can be in different stages of tumor progression, as in the case of glioblastoma (Zacharaki et al., 2009), the network profile can nevertheless still be useful, due to its possible multidimensionality.
4. Clinical applications
The chance to define unique tumor fingerprints through network profiling is probably the most important contribution which is expected from oncology‐driven networks (Ciriello et al., 2013). We report in Figure 4 a reference scheme for our next remarks in both diagnostic and therapeutic terms. At the diagnostic stage, pathological versus non‐pathological conditions may be differentiated, based on specific network profiles. Moreover, the identification of a precise network state through a particular configuration, summarizing all the physio‐pathological conditions of tumor tissue, makes tumor classification more accurate and allows to define a baseline usable as reference point during cancer treatment. The more accurate the classification, and the better is the treatment. Therefore, at a therapeutic stage, the evaluation of tumor fingerprint during treatment and its comparison with the initial state can be used to evaluate in real time its efficacy. This aspect, in principle, makes it possible to enable quick changes in therapy, in case of unsatisfactory preliminary results. A topologically‐characterized network configuration is thus a diagnostic instrument displaying the state of the disease system, normal or altered, and a therapeutic support tool for the assessment of the effects of treatment. In particular, drug‐target interactions may inform on the changes occurring between states, and their propagation effects can be tracked at the network scale by comparing profiles and by detecting the dynamic connectivity patterns which regulate the migrations across them.
Figure 4.
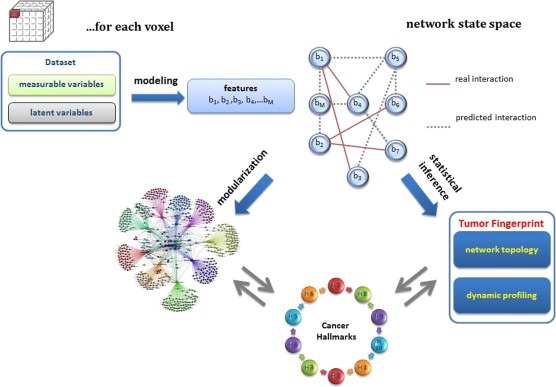
Heading tumor fingerprints. Each voxel of tumor volume can be represented by a set of measurable variables and a set of latent variables defined according to established physiological models. These variables are chosen in order to describe the physiological heterogeneity of tumor tissue. Among them, a specific set of non‐redundant features is selected by means of dimensionality reduction processes. Network architecture is made on the basis of this reduced features dataset (nodes) and on the real or predicted interactions between them (edges). The state space of the network defines configurations as snapshots with topological characterization, and dynamics are at play when transitions between states occur. The combination of the two aspects allows the identification of a fingerprint, which informs on the tumor stage and on the quantification of the expression of each hallmark. Based on tumor fingerprints, voxels that show similar properties, or adhere to the same hallmarks, will be grouped in homogeneous clusters or modules. This way, tissue heterogeneity is split into homogeneous groups of voxels, and the overall complexity is reduced.
4.1. Differential diagnosis
A tumor fingerprint constitutes the ideal marker to differentiate pathological versus non‐pathological conditions, or to distinguish between different stages of the same disease. An example is the differentiation between neoplastic situations as malignant tumor, versus non‐neoplastic ones as benign lesion. This differentiation can be made on the basis of differential network topology, i.e. by observing configurations changes. Then, the study of dynamic behavior can also explain some communication and trafficking occurring between modules, due to movement of fluids, transport of molecules or migration of cells, which can reveal invasion or metastatic potential. A clear advantage of the differential approach supported by networks is reflected by the integrative nature of the clinical evaluation impact derived on the basis of a mix of factors such as tumor features, patient characteristics, gene expression profiles and disease markers, SNPs (single nucleotide polymorphisms), etc.
Among the applications of networks, some refer to tumor classification based also on the integration of omics and clinical information. In his pioneering work, Perou et al. (2000) proposed the classification of invasive breast cancer based on the expression of the oncoprotein HER2 and ER opening the way to the molecular classification of cancer. Many groups extended the approach, then validated (Wirapati et al., 2008) on large groups of datasets with thousands of patients. Consequently, it has been recently demonstrated that breast cancer accounts for ten different disease sub‐types (Curtis et al., 2012). The same approach, based on similar molecular signatures has been also applied in the classification of melanomas (Bittner et al., 2000), lymphomas (Alizadeh et al., 2000) and lung cancer (Bhattacharjee et al., 2001). Clearly enough, the more accurate is the classification, the more specific will be the choice of the therapy. Exerting control on cancer hotspots at network scale (i.e. driver or landmark nodes or modules) can simplify the decision process for the clinician called to personalize the therapy to the patient characteristics.
4.2. Quantifying treatment efficacy
Standard chemotherapeutic drugs, which aim to inhibit replications for fast growing cells, can basically interfere with network topology, which in turn implies that their effects can be monitored. Drug‐dependent topological characterizations are particularly useful for assessing normal versus abnormal network structures, implying that a certain connectivity map among molecules may constitute a marker of the presence of tumor.
Anti‐angiogenic drugs interact with the network at a topological level, but affect also its dynamical behavior due to the fact that the suppression of part of the vascular network alters the way to transport fluids, proteins and nutrients through different parts of the tumors. The anti‐angiogenic effect can be therefore evaluated in a detailed manner including the study of dynamic behavior in space and time. Network topology configurations induced by a tumor fingerprint can also be informative in terms of response to therapy in a contextual way (single versus combinatorial therapy), allowing for the quantification of the possibility of controlling treatment's outcomes based on parameters, i.e. VEGF expression, vascular permeability, vessel density, etc.
4.3. Predicting treatment efficacy
One of the critical points for the assessment of treatment efficacy is the wide range of responses observed among different patients with the same kind of tumor, which is a manifestation of tumor heterogeneity. The latter is responsible for the heterogeneous distribution of the target molecules, making the interaction with the drug sometimes non‐specific. As an example, we can consider the anti‐angiogenic treatment, which aims to destroy tumor vessel network, or equivalently blocking the 5th hallmark. One of the common targets for these drugs is VEGF, which is mainly expressed during the formation of the new vessels and less during the maturation process. The result is a strong concentration of VEGF at capillary level and a weak expression at bigger vessels. Since vascular tumor networks consist of a non‐hierarchical agglomeration of vessels with different dimensions, VEGF will be expressed heterogeneously through the network. It is therefore evident that any specific treatment, which aims to block angiogenesis inhibiting VEGF, will be only partially efficient.
Despite this situation, the goal of modeling for determination of treatment might involve the quantification of the chances that it will succeed or fail, which is a complex task. A network approach can be particularly valuable, due to built‐in characteristics such as phenotype‐responsiveness and occurrence of state transitions. Furthermore, the network fingerprint could in principle work as a prognostic index of the treatment efficacy. In combination with machine‐learning techniques, it is possible to define and train specific algorithms that, based on the initial conditions defined by the network profile, are able to guess the evolution or quantify the chances of events following the treatment. Figure 5 sketches this idea, in terms of differential analysis allowed by networks. Coming back to the example of the anti‐angiogenic treatment, the knowledge of the distribution of VEGF and the architecture of the vascular network, which is embedded in the network profile, constitute the starting point. Based on these factors and the performance over the training dataset, it is possible to train a machine‐learning algorithm in terms of predictive inference towards the system, and with consideration of only the initial fingerprint.
Figure 5.
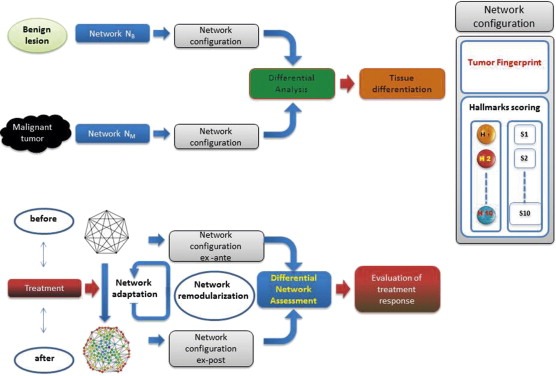
Two complex application settings. In differential diagnosis (top panel), the goal is to distinguish between malignant tumors versus benign lesions. The configuration of the network, which is used as fingerprint of the physiological situation measured in‐vivo, allows for the differential quantification of the presence of each phenotypic hallmark. The quantification is performed by the assignment of a score to each hallmark. The differentiation between the two tissues is therefore made on the basis of score results. The quantification of treatment efficacy (bottom panel) aims to evaluate the effects caused by a perturbation (i.e. cytotoxic drug treatment) on the configuration of the network. In this case, the comparison occurs simply as evaluation between treatment scenarios, ex ante and ex post, leading to the assessment of therapy effects.
4.4. Prognostic inference
Prognostic inference is linked to advancement in medicine, and networks are increasingly providing support to such achievements (Kim et al., 2012); Wu and Stein, 2012; Shi et al., 2012; Li et al., 2010). Given patient groups and parameters specific to them, prognostic models should be selected to predict the future occurrence of outcomes. Examples are a specific medical condition or disease, but also events such as medical interventions and death. Traditionally, inference models are derived from historical data by applying supervised methods of analysis. There are limitations in this approach, and a first one is that variable selection may cause loss of clinical information, while another one comes from the static nature of such models, which partially adapt to new measurable patient information occurring during disease or treatment. Finally, the innermost methodological rationale of such models is to build relationships between dependent (outcome) and independent (predictor) variables, and this model choice prevents the learning process from optimally dealing with highly convoluted dynamics.
The clinical use of network prognostic models is generally targeted to the estimation of the distribution of variables that represent future events related to conditions observed for patients. This is the case occurring during the disease process, where the observational time window reaches the endpoints. In particular, networks can act as the principal inference tools for the generation of clusters and modules whose linear or non‐linear combinations can be significantly correlated with patient survival. Network modularization is particularly valuable in establishing communities whose association strength arise from the patient's characteristics. This way, a novel type of patient stratification can be produced from networks, which in turn can be used for improving the management of therapeutic options though better scenario prediction and monitoring.
5. Outlook
Understanding tumor heterogeneity by complex network approaches opens two important scenarios in pre‐clinical and clinical stages, respectively. At a pre‐clinical stage, the idea to incorporate all the variables that can be measured in a network goes in the direction of developing treatment according to a systems medicine approach (Auffray et al., 2009; Capobianco, 2012). Consequently, all tumor components are considered not as single entities but instead together, indeed interacting according to a “chaotic” orchestra of instruments designed to play a piece of music. However, it is the music that counts! Similarly, tumors would not be considered as an anomaly, rather an active part of the body with some characteristics that interact with other physiological systems endowed with other characteristics, primarily with the immune system. It is the result of this complex exchange that makes a difference at the clinical level, and particularly in prognostic terms.
This new paradigm implies, for instance, that tumor progression goes into two directions. On one hand, the tumor promotes oncogene expressions and growing factors in order to invade the hosting tissue. On the other hand, the host tissue is not able to work against this invasion, and the immune system's response which tries to infiltrates the tumor with macrophages is not effective. Evidence of the tumor invading the neighbor organs implies that such organs also allow the tumor to do it.
From a methodological side, we promoted a complex network approach for modeling the hallmarks of cancer, but an important remark is that, in reality, it is not possible to measure all the variables and parameters relevant for tumors, and then represent them in a network. Some degree of latency of information remains in such systems, depending on variables for which experimental measurement is lacking, and/or computational quantification not accurate enough. To overcome this problem arising in any kind of physiological system, statistical inference methods or differential networks (Cabusora et al., 2005; Ideker and Krogan, 2012) can be applied to optimize an efficient use of the available information.
Substantial intra‐tumor heterogeneity is visible through anomalies which appear to vary within the tumor, and they refer to genomic aberrations, for instance. In our context, MI features could be informative as well, and their variation would be investigated at the network scale reflecting the spatial distribution of nodes. We know that from the various metrics underlying network topology, we can infer about the relevance of particular patterns which are observed to differentiate. For instance, multiple mechanisms of drug resistance or sensitivity may be present within the same tumor, and dictate the response to therapy, determining deviation from linearity with regard to tumor evolution, as evidenced in hematological cancers, renal and breast cancer, and also medulloblastoma (Yap et al., 2012). The outcome of this process is that clonal sub‐populations of cancer cells modularize both genotypically and phenotypically within the same cancer. In particular, even at single biopsy level, a similar intra‐heterogeneity in genomic number and chromosome numbers has been observed in breast cancer (Navin et al., 2010; Navin et al., 2011).
One of the interesting factors that networks could elucidate when presented in their differential configurations, is the scenario in which somatic events present in cancer before drug treatment, and being suspected to determine the outcome of it, could be mapped before and after treatment to verify the contribution to the identification of key biomarker and to the characterization of the influence coming from each type of mutation at different times, whether driver or passenger, in terms of effects on prediction of drug response. The clonal cell population hierarchies characterizing intra‐tumor heterogeneity have particular relevance for metastasis. Biological networks are modular objects, and it's natural to expect that detecting associations between modules and sub‐structures induced by special signatures could be achieved. Whether the signal‐to‐noise ratio can be a criterion for modularization is something that can be either investigated on the basis of the available data (and in this case, an integrative data approach would set more power available for signal deconvolution) or modeled by some approaches providing multiresoluted modular stratification.
We conclude by proposing that time has arrived to leverage the information that we have available on cancer from MI modalities, and when this is integrated within a complex network framework, new opportunities can arise for characterizing the personalization of treatment, not much as the best possible one according to the clinical assessment of the disease or pathological condition, but rather as a reflection of the entire physiological condition of the patient, including primarily the genetic background.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgments
EC thanks CCS (University of Miami) for support. MD thanks Markus Rudin for advice and discussions.
The authors thank two referees for their stimulating questions and important remarks, whose consideration and treatment have led to an improved manuscript.
Dominietto Marco, Tsinoremas Nicholas, Capobianco Enrico, (2015), Integrative analysis of cancer imaging readouts by networks, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.08.013.
References
- Ahrens, E.T. , Bulte, J.W.M. , 2013. Tracking immune cells in vivo using magnetic resonance imaging. Nat. Rev. Immunol. 13, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh, A.A. , Eisen, M.B. , Davis, R.E. , Ma, C. , 2000. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 403, 503–511. [DOI] [PubMed] [Google Scholar]
- Anderson, A.A. , 2007. A hybrid multiscale model of solid tumour growth and invasion: evolution and the microenvironment. In Anderson A.A., Chaplain M.J., Rejniak K.(Eds.), Single-ell-ased Models in Biology and. Medicine. Birkhäuser Basel; 3–28. [Google Scholar]
- Auffray, C. , Chen, Z. , Hood, L. , 2009. Systems medicine: the future of medical genomics and healthcare. Genome Med. 1, 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakal, C. , Aach, J. , Church, G. , Perrimon, N. , 2007. Quantitative morphological signatures define local signaling networks regulating cell morphology. Science. 316, 1753–1756. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay, S. , Chiang, C.Y. , Srivastava, J. , Gersten, M. , 2010. A human MAP kinase interactome. Nat. Meth. 7, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási, A.L. , Albert, R. , 1999. Emergence of scaling in random networks. Science. 286, (5439) 509–512. [DOI] [PubMed] [Google Scholar]
- Barabasi, A.L. , Gulbahce, N. , Loscalzo, J. , 2011. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 12, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett, T. , Brechbiel, M. , Bernardo, M. , Choyke, P.L. , 2007. MRI of tumor angiogenesis. J. Magn. Reson. Imaging. 26, (2) 235–249. [DOI] [PubMed] [Google Scholar]
- Bedard, P.L. , Hansen, A.R. , Ratain, M.J. , Siu, L.L. , 2013. Tumour heterogeneity in the clinic. Nature. 501, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Hamo, R. , Efroni, S. , 2013. Network as biomarker: quantifying transcriptional co-expression to stratify cancer clinical phenotypes. Syst. Biomed. 1, 35–41. [Google Scholar]
- Berry, L.R. , Barck, K.H. , Go, M.A. , Ross, J. , 2008. Quantification of viable tumor microvascular characteristics by multispectral analysis. Magn. Reson. Med. 60, (1) 64–72. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee, A. , Richards, W.G. , Staunton, J. , Li, C. , 2001. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl. Acad. Sci. U S A. 98, 13790–13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner, M. , Meltzer, P. , Chen, Y. , Jiang, Y. , 2000. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 406, 536–540. [DOI] [PubMed] [Google Scholar]
- Blankenberg, F.G. , 2008. In vivo detection of apoptosis. J. Nucl. Med. 49, (2) 81S–95S. [DOI] [PubMed] [Google Scholar]
- Bulte, J.W. , 2009. In vivo MRI cell tracking: clinical studies. AJR Am. J. Roentgenol. 193, (2) 314–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell, R.A. , McGranahan, N. , Bartek, J. , Swanton, C. , 2013. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 501, 338–345. [DOI] [PubMed] [Google Scholar]
- Cabusora, L. , Sutton, E. , Fulmer, A. , Forst, C.V. , 2005. Differential network expression during drug and stress response. Bioinformatics. 21, 2898–2905. [DOI] [PubMed] [Google Scholar]
- Capobianco, E. , 2012. Ten challenges for systems medicine. Front Genet. 3, 193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capobianco, E. , 2013. Protein networks tomography: targeting cancer and associated morbidities. Syst. Biomed. 1, 0–17. [Google Scholar]
- Capobianco, E. , Marras, E. , Travaglione, A. , 2011. Multiscale characterization of signaling network dynamics through features. Stat. Appl. Genet. Mol. Biol. 10, (1) [DOI] [PubMed] [Google Scholar]
- Chen, W. , 2007. Clinical applications of PET in brain tumors. J. Nucl. Med. 48, 1468–1481. [DOI] [PubMed] [Google Scholar]
- Chitneni, S.K. , Palmer, G.M. , Zalutsky, M.R. , Dewhirst, M.W. , 2011. Molecular imaging of hypoxia. J. Nucl. Med. 52, 165–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello, G. , Miller, M.L. , Aksoy, B.A. , Senbabaoglu, Y. , 2013. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 45, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway, J.R.W. , Carragher, N.O. , Timpson, P. , 2014. Developments in preclinical cancer imaging: innovating the discovery of therapeutics. Nat. Rev. Cancer. 10.1038/nrc3714 online April 17, 2014 [DOI] [PubMed] [Google Scholar]
- Cox, T.R. , Erler, J.T. , 2012. Network biology and the 3-Dimensional tumor microenvironment: personalizing medicine for the future. Tumour Microenv. Ther. 1, 14–18. [Google Scholar]
- Creixell, P. , Schoof, E.M. , Erler, J.T. , Linding, R. , 2012. Navigating cancer network attractors for tumor-specific therapy. Nat. Biotechnol. 30, 842–848. [DOI] [PubMed] [Google Scholar]
- Curtis, C. , Shah, S.P. , Chin, S.F. , Turashvili, G. , 2012. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao, P. , Wang, K. , Collins, C. , Ester, M. , 2011. Optimally discriminative subnetwork markers predict response to chemotherapy. Bioinformatics. 27, i205–i213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Sol, A. , Balling, R. , Hood, L. , Galas, D. , 2010. Diseases as network perturbations. Curr. Opin. Biotechnol. 21, 566–571. [DOI] [PubMed] [Google Scholar]
- Drevelegas, A. , 2011. Imaging of Brain Tumors with Histological Correlations Springer; 13–33. [Google Scholar]
- Erler, J.T. , Linding, R. , 2010. Network-based drugs and biomarkers. J. Pathol. 220, 290–296. [DOI] [PubMed] [Google Scholar]
- Farnsworth, R.H. , Lackman, M. , Achen, M.G. , Stacker, S.A. , 2014. Vasucalr remodeling in cancer. Oncogene. 33, 3496–3505. [DOI] [PubMed] [Google Scholar]
- Goh, K.I. , Cusick, M.E. , Valle, D. , Childs, B. , 2007. The human disease network. Proc. Natl. Acad. Sci. U S A. 104, 8685–8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göke, J. , Chan, Y.S. , Yan, J. , Vingron, M. , 2013. Genome-wide kinase-chromatin interactions reveal the regulatory network of ERK signaling in human embryonic stem cells. Mol. Cell. 50, (6) 844 [DOI] [PubMed] [Google Scholar]
- Haacke, E. Mark , 1999. Magnetic Resonance Imaging: Physical Principles and Sequence Design Wiley-Liss; Hoboken, NJ: [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2000. The hallmarks of cancer. Cell. 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2011. Hallmarks of cancer: the next generation. Cell. 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hartung, M.P. , Grist, T.M. , François, C.J. , 2011. Magnetic resonance angiography: current status and future directions. J. Cardiovasc. Magn. Reson. 13, 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson, K. , Phillips, M. , Smith, W. , Peterson, L. , 2011. Hypoxia imaging with [F-18] FMISO-PET in head and neck cancer: potential for guiding intensity modulated radiation therapy in overcoming hypoxia-induced treatment resistance. Radiother. Oncol. 101, (3) 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan, C. , Van Schaeybroeck, S. , Longley, D.B. , Johnston, P.G. , 2013. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer. 13, 714–726. [DOI] [PubMed] [Google Scholar]
- Huang, S.S. , Fraenkel, E. , 2009. Integrating proteomic, transcriptional, and interactome data reveals hidden components of signaling and regulatory networks. Sci. Signal. 2, ra40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S. , Ernberg, I. , Kauffman, S. , 2010. Cancer attractors: a systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 20, 869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker, T. , Krogan, N.J. , 2012. Differential network biology. Mol. Syst. Biol. 8, 565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes, K.A. , Albeck, J.G. , Gaudet, S. , Sorger, P.K. , 2005. A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science. 310, (5754) 1646–1653. [DOI] [PubMed] [Google Scholar]
- Johansson, A. , Hamzah, J. , Ganss, R. , 2014. License for destruction: tumor-specific cytokine targeting. Trends Mol. Med. 20, 16–24. [DOI] [PubMed] [Google Scholar]
- Kauffman, S. , 1969. Homeostasis and differentiation in random genetic control networks. Nature. 224, 177–178. [DOI] [PubMed] [Google Scholar]
- Kickingereder, P. , Sahm, F. , Wiestler, B. , Roethke, M. , 2014. Evaluation of microvascular permeability with dynamic contrast-enhanced MRI for the differentiation of primary CNS lymphoma and glioblastoma: radiologic-pathologic correlation. AJNR Am. J. Neuroradiol. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. , Gao, L. , Tan, K. , 2012. Multi-analyte network markers for tumor prognosis. PLoS One. 7, e52973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.G. , Harel, N. , TaeKim, T. , Lee, P. , 2013. Cerebral blood volume MRI with intravascular superparamagnetic iron oxide nanoparticles. NMR Biomed. 26, (8) 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirouac, D.C. , Onsum, M.D. , 2013. Using network biology to bridge pharmacokinetics and pharmacodynamics in oncology. Cpt: Pharmacometrics Syst. Pharmacol. 2, e71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleijn, A. , Chen, J.W. , Buhrman, J.S. , Wojtkiewicz, G.R. , 2011. Distinguishing inflammation from tumor and peritumoral edema bymyeloperoxidase magnetic resonance imaging. Clin. Cancer Res. 17, (13) 4484–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurhanewicz, J. , Vigneron, D.B. , Brindle, K. , Chekmenev, E.Y. , 2011. Analysis of cancer metabolism by imaging hyperpolarized nuclei: prospects for translation to clinical research. Neoplasia. 13, (2) 81–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M.J. , Ye, A.S. , Gardino, A.K. , Heijink, A.M. , 2012. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 149, (4) 780–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Lenferink, A.E. , Deng, Y. , Collins, C. , 2010. Identification of high-quality cancer prognostic markers and metastasis network modules. Nat. Comm. 1, 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar, J.C. , Quackenbush, J. , 2009. Decomposition of gene expression state space trajectories. PLoS Comput. Biol. 5, (12) e1000626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marras, E. , Travaglione, A. , Capobianco, E. , 2010. Sub-modular resolution analysis by network mixture models. Stat. Appl. Genet. Mol. Biol. 9, Art.19 [DOI] [PubMed] [Google Scholar]
- Marusyk, A. , Polyak, K. , 2010. Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta. 1805, 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumotoa, M. , Kodamaa, N. , Endoa, Y. , Sakumaa, J. , 2007. Dynamic 3D-CT angiography. AJNR Am. J. Neuroradiol. 28, (2) 299–304. [PMC free article] [PubMed] [Google Scholar]
- McCann, T.E. , Kosaka, N. , Turkbey, B. , Mitsunaga, M. , 2011. Molecular imaging of tumor invasion and metastases: the role of MRI. NMR Biomed. 24, (6) 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham, C.E. , Morrison, S.J. , 2013. Tumour heterogeneity and cancer cell plasticity. Nature. 501, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, M.K. , Saez-Rodriguez, J. , Sorger, P.K. , Lauffenburger, D.A. , 2010. Logic-based models for the analysis of cell signaling networks. Biochemistry. 49, 3216–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin, N. , Krasnitz, A. , Rodgers, L. , Cook, K. , 2010. Inferring tumor progression from genomic heterogeneity. Genome Res. 20, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin, N. , Kendall, J. , Troge, J. , Andrews, P. , 2011. Tumour evolution inferred by single-cell sequencing. Nature. 472, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibbe, R.K. , Koyuturk, M. , Chance, M.R. , 2010. An integrative-omics approach to identify functional sub-networks in human colorectal cancer. PLoS Comput. Biol. 6, e1000639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilesh, M. , Quarles, C.C. , 2011. Imaging Tissue Oxygenation Status with MRI. Quantitative MRI in Cancer, Imagingin Medical Diagnosis and Therapy Taylor & Francis; Oxford: [Google Scholar]
- Paulmurugan, R. , Oronsky, B. , Brouse, C.F. , Reid, T. , 2013. Real time dynamic imaging and current targeted therapies in the war on cancer: a new paradigm. Theranostics. 3, (6) 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pe'er, D. , Hacohen, N. , 2011. Principles and strategies for developing network models in cancer. Cell. 144, 864–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou, C.M. , Sorlie, T. , Eisen, M.B. , van de Rijn, M. , 2000. Molecular portraits of human breast tumours. Nature. 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Qutub, A.A. , Mac Gabhann, F. , Karagiannis, E.D. , Vempati, P. , 2009. Multiscale model of angiogenesis. IEEE Eng. Med. Biol. Mag. 28, (2) 14–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunand, N. , 2006. Tissue pH measurement by magnetic resonance spectroscopy and imaging In Prasad P., Magnetic Resonance Imaging. Methods in Molecular Medicine. vol. 124, Humana Press; Totowa, NJ: 347–364. [DOI] [PubMed] [Google Scholar]
- Rangarajan, R. , Zaman, M.H. , 2008. Modeling cell migration in 3D: status and challenges. Cell Adh. Migr. 2, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, C.J. , Cimermancic, P. , Szpiech, Z.A. , Sali, A. , 2013. High-resolution network biology: connecting sequence with function. Nat. Rev. Genet. 14, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Musach, J. , Aguilar, H. , Iorio, F. , Comellas, F. , 2012. Cancer develops, progresses and responds to therapies through restricted perturbation of the protein-protein interaction network. Integr. Biol. 4, (9) 1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, M. , Beauchamp, R.D. , Zhang, B. , 2012. A network-based gene expression signature informs prognosis and treatment for colorectal cancer patients. PLoS One. 7, e41292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi, U. , Gamez, C. , Dawood, S. , Yeung, H.W.D. , 2008. Bone metastases in patients with metastatic breast cancer: morphologic and metabolic monitoring of response to systemic therapy with integrated PET/CT. Radiology. 247, (1) 189–196. [DOI] [PubMed] [Google Scholar]
- Troprès, I. , Grimault, S. , Vaeth, A. , Grillon, E. , 2011. Vessel size imaging. Magn. Reson. Med. 45, (3) 397–408. [DOI] [PubMed] [Google Scholar]
- Ungefroren, H. , Sebens, S. , Seidl, D. , Lehnert, H. , 2011. Interaction of tumor cells with the microenvironment. Cell Comm. Signal. 9, 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayagam, A. , Stelzl, U. , Foulle, R. , Plassmann, S. , 2011. A directed protein interaction network for investigating intracellular signal transduction. Sci. Signal. 4, (189) rs8 [DOI] [PubMed] [Google Scholar]
- Weissleder, R. , Ross, B.D. , Rehemtulla, A. , Gambhir, S.S. , 2010. Molecular Imaging: Principles and Practice People's Medical Publishing House; [Google Scholar]
- Welch, C.M. , Elliott, H. , Danuser, G. , Hahn, K.M. , 2011. Imaging the coordination of multiple signalling activities in living cells. Nat. Rev. Mol. Cell Biol. 12, 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen, Z. , Liu, Z.P. , Liu, Z. , Zhang, Y. , 2013. An integrated approach to identify causal network modules of complex diseases with application to colorectal cancer. J. Am. Med. Inform. Assoc. 20, 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzl, T. , Wilkens, J.J. , 2011. Theoretical analysis of the dose dependence of the oxygen enhancement ratio and its relevance for clinical applications. Radiat. Oncol. 6, 171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirapati, P. , Sotiriou, C. , Kunkel, S. , Farmer, P. , 2008. Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 10, R65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G. , Stein, L. , 2012. A network module-based method for identifying cancer prognostic signatures. Genome Biol. 13, R112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G. , Feng, X. , Stein, L. , 2010. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 11, R53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. , Zhang, W. , Li, J. , Zhang, Y. , 2013. Optical imaging of tumor microenvironment. Am. J. Nucl. Med. Mol. Imaging. 3, (1) 1–15. [PMC free article] [PubMed] [Google Scholar]
- Yap, T.A. , Gerlinger, M. , Futreal, P.A. , Pusztai, L. , 2012. Intratumor heterogeneity: seeing the wood for the trees. Sci. Transl. Med. 4, (127) 1–4. [DOI] [PubMed] [Google Scholar]
- Young, G.S. , 2007. Advanced MRI of adult brain tumors. Neurol. Clin. 25, (4) 947–973. viii [DOI] [PubMed] [Google Scholar]
- Zacharaki, E.I. , Wang, S. , Chawla, S. , Soo Yoo, D. , 2009. Classification of brain tumor type and grade using MRI texture and shape in a machine learning scheme. Magn. Reson. Med. 62, 1609–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman, M.H. , 2006. Multiscale modeling of tumor cell migration. AIP Conf. Proc. 851, 117–122. [Google Scholar]
- Zaman, M.H. , 2007. A multiscale probabilisitic framework to model early steps in tumor metastasis. Mol. Cell Biomech. 4, 133–141. [PubMed] [Google Scholar]
- Zanzoni, A. , Soler-Lopez, M. , Aloy, P. , 2009. A network medicine approach to human disease. FEBS Lett. 583, 1759–1765. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Lin, Y. , Gillies, R.J. , 2010. Tumor pH and its measurement. J. Nucl. 51, 1167–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, Q. , Simonis, N. , Li, Q.R. , Charloteaux, B. , 2009. Edgetic perturbation models of human inherited disorders. Mol. Syst. Biol. 5, 321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, A. , Lee, D. , Shim, H. , 2011. Metabolic PET imaging in cancer detection and therapy response. Semin. Oncol. 38, (1) 55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]