

Abstract
Modern cancer research on prognostic and predictive biomarkers demands the integration of established and emerging high‐throughput technologies. However, these data are meaningless unless carefully integrated with patient clinical outcome and epidemiological information. Integrated datasets hold the key to discovering new biomarkers and therapeutic targets in cancer. We have developed a novel approach and set of methods for integrating and interrogating phenomic, genomic and clinical data sets to facilitate cancer biomarker discovery and patient stratification. Applied to a known paradigm, the biological and clinical relevance of TP53, PICan was able to recapitulate the known biomarker status and prognostic significance at a DNA, RNA and protein levels.
Keywords: Pathology integromics, Biomarker discovery, Digital pathology
Highlights
Novel methodology for integrating and interrogating phenomic, genomic and clinical data.
Facilitates cancer biomarker discovery, patient stratification and digital pathology.
Flexible and dynamic to accommodate large data cohorts and high‐throughput data.
Applied to demonstrate utility on the biological and clinical relevance of TP53.
PICan could recapitulate the known biomarker status and prognostic significance at a DNA, RNA and protein levels.
1. Introduction
High‐throughput (HT) techniques elevate the volume of genomic information in molecular pathology, with increasing cohort sizes offering molecular and phenotypic sub‐classifications. This is driving a more holistic understanding of the phenotypic traits that underpin disease. Such data generation, its convergence with other data and the need to fast‐track biomarker discovery and evaluation is causing a major bottleneck in interpretation. Diagnostic pathology departments globally store many millions of tissue samples across different cancer types and patient populations in the form of formalin‐fixed paraffin‐embedded (FFPE) blocks. This in itself provides an enormously rich cohort of samples which can be used in molecular discovery and translational medicine in solid tumours. For example, many studies utilise pathology archives to generate tissue microarrays (TMAs) for the HT‐analysis of tissue biomarkers across multiple patient samples in a single assay. However, the handling, preparation and storage of tissue samples can be highly variable, potentially impacting on the quality of downstream molecular evaluation that is sensitive to these pre‐analytical variables. The goal of biobanks or bio‐repositories is to overcome variability by planned prospective collection of samples where several samples per patient are collected concomitantly with fully controlled variables, specifically for the purposes of research. Collating clinical and analytical data on these samples using dedicated informatics‐based methods provides us with unique opportunities to understand these complex clinico‐genomic data sets. What impact this HT‐integrative approach has on treatment has been the focus of many recent seminal papers and is the foundation of stratified medicine (Garnett et al. 2012; Misale, S. et al. 2012; Diaz et al. 2012).
In an interpretative context, clinical/genomic resources are becoming increasingly conspicuous, such as the cancer genome atlas (Cancer Genome Atlas Research Network, 2008) offering large data collections on multiple cancer types. Kristensen and colleagues discuss in their recent review the tools and complexity required in order to analyse integrative genomic methodologies to discover patient subgroups for prognostic and diagnostic insight (Kristensen et al., 2014). Recent publications in cancer research from an integrative context are starting to depict a landscape of increased scrutiny, complexity and discovery (Ding et al. 2014; Schroeder et al. 2013; Wang et al. 2013). Synergistically, it is also possible to analyse HT‐tissue biomarker data by uploading and analysing with TMANavigator (Lubbock et al. 2013) as well as cohort data analysis by designing an open platform for web based biomedical queries (Pennington et al. 2014). However, we still have a dearth of methods supporting the merger of disparate data sets spanning patients, their samples, tissue and genomic biomarkers and the interrogation of these complex data sets for biomarker discovery, translational research and patient stratification.
An additional issue with developing an integrated data analysis system is that clinical phenogenomic data tends to be dynamic. Most studies are longitudinal, requiring on‐going patient follow‐up where new data is constantly being added to the system. Digital image evaluation is also an important component in tissue‐based research, particularly in heterogeneous biomarkers requiring HT‐evaluation where data integration and concordance are of particular interest in biomarker discovery, adding performance, robustness and reproducibility. In addition, new analytical methods are constantly evolving, requiring analysis and re‐analysis of the sample cohort. As such, the statistical approaches to data‐mining would also need to be updated constantly and with a modern adaptable framework designed to support it.
2. Materials and methods
We have adopted an integrative “omics” (‘integromics’) method that allows the streamlined merging of data from diverse sources (Searles, 2005; Lê Cao et al. 2009). This we have called PICan (Pathology Integromics in Cancer). PICan is a comprehensive clinical genomics data management approach for molecular tissue pathology using MySQL and ASP.Net (supp methods 1), allowing seamless integration and analysis of carefully defined, cancer specific clinical data sets with associated tissue biomarker data and molecular genomics (Figure 1). With ethical approval in place, patient clinical and pathological information can be collated from biobanks, where data‐exchange is possible between platforms through unique identifiers (Supp Methods 1). Upon data‐exchange completion, we have a collection that we integrate with the digital and molecular derived information generated from on‐going studies using HT‐data and singular biomarker tests (Figure 2A) (Supp Methods 3 & 4). With this, we have a scaffold to support the analysis of high‐dimensional data for expression signatures, which are shifting the current paradigm in cancer research (Sadanandam et al. 2013; Mulligan et al. 2014).
Figure 1.

Overarching view of the PICan system. Resolving data to minimal information templates allows the creation of a knowledge base we implement into database tables which can be resolved to hold molecular and digital pathology information. We can then, in turn, statistically analyse data using stratified selections. These are streamlined and instantly expandable both on the cohort level as well as at a data entry level, in order to place new biomarkers in a molecular pathology context.
Figure 2.
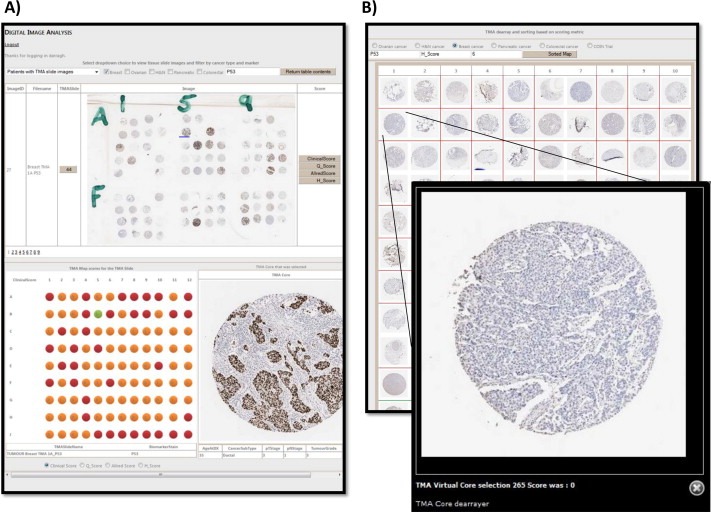
Digitally scanned IHC TMA information can be seamlessly matched to the patient clinical and pathological information. A) Users can select cores from the map interface or scoring interface to retrieve the core in higher resolution and supporting information, progress is tracked on both maps. B) Also, as cores are virtually de‐arrayed we can rank the cores by their associated scoring metrics across multiple maps, an important resource in collaboration, consistency and training.
With PICan offering a browser based interface, we segregate the data collections into separate navigational browser pages for data interpretation and digital image assessment (Figure 3B) (supp methods 2). PICan's utility differentiates it from statistical software packages in its capacity to integrate different types of patient, sample and biomarker analytic data, facilitating the concatenation of data into a single searchable interface where molecular and phenotypic data can be combined to generate new signatures of disease. The system will not only run routine statistical tests, but with the added HT‐data we can perform supervised and unsupervised analytical techniques factoring in other known molecular information. This approach will tolerate research flexibility in prognostic evaluation and the selection of a ‘best scoring metric criteria’ in IHC either using receiver operator characteristic curves, Kaplan Meier curves or Cox proportional hazards models (Supp Methods 5). The integration of digital pathology technology within PICan allows us to review tissue morphology and protein expression profiles within the system. Patient stratification will resolve the difference in frequencies in protein biomarkers and transcriptomic signatures integrating in additional mutational knowledge.
Figure 3.
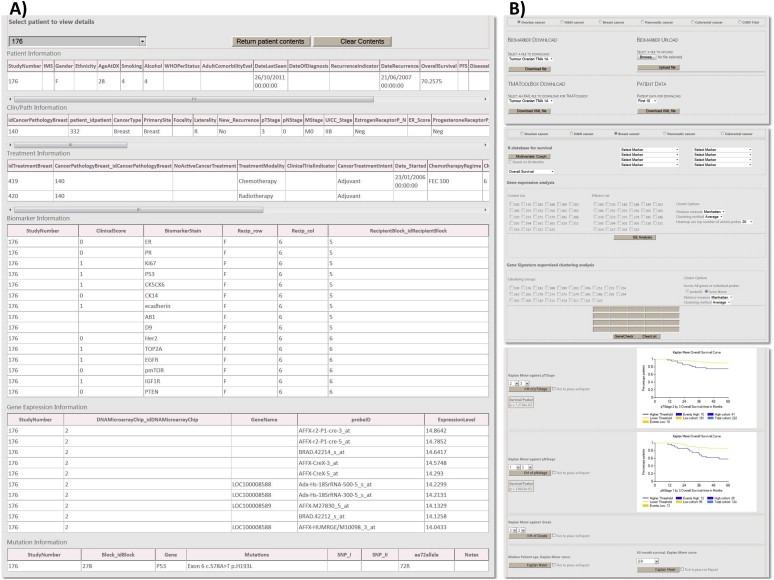
A) Resolved patient information, includes clinical and pathological data matched to genomic and transcriptomic information with interactive graphical functionality (inset). B) Navigational pages for: data‐exchange/upload (Top); HT‐analysis for differential expression and supervised analysis (Middle); and single biomarker statistical analyses (Bottom).
3. Results and discussion
As an example of the power and the utility of the PICan method we used this approach as the primary data integration and analysis platform for a large breast cancer tissue biomarker study (Boyle et al. 2014). Taking one of the best known genes and proteins in cancer biology, p53, we address two main test hypotheses. Firstly, that a PICan driven analysis of image related information in TMAs is able to achieve the same biological and clinical relevance in breast cancer, after the IHC results are digitally scanned, de‐arrayed and information resolved so that it can be analysed against other baseline biomarkers (Figure 3A) and from here it becomes a marker that can be used in other browser pages (i.e. stratifying HT‐data) or downloaded for the researcher to use elsewhere (Figure 3B). Secondly, we explore if known HT‐genomic signatures of p53 biology hold their statistical relevance when analysing, in the PICan context, on independent sets of HT‐generated results in breast cancer. The latter is one of the most important uses of this method, which we demonstrate here by way of understanding the p53 correlations at the 3 levels of the central dogma of molecular biology (DNA mutation, RNA gene amplification and protein expression), and understand the clinical relevance of such holistic analysis for individual cases and for the whole cohort.
With the number of scoring metrics (H Score, percentage, quick score, allred score, intensity scores or user defined scores) being easily integrated into the platform (because we can resolve TMA maps against the patient information) we obtain in this instance 288 p53 IHC cores from the 293 patients. The cores digitally de‐arrayed and matched to the individual can be visualised, integrating the p53 IHC data by direct data‐exchange and linked by unique identifiers to the individual. It can then seamlessly integrate our associated scoring metrics against the clinical cohort and across TMA maps as can be achieved with leading digital pathology software (Leica http://www.leicabiosystems.com/, PathXL http://www.pathxl.com/, Definiens http://www.definiens.com/or Visiopharm http://www.visiopharm.com/). All de‐arrayed cores can be virtually remapped into ascending/descending order to ensure consistency in scoring (Figure 2B) allowing virtual de‐arraying as demonstrated by Quintayo and colleagues (Quintayo et al. 2014). From here, ROC curve, Kaplan Meier survival analysis and digital image immunohistochemistry thresholding is performed, which allows us to directly interpret the best settings for IHC analysis statistically. This allows us to assess the diagnostic threshold for use in downstream analysis (Figure 4C). In this instance, we corroborate the findings by Boyle et al. of low aberrant expression adding to the prognostic interpretation of p53 in breast cancer and include this cohort with the more traditional high aberrant expression (Boyle et al. 2014). Utilising the streamlined analyses we can further corroborate the kappa concordance between original baseline marker clinical assessment and TMA baseline makers, and correlate with histopathological parameters and Fisher's and chi‐square tests obtained in the original study. At this point, it is possible to measure the frequency of this marker in sub‐classifications (such as triple negative cancers) against other markers using our marker charting functionality, seen in Figure 1, see ‘Clinical relevance’. In turn, we can then evaluate its prognostic relevance (Figure 4C). This can be concluded with an exploratory analysis using Cox regression proportional hazards of the new marker against histopathological parameters and other established markers on the system.
Figure 4.
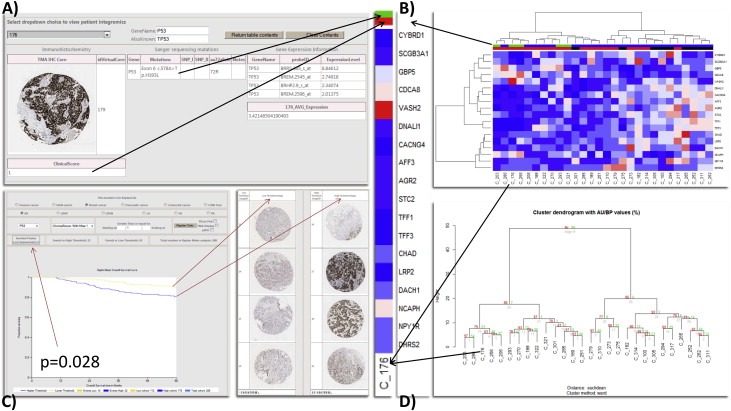
A) We use the patient integromics analysis to reveal candidates with specific mutations of interest. B) In line with the seminal Miller et al. paper in 2005, we examined the 18/20 identifiable genes from their signature in our microarrays that survived a median filter, using the probe with the highest variance of each gene (if more than one) to examine the integrated information clustered in our data. Top column bar P53 mutation: Green – Mt; Blue – Wt; second column bar IHC: Red – Aberrant extreme (by Boyle et al.); Black – non‐extreme (intermediate patterns in IHC). C) The clinical significance of this data is obtained demonstrating a significant prognostic IHC threshold for aberrant extreme p53, which can be visualised by the thresholded de‐arrayed cores. D) Pvclust bootstrap resampling for the estimation of uncertainty in the data evaluated in B. with bootstrap n = 1000.
This new ‘integromic’ framework allows us to test HT‐signatures and confirm their biological value when correlating the resulting taxonomy with the mutation and aberrant protein status for the same biomarker. To do so, we query which patients in our breast cancer cohort had mutational aberrations in TP53 (Figure 4A) based on the TP53 signature hypothesis and mutational analysis by Miller et al. (Miller et al. 2005). This summarises patients individually by their genomic and clinical/pathological/treatment profiles and call upon mutational information such as Sanger sequencing or NGS variants. Employing this additional information we can create a new analysis framework by A) stratifying and clustering the expression data deriving differential expression signatures and B) submitting gene lists for unsupervised signature validation (Figure 4B and D). Our analysis, using the signature stated above was able to differentiate p53 mutant versus wild‐type cases, which also correlated with the aberrant protein status and with patient survival.
The PICan method has a wide range of emerging applications, improving the resolution of the complex, multivariate, multiplex analysis of modern tissue‐based research and its role in the delivery of personalised medicine. Over time, the expansion of PICan will demand the incorporation of new technologies and their associated datatypes, as and when these techniques become available. In our setting, this has led to the need to expand our translational bioinformatics team. In addition, safeguarding and expanding high quality curated clinical and pathological data has required dedicated resource to support expansion. Ensuring high quality data expansion and well defined ontologies are going to be in high demand for data integromics in the future, and where lack of control across different analytical platforms may decrease analytical sensitivity. In our setting, the fact that the clinical/pathological data is collected and curated by in‐house pathologists and clinicians, allows us to control statistical selection and safe‐guard data analysis. However, the system is designed for exploratory mining of the multivariate data where findings will hopefully enhance discovery, but where validation and more sophisticated statistical interrogation will be required. This approach is, however, flexible across all cancer types and analytical tools and will allow researchers to statistically analyse complex datasets within or across different diseases. This novel approach would be beneficial to other molecular pathology laboratories and clinical trials facilities as the method to support data integration, improve tissue‐based analysis and fast‐track biomarker discovery and validation (Salto‐Tellez et al., 2014).
Author contributions
MST and PH lead the project, designed the concept and guided its development from a digital pathology and molecular pathology context. DMA developed the system and undertook the validation. JB, YW and DK constructed table entities and statistical methods to interpret the data. DB, GI, MM, PD designed the templates, in this instance for Breast, in the focus of this manuscript. PB and RH helped in TMA de‐arraying and digital image interpretation. RK, PM, PH provided insight and interpretation on HT‐data. MC provided the data on mutational analysis. JJ governed the ethical framework and integrity of the system surrounding clinical and pathological collections.
Conflict of interest
Prof. D. Paul Harkin, president and managing director, Prof. Richard D. Kennedy, VP and clinical director, currently at Almac diagnostics, manufacturers of the Breast DSA used, in part, in this methodology. Prof. Manuel Salto‐Tellez was a Consultant for Almac during part of the duration of this project, and is currently member of the Advisory Panel of PathXL. Prof. Peter Hamilton is founder board member at PathXL.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgements
The authors wish to thank the NI Biobank for help in constructing the necessary framework and ethics involved in data collection. We would also like to thank the Northern Ireland molecular pathology laboratory for tissue microarray construction and also to Anne Carson for digital image scanning and hosting on dedicated servers along with the excellent team at PathXL. The clinicians deserve special mention for devising minimal information templates for their particular focus groups and dedicated teams. The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007‐2013/ under REA grant agreement no [285910].
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.02.002.
McArt Darragh G., Blayney Jaine K., Boyle David P., Irwin Gareth W., Moran Michael, Hutchinson Ryan A., Bankhead Peter, Kieran Declan, Wang Yinhai, Dunne Philip D., Kennedy Richard D., Mullan Paul B., Harkin D. Paul, Catherwood Mark A., James Jacqueline A., Salto-Tellez Manuel, Hamilton Peter W., (2015), PICan: An integromics framework for dynamic cancer biomarker discovery, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.02.002.
Contributor Information
Manuel Salto-Tellez, Email: m.salto-tellez@qub.ac.uk.
Peter W. Hamilton, Email: p.hamilton@qub.ac.uk
References
- Boyle, D.P. , McArt, D.G. , Irwin, G. , Wilhelm-Benartzi, C.S. , Lioe, T.F. , Sebastian, E. , McQuaid, S. , Hamilton, P.W. , James, J.A. , Mullan, P.B. , Catherwood, M.A. , Harkin, D.P. , Salto-Tellez, M. , 2014. The prognostic significance of the aberrant extremes of p53 immunophenotypes in breast cancer. Histopathology 65, (3) 340–352. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz, L.A. , Williams, R.T. , Wu, J. , Kinde, I. , Hecht, J.R. , Berlin, J. , Allen, B. , Bozic, I. , Reiter, J.G. , Nowak, M.A. , Kinzler, K.W. , Oliner, K.S. , Vogelstein, B. , 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, H. , Wang, C. , Huang, K. , Machiraju, R. , 2014. iGPSe: a visual analytic system for integrative genomic based cancer patient stratification. BMC Bioinformatics 15, 203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett, M.J. , Edelman, E.J. , Heidorn, S.J. , Greenman, C.D. , Dastur, A. , Lau, K.W. , Greninger, P. , Thompson, I.R. , Luo, X. , Soares, J. , Liu, Q. , Iorio, F. , Surdez, D. , Chen, L. , Milano, R.J. , Bignell, G.R. , Tam, A.T. , Davies, H. , Stevenson, J.A. , Barthorpe, S. , Lutz, S.R. , Kogera, F. , Lawrence, K. , McLaren-Douglas, A. , Mitropoulos, X. , Mironenko, T. , Thi, H. , Richardson, L. , Zhou, W. , Jewitt, F. , Zhang, T. , O'Brien, P. , Boisvert, J.L. , Price, S. , Hur, W. , Yang, W. , Deng, X. , Butler, A. , Choi, H.G. , Chang, J.W. , Baselga, J. , Stamenkovic, I. , Engelman, J.A. , Sharma, S.V. , Delattre, O. , Saez-Rodriguez, J. , Gray, N.S. , Settleman, J. , Futreal, P.A. , Haber, D.A. , Stratton, M.R. , Ramaswamy, S. , McDermott, U. , Benes, C.H. , 2012. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen, V.N. , Lingjærde, O.C. , Russnes, H.G. , Vollan, H.K. , Frigessi, A. , Børresen-Dale, A.L. , 2014. Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer 14, (5) 299–313. [DOI] [PubMed] [Google Scholar]
- Lê Cao, K.-A. , González, I. , Déjean, S. , 2009. integrOmics: an R package to unravel relationships between two omics datasets. Bioinformatics 25, 2855–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubbock, A.L.R. , Katz, E. , Harrison, D.J. , Overton, I.M. , 2013. TMA Navigator: Network inference, patient stratification and survival analysis with tissue microarray data. Nucl. Acids Res. 41, W562–W568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, L.D. , Smeds, J. , George, J. , Vega, V.B. , Vergara, L. , Ploner, A. , Pawitan, Y. , Hall, P. , Klaar, S. , Liu, E.T. , Bergh, J. , 2005. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Natl. Acad. Sci. USA 102, 13550–13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale, S. , Yaeger, R. , Hobor, S. , Scala, E. , Janakiraman, M. , Liska, D. , Valtorta, E. , Schiavo, R. , Buscarino, M. , Siravegna, G. , Bencardino, K. , Cercek, A. , Chen, C.T. , Veronese, S. , Zanon, C. , Sartore-Bianchi, A. , Gambacorta, M. , Gallicchio, M. , Vakiani, E. , Boscaro, V. , Medico, E. , Weiser, M. , Siena, S. , Di Nicolantonio, F. , Solit, D. , Bardelli, A. , 2012. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan, J.M. , Hill, L.A. , Deharo, S. , Irwin, G. , Boyle, D. , Keating, K.E. , Raji, O.Y. , McDyer, F.A. , O'Brien, E. , Bylesjo, M. , Quinn, J.E. , Lindor, N.M. , Mullan, P.B. , James, C.R. , Walker, S.M. , Kerr, P. , James, J. , Davison, T.S. , Proutski, V. , Salto-Tellez, M. , Johnston, P.G. , Couch, F.J. , Paul Harkin, D. , Kennedy, R.D. , 2014. Identification and validation of an anthracycline/cyclophosphamide-based chemotherapy response assay in breast cancer. J. Natl. Cancer Inst. 106, djt335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington, J.W. , Ruth, B. , Italia, M.J. , Miller, J. , Wrazien, S. , Loutrel, J.G. , Crenshaw, E.B. , White, P.S. , 2014. Harvest: an open platform for developing web-based biomedical data discovery and reporting applications. J. Am. Med. Inform Assoc. 21, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintayo, M.A. , Starczynski, J. , Yan, F.J. , Wedad, H. , Nofech-Mozes, S. , Rakovitch, E. , Bartlett, J.M. , 2014 Jul. Virtual tissue microarrays: a novel and viable approach to optimizing tissue microarrays for biomarker research applied to ductal carcinoma in situ. Histopathology 65, (1) 2–8. [DOI] [PubMed] [Google Scholar]
- Sadanandam, A. , Lyssiotis, C.A. , Homicsko, K. , Collisson, E.A. , Gibb, W.J. , Wullschleger, S. , Ostos, L.C. , Lannon, W.A. , Grotzinger, C. , Del Rio, M. , Lhermitte, B. , Olshen, A.B. , Wiedenmann, B. , Cantley, L.C. , Gray, J.W. , Hanahan, D. , 2013. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 19, 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salto-Tellez, M. , James, J.A. , Hamilton, P.W. , 2014. Molecular pathology – the value of an integrative approach. Mol. Oncol. 8, (7) 1163–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder, M.P. , Gonzalez-Perez, A. , Lopez-Bigas, N. , 2013. Visualizing multidimensional cancer genomics data. Genome Med. 5, (1) 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searles, D.B. , 2005. Data integration: challenges for drug discovery. Nat. Rev. Drug Discov. 4, 45–58. [DOI] [PubMed] [Google Scholar]
- Wang, C. , Pécot, T. , Zynger, D.L. , Machiraju, R. , Shapiro, C.L. , Huang, K. , 2013. Identifying survival associated morphological features of triple negative breast cancer using multiple datasets. J. Am. Med. Inform Assoc. 20, (4) 680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data