

Abstract
Radiation‐induced DNA damage activates the DNA damage response (DDR). DDR up‐regulation may predict radio‐resistance and increase the risk of early local recurrence despite radiotherapy in early stage breast cancers. In 1755 early stage breast cancers, DDR signalling [ATM, ATR, total Ckh1, Chk1 phosphorylated at serine345 (pChk1), Chk2, p53], base excision repair [PARP1, POLβ, XRCC1, FEN1, SMUG1], non‐homologous end joining (Ku70/Ku80, DNA‐PKcs) and homologous recombination [RAD51, BRCA1, γH2AX, BLM, WRN, RECQL5, PTEN] protein expression was correlated to time to early local recurrence. Pre‐clinically, radio‐sensitization by inhibition of Chk1 activation by ATR inhibitor (VE‐821) and inhibition of Chk1 (V158411) were investigated in MDA‐MB‐231 (p53 mutant) and MCF‐7 (p53 wild‐type) breast cancer cells. In the whole cohort, 208/1755 patients (11.9%) developed local recurrence of which 126 (61%) developed local recurrence within 5 years of initiation of primary therapy. Of the 20 markers tested, only pChk1 and p53 significantly associated with early local recurrence (p value = 0.015 and 0.010, respectively). When analysed together, high cytoplasmic pChk1‐nuclear pChk1 (p = 0.039), high cytoplasmic pChk1‐p53 (p = 0.004) and high nuclear pChk1‐p53 (p = 0.029) co‐expression remain significantly linked to early local recurrence. In multivariate analysis, cytoplasmic pChk1 level independently predicted early local recurrence (p = 0.025). In patients who received adjuvant local radiotherapy (n = 949), p53 (p = 0.014) and high cytoplasmic pChk1‐p53 (p = 0.017) remain associated with early local recurrence. Pre‐clinically, radio‐sensitisation by VE‐821 or V158411 was observed in both MCF‐7 and MDA‐MB‐231 cells and was more pronounced in MCF‐7 cells. We conclude that pChk1 is a predictive biomarker of radiotherapy resistance and early local recurrence.
Keywords: Breast cancer, Local recurrence, Radiotherapy, Resistance, p53, Chk1, ATR inhibitor, Chk1 inhibitor
Highlights
Radiation‐induced DNA damage activates the DNA damage response (DDR).
1755 breast cancers were profiled for DDR and correlated to local recurrence.
High p‐Chk1 was significantly associated with local recurrence.
ATR‐Chk1 pathway inhibition increased radio sensitivity in breast cancer cells.
1. Introduction
Despite advances in surgery, adjuvant radiation and systemic therapies, 10–20% of early stage breast cancer patients will develop local recurrence (della Rovere and Benson, 2002; Gieni et al., 2012; Schnitt, 2003; van der Leij et al., 2012). Established risk factors for early local recurrence include young age at diagnosis (≤40 years), larger tumour size, multifocal disease, axillary nodal involvement, extra‐capsular tumour extension, positive margin, high grade, definite positive lympho‐vascular invasion, HER‐2 overexpression, ER negativity, and extensive intra‐ductal component (della Rovere and Benson, 2002; Gieni et al., 2012; Schnitt, 2003; van der Leij et al., 2012). Local recurrence may herald the emergence of radio‐resistant cancer clones with aggressive biology that may adversely impact upon patient outcomes (Clarke et al., 2005). Therefore, mining for predictive biomarkers for development of radio‐resistance is a high priority. Ionizing radiation (IR) induced DNA damage is a key mechanism for cytotoxicity and therapeutic efficacy in tumours (Derks et al., 2014; Santivasi and Xia, 2014). However, the ability of tumour cells to initiate an effective DNA damage response (DDR), by activating multiple DNA damage signalling and repair pathways, may result in resistance to radiotherapy and ultimately influence the emergence of local recurrence in patients (Derks et al., 2014; Santivasi and Xia, 2014).
In the current study, we have comprehensively investigated the expression of a panel of DNA repair factors involved in the DDR: DNA damage signalling (ATM, ATR, total Chk1, Chk1 phosphorylated at serine345 (here in referred to as pChk1) (Tian et al., 2015), CHK2, p53, base excision repair (PARP1, POLβ, XRCC1, FEN1, SMUG1) (Wallace, 2014), non‐homologous end joining (Ku70/Ku80, DNA‐PKcs) (Williams et al., 2014) and homologous recombination (RAD51, BRCA1, γH2AX, BLM, WRN, RECQL5, PTEN) (Liu and Huang, 2014) for local recurrence prediction in early stage breast cancers. In addition, work was also undertaken to test the blockade of CHK1 activation through ATR inhibition (VE‐821, Vertex pharmacuticals) (Josse et al., 2014) and direct CHK1 inhibition (V158411, Vernalis R&D Ltd) (Bryant et al., 2014; Stokes et al., 2009) in MDA‐MB‐231 and MCF‐7 breast cancer cell lines.
2. Methods
2.1. Clinical study
2.1.1. Patients
Demographics of the study population are summarised in Table 1. The cohort comprised of primary operable early‐stage (stage I–III) invasive breast cancers from patients treated by breast‐conserving surgery (wide local excision) and radiotherapy at Nottingham University Hospitals. Information on clinical history and outcome is prospectively maintained and patients were assessed in a standardised manner for clinical history and tumour characteristics. Local recurrence‐free survival was defined as the time interval (in months) between the start of primary treatment and date of first histological confirmation of recurrent cancer (invasive or in‐situ) at the vicinity of the treated breast. Distant metastasis‐free survival was defined as the time interval (in months) between the start of primary treatment and date of distant disease relapse. Breast Cancer Specific Survival (BCSS) is defined as the time (in months) from the date of primary surgery to the date of breast cancer related death.
Table Table 1.
Multivariate analysis (Time to local recurrence).
| P value | Exp(B) | 95.0% CI for Exp(B) | ||
|---|---|---|---|---|
| Lower | Upper | |||
| Time to local recurrence | ||||
| Lymph node status | 0.710 | 0.920 | 0.591 | 1.431 |
| Tumour size | 0.592 | 0.888 | 0.575 | 1.371 |
| Tumour Grade | 0.396 | 1.165 | 0.818 | 1.659 |
| ER | 0.410 | 0.791 | 0.453 | 1.381 |
| Vascular Invasion | 0.216 | 1.488 | 0.793 | 2.794 |
| p53 | 0.315 | 1.278 | 0.792 | 2.062 |
| pChk1 (cytoplasmic) | 0.025 | 1.639 | 1.065 | 2.522 |
Bold = statistically significant.
In this early breast cancer cohort, 949 patients had received adjuvant local radiotherapy. Patients were managed in accordance to a uniform protocol, where all underwent wide local excision followed by radiotherapy which was given in daily 2 Gray (Gy) fractions, to a total dose of 50–55 Gy over 5 weeks. Thirty one patients received radiotherapy in the context of clinical trials investigating altered fractionation schedules. Radiotherapy was delivered to the whole breast. During the treatment period a “boost” to the tumour bed was not routinely given, however a “16 Gy boost” was given to six patients. Patients received systemic adjuvant treatment on the basis of Nottingham Prognostic Index (NPI), estrogen receptor (ER) status and menopausal status. Patients with an NPI score <3.4 did not receive adjuvant treatment. ER negative or premenopausal cases with an NPI score of 3.4 or more were candidates for cyclophosphamide, Methotrexate and 5‐Flourouracil (CMF) combination chemotherapy; ER positive cases received Tamoxifen for a period of 5 years.
This study is reported according to REMARK (Reporting Recommendations for Tumour Marker Prognostic Studies) criteria (McShane et al., 2005). Ethical approval for the study was granted by Nottingham Research Ethics committee 2 under the title “Development of a molecular genetic classification of breast cancer” (C202313).
2.1.2. Tissue microarrays (TMAs) and immunohistochemistry (IHC)
Tumours were arrayed in tissue microarrays (TMAs) constructed with 0.6 mm cores from the periphery of the tumours. The TMAs were immunohistochemically stained for a panel of DNA repair markers (Supplementary Table S2) as previously described (Abdel‐Fatah et al., 2014, 2015, 2015, 2015). Immunohistochemical staining was performed using the Novolink Max Polymer Detection System (RE7280‐K: 1250 tests), and the Leica Bond Primary Antibody Diluent (AR9352), each used according to the manufacturer's instructions (Leica Microsystems). The tissue sections were deparaffinised with xylene and rehydrated through five descending concentrations of alcohol (100%, 90%, 70%, 50% and 30%), two minutes each. Pre‐treatment antigen retrieval was performed using sodium citrate buffer (pH 6.0) and heated for 20 min at 95 °C in a microwave (Whirpool JT359 Jet Chef 1000W). Negative and positive (by omission of the primary antibody and IgG‐matched serum) controls were included in each run. The negative control ensured that all the staining was produced from the specific interaction between antibody and antigen.
2.1.3. Evaluation of immunostaining
The tumour cores were evaluated by two scorers (TAF and AA) and the concordance between the two scorer was excellent (k = 0.79). Only TMA cores with adequate invasive tumour tissue (>15% tumour) were scored, leaving out those with inadequate invasive tissue. Whole field inspection of the core was scored and intensities of nuclear staining were grouped as follows: 0 = no staining, 1 = weak staining, 2 = moderate staining, 3 = strong staining. The percentage of each category was estimated (0–100%). H‐score (range 0–300) was calculated by multiplying intensity of staining and percentage staining. Cut‐offs for individual markers are summarised in Supplementary Table 2.
2.1.4. Statistical analysis
Data analysis was performed using SPSS (SPSS, version 17 Chicago, IL). Cumulative local recurrence, distant metastasis and survival probabilities were estimated using the Kaplan–Meier method, and differences between survival rates were tested for significance using the log‐rank test. Multivariate analysis for survival was performed using the Cox proportional hazard model. The proportional hazards assumption was tested using standard log–log plots. Hazard ratios (HR) and 95% confidence intervals (95% CI) were estimated for each variable. All tests were two‐sided with a 95% CI and a p value < 0.05 considered significant.
2.2. Pre‐clinical study
2.2.1. Tissue culture and reagents
MCF7 and MDA‐MB‐231 breast cancer cell lines were obtained from ATCC (Manassas, VA, USA)and cultured with Gibco® RPMI 1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Sigma–Aldrich, St. Louis, MO, USA), and 10 mg/ml streptomycin/penicillin (Gibco) in an atmosphere of 20% O2 and 5% CO2. Cells were authenticated by STR profiling by LGC standards (Teddington, UK) and confirmed to be mycoplasma free (MycoAlert, Lonza, Basel, Switzerland) and used within 30 passages of authentication. VE‐821, a specific ATR inhibitor was provided by Vertex pharmaceuticals (Josse et al., 2014). V158411, a specific CHK1 inhibitor was provided by Vernalis R&D Ltd (Bryant et al., 2014; Stokes et al., 2009). VE‐821 and V158411 were dissolved in DMSO as stock solutions at 10 mM stored at −20 °C.
2.2.2. Chk1 knockdown
For Chk1 knockdown, Chk1 siRNA targeting Chk1 and the mock siRNA control (Dharmacon, GE Biosciences, Pittsburgh, PA, USA) were transfected into cells by InterferIN (Polyplus transfection, Illkirch, France) according to manufacturer's protocol. Western blotting was performed to confirm CHK1 knockdown.
2.2.3. Cell growth assay
MCF7 cells or MDA‐MB‐231 cells were seeded into 96‐well tissue culture plates and allowed to adhere for 24 h. Cells were treated with increasing concentrations of VE‐821 for 24 h before being allowed to grow in fresh media for 5 more days. Cells were fixed using Carnoy's fixative and stained with DAPI before being solubilised and the DAPI fluorescence measured. Data is mean data from 3 individual replicates. Mean GI50 values ± S.E.M. were calculated using GraphPad Prism version 6.0.
2.2.4. Clonogenic cell survival assays
MCF7 and MDA‐MB‐231 cells were seeded into 5 cm tissue culture plates and allowed to adhere for 24 h. Following 1 h pre‐treatment with 1 μM VE‐821 or 50 nM or 150 nM V185411 or a vehicle control of 0.1% DMSO, cells were irradiated. Pre‐treatment was to allow drugs to enter cell prior to irradiation. Since activation of checkpoints would come after IR damage the cells were co incubated for a further 24 h in the presence of drug prior to seeding for colony formation. Ionising radiation (X‐irradiation; IR) was administered using a Gulmay Medical Xstrahl RS320 X‐irradiator (Gulmay Medical, Chertsey, UK) at a rate of 3.15 Gy/min. After 24 h, cells were harvested, counted and re‐seeded at low density for colony formation in drug‐free medium. After 2 weeks, colonies were fixed using Carnoy's fixative, stained using 1% crystal violet, and counted. All experiments were performed atleast thrice. LD50 values (dose of IR where 50% of cells no longer survive) were calculated from each experiment and the potentiation factor at 50% cell kill (PF50) was calculated.
2.2.5. Cell cycle progression by FACS
Exponentially growing MCF7 or MDA‐MB‐231 were seeded into 10 cm tissue culture dishes and allowed to adhere for 24 h. Where indicated, cells were pre‐treated with 1 μM VE‐821, 50 nM V158411 or 150 nM V158411 for 1 h before IR (2 Gys) or mock treatment. After 24 h, media from cells was collected, the cells washed and harvested and fixed and frozen in ice cold methanol overnight. Cells were stained with 200 μg/ml PI and 200 μg/ml RNAase A in PBS and samples run using a FACScalibur counting a minimum of 20000 events. Single cell populations were then gated into phases of the cell cycle by DNA content (A – one represtentative example from each cell line) and the resulting profiles analysed using Cyflogic software (B – mean ± S.D. of three individual experiments).
3. Results
3.1. DNA repair and local recurrence (whole cohort)
We initially investigated local recurrence in the whole cohort irrespective of whether they received adjuvant radiotherapy or not. Within the studied patients' cohort, 208/1755 patients (11.9%) developed local recurrence. Among patients who developed local recurrence, 126/208 (61%) patients developed early local recurrence within 5‐years of primary treatment, indicating the presence of an aggressive tumour. We proceeded to investigate DNA repair expression and time to early local recurrence in this cohort. Kaplan–Meier plots for time to local recurrence for all the DNA repair markers are summarised in Supplementary Figure. S1A–S1W. Of the twenty markers investigated (Supplementary Figure S1A–S1X), only pChk1 and p53 protein levels were significantly associated with shorter time to local recurrence (1, 2).
Figure 1.
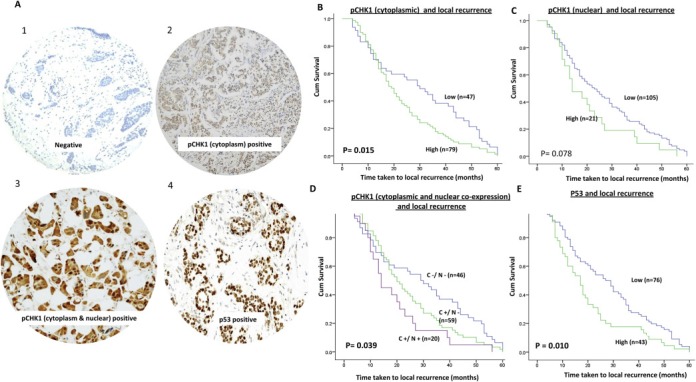
A. Microphotograph of pCHK1 and p53 protein expression in breast tumours [1 = negative, 2 = pCHK1 cytoplasmic staining, 3 = pCHK1 cytoplasmic & nuclear staining and 4 = p53 staining]. Kaplan Meier curves showing time taken to local recurrence based on B. pChk1 cytoplasmic expression; C. pChk1 nuclear expression D. pChk1 cytoplasmic & pChk1 nuclear co‐expression; D. p53 expression.
Figure 2.
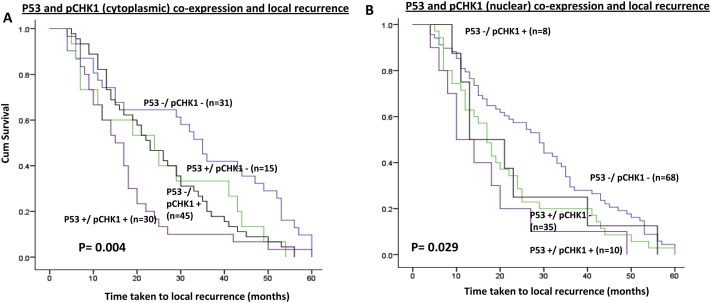
Kaplan Meier curves showing time taken to local recurrence based on A. p53 & pChk1 cytoplasmic co‐expression; B. p53 & pChk1 nuclear co‐expression.
High levels of cytoplasmic pChk1 (p = 0.015) (Figure 1B) was significantly associated with shorter time to local recurrence. However, nuclear pChk1 level was not significantly associated with local recurrence (Figure 1C). When nuclear and cytoplasmic expression of pChk1 were combined together, we observed that tumours with a high nuclear/cytoplasmic pChk1 level were more likely to have shorter time to local recurrence, compared to tumours with low nuclear/cytoplasmic pChk1 level (Figure 1D). We also investigated total non‐phosphorylated form of Chk1. Tumours exhibited only nuclear staining. There was no cytoplasmic expression. Chk1 expression did not influence time to local recurrence (Supplementary Fig. S1X).
High nuclear p53 was associated with early local recurrence compared to tumours with low p53 expression (p = 0.010) (Figure 1E). When p53 and pChk1 were combined, high p53/high cytoplasmic pChk1 level were more likely to have shorter time to local recurrence compared to tumours with low p53/low cytoplasmic pChk1 level (Figure 2A) (p = 0.004). In addition, high p53/high nuclear pChk1 level were more likely to have shorter time to local recurrence compared to tumours with low p53/low nuclear pChk1 level (Figure 2B) p = 0.029).
In multivariate analysis, high cytoplasmic pChk1 was an independent predictor of early local recurrence (Table 1).
Taken together, these data suggest that cytoplasmic pChk1 is a key predictor of early local recurrence in patients with early breast cancers. In addition, p53 status could further stratify patients into two subsets; those who have local recurrence or not. We then proceeded to investigate if p53 and pChk1 expression status also has an impact on distant metastasis and BCSS.
3.2. p53, pChk1, time to distant metastasis and survival (whole cohort)
Neither cytoplasmic pChk1 alone nor nuclear pChk1 alone influenced distant metastasis (Supplementary Figure S2A, S2C). High cytoplasmic pChk1 alone was associated with poor survival (p = 0.040) (Supplementary Figure S2B) but nuclear pChk1 alone was not associated with BCSS survival (Supplementary Figure S2D). Moreover, when combined together, cytoplasmic pChk1/nuclear pChk1 did not influence metastasis (Supplementary Figure S2E). However, BCSS was poor in patients with tumours showing high cytoplasmic pChk1 and low nuclear pChk1 expression (Supplementary Figure S2F) (p = 0.042).
High p53 was significantly associated with time to distant metastasis (p < 0.001) and BCSS (p < 0.001) (Supplementary Figure S2G, S2H). When p53 and CHK1 were combined together, we observed that tumours with high p53 and high pChk1 (nuclear as well as cytoplasmic) were significantly associated with shorter time to distant metastasis and BCSS (Supplementary Figure S2I–S2L) (p < 0.005).
Together, the data suggest that p53 and pChk1 expression result in aggressive breast cancer phenotype with adverse prognostic significance.
3.3. p53 and pChk1 predict resistance to adjuvant radiotherapy
To investigate whether p53 and pChk1 also predict resistance to adjuvant radiotherapy we proceeded to sub‐group analysis in 949 (out of 1755) patients who had received adjuvant radiotherapy. 92/949 (9.69%) had local recurrence. Tumours with high p53 was associated with early local recurrence compared to tumours with low p53 expression (p = 0.014) (Supplementary Figure S3A). When combined with pChk1, tumours with high p53/high cytoplasmic pChk1 was associated with early local recurrence (p = 0.017) (Supplementary Figure S3D). p53/pChk1 (nuclear) (Supplementary Figure S3E), pChk1 (nuclear alone) (Supplementary Figure S3B) or pChk1 (cytoplasmic alone) (Supplementary Figure S3C) alone did not influence early local recurrence.
Taken together the data provides evidence that p53 as well as p53/pChk1 (cytoplasmic) co‐expression are promising biomarkers of radiotherapy response.
3.4. Targeting the CHK1 pathway for radio‐sensitization
The clinical data presented here provides evidence that pChk1 has prognostic and predictive significance in breast cancer. Therefore Chk1 pathway targeting may be a promising radio‐sensitising approach. To address this possibility, we followed two pharmacological approaches; ATR inhibition by VE‐821 which prevents Chk1 activation (by blocking phosphorylation on serine345) (Josse et al., 2014) and direct Chk1 inhibition by V158411 which blocks auto‐phosphorylation of Chk1 on Serine296 (Bryant et al., 2014; Stokes et al., 2009).
3.4.1. VE‐821 mediated inhibition of Chk1 phosphorylation at serine345 and radio‐sensitivity
ATR is activated by single stranded (ss)‐double stranded (ds) DNA junctions generated at sites of DNA damage. Activated ATR in turn phosphorylates Chk1 at Ser 345 (Fokas et al., 2014; Marechal and Zou, 2013). VE‐821 is a highly specific small molecule inhibitor of ATR (Josse et al., 2014). We have previously shown that VE‐821 blocks phosphorylation of Chk1 at serine345 and is a biomarker of ATR inhibition (Abdel‐Fatah et al., 2015; Chen et al., 2015). We initially investigated VE‐821 treatment alone in MCF‐7 and MDA‐MB‐231 cells. VE‐821 was growth inhibitory in both breast cancer cells (Figure 3A) and this effect was greater in p53 mutant MDA‐MB‐231 cells compared to p53 wild type MCF7 cells [GI50 (concentration required to reduce 50% of growth) = 1.1 ± 0.2 μM and 2.8 ± 0.1 μM, respectively]. We then proceed to IR combination studies. As shown in Figure 3B and 3C, VE‐821 significantly potentiated IR in MCF7 cells and MDA‐MB‐231 cells. The dose of IR at which 50% of cells were killed (LD50) was interpolated in the presence and absence of 1 μM VE‐821 and the fold potentiation calculated. VE‐821 significantly potentiated IR in MCF7 cells by almost 3‐fold (2‐way ANOVA, p < 0.0001) and in MDA‐MB‐231 by almost 2‐fold (2‐way ANOVA, p = 0.0012; Figure 3D; Supplementary Table S3).
Figure 3.
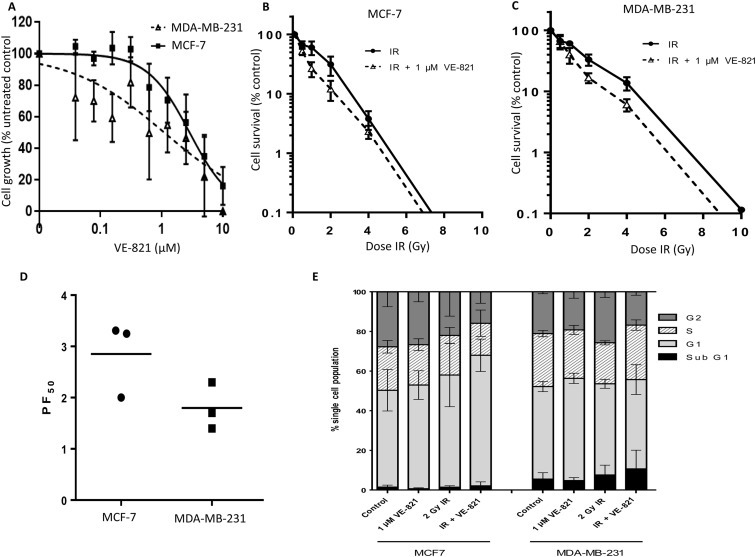
Blockade of CHK1 phosphorylation by VE‐821 (ATR inhibitor) and radio‐potentiation. A. Effect of VE‐821 on breast cancer cell growth. For clonogenic assays, data are mean ± standard deviation from three separate experiments in MCF7 (B) and MDA‐MB‐231 (C). D. LD50 values (dose of IR where 50% of cells no longer survive) were calculated from each experiment and the potentiation factor at 50% cell kill (PF50) was calculated (see Supplementary Table S3 for further details). E. Exponentially growing MCF7 or MDA‐MB‐231 were seeded into 10 cm tissue culture dishes and allowed to adhere for 24 h. Where indicated, cells were pre‐treated with 1 μM VE‐821 for 1 h before IR (2 Grays) or mock treatment. After 24 h, media from cells was collected, the cells washed and harvested and fixed and frozen in ice cold methanol overnight. Cells were stained with 200 μg/ml PI and 200 μg/ml RNAase A in PBS and samples run using a FACScalibur counting a minimum of 20000 events. Single cell populations were then gated into phases of the cell cycle by DNA content. The resulting FACS profiles analysed using Cyflogic software (mean ± S.D. of three individual experiments) is show here.
As both p53 and ATR are implicated in different cell cycle checkpoints (Gudkov and Komarova, 2003; Zhang and Hunter, 2014), radiopotentiation by VE‐821 may be linked to changes in cell cycle profiles. To establish whether any differences in radiopotentiation were due to cell cycle effects, each cell line was treated with a clinically relevant (2 Gy) dose of IR with or without VE‐821 and cell cycle profiles were assessed (Figure 3E and Supplementary Figure S4). VE‐821 caused a modest increase in the G1 population in MCF7 (8.2 ± 7.8%) and MDA‐MB‐231 cells (10.5 ± 1.0%), but reduced S‐phase arrest by approximately 8% in both lines which would be consistent with the growth inhibitory effect seen in (Figure 3C and D). This reduction in S arrest accompanied a small reduction in the G2 fraction. In contrast, and consistent with mutated p53, IR did not induce any G1 arrest in MDA‐MB‐231 cells but induced a substantial (22.1 ± 18.6%) increase in G2 arrest which was completely abrogated by VE‐821. IR did not induce G2 arrest in MCF7 cells, but VE‐821 did cause a substantial reduction in the S and G2 fractions of irradiated cells suggesting that there may be an element of S/G2 checkpoint activation by IR that was abrogated by VE‐821 in these cells. Interestingly, the combination of IR and VE‐821 in MCF7 cells caused a substantial (37.5 ± 30%) increase in G1 arrested cells which would be consistent with the induction of p53 by this combination.
Activated ATR not only phosphorylates Chk1 at Ser 345 but also several other target proteins involved in homologous recombination repair and DNA cross link repair (Fokas et al., 2014; Marechal and Zou, 2013). Therefore, to clarify whether the radio‐potentiation is contributed by CHK1 mediated events, we investigated V185411, a highly specific CHK1 inhibitor.
3.4.2. Chk1 inhibition and radio‐sensitivity
We initially attempted to generate Chk1 knockdown (KD) breast cancer cell lines (Figure 4A). Figure 4B quantifies this knockdown of Chk1 by siRNA in comparison to cell line samples treated with control scrambled siRNA. There was 80% knockdown after 48 h and more than 98% knockdown after 5 days. In parallel with the samples prepared for western blotting a clonogenic assay was performed in control cells, those treated with scrambled siRNA and with Chk1 siRNA for 48 h. This shows that there was no clonogenic survival after 2 weeks in cells treated for 48 h with Chk1 siRNA (Figure 4C). Therefore it was not possible to test radio‐sensitisation in Chk1 KD cells.
Figure 4.
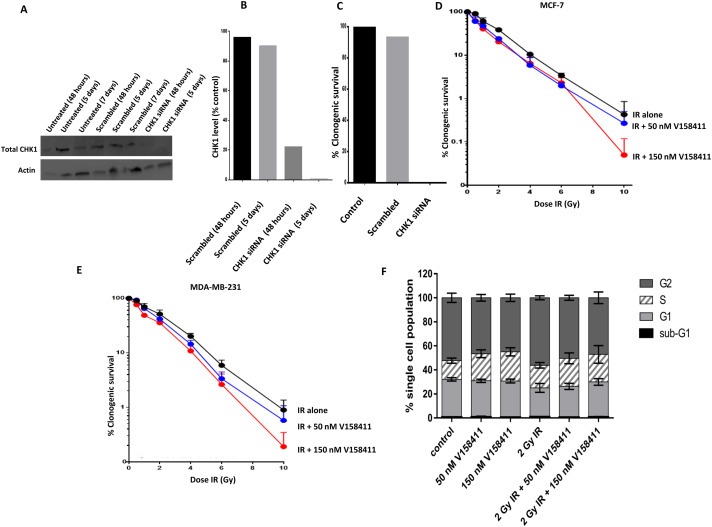
Blockade of CHK1 activation by V158411 and radio‐potentiation. A. Representative western blot in MCF7 breast cancer cell line. Expression of Chk1 and actin in untreated controls treated for 48 h, 5 days and 7 days, cells treated with scrambled siRNA for 48 h, 5 days and 7 days, and cells treated with Chk1 siRNA for 48 h, 5 days. Note no 7 day sample with Chk1 siRNA as insufficient cells alive to extract protein for western blot analysis. B. Quantification CHK1 KD by siRNA in comparison to cell line samples treated with control scrambled siRNA. There was 80% KD after 48 h and more than 98% KD after 5 days. C. Clonogenic assays. In parallel with the samples prepared for western blotting a clonogenic assay was performed in control cells, those treated with scrambled siRNA and with CHK1 siRNA for 48 h. This shows that there was no clonogenic survival after 2 weeks in cells treated for 48 h with CHK1 siRNA. D. Dose dependent radiosensitisation of V158411in MCF‐7 cells. E. Dose dependent radiosensitisation of V158411in MDA‐MB‐231 cells. For all clonogenic assays, data are mean ± standard deviation from three separate experiments in breast cancer cells. F. FACS profiles in MDA‐MB‐231 cells treated with VE158411 either alone or in combination with IR.
We proceeded to investigate V158411, a highly specific, and potent inhibitor of Chk1 [in vitro with IC50 of 3.5 nM and in vivo V158411 inhibited the etoposide induced auto‐phosphorylation of Chk1 on Serine296 with an IC50 of 48 nM] (Bryant et al., 2014; Stokes et al., 2009). As shown in Figure 4D and E, a concentration dependent potentiation of IR toxicity was seen in both MCF‐7 and MDA‐MB‐231 cells. In MCF‐7 cells, potentiation at 2 Gy was 1.6 fold and 1.9 fold with 50 nM or 150 nM V158411, respectively. In MCF‐7 cells, potentiation at 10 Gy was 1.5 fold and 6 fold with 50 nM or 150 nM V158411, respectively. In MDA‐MB‐231 cells, potentiation at 2 Gy was 1.2 fold and 1.4 fold with 50 nM or 150 nM V158411, respectively. In MDA‐MB‐231 cells, potentiation at 10 Gy was 1.6 fold and 5 fold with 50 nM or 150 nM V158411, respectively. Previous studies have shown that G1 check point does not operate in p53 mutant cells and cells are reliant upon S and G2 checkpoints mediated through Chk1 for maintaining genomic stability (Levesque et al., 2005). We therefore investigated cell cycle progression in V158411 (50 nM or 150 nM) treated MDA‐MB‐231 cells either alone or in combination with radiotherapy. As shown in Figure 4E, there was a reduction in cells in G2‐phase in VE158411 treated cells compared to untreated control cells. Whereas in IR treated cells, as expected, the cell accumulated in G2‐phase of the cell cycle. However, with IR + VE158411 combination therapy, there was a reduction in cells in G2‐phase. Taken together, the data provides evidence that VE158411 blocks Chk1 mediated G2 arrest in p53 deficient breast cancer cell.
4. Discussion
IR induces a wide variety of DNA lesions including oxidative base damage, abasic (AP) sites, deoxyribose damage, single strand breaks (SSBs) and double strand breaks (DSBs). An additional feature of IR damage is the introduction of clustered DNA lesions through reactive species generation (Kavanagh et al., 2013; Santivasi and Xia, 2014; Willers et al., 2004). These DNA lesions would require initiation of multiple signal transduction cascades that will dictate the final fate of a cell; whether to tolerate DNA damage, initiate DNA repair or undergo apoptosis (Kavanagh et al., 2013; Santivasi and Xia, 2014; Willers et al., 2004). In the current study, we have comprehensively investigated DNA damage signalling and repair in a cohort of invasive breast cancer patients who developed early local recurrence which is a feature of radio‐resistant breast cancers. We have observed that p53 and pChk1 are key determinants of radio‐sensitivity in breast cancer patients receiving adjuvant radiotherapy. Interestingly, we did not observe any significant associations with other key DNA repair markers including ATM, ATR, total Chk1, Chk2, PARP1, POLβ, XRCC1, FEN1, SMUG1, Ku70/Ku80, DNA‐PKcs, RAD51, BRCA1, γH2AX, BLM, WRN, RECQL5 and PTEN.
The tumour suppressor p53 is centrally involved in the coordination of cell cycle regulation, DNA repair and apoptosis in response to IR (Fei and El‐Deiry, 2003; Gudkov and Komarova, 2003). p53 is typically involved in IR‐induced G1 arrest and may also have roles in S and G2/M phases of the cell cycle (Fei and El‐Deiry, 2003; Gudkov and Komarova, 2003). P53 mediated apoptosis is a central determinant of radio‐sensitivity (Fei and El‐Deiry, 2003; Gudkov and Komarova, 2003). In addition, p53 is involved in IR‐induced growth arrest. Mutation in p53 is seen in up to 30% of all breast cancers and is enriched in certain phenotypes including basal‐like (88%), HER‐2 amplified (50%) and luminal B (41%) (Bertheau et al., 2013). In the luminal A sub‐type, the incidence of p53 mutation is low (17%) (Bertheau et al., 2013). Existing pre‐clinical evidence confirm that mutation in p53 is associated with resistance to radiotherapy (Fei and El‐Deiry, 2003; Gudkov and Komarova, 2003). As expected, we observed that p53 accumulation by IHC, a feature of mutant p53, was significantly associated with early local recurrence. Taken together, this provides clinical evidence that p53 is a marker of radio‐resistance in breast cancer.
Chk1 is a serine threonine protein kinase with critical roles in genomic stability (Bartek and Lukas, 2003; Enders, 2008; Zhang and Hunter, 2014). Under normal conditions, Chk1 is expressed predominantly in the nucleus. Recent evidence suggests that the sub‐cellular localisation of CHK1 is tightly regulated (Wang et al., 2012a). A key activator of Chk1 is ATR which phosphorylates CHK1 at Serine345 and Serine317 (Cimprich and Cortez, 2008; Nam and Cortez, 2011; Smits et al., 2010). Phosphorylated Chk1 is released from the chromatin‐enriched fractions into the soluble nucleus and then to the cytoplasm. In addition, phosphorylation of Chk1 at Serine345 (pChk1) leads to its activation which not only promotes further autophosphorylation at Serine296, but also results in phosphorylation and inactivation of Cdc25A and Cdc25C. Whereas Cdc25A activates S‐phase progression, Cdc25C regulates mitotic entry through CDK1 (Cdc2) (Cimprich and Cortez, 2008; Nam and Cortez, 2011; Smits et al., 2010). In addition, pChk1 also targets many other proteins involved in cell cycle regulation and DNA repair. For example, phosphorylated Chk1 can activate Rad51‐dependent DNA damage repair by the high‐fidelity homologous recombination pathway in the nucleus and inhibit centrosomal cyclin B/Cdk1 activity and stabilize the mRNA in the cytoplasm (Bartek and Lukas, 2003; Enders, 2008; Zhang and Hunter, 2014). Chk1 is an important factor in determining radio‐sensitivity and Chk1 knockdown results in radio‐sensitisation (Wang et al., 2012b). In addition, Chk1 inhibition by small molecule inhibitors potentiates cytotoxicity of radiotherapy in pancreatic and nasopharyngeal cancer xenografts (Feng et al., 2010; Morgan et al., 2010). In the current study, we demonstrate that high levels of pChk1; a marker of functional ATR‐CHK1 pathway (Abdel‐Fatah et al., 2015; Chen et al., 2015), was significantly associated with shorter time to local recurrence both in univariate and multivariate analyses. Variations in the levels of pChk1 could not only relate to differences in the activity of ATR but could also potentially reflect variations in the expression of total Chk1 bearing a basal level of serine345 phosphorylation. We therefore also investigated total Chk1 expression but observed no significant associations with time to local recurrence. Taken together the data suggest that that pChk1 is a promising predictive biomarker. Emerging data suggests that p53 and CHK1 link with each other. CHK1 can phosphorylate p53 and p53 can down‐regulate CHK1 expression (Gottifredi et al., 2001; Ou et al., 2005). Accordingly, in the current study we have shown that patients with tumours expressing high levels of pChk1 and p53 were significantly more at risk of local recurrence compared to those with low levels of pChk1 and p53. This clinical data provides evidence that P53–CHK1 expression status could be utilised for radiotherapy stratification. In addition, we have also shown that high levels of pChk1 and p53 was also associated with shorter time to distant metastasis and BCSS implying an aggressive biological phenotype. CHK1 targeting in p53 mutant cancer has recently emerged as a promising strategy in breast cancer (Ma et al., 2012a). As the G1 check point does not operate in p53 mutant tumours, cells are reliant upon S and G2 checkpoints mediated through CHK1 for maintaining genomic stability. When S and G2 checkpoints are blocked by pharmacological inhibition of CHK1, p53 mutant cells undergo mitotic catastrophe and apoptosis. This targeted approach was demonstrated recently in p53 mutant triple negative breast cancer xenografts treated with CHK1 inhibitor in combination with irinotecan chemotherapy (Ma et al., 2012a). CHK1 inhibitors potentiated apoptosis and abrogated cell‐cycle arrest in irinotecan treated p53 mutant breast cancer xenografts and improved mice survival in that study (Ma et al., 2012a). We have previously demonstrated that CHK1 mRNA overexpression is significantly linked to basal and HER2+ phenotype (Abdel‐Fatah et al., 2015). In addition, high level of cytoplasmic pChk1 was also associated with basal phenotype in that study (Abdel‐Fatah et al., 2015). The data in its entirety provides evidence that CHK1 and p53 are important predictive biomarkers of early local recurrence, reflecting breast cancer radio‐resistance. However, a limitation to the current clinical study is that it is retrospective. Patient in this historical cohort received CMF chemotherapy and radiation boost was not routinely delivered to most patients. A prospective study in the context of modern adjuvant chemotherapy and radiotherapy is warranted to confirm our clinical observations.
To provide additional preclinical evidence, we investigated the ability of ATR‐CHK1 pathway modulation for radio‐sensitisation. First, ATR inhibition that prevents CHK1 activation lead to increased radio‐sensitivity. Unexpectedly this was more evident in p53 wild‐type MCF‐7 cells compared to p53 mutant MDA‐MB‐231 cells but consistent with previous observations in leukemia cell lines (Salovska et al., 2014; Vavrova et al., 2013). Second, we observed radio‐sensitisation using VE158411, a CHK1 inhibitor. A previous study also reported radiosensitisation in p53 mutant breast cancer cells using AZD7762, another CHK1 inhibitor (Ma et al., 2012b). Although radio‐potentiation was more evident in MCF‐7 cells, the extent of radio‐sensitisation by CHK1 inhibitor was comparatively less compared to ATR inhibitor. ATR inhibition besides CHK1 activation could also block other HR related events, contributing to additional IR toxicity. Taken together, these data provide evidence that ATR‐CHK1 blockade is a promising radio‐sensitisation strategy.
In conclusion, we have provided evidence that CHK1 and p53 are predictive biomarker of radiotherapy resistance and early local recurrence in breast cancer. ATR‐CHK1 pathway targeting is a promising personalised radiotherapy strategy.
Conflict of interest
The authors disclose no potential conflicts of interest.
Supporting information
The following are the supplementary data related to this article:
Supplementary Figure S1. Kaplan Meier curves showing time taken to local recurrence based on A. ATM (nuclear), B. ATR (nuclear), C. CHK2 (nuclear), D. RAD51 (nuclear), E. RAD51 (cytoplasmic), F. BRCA1 (nuclear), G. γH2AX (nuclear), H. γH2AX, I. DNA‐PKcs (nuclear), J. Ku70/Ku80 (nuclear), K. RECQL5 (nuclear), L. WRN (nuclear), M. WRN (cytoplasmic), N. BLM (nuclear), O. BLM (cytoplasmic), P. PTEN (nuclear), Q. PTEN, R. SMUG1 (nuclear), S. XRCC1 (nuclear), T. PARP1 (nuclear), U. FEN1 (nuclear), V. FEN1 (cytoplasmic) W. Pol β (nuclear) and X. total Chk1 expression.
Supplementary Figure S2. A. Kaplan Meier curves showing time taken to metastasis based on p CHK1 cytoplasmic expression. B. Kaplan Meier curves showing BCSS based on pCHK1 cytoplasmic expression. C. Kaplan Meier curves showing time taken to metastasis based on pCHK1 nuclear expression. D. Kaplan Meier curves showing BCSS based on pCHK1 nuclear expression. E. Kaplan Meier curves showing time taken to metastasis based on pCHK1 cytoplasmic & nuclear co‐expression. F. Kaplan Meier curves showing BCSS based on pCHK1 cytoplasmic & nuclear co‐expression. G. Kaplan Meier curves showing time taken to metastasis based on p53 expression. H. Kaplan Meier curves showing BCSS based on p53 & pCHK1 cytoplasmic co‐expression. I. Kaplan Meier curves showing time taken to metastasis based on p53 & pCHK1 cytoplasmic co‐expression. J. Kaplan Meier curves showing BCSS based on p53 & pCHK1 cytoplasmic co‐expression. K. Kaplan Meier curves showing time taken to metastasis based on p53 & pCHK1 nuclear co‐expression. L. Kaplan Meier curves showing BCSS based on p53 & pCHK1 nuclear co‐expression.
Supplementary Figure S3. Kaplan Meier curves showing time taken to local recurrence in RT treated cohort based on A. p53 nuclear expression. B. pCHK1 cytoplasmic expression. C. pCHK1 nuclear expression. D. p53‐pCHK1 cytoplasmic co‐expression. E. p53‐pCHK1 nuclear co‐expression.
Supplementary Figure S4 Cell‐cycle analysis. Exponentially growing MCF7 or MDA‐MB‐231 were seeded into 10 cm tissue culture dishes and allowed to adhere for 24 h. Where indicated, cells were pre‐treated with 1 μM VE‐821 for 1 h before IR (2 Grays) or mock treatment. After 24 h, media from cells was collected, the cells washed and harvested and fixed and frozen in ice cold methanol overnight. Cells were stained with 200 μg/ml PI and 200 μg/ml RNAase A in PBS and samples run using a FACScalibur counting a minimum of 20000 events. Single cell populations were then gated into phases of the cell cycle by DNA content. One represtentative example from each cell line is shown here.
Supplementary data
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.09.009.
Alsubhi Nouf, Middleton Fiona, Abdel-Fatah Tarek M.A., Stephens Peter, Doherty Rachel, Arora Arvind, Moseley Paul M., Chan Stephen Y.T., Aleskandarany Mohammed A., Green Andrew R., Rakha Emad A., Ellis Ian O., Martin Stewart G., Curtin Nicola J., Madhusudan Srinivasan, (2016), Chk1 phosphorylated at serine345 is a predictor of early local recurrence and radio‐resistance in breast cancer, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.09.009.
Contributor Information
Nicola J. Curtin, Email: nicola.curtin@ncl.ac.uk
Srinivasan Madhusudan, Email: srinivasan.madhusudan@nottingham.ac.uk.
References
- Abdel-Fatah, T.M. , Arora, A. , Alsubhi, N. , Agarwal, D. , Moseley, P.M. , Perry, C. , Doherty, R. , Chan, S.Y. , Green, A.R. , Rakha, E. , Ball, G. , Ellis, I.O. , Madhusudan, S. , 2014. Clinicopathological significance of ATM-Chk2 expression in sporadic breast cancers: a comprehensive analysis in large cohorts. Neoplasia. 16, 982–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Fatah, T.M. , Middleton, F.K. , Arora, A. , Agarwal, D. , Chen, T. , Moseley, P.M. , Perry, C. , Doherty, R. , Chan, S. , Green, A.R. , Rakha, E. , Ball, G. , Ellis, I.O. , Curtin, N.J. , Madhusudan, S. , 2015. Untangling the ATR-CHEK1 network for prognostication, prediction and therapeutic target validation in breast cancer. Mol. Oncol. 9, 569–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albarakati, N. , Abdel-Fatah, T.M. , Doherty, R. , Russell, R. , Agarwal, D. , Moseley, P. , Perry, C. , Arora, A. , Alsubhi, N. , Seedhouse, C. , Rakha, E.A. , Green, A. , Ball, G. , Chan, S. , Caldas, C. , Ellis, I.O. , Madhusudan, S. , 2015. Targeting BRCA1-BER deficient breast cancer by ATM or DNA-PKcs blockade either alone or in combination with cisplatin for personalized therapy. Mol. Oncol. 9, 204–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora, A. , Abdel-Fatah, T.M. , Agarwal, D. , Doherty, R. , Moseley, P.M. , Aleskandarany, M.A. , Green, A.R. , Ball, G. , Alshareeda, A.T. , Rakha, E.A. , Chan, S.Y. , Ellis, I.O. , Madhusudan, S. , 2015. Transcriptomic and protein expression analysis reveals clinicopathological significance of bloom syndrome helicase (BLM) in breast Cancer. Mol. Cancer Ther. 14, 1057–1065. [DOI] [PubMed] [Google Scholar]
- Bartek, J. , Lukas, J. , 2003. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 3, 421–429. [DOI] [PubMed] [Google Scholar]
- Bertheau, P. , Lehmann-Che, J. , Varna, M. , Dumay, A. , Poirot, B. , Porcher, R. , Turpin, E. , Plassa, L.F. , de Roquancourt, A. , Bourstyn, E. , de Cremoux, P. , Janin, A. , Giacchetti, S. , Espie, M. , de The, H. , 2013. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast. 22, (Suppl. 2) S27–S29. [DOI] [PubMed] [Google Scholar]
- Bryant, C. , Rawlinson, R. , Massey, A.J. , 2014. Chk1 inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC Cancer. 14, 570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T. , Middleton, F.K. , Falcon, S. , Reaper, P.M. , Pollard, J.R. , Curtin, N.J. , 2015. Development of pharmacodynamic biomarkers for ATR inhibitors. Mol. Oncol. 9, 463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich, K.A. , Cortez, D. , 2008. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, M. , Collins, R. , Darby, S. , Davies, C. , Elphinstone, P. , Evans, E. , Godwin, J. , Gray, R. , Hicks, C. , James, S. , MacKinnon, E. , McGale, P. , McHugh, T. , Peto, R. , Taylor, C. , Wang, Y. , 2005. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet. 366, 2087–2106. [DOI] [PubMed] [Google Scholar]
- della Rovere, G.Q. , Benson, J.R. , 2002. Ipsilateral local recurrence of breast cancer: determinant or indicator of poor prognosis?. Lancet Oncol. 3, 183–187. [DOI] [PubMed] [Google Scholar]
- Derks, K.W. , Hoeijmakers, J.H. , Pothof, J. , 2014. The DNA damage response: the omics era and its impact. DNA Repair (Amst). 19, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enders, G.H. , 2008. Expanded roles for Chk1 in genome maintenance. J. Biol. Chem. 283, 17749–17752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei, P. , El-Deiry, W.S. , 2003. P53 and radiation responses. Oncogene. 22, 5774–5783. [DOI] [PubMed] [Google Scholar]
- Feng, Z. , Xu, S. , Liu, M. , Zeng, Y.X. , Kang, T. , 2010. Chk1 inhibitor Go6976 enhances the sensitivity of nasopharyngeal carcinoma cells to radiotherapy and chemotherapy in vitro and in vivo. Cancer Lett. 297, 190–197. [DOI] [PubMed] [Google Scholar]
- Fokas, E. , Prevo, R. , Hammond, E.M. , Brunner, T.B. , McKenna, W.G. , Muschel, R.J. , 2014. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat Rev. 40, 109–117. [DOI] [PubMed] [Google Scholar]
- Gieni, M. , Avram, R. , Dickson, L. , Farrokhyar, F. , Lovrics, P. , Faidi, S. , Sne, N. , 2012. Local breast cancer recurrence after mastectomy and immediate breast reconstruction for invasive cancer: a meta-analysis. Breast. 21, 230–236. [DOI] [PubMed] [Google Scholar]
- Gottifredi, V. , Karni-Schmidt, O. , Shieh, S.S. , Prives, C. , 2001. p53 down-regulates CHK1 through p21 and the retinoblastoma protein. Mol. Cell Biol. 21, 1066–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudkov, A.V. , Komarova, E.A. , 2003. The role of p53 in determining sensitivity to radiotherapy. Nat. Rev. Cancer. 3, 117–129. [DOI] [PubMed] [Google Scholar]
- Josse, R. , Martin, S.E. , Guha, R. , Ormanoglu, P. , Pfister, T.D. , Reaper, P.M. , Barnes, C.S. , Jones, J. , Charlton, P. , Pollard, J.R. , Morris, J. , Doroshow, J.H. , Pommier, Y. , 2014. ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res. 74, 6968–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh, J.N. , Redmond, K.M. , Schettino, G. , Prise, K.M. , 2013. DNA double strand break repair: a radiation perspective. Antioxid. Redox Signal. 18, 2458–2472. [DOI] [PubMed] [Google Scholar]
- Levesque, A.A. , Kohn, E.A. , Bresnick, E. , Eastman, A. , 2005. Distinct roles for p53 transactivation and repression in preventing UCN-01-mediated abrogation of DNA damage-induced arrest at S and G2 cell cycle checkpoints. Oncogene. 24, 3786–3796. [DOI] [PubMed] [Google Scholar]
- Liu, T. , Huang, J. , 2014. Quality control of homologous recombination. Cell Mol Life Sci. 71, 3779–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, C.X. , Cai, S. , Li, S. , Ryan, C.E. , Guo, Z. , Schaiff, W.T. , Lin, L. , Hoog, J. , Goiffon, R.J. , Prat, A. , Aft, R.L. , Ellis, M.J. , Piwnica-Worms, H. , 2012. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J. Clin. Invest. 122, 1541–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Z. , Yao, G. , Zhou, B. , Fan, Y. , Gao, S. , Feng, X. , 2012. The Chk1 inhibitor AZD7762 sensitises p53 mutant breast cancer cells to radiation in vitro and in vivo. Mol. Med. Rep. 6, 897–903. [DOI] [PubMed] [Google Scholar]
- Marechal, A. , Zou, L. , 2013. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 5, [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane, L.M. , Altman, D.G. , Sauerbrei, W. , Taube, S.E. , Gion, M. , Clark, G.M. , 2005. Reporting recommendations for tumor marker prognostic studies (REMARK). J. Natl. Cancer Inst. 97, 1180–1184. [DOI] [PubMed] [Google Scholar]
- Morgan, M.A. , Parsels, L.A. , Zhao, L. , Parsels, J.D. , Davis, M.A. , Hassan, M.C. , Arumugarajah, S. , Hylander-Gans, L. , Morosini, D. , Simeone, D.M. , Canman, C.E. , Normolle, D.P. , Zabludoff, S.D. , Maybaum, J. , Lawrence, T.S. , 2010. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 70, 4972–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam, E.A. , Cortez, D. , 2011. ATR signalling: more than meeting at the fork. Biochem. J. 436, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, Y.H. , Chung, P.H. , Sun, T.P. , Shieh, S.Y. , 2005. p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Mol. Biol. Cell. 16, 1684–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salovska, B. , Fabrik, I. , Durisova, K. , Link, M. , Vavrova, J. , Rezacova, M. , Tichy, A. , 2014. Radiosensitization of human leukemic HL-60 cells by ATR kinase inhibitor (VE-821): phosphoproteomic analysis. Int. J. Mol. Sci. 15, 12007–12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santivasi, W.L. , Xia, F. , 2014. Ionizing radiation-induced DNA damage, response, and repair. Antioxid. Redox Signal. 21, 251–259. [DOI] [PubMed] [Google Scholar]
- Schnitt, S.J. , 2003. Risk factors for local recurrence in patients with invasive breast cancer and negative surgical margins of excision. Where are we and where are we going?. Am. J. Clin. Pathol. 120, 485–488. [DOI] [PubMed] [Google Scholar]
- Smits, V.A. , Warmerdam, D.O. , Martin, Y. , Freire, R. , 2010. Mechanisms of ATR-mediated checkpoint signalling. Front Biosci. (Landmark Ed.). 15, 840–853. [DOI] [PubMed] [Google Scholar]
- Stokes, S. , Nicolas, F. , Fiumana, A. , Drysdale, M. , Bedford, S. , Webb, P. , 2009. Indolyl- pyridone derivatives having checkpoint kinase 1 inhibitory activity. World Intellect. Property Organ. [WO/2009/093012] [Google Scholar]
- Tian, H. , Gao, Z. , Li, H. , Zhang, B. , Wang, G. , Zhang, Q. , Pei, D. , Zheng, J. , 2015. DNA damage response–a double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 358, 8–16. [DOI] [PubMed] [Google Scholar]
- van der Leij, F. , Elkhuizen, P.H. , Bartelink, H. , van de Vijver, M.J. , 2012. Predictive factors for local recurrence in breast cancer. Semin. Radiat. Oncol. 22, 100–107. [DOI] [PubMed] [Google Scholar]
- Vavrova, J. , Zarybnicka, L. , Lukasova, E. , Rezacova, M. , Novotna, E. , Sinkorova, Z. , Tichy, A. , Pejchal, J. , Durisova, K. , 2013. Inhibition of ATR kinase with the selective inhibitor VE-821 results in radiosensitization of cells of promyelocytic leukaemia (HL-60). Radiat. Environ. Biophys. 52, 471–479. [DOI] [PubMed] [Google Scholar]
- Wallace, S.S. , 2014. Base excision repair: a critical player in many games. DNA Repair (Amst). 19, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Han, X. , Feng, X. , Wang, Z. , Zhang, Y. , 2012. Coupling cellular localization and function of checkpoint kinase 1 (Chk1) in checkpoints and cell viability. J. Biol. Chem. 287, 25501–25509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Ma, Z. , Xiao, Z. , Liu, H. , Dou, Z. , Feng, X. , Shi, H. , 2012. Chk1 knockdown confers radiosensitization in prostate cancer stem cells. Oncol. Rep. 28, 2247–2254. [DOI] [PubMed] [Google Scholar]
- Willers, H. , Dahm-Daphi, J. , Powell, S.N. , 2004. Repair of radiation damage to DNA. Br. J. Cancer. 90, 1297–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, G.J. , Hammel, M. , Radhakrishnan, S.K. , Ramsden, D. , Lees-Miller, S.P. , Tainer, J.A. , 2014. Structural insights into NHEJ: building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair (Amst). 17, 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Hunter, T. , 2014. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer. 134, 1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary Figure S1. Kaplan Meier curves showing time taken to local recurrence based on A. ATM (nuclear), B. ATR (nuclear), C. CHK2 (nuclear), D. RAD51 (nuclear), E. RAD51 (cytoplasmic), F. BRCA1 (nuclear), G. γH2AX (nuclear), H. γH2AX, I. DNA‐PKcs (nuclear), J. Ku70/Ku80 (nuclear), K. RECQL5 (nuclear), L. WRN (nuclear), M. WRN (cytoplasmic), N. BLM (nuclear), O. BLM (cytoplasmic), P. PTEN (nuclear), Q. PTEN, R. SMUG1 (nuclear), S. XRCC1 (nuclear), T. PARP1 (nuclear), U. FEN1 (nuclear), V. FEN1 (cytoplasmic) W. Pol β (nuclear) and X. total Chk1 expression.
Supplementary Figure S2. A. Kaplan Meier curves showing time taken to metastasis based on p CHK1 cytoplasmic expression. B. Kaplan Meier curves showing BCSS based on pCHK1 cytoplasmic expression. C. Kaplan Meier curves showing time taken to metastasis based on pCHK1 nuclear expression. D. Kaplan Meier curves showing BCSS based on pCHK1 nuclear expression. E. Kaplan Meier curves showing time taken to metastasis based on pCHK1 cytoplasmic & nuclear co‐expression. F. Kaplan Meier curves showing BCSS based on pCHK1 cytoplasmic & nuclear co‐expression. G. Kaplan Meier curves showing time taken to metastasis based on p53 expression. H. Kaplan Meier curves showing BCSS based on p53 & pCHK1 cytoplasmic co‐expression. I. Kaplan Meier curves showing time taken to metastasis based on p53 & pCHK1 cytoplasmic co‐expression. J. Kaplan Meier curves showing BCSS based on p53 & pCHK1 cytoplasmic co‐expression. K. Kaplan Meier curves showing time taken to metastasis based on p53 & pCHK1 nuclear co‐expression. L. Kaplan Meier curves showing BCSS based on p53 & pCHK1 nuclear co‐expression.
Supplementary Figure S3. Kaplan Meier curves showing time taken to local recurrence in RT treated cohort based on A. p53 nuclear expression. B. pCHK1 cytoplasmic expression. C. pCHK1 nuclear expression. D. p53‐pCHK1 cytoplasmic co‐expression. E. p53‐pCHK1 nuclear co‐expression.
Supplementary Figure S4 Cell‐cycle analysis. Exponentially growing MCF7 or MDA‐MB‐231 were seeded into 10 cm tissue culture dishes and allowed to adhere for 24 h. Where indicated, cells were pre‐treated with 1 μM VE‐821 for 1 h before IR (2 Grays) or mock treatment. After 24 h, media from cells was collected, the cells washed and harvested and fixed and frozen in ice cold methanol overnight. Cells were stained with 200 μg/ml PI and 200 μg/ml RNAase A in PBS and samples run using a FACScalibur counting a minimum of 20000 events. Single cell populations were then gated into phases of the cell cycle by DNA content. One represtentative example from each cell line is shown here.
Supplementary data