

Abstract
CBP (CREB‐binding protein) is a transcriptional co‐activator which possesses HAT (histone acetyltransferases) activity and participates in many biological processes, including embryonic development, growth control and homeostasis. However, its roles and the underlying mechanisms in the regulation of carcinogenesis and tumor development remain largely unknown. Here we investigated the molecular mechanisms and potential targets of CBP involved in tumor growth and survival in lung cancer cells. Elevated expression of CBP was detected in lung cancer cells and tumor tissues compared to the normal lung cells and tissues. Knockdown of CBP by siRNA or inhibition of its HAT activity using specific chemical inhibitor effectively suppressed cell proliferation, migration and colony formation and induced apoptosis in lung cancer cells by inhibiting MAPK and activating cytochrome C/caspase‐dependent signaling pathways. Co‐immunoprecipitation and immunofluorescence analyses revealed the co‐localization and interaction between CBP and CPSF4 (cleavage and polyadenylation specific factor 4) proteins in lung cancer cells. Knockdown of CPSF4 inhibited hTERT transcription and cell growth induced by CBP, and vice versa, demonstrating the synergetic effect of CBP and CPSF4 in the regulation of lung cancer cell growth and survival. Moreover, we found that high expression of both CBP and CPSF4 predicted a poor prognosis in the patients with lung adenocarcinomas. Collectively, our results indicate that CBP regulates lung cancer growth by targeting MAPK and CPSF4 signaling pathways.
Keywords: CBP, CPSF4, hTERT, Lung cancer
Highlights
Knockdown of CBP or inhibition of its HAT activity inhibits lung cancer cell growth and induces apoptosis.
Knockdown of CBP or inhibition of its HAT activity inactivates MAPK signaling pathway.
CBP interacts with and acetylates CPSF4 to promote hTERT expression and tumor growth in lung cancer cells.
Overexpression of both CBP and CPSF4 predicted poor prognosis of the patients with lung adenocarcinomas.
1. Introduction
Lung cancer, a malignant lung tumor with uncontrolled cell growth in lung tissue, remains the most frequent solid tumor worldwide and also a leading cause of cancer‐related mortality in men and women (Allemani et al., 2015; Siegel et al., 2014). Although surgery, chemotherapy, and radiotherapy are applied as common treatments, the average survival time from the time of diagnosis is still short for patients with lung cancer, usually measured in months, and the outcomes are even worse in the developing countries (Provencio and Sanchez, 2014; Slavik et al., 2014). Lung carcinogenesis and development is a multistep process, involving genetic mutations, epigenetic changes, abnormal events of stem cells, and activation of signaling pathways associated with metastasis that accumulate to initiate and worsen this disease (Kratz et al., 2010; Liu et al., 2015; Lundin and Driscoll, 2013; Mitsudomi, 2014; Van Breda et al., 2014; Wang et al., 2013b; Yang and Qi, 2012; Zajkowicz et al., 2015). Such complexity and variation in real time reversely limits therapeutic options, weakens treatment effects, and leads to poor prognosis for patients with this tumor. Therefore, the uncovering of the accurate molecular mechanisms and the further identification of new candidate therapeutic targets are urgently required to improve lung cancer treatment.
The current research focusing on the identification and development of new anti‐tumor drugs is to explore and reveal the particular characteristics or hallmarks involved in cancer development. CBP, a CREB‐binding protein, has been reported to be participated in many biological processes, including embryonic development, growth control, and homeostasis (Goodman and Smolik, 2000; Liu et al., 2014; Stachowiak et al., 2015; Turnell and Mymryk, 2006; Valor et al., 2013). It shares regions of very high‐sequence similarity with protein p300 and is involved in the transcriptional coactivation of many different transcriptional factors by interacting with them and increase the expression of their target genes (Gray et al., 2005; Jansma et al., 2014; Jia et al., 2014; Kasper et al., 2006; Lin et al., 2014; Vo and Goodman, 2001; Wang et al., 2013a; Xiao et al., 2015). Meanwhile, as a histone acetyltransferase, CBP is also involved in gene transactivation or repression by mediating the acetylation of both histone and non‐histone proteins (Cai et al., 2014; Cazzalini et al., 2014; Chen et al., 2014a; Dancy and Cole, 2015; Ferrari et al., 2014; Jin et al., 2011; Kim et al., 2012; Tie et al., 2009). Together with p300, gene mutation or chromosomal translocation within CBP gene or its aberrant recruitment at chromatin structure has been identified to be associated with several types of cancer, including tumors arising from colon and rectum, stomach, breast, pancreas cancers, ovarian and acute myeloid leukemia (Mullighan et al., 2011; Pasqualucci et al., 2011). Moreover, the inhibition of histone acetyltransferase activity of CBP/p300 or the inhibition of CBP's activity as transcriptional co‐activator have been found to be able to block cancer cell growth in vitro and in vivo in neuroblastoma, pancreatic cancer, acute myeloid leukemia (Arensman et al., 2014; Gajer et al., 2015; Giotopoulos et al., 2015). In lung cancer, the patients with CBP‐positive expression had shown significantly lower OS (Overall Survival) and DFS (Disease Free Survival) than those with CBP‐negative tumors (Gao et al., 2014). Furthermore, the high expression of CBP was found in different lung carcinoma cell lines and was positively correlated with the expression of hTERT in lung tumor cells and tissues (Guo et al., 2014). Nevertheless, the accurate and in‐depth molecular mechanisms of CBP on lung carcinoma remain not fully understood.
CPSF4, cleavage and polyadenylation specificity factor subunit 4, is known to be an essential component responsible for the 3′ end processing of cellular pre‐mRNAs (Barabino et al., 1997). When cells were infected by influenza virus, the virus NS1 protein was physically associated with CPSF4 to prevent its binding to the RNA substrate and further inhibited the nuclear export of cellular mRNAs (Nemeroff et al., 1998). However, its other biological functions have been rarely reported and even nearly unknown besides mediating the maturation of pre‐mRNA. We previously have shown that CPSF4 was overexpressed in lung adenocarcinomas and was participated in lung cancer cell growth (Chen et al., 2013). In addition, we demonstrated its new function as transcriptional factor in activating hTERT in lung cancer cells (Chen et al., 2014b). These evidences, together with the reported correlation between CBP and hTERT in lung tumor and the important role of hTERT in carcinogenesis and development, prompted us to test the possibly potential relationship between CBP and CPSF4, and their possible synergistic regulation on hTERT expression and lung cancer survival.
In this study, we investigated the exact functions and molecular mechanisms of CBP involved in lung adenocarcinoma growth and provided the direct evidence from both in vitro experiments and clinical data analyses that CBP actually functionalized as oncoprotein to promote lung cancer progression by modulating MAPK and cytochrome C/caspase signaling pathways. More interestingly, we found the cooperation between CBP and CPSF4 and their synergistic regulation on lung cancer cell survival, indicating this promoting role of CBP in lung cancer was at least partially realized through synergistic interaction with CPSF4.
2. Materials and methods
2.1. Cell lines and culture
All cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Normal human bronchial epithelial cell line (HBE) and human lung fibroblast cell line (HLF) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Human lung cancer cell lines (H1299, A549, H322, H460) were cultured in RPMI 1640 medium containing 10% fetal calf serum (FCS). All the cells were maintained in a humidified atmosphere with 5% CO2 at 37 °C.
2.2. Plasmid vector
A fragment of the hTERT promoter (−459 to +9) was amplified by PCR and inserted into the SacI and SmaI sites of the luciferase reporter vector pGL3‐Basic (Promega Corp., Madison, WI) to generate the hTERT promoter luciferase plasmid pGL3‐hTERT‐400 (Deng et al., 2007). The CPSF4 overexpression vectors pcDNA3.1‐CPSF4, the CBP overexpression vector pcDNA3.1‐CBP or control vector pcDNA3.1‐Lac Z plasmids were designed and synthesized by Cyagen (Cyagen Biosciences Inc., United States).
2.3. Immunoblotting
Proteins from cell and tissue lysate were separated by 10% SDS‐PAGE, transferred to Polyvinylidene Fluoride membrane, and immunoblotted respectively with antibodies against CBP(CST), CPSF4(proteintech), hTERT (Millipore), GAPDH (proteintech), beta‐Actin (proteintech), Bcl‐2 (proteintech), cleaved‐Caspase3(CST), cleaved‐PARP(CST), P38(CST), p‐P38(CST), Erk(CST), p‐Erk(CST), p‐Mek (CST), p‐C‐Raf(CST), and pan‐Acetylation (SANTA CRUZ). Immunoreactive protein bands were detected using ECL (Electro‐Chemi‐Luminescence) substrates.
2.4. MTT assay
Cell viability was determined using an MTT Reagent. Briefly, the cells plated in 96‐well plates (5000 cells/well) were treated with the designed protocol. 48 h after treatment, MTT was added to the cells with continuous culture for another 4 h. Then the absorbance value at OD490 was detected.
2.5. Wound scratch assay
Cells were plated in a 6‐well plate and grown to nearly 70–80% confluence. Then the cells were treated with plasmids, or siRNA, or inhibitor for 24 h and scraped in a straight line to create a “scratch”. The images of the cells at the beginning and at regular intervals during cell migration to close the scratch were captured and compared through quantifying the migration rate of the cells.
2.6. Colony formation assay
H1299 and H322 cells were plated in 6‐well plates overnight and treated with C646 for 24 h. The cells were then trypsinized into single cells and were seeded into a 6‐well plate at 1000 cells/well with continuous incubation at 5% CO2 at 37 °C for 14 days. The cells were washed with PBS and fixed with the mixture (methanol:glacial:acetic 1:1:8) for 10 min, and stained with 0.1% crystal violet for 30 min. The clones with more than 50 cells were counted under an optical microscope.
2.7. Apoptosis assay
Detection of cell apoptosis was based on FACS analysis by FITC‐AV/PI staining. The cells were grown in 6‐well plates and then cells were transfected with CBP‐specific siRNA and negative control siRNA alone or treated with C646 or DMSO alone or co‐transfected with Lac Z plasmids, or CBP plasmids, or CBP plasmids and CPSF4‐specific siRNA or CBP plasmids and negative control siRNA for 48 h. Then the cells were trypsinized, washed twice with cold PBS and centrifuged. The cell pellet was resuspended in 500 μl cold Binding buffer, and 5ul AnnexinV‐FITC was added, mixed and 5 μl Propidium Iodide was added, mixed. The mixture was incubated at room temperature and kept away from light response for 15 min. Detection of cell apoptosis was based on FACS analysis.
2.8. Confocal immunofluorescence assay
Cells were cultured onto glass slides located into the 6‐well plate. After treatment for the desired time, the cells were fixed with 4% paraformaldehyde in PBS for 10 min, permeabilized with 0.2% Triton X‐100 in PBS for 5 min, and blocked with blocking buffer (10% BSA) for one hour. Following this, cells were incubated with target antibodies overnight. After washes with PBS, the slides were stained with DAPI and incubated for 1 h at room temperature. The slides were then washed three times with PBS and incubated with secondary antibodies conjugated with fluorescein isothiocyanate or rhodamine for 1 h and washed with PBS. The cells were detected with Leica confocal microscope and the images were processed with Image‐Pro Plus 5.1 software.
2.9. Co‐Immunoprecipitation
The nuclear lysate was mixed with the antibodies against CBP or CPSF4 and kept rotating at 4 °C for 3.5 h. Then the protein‐A/G agarose beads were added and continuously incubated at 4 °C overnight. After washing with pre‐cold PBSI buffer, the beads were mixed with loading buffer and boiled at 100 °C. After centrifugation, the precipitated proteins existing in the supernatant were separated by SDS‐PAGE and detected by Western blot analysis.
2.10. siRNA design and transfection
The siRNAs targeting CPSF4 (siRNA1: 5′‐GGUCACCUGUUACAAGUGUTT‐3′; 5′‐ACACUUGUAACAGGUGACCTT‐3′. siRNA2:5′‐CAUGCACCC UCGAUUUGAATT‐3′; 5′‐UUCAAAUCGAGGGUGUAUGTT‐3′), siRNA targeting CBP (5′‐GAGGUCGUUUACAUAAATT‐3′; 5′‐UUUAUGUAAACGCGACCUCTT‐3′), and negative control siRNA (5′‐UUCUCCGAACGUGUCACGUTT‐3′; 5′‐ACGUGACACGUUCGGAGAATT‐3′) were purchased from Shanghai GenePharma Co (Shanghai, China). Cells plated in 96‐well plates (5000 cells/well) or six‐well plates (200,000 cells/well) were transfected with siRNA duplexes (0.05–0.1 μg or 1–2 μg) encapsulated by DC‐nanoparticles. 48 h after treatment, protein expression and cell viability were tested by Western blot and MTT analysis, respectively.
2.11. Detection of hTERT promoter activity
Cells (200,000 cells/well) plated in six‐well plates were transfected with the hTERT promoter‐driven luciferase plasmids encapsulated with DC‐nanoparticles. Meanwhile, cells were co‐transfected with Lac Z plasmids, or CBP plasmids, or CBP plasmids and CPSF4‐specific siRNA or CBP plasmids and negative control siRNA used as control for 48 h. Then the luciferase activity was measured as described using a DUAL‐luciferase reporter assay kit (Promega, E1910). The ratio of firefly luciferase to Renilla luciferase activity (relative luciferase activity) was calculated to correct the variations in the transfection process.
2.12. Mathematical statistics and data analysis
All values were expressed as mean ± SD (standard deviation of the mean). Statistical significance between groups was measured by Student's t‐test with statistical significance defined as: *P < 0.05; **P < 0.01 and ***P < 0.001.
3. Results
3.1. CBP was highly expressed in lung cancer cells and tumor tissues
We first detected the expression of CBP in lung cancer cells by Western blot assay. As shown in Figure 1A, CBP was highly expressed in various lung cancer cell lines and immortalized lung cell line (HBE) compared to the normal lung cell line (HLF). We also tested its expression and localization by immunofluorescent imaging assay. The overexpression of CBP was similarly found in lung cancer cells but not in normal cells, and CBP was shown to be mainly localized in nucleus (Figure 1B). Furthermore, we examined the expression of CBP in lung cancer tissues and corresponding adjacent non‐cancer tissues. The protein samples extracted from seven couples of human lung carcinoma tissues and adjacent tissues were used to detect the expression of CBP by Western blot analysis. As shown in Figure 1C, CBP was highly expressed in lung cancer tissues compared with their adjacent non‐cancer tissues in 5 cases of patients (Case 1, 2, 3, 6, 7). Similarly, immunohistochemical staining showed that lung tumor tissues, but not their adjacent non‐cancer tissues, showed high expression of CBP (Figure 1D). These results indicated that CBP was over‐expressed in lung tumor and its high expression might participate in the development of lung cancer.
Figure 1.
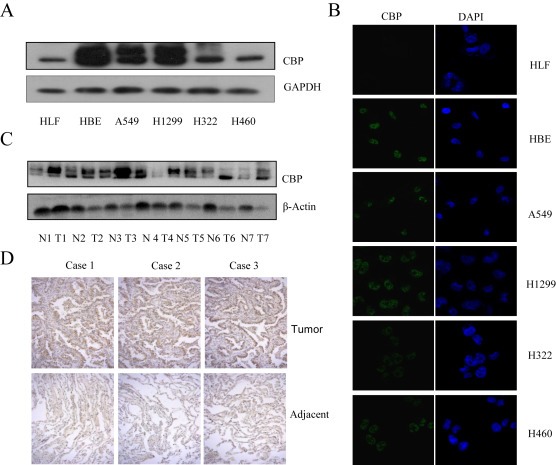
The high expression of CBP in lung cancer cells and tissues. (A) The expression of CBP protein in various lung cancer cell lines and normal cells was determined by western blot analysis. (B) The expression and localization of CBP were detected by an immunofluorescent staining in lung cancer cell lines and normal cells. (C) The protein samples were extracted from five couple of human lung carcinoma tissues and adjacent normal tissues and the expression of CBP was examined by western blot. (D) The expression of CBP protein in tumor tissues from patients with lung adenocarcinomas and corresponding adjacent normal lung tissues was detected by immunohistochemistry analysis.
3.2. CBP regulated the proliferation and migration of lung cancer cells
The role of CBP in lung cancer progression was initially assessed by evaluating its effects on lung cancer cell proliferation and migration. We transfected lung cancer cells with the specific siRNA targeting CBP, or treated them with C646, a selective inhibitor of CBP HAT (histone acetyltransferases) activity. At 48 h after treatment, the cell viability or migration ability was tested respectively. As shown in Figure 2A and C, the silencing of CBP expression or its activity inhibition resulted in the significant suppression of tumor cell viability and migration in cells transfected with si‐CBP or treated with C646, compared to those transfected with the control siRNA or treated with DMSO. Consistent with this, lung cancer cells treated with C646 also had lower colony‐forming ability compared with the control group (Figure 2B). We also detected the effect of CBP on the expression of MMP‐9, a major protein associated with cell migration, and found that CBP knockdown or its activity inhibition markedly attenuated MMP‐9 expression (Figure 2D). Furthermore, we tested the effect of CBP on the proliferation, colony formation and migration ability in HBE cells, as it similarly showed high expression of CBP. As shown in Figure S1 A–C, the silencing of CBP by its specific siRNA or its HAT activity inhibition by using C646 did not cause obvious changes, compared to the control groups. All these results proved that CBP was involved in the proliferation and migration control of lung cancer cells.
Figure 2.
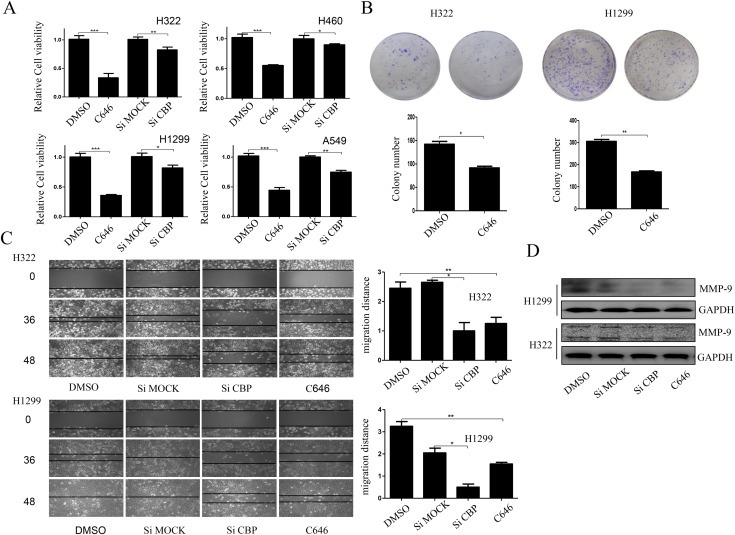
CBP promoted the proliferation and migration of lung cancer cells. (A) Cell viability measured by MTT assay in different lung cancer cell lines following CBP knockdown or HAT activity inhibition. (B) Colony formation assay of H1299 and H322 cells treated with C646 or DMSO twice a week for two weeks. The quantification assay of the number of the colonies was also shown. (C) Cell migration assay in H1299 and H322 cells following CBP knockdown or activity inhibition, and the migration rate was calculated. (D) Western blot analysis of the expression of MMP‐9 protein in H1299 and H322 cells following CBP knockdown or activity inhibition. Both the colony formation assay and migration assay were done 3 times independently, and we selected the images from one time experiment in the result part. The mean ± SD in the quantitative analysis was calculated based on different counting and measurement for the colony number and migration distance from 3 different experiments. (*P < 0.05, **P < 0.01).
3.3. CBP regulated apoptosis of lung cancer cells by regulating Bcl‐2 and cytochrome C/caspase pathway
The role of CBP in lung cancer progression was next assessed by observing its effects on cell apoptosis. By using Annexin V‐FITC/PI staining‐based FACS analysis, we found that knockdown of CBP by its specific siRNA or activity suppression with C646 exerted a significant induction of cell apoptosis in H1299 and H322 cells, resulting in more apoptotic cell populations (Figure 3A), but not in HBE cells (Figure S1D). In addition, Western blot analysis indicated that knockdown of CBP by siRNA or inhibition of its HAT activity by C646 significantly down‐regulated the expression of Bcl‐2, an important anti‐apoptotic protein, in H1299 cells, and meanwhile, up‐regulated the cleavage of two key pro‐apoptotic proteins, caspase‐3 and PARP (Figure 3B).
Figure 3.
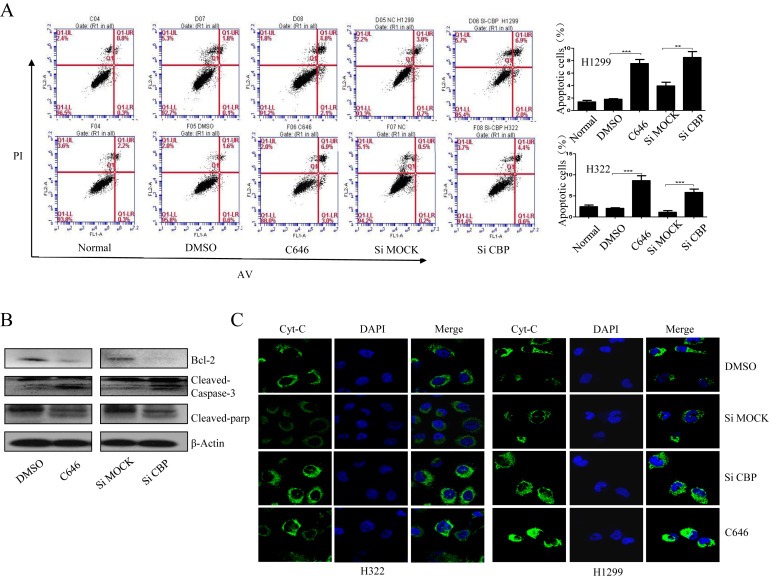
CBP mediated the apoptosis of lung cancer cells through regulating Cyt C/Caspase 3/PARP pathway. (A) Apoptosis assay in H1299 and H322 cells by flow cytometry following CBP knockdown or activity inhibition. (B) Western blot analysis of the expression of Bcl‐2, cleaved caspase‐3 and cleaved PARP proteins in H1299 cells following CBP knockdown or activity inhibition. (C)The immunofluorescent assay of the distribution of Cytochrome C in H1299 and H322 cells following CBP knockdown or activity inhibition.
We next examined the upstream mitochondrial events that contribute to caspase activation‐dependent apoptosis. Apoptotic signal stimulation could result in the release of cytochrome C from mitochondria to cytosol, where it binds to Apaf‐1 and initiate caspase activation. We therefore examined the translocation of cytochrome C mediated by CBP in lung cancer cells using IF assay. As shown in Figure 3C, more cytochrome C was released from mitochondria to cytosol in lung cancer cells upon treatment with CBP specific siRNA or its HAT activity inhibitor, compared to the untreated or nsp‐siRNA treated cells, suggesting that lung cancer cell apoptosis caused by CBP knockdown or HAT activity inhibition might be mediated by the Bcl‐2 and cytochrome C/caspase signaling pathway.
3.4. MAPK pathway was involved in the proliferation regulation mediated by CBP
To further identify the underlying molecular mechanisms by which CBP promoted lung cancer cell growth, we then analyzed the signaling pathways activated by CBP in its regulation on tumor cell proliferation. We initially examined the effects of CBP on MAPK pathway, given its key role in mediating tumor progression. As shown in Figure 4A, CBP knockdown or its HAT activity inhibition by C646 was accompanied by a marked reduction in the levels of the phosphorylated ERK1/2 and MEK1/2, and a slight increase in the expression of the phosphorylated p38. As the upstream signaling molecule of MAPK pathway, we further assessed the effect of CBP on the levels of p‐C‐Raf. CBP knockdown or its HAT activity inhibition similarly decreased the levels of p‐C‐Raf (Figure 4A).
Figure 4.
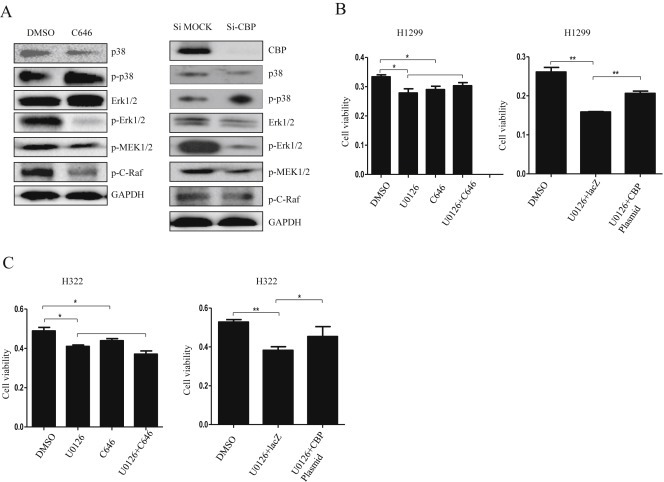
MAPK/ERK signaling pathway was affected by CBP in lung cancer cells. (A) Western blot analysis of the expression of the total and phosphorylated p38, ErK, MEK1/2, and C‐raf proteins in H1299 cells treated with CBP specific siRNA or its inhibitor. (B) Cell viability affected by inhibition of CBP activity or overexpression of CBP in H1299 cells treated with MEK1/2 specific inhibitor U0126. (C) Cell viability affected by inhibition of CBP activity or overexpression of CBP in H322 cells treated first with MEK1/2 specific inhibitor U0126.
To further confirm the involvement of MAPK pathway in lung tumor cell viability regulated by CBP, we evaluated the effect of U0126, the selective inhibitor of its core component, MEK1/2, in MAPK pathway, on CBP in its regulation of lung cancer cell viability. The treatment of C646 did not obviously synergize U0126‐mediated suppression of lung tumor cell viability. In other words, CBP had no effect or displayed compromised effect when the MAPK pathway was inhibited by U0126. By contrast, the overexpression of CBP in H1299 and H322 cells significantly reversed the U0126‐mediated cell viability inhibition (Figure 4B and C). As such, the effect of CBP appeared to be dependent on MAPK. These results therefore showed that the function of CBP in promoting cell viability is mediated, at least in part, through the activation of the MAPK/ERK signaling in lung cancer cells.
3.5. CBP interacted with CPSF4 and mediated the acetylation of CPSF4
Our previous studies respectively indicated the role of CBP and CPSF4 as transcriptional factors in regulating hTERT expression in lung adenocarcinomas (Jia et al., 2014; Chen et al., 2014b). Given the basic function of CBP as a co‐transactivator and the similarly high expression of CPSF4 in lung cancer cells and tissues (Figure S2), we hypothesized the interaction between CBP and CPSF4 and their possible synergy in regulating hTERT expression in lung cancer. In order to address this hypothesis, the co‐immunoprecipitation experiments were first done to analyze their direct interaction. The nuclear extracts from lung cancer cell lines and normal cell lines were immunoprecipitated using antibody against CBP or CPSF4 or the control non‐specific IgG, respectively, and the eluted proteins were detected by Western blot using antibody against CPSF4, or CBP, or the acetylated antibody, respectively. CBP was co‐immunoprecipitated in the complexes pulled down by anti‐CPSF4 antibody, but not by the non‐specific IgG (Figure 5A), indicating that CPSF4 indeed interacted with CBP directly in the nucleus of lung cancer cell lines. Similarly, dual‐immunofluorescent assay showed the co‐localization of CBP and CPSF4 in the nucleus of lung cancer cells (Figure 5B), further confirming the possibility of their interaction.
Figure 5.
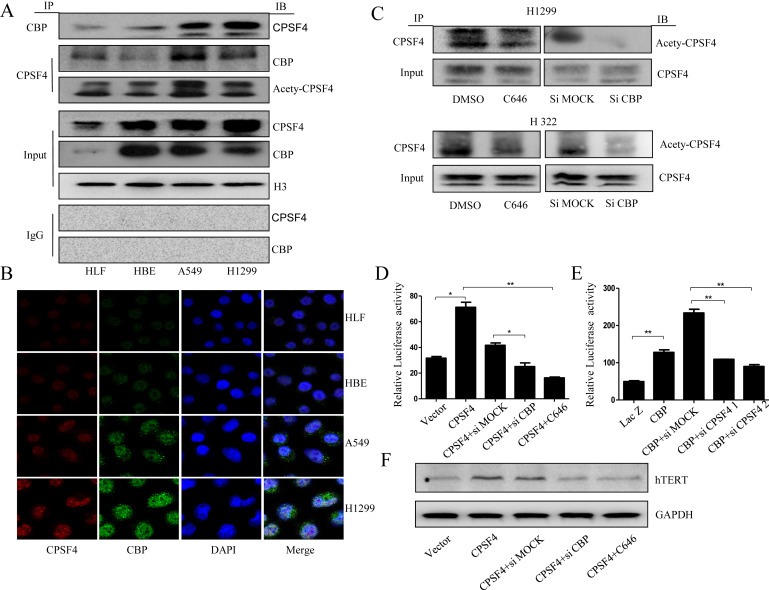
CBP interacted with CPSF4 and mediated the acetylation of CPSF4 and their synergistic regulation on hTERT expression in lung cancer cells. (A) The extracted proteins from nuclear of HLF, HBE, A549 and H1299 cells were immunoprecipitated by antibody against CBP or CPSF4 or IgG as control. The complex was detected with anti‐CBP or CPSF4 antibody. In put represents the whole nuclear extracts. (B) The co‐localization of CBP and CPSF4 in human lung normal and cancer cells through immunofluorescence analysis. (C) The expression analysis of the acetylated CPSF4 in H322 and H1299 cells following CBP knock down or activity inhibition through IP assay. In put represents the whole nuclear extracts. (D) The hTERT promoter‐driven luciferase activity in H1299 cells stably expressing CPSF4 after co‐transfection with hTERT promoter (−459/+9)‐driven luciferase plasmids and CBP siRNA or C646. (E) The hTERT promoter‐driven luciferase activity in H1299 cells after co‐transfection with hTERT promoter (−459/+9)‐driven luciferase plasmids, CBP‐overexpressing plasmids and CPSF4 siRNAs. (F) The hTERT expression in H1299 cells stably expressing CPSF4 after transfection with CBP siRNA or treatment with C646.
The possible acetylation of CPSF4 mediated by CBP was then examined in lung cancer cells, given the activity of CBP as a histone acetylase to acetylate histones or other transcriptional factors (Cai et al., 2014; Cazzalini et al., 2014; Chen et al., 2014a; Dancy and Cole, 2015; Ferrari et al., 2014; Jin et al., 2011; Kim et al., 2012; Tie et al., 2009.) We found that CBP knockdown or its HAT activity inhibition by C646 was accompanied by a reduction of the acetylated CPSF4 levels in lung cancer cells (Figure 5C). Additionally, the same trend for the expression of the acetylated CPSF4 and CBP was observed in different lung cancer cells (Figure 5A). These results collectively demonstrate that CBP interacted with CPSF4 most possibly through acetylating the latter in lung cancer cells.
3.6. CBP and CPSF4 synergistically regulated the transcription and expression of hTERT
Based on the direct interaction between CBP and CPSF4, and their respective regulation on hTERT, we then evaluated the synergetic regulation of hTERT transcription and expression by CBP and CPSF4. The H1299 cells stably overexpressing CPSF4 were co‐transfected with hTERT promoter‐driven luciferase plasmids and CBP‐specific siRNA or treated with CBP inhibitor C646. At 48 h after treatment, the expression of luciferase was assayed. CBP knockdown or its HAT activity inhibition significantly suppressed the expression of hTERT promoter‐driven luciferase (Figure 5D). On the contrary, CPSF4 knockdown using its specific siRNA in the CBP‐overexpressed H1299 cells reversed the CBP‐mediated up‐regulation of hTERT promoter‐driven luciferase expression (Figure 5E). Furthermore, CBP knockdown or its HAT activity inhibition similarly reversed the increased expression of hTERT mediated by CPSF4 overexpression in H1299 cells (Figure 5F), suggesting that CBP indeed synergized with CPSF4 in regulating hTERT transcription and expression in lung cancer cells.
3.7. CBP and CPSF4 synergistically regulated lung cancer cell growth and apoptosis
Since CBP synergized with CPSF4 in controlling hTERT expression, and hTERT has been shown to be participated in the growth of lung cancer cells, we then examined the effect of CBP and CPSF4 synergy on cell viability in H1299 and H322 cells. H1299 cells stably overexpressing CPSF4 were transfected with the CBP‐specific siRNA or treated with CBP inhibitor C646. At 48 h later, the cell viability was assayed. We found that CBP knockdown or its HAT activity inhibition significantly reversed the increased cell viability mediated by CPSF4 overexpression (Figure 6A). Conversely, silencing of CPSF4 using its specific siRNA in H1299 cells with overexpressed CBP reversed the CBP‐mediated up‐regulation of cell proliferation (Figure 6B). Similar results were observed in H322 cells (Figure 6C, D). Further molecular mechanisms assay indicated that CBP knockdown or activity inhibition reversed the CPSF4 overexpression‐mediated increase of p‐Erk, while nearly had no effects on the levels of total Erk (Figure 6E).
Figure 6.
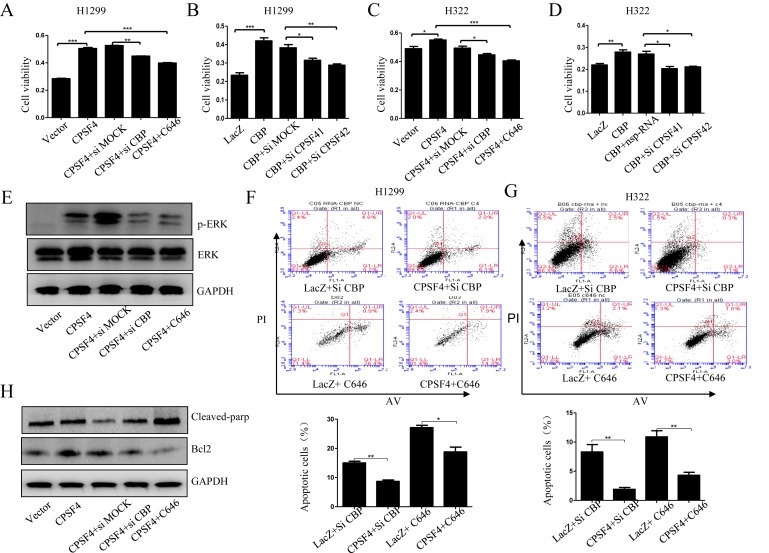
The synergistic regulation of lung cancer cell growth and apoptosis by CBP and CPSF4. (A, C) Cell viability analysis in H1299 cells stably expressing CPSF4 after transfection with CBP siRNA or treatment with C646. (B, D) Cell viability analysis in H322 cells after co‐transfection with CBP‐overexpressing plasmids and CPSF4 siRNAs. (E) Western blot analysis of the ErK and p‐ErK expression in H1299 cells stably expressing CPSF4 after transfection with CBP siRNA or treatment with C646. (F–G) Apoptosis assay in H1299 and H322 cells after co‐treatment respectively with Lac Z plasmids and CBP siRNA, or Lac Z plasmids and C646, or CPSF4 plasmids and CBP siRNA, or CPSF4 plasmids and C646. The corresponding quantitative analysis of the apoptotic cell numbers was given below. (H) Western blot analysis of the Bcl‐2 and cleaved PARP expression in H1299 cells stably expressing CPSF4 after transfection with CBP siRNA or treatment with C646. Data In panel (A–D) are all represented as mean ± SD of three separate experiments with statistic significance calculated from the two‐tailed student's t test. (*P < 0.05, **P < 0.01).
The synergistic effects of CBP and CPSF4 on lung cancer cell apoptosis were then determined using FACS assay in H1299 and H322 cells. CPSF4 overexpression significantly reversed the increased cell apoptosis caused by CBP knockdown or activity inhibition (Figure 6F, G). In addition, CBP knockdown or activity inhibition obviously suppressed Bcl‐2 protein expression and increased cleaved PARP expression compared with the NSP‐siRNA treatment group in H1299 cells with CPSF4 overexpression (Figure 6H), suggesting the cooperative roles of CBP and CPSF4 in mediating lung cancer cell apoptosis. All the results above demonstrated that CBP regulated lung cancer cell proliferation and apoptosis at least in part through its cooperation with CPSF4.
3.8. CBP and CPSF4 overexpression was positively correlated with poor prognosis of patients with lung adenocarcinomas
To further confirm the involvement of CPSF4 in CBP‐mediated lung cancer survival, we examined the expression of CBP and CPSF4 in clinical lung tumor tissue samples and analyzed their relationship with the prognosis of patients with lung adenocarcinomas. The expression of CBP and CPSF4 in lung tumor tissues from 75 cases lung carcinoma patients were tested through IHC assay, and 34 cases showed simultaneous high expression of CBP and CPSF4, accounting for 45% of all the tested cases (Figure 7A). Additionally, the relationship between CBP or CPSF4 expression and clinicopathologic variables were assayed and summarized in Figure 7B–D. As shown in Figure 7C, the expression of CBP was significantly associated with lung tumor differentiation, while the expression of CPSF4 was respectively associated with lymphatic metastasis, or distant metastasis. Moreover, the overall survival (OS) analysis indicated the patients with low CBP and CPSF4 expression owned significantly higher 5‐OS and the extended survival rate compared to the patients with both high expression of these two proteins (Figure 7D, E). These results demonstrated a potentially synergetic involvement of CPSF4 in CBP‐mediated lung cancer progression and their indication for the poor prognosis of patients with lung cancer.
Figure 7.
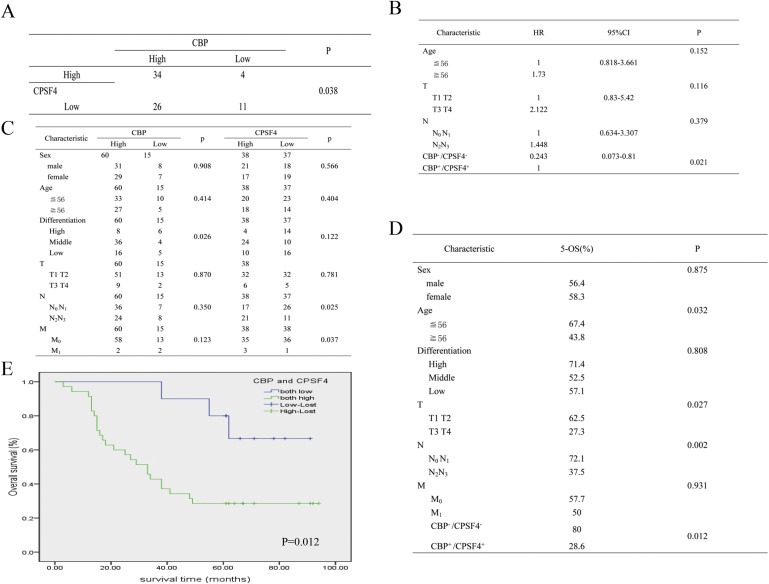
The positive correlation between CBP and CPSF4 expression in clinical lung tumor tissue samples and their prediction for the poor prognosis of patients with lung adenocarcinoma. (A) The protein level of CPSF4 correlates positively with the protein level of CBP in lung adenocarcinoma tissues from 75 patients. (B) Cox‐regression analyses for prognosis of 75 lung carcinoma patients. (C) Correlation analyses of CBP or CPSF4 protein expression in relation to clinicopathologic variables of 75 lung carcinoma patients. (D) Correlation analyses of CBP or CPSF4 protein expression in relation to 5‐OS of 75 lung carcinoma patients. (E) Kaplan–Meier analysis of overall survival of lung cancer patients with different CBP and CPSF4 expression (P < 0.05, log‐rank test).
4. Discussion
Most previous studies about the roles and the related molecular mechanisms of CBP participated in carcinogenesis and development focused on its gene mutation or chromosome translocation (Lin et al., 2014; Mullighan et al., 2011; Pasqualucci et al., 2011). Only rare researches recently revealed the anti‐tumor effects of CBP inhibition by suppressing its HAT activity or function as transcriptional co‐activator (Arensman et al., 2014; Gajer et al., 2015; Giotopoulos et al., 2015). Nevertheless, the accurate functions and molecular mechanisms by which CBP was involved in tumor progression have still been largely unknown. In this study, we reported that, in addition to inhibition of HAT activity, the knockdown of CBP itself consistently exhibited anti‐tumor activity in lung tumor cells, but not in the normal cells or immortalized cells. Furthermore, this anti‐tumor effect was found to be realized through suppressing C‐Raf/MEK/Erk signaling pathway and activating the cytochrome C/caspase 3/PARP signaling pathway. As CBP was reported to be able to regulate the expression of many key target genes involved in cancer, including COX‐2 and hTERT (Xiao et al., 2015; Guo et al., 2014) independently or through synergizing with other transcriptional factors, and C‐Raf/MEK/Erk and cytochrome C/caspase 3/PARP signaling pathways usually function as the downstream signaling molecules of these target genes (Wang et al., 2005; Lee et al., 2009), we hypothesized that CBP played its anti‐tumor effect through controlling some key genes expression to further affect C‐Raf/MEK/Erk and cytochrome C/caspase 3/PARP signaling pathways in lung cancer. To our knowledge, this might be the first comprehensive evaluation of the effects of CBP on lung cancer cells.
As a transcriptional co‐activator, CBP is thought to increase gene expression through recruiting basal transcriptional machinery to the promoter by interacting with other transcriptional factors (Gray et al., 2005; Jansma et al., 2014; Jia et al., 2014). Hereby we hypothesized the existence of one or more other transcriptional factors interacting and co‐anchoring with CBP at gene promoter to synergistically regulate gene expression and further to control lung tumor cell growth. In our previous study (Guo et al., 2014), we demonstrated that CBP promoted hTERT expression as a transcriptional co‐activator by interacting with SP1 transactivator in lung cancers. In this study, we aimed to uncover the potential molecular mechanisms of CBP in mediating lung cancer survival. Since CPSF4 also has been proved to up‐regulate hTERT expression as a transcriptional factor in lung cancers (Chen et al., 2014b), we hypothesized their synergistic effects in regulating lung cancer progress and tried to test this hypothesis in this paper. Furthermore, as a transcriptional coactivator, CBP plays its transcriptional regulatory role through recruiting and co‐anchoring with many other transcriptional factors. Therefore, it is not contradictory that besides SP1, CBP also interacts with and recruit CPSF4 to synergistically regulate hTERT expression. To clarify the interaction between CBP and CPSF4 and their possibly synergistic effects on lung cancer proceeding, we performed IP and IF analyses and showed their co‐localization in nucleus and direct interaction. We also showed that the inhibition of HAT activity of CBP or its knockdown attenuated the levels of acetylated CPSF4, suggesting that the acetylation of CPSF4 mediated by CBP might be the prerequisite for its recruitment by CBP at gene promoters. Further analyses showed that the activity inhibition or knockdown of one of these two proteins reversed the proliferative promotion or apoptosis suppression caused by the over‐expression of the other one. This fully demonstrated the synergistic regulation of lung cancer survival by CBP and CPSF4, and also demonstrated the CPSF4‐dependent growth regulation by CBP in lung cancer. Combined with the previous findings that hTERT was transcriptionally controlled by CBP and CPSF4 individually, we supposed the potential of these two proteins in coordinative anchoring at hTERT promoter elements and synergistic regulation on hTERT expression. This hypothesis was further supported when we found that the down‐regulation of one of these two proteins attenuated the improved transcriptional activity and expression of hTERT mediated by the up‐regulation of the other one. All the findings suggest that CBP promoted lung cancer progress at least in part through cooperating with CPSF4, and also suggest this cooperative control on lung tumor cell growth was presumably through stimulation of their co‐downstream responsive elements, hTERT.
In our study, we found that CBP knockdown or inhibition increased the level of activated p38, p‐p38. On the one hand, the activation of p38‐MAPK signaling pathways may have anti‐apoptotic and proliferative effects. But on the other hand, p38 can also function as a tumor suppressor (Koul et al., 2013). Thus, the increase of p‐p38 level possibly led to apoptosis induction and proliferation inhibition, which was consistent with our findings about the effect caused by CBP silencing.
CBP was observed to be highly expressed in lung cancer cells and tissues in our study. In this study, we explored its downstream molecular mechanisms involved in lung cancer progression. Based on the previous studies that gene mutation or chromosomal translocation within CBP gene or its aberrant recruitment at chromatin structure is associated with several types of cancers, we deduced that copy number increase caused by some carcinogenic factors might contribute its high expression in lung cancer. The detailed molecular mechanisms deserve further investigation in our future study.
Our clinical data analyses also showed that the cases of patients with the simultaneous high expression of CBP and CPSF4 occupied a large proportion among all the tested cases, and such patients displayed much shorter OS compared to those with both low expression of these two proteins, which agreed with the in vitro findings that CBP and CPSF4 could synergistically accelerate tumor progression. Although this cooperativity was not obviously represented based on the analyses of the relationship between CBP or CPSF4 expression and a series of parameters of clinical pathology, the individual correlation, between CBP expression and tumor differentiation, and between CPSF4 expression and tumor lymph node metastasis or distant metastasis, was clearly shown. Their functional changes might also contribute to the correlation between their expression and poor prognosis, and such linkage between their functional changes and prognosis, such as the change of HAT activity of CBP, the involvement of CPSF4 in RNA splicing and maturation, needs more in‐depth investigation in our future study. Nevertheless, our current data at least indicate the significance of the simultaneous silencing or inhibition of these two proteins in lung cancer treatment.
In summary, we found that CBP induced proliferative growth of lung tumor cells by affecting C‐Raf/MEK/Erk and cytochrome C/caspase signaling pathways. In addition, we showed that such induction happened in a process requiring CPSF4, whereby CBP recruited, interacted with and acetylated CPSF4 at gene promoter regions to synergistically regulate downstream gene transcription and tumor cell proliferation. This association between CBP and CPSF4 was synergistically responsible for the activation of hTERT expression and may, in part, contributed to the mechanisms by which CBP is involved in growth promotion of lung cancer. Since both CBP and CPSF4 are highly expressed in lung cancer tissues, our study might provide a very potential therapeutic strategy to treat this cancer by dual blocking these two proteins simultaneously.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
The following are the supplementary data related to this article:
Figure S1. CBP caused nearly no effects on the proliferation, migration and apoptosis of HBE cells. (A) Cell viability measured by MTT assay in HBE cell lines following CBP knockdown or activity inhibition. (B) Colony formation assay of HBE cells treated with C646 or DMSO twice a week for two weeks. The quantification assay of the number of the formed colonies was also shown. (C) Cell migration assay in HBE cells following CBP knockdown or activity inhibition, and the migration rate was calculated. (D) Apoptosis assay in HBE cells by flow cytometry following CBP knockdown or activity inhibition.
Figure S2. The high expression of CPSF4 in lung cancer cells and tissues. (A) The expression of CPSF4 protein in lung cancer cell lines and normal lung cells through western blot analysis. (B) The protein samples were extracted from seven s of human lung carcinoma tissues and adjacent normal tissues and the expression of CPSF4 were examined by western blot.
Acknowledgments
This work was supported by the funds from the National Natural Science Foundation of China (81301721, 81470337, 81472178, 81071687, 81272195), the State “863 Program” of China (SS2012AA020403), the State “973 Program” of China (2014CB542005), the Education Department of Liaoning Province, China (“the Program for Pan‐Deng scholars”), the Scientific Research Project from Education Department of Liaoning Province, China (L2015142).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.10.015.
Tang Zhipeng, Yu Wendan, Zhang Changlin, Zhao Shilei, Yu Zhenlong, Xiao Xiangsheng, Tang Ranran, Xuan Yang, Yang Wenjing, Hao Jiaojiao, Xu Tingting, Zhang Qianyi, Huang Wenlin, Deng Wuguo, Guo Wei, (2016), CREB‐binding protein regulates lung cancer growth by targeting MAPK and CPSF4 signaling pathway, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.10.015.
Contributor Information
Wuguo Deng, Email: dengwg@sysucc.org.cn.
Wei Guo, Email: wei1015@msn.com.
References
- Allemani, C. , Weir, H.K. , Carreira, H. , Harewood, R. , Spika, D. , Wang, X.S. , Bannon, F. , Ahn, J.V. , Johnson, C.J. , Bonaventure, A. , Marcos-Gragera, R. , Stiller, C. , Azevedo e Silva, G. , Chen, W.Q. , Ogunbiyi, O.J. , Rachet, B. , Soeberg, M.J. , You, H. , Matsuda, T. , Bielska-Lasota, M. , Storm, H. , Tucker, T.C. , Coleman, M.P. , Group, C.W.2015. Global surveillance of cancer survival 1995-2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet. 385, 977–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arensman, M.D. , Telesca, D. , Lay, A.R. , Kershaw, K.M. , Wu, N. , Donahue, T.R. , Dawson, D.W. , 2014. The CREB-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol. Cancer Ther. 13, 2303–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabino, S.M. , Hubner, W. , Jenny, A. , Minvielle-Sebastia, L. , Keller, W. , 1997. The 30-kD subunit of mammalian cleavage and polyadenylation specificity factor and its yeast homolog are RNA-binding zinc finger proteins. Genes Dev. 11, 1703–1716. [DOI] [PubMed] [Google Scholar]
- Cai, K. , Wan, Y. , Wang, Z. , Wang, Y. , Zhao, X. , Bao, X. , 2014. C5a promotes the proliferation of human nasopharyngeal carcinoma cells through PCAF-mediated STAT3 acetylation. Oncol. Rep. 32, 2260–2266. [DOI] [PubMed] [Google Scholar]
- Cazzalini, O. , Sommatis, S. , Tillhon, M. , Dutto, I. , Bachi, A. , Rapp, A. , Nardo, T. , Scovassi, A.I. , Necchi, D. , Cardoso, M.C. , Stivala, L.A. , Prosperi, E. , 2014. CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res. 42, 8433–8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. , Ruiz, P.D. , Novikov, L. , Casill, A.D. , Park, J.W. , Gamble, M.J. , 2014. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat. Struct. Mol. Biol. 21, 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Guo, W. , Li, M. , Shi, D. , Tian, Y. , Li, Z. , Wang, J. , Fu, L. , Xiao, X. , Liu, Q.Q. , Wang, S. , Huang, W. , Deng, W. , 2013. Upregulation of cleavage and polyadenylation specific factor 4 in lung adenocarcinoma and its critical role for cancer cell survival and proliferation. PLoS One. 8, e82728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Qin, L. , Wang, S. , Li, M. , Shi, D. , Tian, Y. , Wang, J. , Fu, L. , Li, Z. , Guo, W. , Yu, W. , Yuan, Y. , Kang, T. , Huang, W. , Deng, W. , 2014. CPSF4 activates telomerase reverse transcriptase and predicts poor prognosis in human lung adenocarcinomas. Mol. Oncol. 8, 704–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancy, B.M. , Cole, P.A. , 2015. Protein lysine acetylation by p300/CBP. Chem. Rev. 115, 2419–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, W.G. , Jayachandran, G. , Wu, G. , Xu, K. , Roth, J.A. , Ji, L. , 2007. Tumor-specific activation of human telomerase reverses transcriptase promoter activity by activating enhancer-binding protein-2beta in human lung cancer cells. J. Biol. Chem. 282, 26460–26470. [DOI] [PubMed] [Google Scholar]
- Ferrari, R. , Gou, D. , Jawdekar, G. , Johnson, S.A. , Nava, M. , Su, T. , Yousef, A.F. , Zemke, N.R. , Pellegrini, M. , Kurdistani, S.K. , Berk, A.J. , 2014. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe. 16, 663–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajer, J.M. , Furdas, S.D. , Grunder, A. , Gothwal, M. , Heinicke, U. , Keller, K. , Colland, F. , Fulda, S. , Pahl, H.L. , Fichtner, I. , Sippl, W. , Jung, M. , 2015. Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo. Oncogenesis. 4, e137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Y. , Geng, J. , Hong, X. , Qi, J. , Teng, Y. , Yang, Y. , Qu, D. , Chen, G. , 2014. Expression of p300 and CBP is associated with poor prognosis in small cell lung cancer. Int. J. Clin. Exp. Pathol. 7, 760–767. [PMC free article] [PubMed] [Google Scholar]
- Giotopoulos, G. , Chan, W.I. , Horton, S.J. , Ruau, D. , Gallipoli, P. , Fowler, A. , Crawley, C. , Papaemmanuil, E. , Campbell, P.J. , Gottgens, B. , Van Deursen, J.M. , Cole, P.A. , Huntly, B.J. , 2015. The epigenetic regulators CBP and p300 facilitate leukemogenesis and represent therapeutic targets in acute myeloid leukemia. Oncogene. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman, R.H. , Smolik, S. , 2000. CBP/p300 in cell growth, transformation, and development. Genes Dev. 14, 1553–1577. [PubMed] [Google Scholar]
- Gray, M.J. , Zhang, J. , Ellis, L.M. , Semenza, G.L. , Evans, D.B. , Watowich, S.S. , Gallick, G.E. , 2005. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 24, 3110–3120. [DOI] [PubMed] [Google Scholar]
- Guo, W. , Lu, J. , Dai, M. , Wu, T. , Yu, Z. , Wang, J. , Chen, W. , Shi, D. , Yu, W. , Xiao, Y. , Yi, C. , Tang, Z. , Xu, T. , Xiao, X. , Yuan, Y. , Liu, Q. , Du, G. , Deng, W. , 2014. Transcriptional coactivator CBP upregulates hTERT expression and tumor growth and predicts poor prognosis in human lung cancers. Oncotarget. 5, 9349–9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansma, A.L. , Martinez-Yamout, M.A. , Liao, R. , Sun, P. , Dyson, H.J. , Wright, P.E. , 2014. The high-risk HPV16 E7 oncoprotein mediates interaction between the transcriptional coactivator CBP and the retinoblastoma protein pRb. J. Mol. Biol. 426, 4030–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, Y. , Nie, F. , Du, A. , Chen, Z. , Qin, Y. , Huang, T. , Song, X. , Li, L. , 2014. Thymine DNA glycosylase promotes transactivation of beta-catenin/TCFs by cooperating with CBP. J. Mol. Cell Biol. 6, 231–239. [DOI] [PubMed] [Google Scholar]
- Jin, Q. , Yu, L.R. , Wang, L. , Zhang, Z. , Kasper, L.H. , Lee, J.E. , Wang, C. , Brindle, P.K. , Dent, S.Y. , Ge, K. , 2011. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper, L.H. , Fukuyama, T. , Biesen, M.A. , Boussouar, F. , Tong, C. , de Pauw, A. , Murray, P.J. , van Deursen, J.M. , Brindle, P.K. , 2006. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol. Cell Biol. 26, 789–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, W.J. , Rivera, M.N. , Coffman, E.J. , Haber, D.A. , 2012. The WTX tumor suppressor enhances p53 acetylation by CBP/p300. Mol. Cell. 45, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul, H.K. , Pal, M. , Koul, S. , 2013. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer. 4, 342–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz, J.R. , Yagui-Beltran, A. , Jablons, D.M. , 2010. Cancer stem cells in lung tumorigenesis. Ann. Thorac. Surg. 89, S2090–S2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K.S. , Lee, H.J. , Ahn, K.S. , Kim, S.H. , Nam, D. , Kim, D.K. , Choi, D.Y. , Ahn, K.S. , Lu, J. , Kim, S.H. , 2009. Cyclooxygenase-2/prostaglandin E2 pathway mediates icariside II induced apoptosis in human PC-3 prostate cancer cells. Cancer Lett. 280, 93–100. [DOI] [PubMed] [Google Scholar]
- Lin, Z. , Feng, R. , Li, J. , Meng, Y. , Yuan, L. , Fu, Z. , Guo, J. , Bringhurst, F.R. , Yang, D. , 2014. Nuclear translocation of CBP/p300-interacting protein CITED1 induced by parathyroid hormone requires serine phosphorylation at position 79 in its 63-84 domain. Cell Signal. 26, 2436–2445. [DOI] [PubMed] [Google Scholar]
- Liu, W.B. , Han, F. , Jiang, X. , Yin, L. , Chen, H.Q. , Li, Y.H. , Liu, Y. , Cao, J. , Liu, J.Y. , 2015. Epigenetic regulation of ANKRD18B in lung cancer. Mol. Carcinog. 54, 312–321. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Wang, L. , Han, R. , Beier, U.H. , Akimova, T. , Bhatti, T. , Xiao, H. , Cole, P.A. , Brindle, P.K. , Hancock, W.W. , 2014. Two histone/protein acetyltransferases, CBP and p300, are indispensable for Foxp3+ T-regulatory cell development and function. Mol. Cell Biol. 34, 3993–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin, A. , Driscoll, B. , 2013. Lung cancer stem cells: progress and prospects. Cancer Lett. 338, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsudomi, T. , 2014. Molecular epidemiology of lung cancer and geographic variations with special reference to EGFR mutations. Transl. Lung Cancer Res. 3, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan, C.G. , Zhang, J. , Kasper, L.H. , Lerach, S. , Payne-Turner, D. , Phillips, L.A. , Heatley, S.L. , Holmfeldt, L. , Collins-Underwood, J.R. , Ma, J. , Buetow, K.H. , Pui, C.H. , Baker, S.D. , Brindle, P.K. , Downing, J.R. , 2011. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 471, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff, M.E. , Barabino, S.M. , Li, Y. , Keller, W. , Krug, R.M. , 1998. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol. Cell. 1, 991–1000. [DOI] [PubMed] [Google Scholar]
- Pasqualucci, L. , Dominguez-Sola, D. , Chiarenza, A. , Fabbri, G. , Grunn, A. , Trifonov, V. , Kasper, L.H. , Lerach, S. , Tang, H. , Ma, J. , Rossi, D. , Chadburn, A. , Murty, V.V. , Mullighan, C.G. , Gaidano, G. , Rabadan, R. , Brindle, P.K. , Dalla-Favera, R. , 2011. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 471, 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provencio, M. , Sanchez, A. , 2014. Therapeutic integration of new molecule-targeted therapies with radiotherapy in lung cancer. Transl. Lung Cancer Res. 3, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, R. , Ma, J. , Zou, Z. , Jemal, A. , 2014. Cancer statistics, 2014. CA – Cancer J. Clini. 64, 9–29. [DOI] [PubMed] [Google Scholar]
- Slavik, T. , Asselah, F. , Fakhruddin, N. , El Khodary, A. , Torjman, F. , Anis, E. , Quinn, M. , Khankan, A. , Kerr, K.M. , 2014. Diagnosis and predictive molecular analysis of non-small-cell lung cancer in the Africa-Middle East region: challenges and strategies for improvement. Clin. Lung Cancer. 15, 398–404. [DOI] [PubMed] [Google Scholar]
- Stachowiak, M.K. , Birkaya, B. , Aletta, J.M. , Narla, S.T. , Benson, C.A. , Decker, B. , Stachowiak, E.K. , 2015. Nuclear FGF receptor-1 and CREB binding protein: an integrative signaling module. J. Cell Physiol. 230, 989–1002. [DOI] [PubMed] [Google Scholar]
- Tie, F. , Banerjee, R. , Stratton, C.A. , Prasad-Sinha, J. , Stepanik, V. , Zlobin, A. , Diaz, M.O. , Scacheri, P.C. , Harte, P.J. , 2009. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 136, 3131–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnell, A.S. , Mymryk, J.S. , 2006. Roles for the coactivators CBP and p300 and the APC/C E3 ubiquitin ligase in E1A-dependent cell transformation. Br. J. Cancer. 95, 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valor, L.M. , Viosca, J. , Lopez-Atalaya, J.P. , Barco, A. , 2013. Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr. Pharm. Des. 19, 5051–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Breda, S.G. , Claessen, S.M. , Lo, K. , van Herwijnen, M. , Brauers, K.J. , Lisanti, S. , Theunissen, D.H. , Jennen, D.G. , Gaj, S. , de Kok, T.M. , Kleinjans, J.C. , 2014. Epigenetic mechanisms underlying arsenic-associated lung carcinogenesis. Arch. Toxicol. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Vo, N. , Goodman, R.H. , 2001. CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 276, 13505–13508. [DOI] [PubMed] [Google Scholar]
- Wang, F. , Marshall, C.B. , Ikura, M. , 2013. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol. Life Sci. – CMLS. 70, 3989–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Feng, H. , Huang, X.Q. , Xiang, H. , Mao, Y.W. , Liu, J.P. , Yan, Q. , Liu, W.B. , Liu, Y. , Deng, M. , Gong, L. , Sun, S. , Luo, C. , Liu, S.J. , Zhang, X.J. , Liu, Y. , Li, D.W. , 2005. Human telomerase reverse transcriptase immortalizes bovine lens epithelial cells and suppresses differentiation through regulation of the ERK signaling pathway. J. Biol. Chem. 280, 22776–22787. [DOI] [PubMed] [Google Scholar]
- Wang, Z.L. , Fan, Z.Q. , Jiang, H.D. , Qu, J.M. , 2013. Selective Cox-2 inhibitor celecoxib induces epithelial-mesenchymal transition in human lung cancer cells via activating MEK-ERK signaling. Carcinogenesis. 34, 638–646. [DOI] [PubMed] [Google Scholar]
- Xiao, Y. , Wang, J. , Qin, Y. , Xuan, Y. , Jia, Y. , Hu, W. , Yu, W. , Dai, M. , Li, Z. , Yi, C. , Zhao, S. , Li, M. , Du, S. , Cheng, W. , Xiao, X. , Chen, Y. , Wu, T. , Meng, S. , Yuan, Y. , Liu, Q. , Huang, W. , Guo, W. , Wang, S. , Deng, W. , 2015. Ku80 cooperates with CBP to promote COX-2 expression and tumor growth. Oncotarget. 6, 8046–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, D. , Qi, Z. , 2012. Expression and significance of Raf kinase inhibitory protein in lung cancer. Zhongguo Fei Ai Za Zhi – Chin. J. Lung Cancer. 15, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajkowicz, A. , Butkiewicz, D. , Drosik, A. , Giglok, M. , Suwinski, R. , Rusin, M. , 2015. Truncating mutations of PPM1D are found in blood DNA samples of lung cancer patients. Br. J. Cancer. 112, 1114–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Figure S1. CBP caused nearly no effects on the proliferation, migration and apoptosis of HBE cells. (A) Cell viability measured by MTT assay in HBE cell lines following CBP knockdown or activity inhibition. (B) Colony formation assay of HBE cells treated with C646 or DMSO twice a week for two weeks. The quantification assay of the number of the formed colonies was also shown. (C) Cell migration assay in HBE cells following CBP knockdown or activity inhibition, and the migration rate was calculated. (D) Apoptosis assay in HBE cells by flow cytometry following CBP knockdown or activity inhibition.
Figure S2. The high expression of CPSF4 in lung cancer cells and tissues. (A) The expression of CPSF4 protein in lung cancer cell lines and normal lung cells through western blot analysis. (B) The protein samples were extracted from seven s of human lung carcinoma tissues and adjacent normal tissues and the expression of CPSF4 were examined by western blot.