

Abstract
γ-Secretase is a structurally enigmatic multiprotein complex that catalyzes intramembrane proteolysis of a variety of substrates, including the amyloid β-protein precursor of Alzheimer's disease and the Notch receptor essential to cell differentiation. The active site of this transmembrane aspartyl protease apparently lies at the interface between two subunits of presenilin-1 (PS1); however, evidence suggests the existence of an initial substrate-binding site that is distinct from the active site. Here, we report that photoaffinity probes based on potent helical peptide inhibitors and designed to mimic the amyloid β-protein precursor substrate bind specifically to the PS subunit interface, at a site close to the active site. The location of the helical peptide-binding site suggests that substrate passes between the two PS1 subunits to access the active site. An aggressive Alzheimer-causing mutation in PS1 strongly reduced photolabeling by a transition-state analogue but not by helical peptides, providing biochemical evidence that the pathological effect of this PS mutation is due to alteration of the active-site topography.
Keywords: affinity labeling, mechanism, membrane protein, protease
The γ-secretase complex is ostensibly a multicomponent aspartyl protease with presenilin (PS) as the catalytic component (1–3). Three other integral membrane proteins, nicastrin (NCT), Aph-1, and Pen-2, are necessary and, along with PS, sufficient members of the protease complex (4–10). However, despite the elucidation of the identity of γ-secretase, its structure and mechanism remain uncertain. γ-Secretase cleaves amide bonds within the transmembrane regions of its substrates, a poorly understood process of hydrolysis within a hydrophobic environment (3). The study of transition-state analogue inhibitors of γ-secretase led to the suggestion that γ-secretase is an aspartyl protease and that two conserved aspartates in PS are catalytic residues (11–13). PS is processed into an N-terminal fragment (NTF) and C-terminal fragment (CTF). These fragments are metabolically stable and remain associated, and their formation is tightly regulated (14). The direct binding of transition-state analogue γ-secretase inhibitors to these fragments strongly suggested that the active site is at the NTF/CTF heterodimeric interface (15, 16), consistent with the fact that each subunit contributes one of the two critical aspartates (13).
Because of its requirement for water, the active site of γ-secretase is thought to be in the protein interior to avoid the hydrophobic environment of the lipid bilayer (17). As a result, the integral membrane substrates initially should interact on the surface of the protease before entering the internal active site. In fact, an endogenous γ-secretase substrate copurifies with the protease complex isolated from an immobilized transition-state analogue (10, 18), suggesting that substrate can bind at some other site while the immobilized inhibitor occupies the active site. Such substrate-binding sites that are distinct from the active site generally are called exosites (19). Because the cleavage site of a γ-secretase substrate is thought to interact with the exosite before entry into the active site, we dubbed this exosite the initial binding (or docking) site (10).
Recently, we reported the design of new inhibitor prototypes, short peptides derived from the intramembrane domain of amyloid β-protein (Aβ) precursor (APP) (20). Based on evidence that the APP transmembrane domain adopts a helical conformation upon initial interaction with γ-secretase (17, 21), we identified short helical peptides that directly inhibit γ-secretase both in cell-based and cell-free assays. We found that d-enantiomers of these peptides were equally or more potent than their l-counterparts, and a 10-residue helical d-peptide was identified as the most potent first-generation inhibitor. Importantly, we discovered that this molecule does not prevent the labeling of γ-secretase by an active-site-directed photoprobe (22). Other evidence demonstrated that although a transition-state analogue does not interfere with the association between APP substrate and PS, helical peptide inhibitors do (20). These results indicated that the helical peptide inhibitors apparently bind to γ-secretase at the substrate-docking site. However, the γ-secretase component on which this site resides has been unknown. To identify the docking site, we transformed helical peptide inhibitors into photoaffinity reagents and used these molecules to probe γ-secretase. The location of the docking site provides critical insights into the mechanism of γ-secretase, with implications for the entire class of intramembrane proteases.
Methods
Compound Synthesis and Cell-Free Aβ Production. Photoprobe III-63 and analogue III-31-C, N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT), and the benzodiazepine-containing Compound E were synthesized as described in refs. 10 and 22. Ten-residue d-peptides were prepared by solution-phase peptide synthesis using d-amino acids and the achiral α-aminoisobutyric acid (20), and 13-residue d-peptides were made by solid-phase peptide synthesis using Rink resin. Photoprobes were prepared by replacing the sixth residue of the peptides with 4-benzoyl-d-phenylalanine (Bpa; Sigma). Biotin (Sigma) was coupled to the N terminus of the d-13 after it was elongated with the Aun linker (aminoundecanoic acid; Sigma) to yield d-13-Bpa-Bt, and d-10-Bpa-Bt was obtained by coupling of d-10-Bpa to N-hydroxysuccinimide–discrete PEG4–biotin (Quanta Biodesign, Powell, OH). All compounds were HPLC-purified, analyzed by MALDI-TOF, and dissolved in DMSO to make stock solutions. The cell-free assay was performed as described in ref. 10. IC50 values were estimated by plotting the ELISA data on sigmaplot (Systat, Point Richmond, CA) and fitting it to a sigmoidal function.
Preparation of Cell Lysates and Photoaffinity Labeling. HeLa or γ-30 cells (8) were lysed in buffer containing 50 mM Mes (pH 6.0), 150 mM NaCl, 5 mM MgCl2, 5 mM CaCl2, 1% 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate (CHAPSO), and protease inhibitors (Roche). The cell homogenate was centrifuged at 20,000 × g, and the supernatant was spun at 100,000 × g for 1 h. The final supernatant was diluted with Pipes buffer (50 mM Pipes, pH 7.0/150 mM NaCl/5 mM MgCl2/5 mM CaCl2) to a final 0.25% CHAPSO solution. The photolabeling was performed essentially as described in refs. 15 and 22. The labeled proteins were eluted with 2× sample buffer (pH 6.8) and were detected by Western blotting using the following antibodies: Ab14 [presenilin-1 (PS1)-NTF, 1:2,000 dilution; from S. Gandy, Thomas Jefferson University, Philadelphia], 13A11 (PS1-CTF, 5 μg/ml; from D. Selkoe, Harvard Medical School), PC235 (PS2-CTF, 1:1,000; Chemicon), M2 Flag (Flag-Pen2, 1:1,000; Sigma), 3F10 (HA-Aph1, 1:2,000; Roche), and N1660 (NCT, 1:1,000; Sigma).
Results
Design and Activity of Helical Peptide Photoprobes for γ-Secretase. We previously reported the structures and cell-free inhibitory potencies of helical peptide inhibitors, transition-state analogue III-31-C, and two other types of inhibitors used in this study, DAPT and Compound E (22) (Table 1).
Table 1. Chemical structures and inhibitory properties of γ-secretase inhibitors and photoprobes used in this study.
| Name | Structure | In vitro, nM |
|---|---|---|
| d–10 | Boc–Val–Gly–Aib–Val–Val–Ile–Aib–Thr*–Val–Aib–OMe | 100 |
| d–13 | Ac–Val–Gly–Aib–Val–Val–Ile–Aib–Thr*–Val–Aib–Val–Val–Aib–NH2 | 30 |
| III–31–C |  |
20 |
| DAPT | 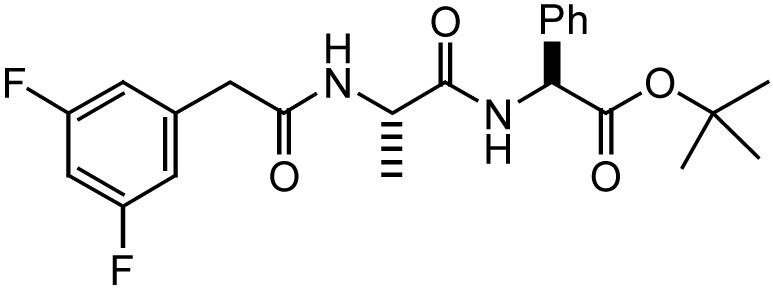 |
10 |
| Compound E |  |
3 |
| d–10–Bpa–Bt | Bt–Link–Val–Gly–Aib–Val–Val–Bpa–Aib–Thr*–Val–Aib–OMe | 100 |
| d–13–Bpa–Bt | Bt–Link–Val–Gly–Aib–Val–Val–Bpa–Aib–Thr*–Val–Aib–Val–Val–Aib–NH2 | 10 |
| III–63 |  |
10 |
To generate photoprobes, we modified a 10-residue helical d-peptide (called compound 11 in ref. 20, but called d-10 in this report for clarity). Bpa was incorporated as the sixth residue, and biotin was installed at the N terminus by means of a linker to yield photoprobe d-10-Bpa-Bt (Table 1). The side chain of the Bpa residue is benzophenone, a commonly used photoreactive moiety that covalently inserts into the closest C–H bond (within 3 Å) upon irradiation at 350 nm (23). Biotin allows isolation of the labeled species with avidin-based beads. A 13-residue helical d-peptide inhibitor, based on a highly potent prototype (24), was modified in a similar way to yield the photoprobe d-13-Bpa-Bt. Active-site-directed photoprobe III-63 is based on a transition-state analogue inhibitor III-31-C, a (hydroxyethyl)urea peptidomimetic, and this probe likewise contains benzophenone and biotin (22).
Photoactivatable Helical 10-Residue d-Peptide Inhibitor of γ-Secretase Covalently Labels PS1 Heterodimer. To detect every component of the γ-secretase complex, we used the CHO cell line γ-30, in which PS1, Flag-Pen-2, and HA-Aph-1 are overexpressed (8). CHO cells contain excess endogenous NCT, and γ-30 cells possess elevated levels of γ-secretase activity. The CHAPSO-solubilized lysates were incubated with the d-10-Bpa-Bt photoprobe in the presence or absence of the parent helical peptide d-10 and irradiated at 350 nm. The photolabeled species carrying biotin were pulled down by absorption to streptavidin beads and analyzed by Western blot. We detected a band at ≈32 kDa, which was identified as PS1-NTF, because this protein was crossreactive with anti-PS1-NTF antibody and was of the appropriate size (Fig. 1a, lane 1). The photoprobe also labeled a small amount of PS1-CTF, identified with anti-PS1-CTF antibody upon a longer exposure. Little or no photolabeled PS1 species were observed in the presence of excess unmodified helical peptide d-10 (Fig. 1a, lane 2). We did not observe labeling of any other γ-secretase components by this photoprobe, even upon longer exposures. These results strongly suggest that the d-10-Bpa-Bt photoprobe directly and specifically binds to the PS1 heterodimer. Labeling of endogenous PS1 by this photoprobe was also seen upon irradiation in HeLa cell lysates (see lane 1 in Fig. 1b, as discussed below), which excludes the possibility that tags and overexpressed protein levels in γ-30 cell lysates lead to artifactual inhibitor binding.
Fig. 1.

Photoactivatable 10-residue helical d-peptide inhibitor of γ-secretase covalently labels PS1 heterodimer at a site distinct from the active site. (a) d-10-Bpa-Bt (500 nM) was incubated with γ-30 lysates in the absence or presence of d-10 (10 μM), and the samples were irradiated at 350 nm. Biotinylated proteins were precipitated with immobilized streptavidin and blotted for NCT, PS1-NTF, PS1-CTF, the Flag-tag of Pen-2, and the HA-tag of Aph-1. Western blotting reveals that PS1-NTF is specifically tagged, but not NCT, APH-1, or Pen-2. Small traces of the labeled PS1-CTF also were observed that were only visible upon longer exposure. (b) d-10-Bpa-Bt (500 nM) was incubated with HeLa lysates in the absence or presence of d-10 (10 μM), L-10 (10 μM), and III-31-C (5 μM). (c) d-10-Bpa-Bt (500 μM) was incubated with HeLa lysates in the absence or presence of Compound E (5 μM) and DAPT (5 μM).
We previously reported that photolabeling by the active-site-directed photoprobe III-63 (Table 1), which specifically binds to PS1 heterodimer, is prevented by a number of structurally diverse γ-secretase inhibitors, including the parent transition-state analogue γ-secretase inhibitor III-31-C (22). Notably, the 10-residue helical peptide inhibitor d-10 (IC50 = 100 nM in vitro) did not prevent the III-63 photoprobe from binding PS1 even at a concentration 1,000-fold above its IC50, which was the first indication that helical peptides bind to a site distinct from the active site (22). This time, we analyzed the effect of III-31-C on the ability of the 10-residue peptide photoprobe to label PS1. In this displacement assay performed with HeLa cell lysates, III-31-C (IC50 = 20 nM) was used at 5 μM to achieve at least 10-fold excess over the concentration of the photoprobe (IC50 = 100 nM). Even under these conditions, compound III-31-C did not prevent the ability of d-10-Bpa-Bt to bind PS1 covalently (Fig. 1b). This result suggests that the transition-state analogue, which is an active-site-directed inhibitor, neither binds to the same site as the d-10 peptide nor allosterically changes the d-10 binding site, at least not in a manner that prevents labeling by the d-10 photoprobe. These observations provide compelling biochemical evidence for the existence of two separate sites on γ-secretase, both of which are pharmacologically relevant and located on PS1. Importantly, an l-peptide l-10 (IC50 = 30 nM) also substantially decreased photolabeling by d-10-Bpa-Bt, although somewhat less efficiently than by parent peptide d-10 (Fig. 1b). This result implies that d- and l-enantiomeric helical peptides bind to the same site on γ-secretase.
In addition, we analyzed the ability of two other highly potent γ-secretase inhibitors, non-transition-state analogues Compound E and DAPT (in vitro IC50 = 3 nM and 10 nM, respectively), to block the labeling of PS1 by photoprobe d-10-Bpa-Bt. For this displacement assay, we used HeLa lysates and concentrations of the competitors at 10-fold excess over the concentration of the probe. Neither of these potent compounds prevented the photolabeling as effectively as the parent d-10 peptide, although the presence of either caused some decrease (Fig. 1c). This result indicates that Compound E and DAPT probably do not bind γ-secretase at the same site as helical peptide d-10, although they might affect helical peptide binding allosterically.
Photoactivatable 13-Residue Helical d-Peptide Inhibitor Labels PS1-NTF and -CTF, but Its Binding Mode Differs from That of the 10-Residue Photoprobe. We next gained an unexpected insight into γ-secretase when we used the longer and more potent photoprobe d-13-Bpa-Bt, which was based on our recently discovered subnanomolar helical d-peptide inhibitor of γ-secretase (24). Surprisingly, although this molecule differs from the first-generation peptide d-10 only by three extra amino acids, this elongation resulted in a sharp increase of inhibitory potency toward γ-secretase (IC50 = 0.140 nM). In the present work, convenience of solid-phase synthesis led us to modify the termini of this subnanomolar helical d-peptide inhibitor, incorporating N-acetyl instead of N-butoxycarbonyl and C-amide instead of methyl ester, and these modifications resulted in some loss of potency (Compound d-13 in Table 1; IC50 = 30 nM). The longer d-13-Bpa-Bt probe (IC50 = 10 nM) was nevertheless 10-fold more potent than d-10-Bpa-Bt (IC50 = 100 nM). Photoactivation of d-13-Bpa-Bt in γ-30 cell lysates yielded biotinylated PS1-NTF and -CTF (Fig. 2a). Little or no photolabeled PS1 subunits were observed in the presence of excess parent peptide d-13, demonstrating specific binding. Notably, the observed bands run a little higher than PS1-NTF and -CTF themselves in the control samples (Fig. 2a, lane 3), indicating that these species carry additional mass from the hydrophobic photoprobe. As with d-10-Bpa-Bt, no other components of γ-secretase were labeled by this photoprobe, and these results strongly suggest that d-13-Bpa-Bt selectively binds to the γ-secretase complex at the interface of PS1 heterodimer. Apparently, no other components of γ-secretase are within 3 Å of the photosensitive side chain of the Bpa residue in the bound peptide (23)
Fig. 2.

Photoactivatable 13-residue helical d-peptide inhibitor of γ-secretase covalently labels PS1-NTF and -CTF, but differently from 10-residue helical photoprobe. (a) d-13-Bpa-Bt (50 nM) was incubated with γ-30 lysates in the absence or presence of d-13 (250 nM), and the samples were treated and analyzed as in Fig. 1a. (b) d-13-Bpa-Bt (50 nM) was incubated with HeLa lysates in the absence or presence of III-31-C (250 nM). (c) d-13-Bpa-Bt (50 nM) was incubated with HeLa lysates in the absence or presence of d-13 (250 nM). Immunoblotting was performed with antibodies directed against the PS2 CTF. (d) d-13-Bpa-Bt (100 nM) was incubated with HeLa lysates in the absence or presence of Compound E (2 μM) and DAPT (2 μM). (e) III-63 (50 nM) was incubated with HeLa lysates in the absence or presence of d-13 (250 nM) and III-31-C (250 nM). (f)(Left) d-13-Bpa-Bt (100 nM) was incubated with HeLa lysates in the absence or presence of d-10 (2 μM). (Right) d-10-Bpa-Bt (500 nM) was incubated with HeLa lysates in the absence or presence of d-13 (5 μM) and d-10 (10 μM).
Photoprobe d-13-Bpa-Bt also labeled the PS1 heterodimer of endogenous γ-secretase, when photolabeling was repeated in HeLa cell lysates (Fig. 2b, lane 1). Additionally, we identified another population of labeled species in HeLa lysates that was crossreactive with antibodies against PS2-CTF (Fig. 2c). The photoprobe labeled PS2-CTF specifically, because excess non-photoreactive d-13 prevented the covalent labeling by d-13-Bpa-Bt. This result and the homology between PS1 and PS2 (67% identity) (25, 26) suggest that PS2 contains a similar helical peptide binding site as PS1.
Surprisingly, in contrast to the results seen with the d-10-Bpa-Bt, the extent of labeled PS species recovered was dramatically reduced when photolabeling with d-13-Bpa-Bt was performed in the presence of excess transition-state analogue III-31-C (Fig. 2b, lane 2). Also in contrast to results with the d-10-Bpa-Bt labeling, Compound E and DAPT substantially reduced d-13-Bpa-Bt labeling of the PS1 heterodimer (Fig. 2d). This observation indicated that III-31-C, Compound E, and DAPT bind either to the same site or overlapping sites as d-13-Bpa-Bt or that their binding allosterically changes the enzyme conformation so that the photoprobe cannot bind to the protease.
To clarify the unexpected relationship between PS1 binding by the transition-state analogue and by the 13-residue helical peptide, we tested d-13 in the displacement assay with III-63. d-13 prevented the ability of III-63 to photolabel PS1 and almost as efficiently as the unmodified parent III-31-C itself (Fig. 2e). Clearly, this observation is consistent with the ability of the transition-state analogue to prevent the photolabeling by d-13-Bpa-Bt. However, this result is strikingly different from observations with d-10, which was unable to affect labeling by the active-site-directed photoprobe III-63, even at a concentration 1,000 times above its IC50 (22). This difference is puzzling but suggests that despite the close structural similarities between d-10 and d-13, these two helical peptides of different length have different binding modes on γ-secretase.
Ten- and 13-Residue Helical Peptide Inhibitors Compete with Each Other for Photolabeling of PS1. To clarify the unexpected difference in observed behavior between the d-10 and d-13, we analyzed their ability to compete with each other for binding to the protease. In HeLa cell lysates, we observed that d-10 (at 2 μM; 20 times its IC50; 40-fold over photoprobe) dramatically reduced labeling by d-13-Bpa-Bt (Fig. 2f Left). d-13 (5 μM; 160 times its IC50, but only 10-fold over photoprobe) also substantially decreased the labeling by d-10-Bpa-Bt, just as efficiently as the unmodified parent d-10 did (Fig. 2f Right). This observation shows that both peptides compete with each other for the labeling of PS1, suggesting that they bind to the same site (e.g., the initial binding site) or to overlapping ones. This finding eliminates the possibility that the 13-residue peptide solely inhibits γ-secretase by directly binding to the active site, bypassing the docking site altogether. If that were the case, then this molecule should not interfere with agents binding to the initial binding site, such as d-10. However, the possibility remains that there are two populations of differently bound d-13, one binding to the active site and another binding to the initial binding site (see below).
Alzheimer-Associated PS1 Mutation L166P Affects Photolabeling by Active-Site-Directed III-63 but Not by Helical Peptide Photoprobes. To resolve this structural puzzle and further test whether the active-site-directed inhibitor and the 13-residue helical peptide share the same site of binding, we analyzed the ability of III-63, d-13-Bpa-Bt, and d-10-Bpa-Bt to photolabel PS mutants linked to familial Alzheimer's disease (FAD). We chose two PS1 mutants, L166P and G384A, that cause the most profound elevation of the proportion of the more aggregation-prone 42-residue form of Aβ (27, 28). CHAPSO-solubilized membranes isolated from HEK cell lines stably expressing L166P-PS1, G384A-PS1, or wild-type PS1 (PS1-wt) were incubated and irradiated with III-63, d-10-Bpa-Bt, or d-13-Bpa-Bt. The samples were normalized by the amount of PS1 fragments (Fig. 3a). We observed that both helical peptide photoprobes labeled PS1 heterodimer to a similar degree in all three samples (Fig. 3 b and c), although d-13-Bpa-Bt appeared to label the CTF of the PS1 mutants somewhat less effectively. This result indicated that neither the L166P-PS1 nor the G384A-PS1 mutation substantially affected the conformation of the sites at which these helical peptides bind, at least not to the degree that dramatically alters labeling by the photoaffinity probes. The labeling by the active-site-directed III-63, however, displayed a very different pattern. Only a very small amount of L166P-PS1 was labeled by this photoprobe, compared with the amount of PS1-wt and G384A-PS1 labeled, although the latter was not labeled as efficiently as PS1-wt (Fig. 3d). Additionally, we tested the enzymatic activity of these mutants in a cell-free γ-secretase assay (Fig. 3e) and found that the activity pattern followed the pattern of photolabeling by III-63. That is, the enzymatic activity of γ-secretase containing L166P-PS1 at its core is dramatically decreased compared with the activity of G384A-PS1 and PS1-wt. The decrease in APP intracellular domain production by these mutants is consistent with previous reports (27–29). Significantly, our observations taken together suggest that the L166P-PS1 mutation, and perhaps G384A-PS1 as well, affects the binding site of the transition-state analogue (that is, the active site of γ-secretase). At the same time, these results imply that the active-site-directed and the helical peptide photoprobes bind to different sites, and they suggest that it is unlikely that a population of 13-residue peptide solely binds to the active site. Otherwise, one would observe a substantially diminished ability of 13-residue photoprobe to label the distorted active site of L166P-PS1.
Fig. 3.

FAD-PS1 mutation L166P affects the photolabeling by the active-site-directed III-63, with little or no effect on both helical peptide photoprobes. (a) CHAPSO-solubilized membranes derived from HEK cells stably expressing PS1-L166P, PS1-G384A, and PS1-wt were normalized by the amount of PS1. (b–d) Membranes were incubated with d-10-Bpa-Bt (500 nM) (b), d-13-Bpa-Bt (500 nM) (c), and III-63 (500 nM) (d). Samples were treated and analyzed as in Fig. 1a. (e) γ-Secretase activity measured in the solubilized membranes. C100-Flag was incubated with membranes for 4 h, and APP intracellular domain (AICD)-Flag (shown), the product of the proteolysis, was detected with anti-Flag antibody.
Discussion
Intramembrane proteases are an emerging class of enzymes (3), and how these proteases interact with their integral membrane substrates is mysterious. Considering that both the site of substrate cleavage and the protease residues implicated in catalysis typically reside within transmembrane domains, these proteases apparently catalyze hydrolysis within the confines of the lipid bilayer. Because the active site requires water and consists of hydrophilic catalytic residues (e.g., aspartates), it should be sequestered away from the hydrocarbon chains of lipids. Indeed, γ-secretase apparently possesses an initial substrate-binding site (“docking site”) that is distinct from the active site (10). By using affinity reagents based on helical peptide substrate mimics, we have identified the location of the initial binding site on γ-secretase, and this finding has clear implications for how substrate accesses the internal active site and the role of PS vis-à-vis its partners in the activity of the protease.
The helical peptide inhibitors of γ-secretase were designed from first principles; the single transmembrane domain of the APP substrate should be in a helical conformation before interaction with the protease, and modeling (11) and mutagenesis (21) support this contention. Indeed, designed helical peptides can be highly potent inhibitors of γ-secretase (20, 24). Prior evidence further demonstrated that 10-residue peptides such as d-10 inhibit the protease by a mechanism different from that of transition-state analogues and in a manner consistent with interaction with the substrate-docking site (20, 22). Intriguingly, we found in the present study that d-10 binds directly to PS, at the interface between the NTF and CTF subunits (Fig. 4a Top). Although transition-state analogues also bind directly to the heterodimeric interface of PS, this site is nevertheless clearly distinct from the site of d-10 binding (Fig. 4a Middle). Transition-state analogue inhibitor III-31-C cannot block labeling by the d-10 photoprobe, and d-10 cannot block labeling by the photoprobe based on III-31-C. Unlike III-31-C, d-10 can prevent the association of APP substrates with PS (20). Thus, the identification of the PS NTF/CTF interface as the direct binding site for d-10 strongly suggests that this part of PS is the location of the substrate-docking site on the γ-secretase complex.
Fig. 4.

Proposed models for inhibitors (a) and substrate (b) interactions with γ-secretase. (a)(Top) Substrate-based d-10 helical peptide interacts with PS1 at the initial binding site. (Middle) Transition-state analogue III-31-C binds to PS1 NTF/CTF heterodimer at the active site, located internally and containing two aspartates (denoted as “D”). (Bottom) d-13 peptide interacts with both active and initial binding sites. (b) Upon docking to the initial binding site, a substrate passes through PS1 subunits fully (path A) or partially (path B) to access the nearby active site.
The identification of the initial substrate-binding site also implies a path for substrate entry into the active site (Fig. 4b). Because the docking and the active sites are both located at the heterodimeric interface of PS, all (path A) or part (path B) of the substrate apparently passes between these two subunits to access the presumptive catalytic aspartates. The longer and more potent 13-residue peptide provides important information about the proximity of the docking site to the active site and offers further insight into how the enzyme might handle substrates. This peptide only differs from d-10 by the extension of three residues to the C terminus, and the two peptides apparently compete for the same site on PS. Thus, d-13 likewise binds to the docking site. However, the ability of d-13 to prevent labeling by the transition-state-mimicking photoprobe suggests that this peptide also can interact with the active site.
Although other explanations are possible, two primary scenarios for 13-residue peptide's binding mode are consistent with all of the results: (i) d-13 binds to the initial binding site but protrudes into the active site because of its three additional amino acids; or (ii) it binds to the initial binding site and allosterically affects the active site by means of the interaction of its three extra amino acids with the protein. We favor the former possibility: The extra three residues in d-13 allow part of this peptide to extend beyond the d-10 binding site into the active site. Thus, the active site is apparently in close proximity to the docking site (i.e., less than or equal to the length of three residues). This notion is consistent with the finding that PS1-CTF contributes to both the active and the docking sites: The CTF is small and, more importantly, has only two transmembrane domains, one of which contains one of the essential aspartates. The close proximity of the docking and active sites suggests that only part of the transmembrane substrate may need to insert into the active site, with the rest remaining in the docking site (Fig. 4b, path B).
The observation that extension of a docking-site-directed peptide inhibitor leads to entry into the active site with improved potency has a precedent in studies on the serine protease FVIIa (30). In this case, elongation of a peptide inhibitor directed to a distinct binding site (i.e., an exosite) in close proximity to the active site yields a much more potent inhibitor with an altered mode of binding. The longer peptide binds to the exosite, with the elongated end inserting into the active site. In this manner, such peptide inhibitors can be considered “molecular rulers” to measure the proximity of the active site to the exosite. Because the addition of only three residues to the helical peptide γ-secretase inhibitor dramatically improves potency and leads to displacement of a transition-state analogue inhibitor, the initial binding site and the active site are apparently in very close proximity and possibly even overlap.
With affinity probes directed to two different sites on PS, we also could determine the effects of AD-causing mutations in PS1 on the active and docking sites. Of the >100 missense mutations in PS1, L166P is the most severe, causing the most dramatic increase in the critical ratio of 42-residue vs. 40-residue Aβ and the development of clinical AD at the youngest age (18 years) (27). As previously reported, this PS1 mutant has substantially reduced total γ-secretase activity. Along with this reduced activity, we observed substantially less labeling by the transition-state analogue photoprobe. Thus, the L166P mutation, located in TM3 of PS1, causes a change in the topography of the γ-secretase active site, which includes aspartates located in TM6 and TM7. TM3 also may be part of the active site, or, alternatively, the L166P mutation may be located distally and lead to a conformational change in the active site through allosteric means. The G384A mutation (28), directly adjacent to the essential D385 on TM7, also causes some reduction in activity and apparently reduces labeling by the transition-state photoprobe. Determining whether a change in the shape of the active site is a common feature of AD-causing PS mutations will require careful examination of many mutations, and the possibility remains that some other mutations may alter labeling by helical peptide photoprobes directed to the docking site. Nevertheless, this study provides, to our knowledge, the first evidence that the most severe of these mutations can result in a structural change in the γ-secretase active site.
This study also strongly suggests that PS is the γ-secretase component that directly interacts with the substrate transmembrane domain. No other components of this protease complex are labeled by either transition-state analogues or substrate-based helical peptides. Consistent with this finding, no AD-causing mutations have yet been identified in NCT, Aph-1, or Pen-2. Moreover, signal peptide peptidase, a distantly related PS homologue, does not require cleavage into two fragments or other protein cofactors (31). Taken together, these findings suggest that the primary role of NCT, Aph-1, and Pen-2 is to render PS competent for proteolysis. How these other components accomplish this goal should be the object of future studies employing molecular, biochemical, and structural approaches.
Acknowledgments
We thank S. Gandy for Ab14 antibody; D. Teplow, S. Urban, and D. Walsh for helpful comments on the manuscript; and J. Stahle J. Sears, and W. Xia (all three of Harvard Medical School) for Aβ ELISAs. This work was supported by National Institutes of Health Grants NS41355 and AG17574 (to M.S.W.), Alzheimer's Association Grant IIRG-02-4047 (to M.S.W.), and a Harvard Medical School Lefler Fellowship (to A.Y.K.).
Author contributions: A.Y.K., F.B., C.D., and M.S.W. designed research; A.Y.K., F.B., and C.D. performed research; A.Y.K., F.B., C.D., and M.S.W. analyzed data; and A.Y.K. and M.S.W. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Aβ, amyloid β-protein; APP, Aβ precursor; Bpa, 4-benzoyl-d-phenylalanine; CHAPSO, 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate; CTF, C-terminal fragment; NTF, N-terminal fragment; DAPT, N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester; NCT, nicastrin; PS, presenilin; PS1, presenilin-1; wt, wild type.
References
- 1.Esler, W. P. & Wolfe, M. S. (2001) Science 293, 1449-1454. [DOI] [PubMed] [Google Scholar]
- 2.De Strooper, B. (2003) Neuron 38, 9-12. [DOI] [PubMed] [Google Scholar]
- 3.Wolfe, M. S. & Kopan, R. (2004) Science 305, 1119-1123. [DOI] [PubMed] [Google Scholar]
- 4.Yu, G., Nishimura, M., Arawaka, S., Levitan, D., Zhang, L., Tandon, A., Song, Y. Q., Rogaeva, E., Chen, F., Kawarai, T., et al. (2000) Nature 407, 48-54. [DOI] [PubMed] [Google Scholar]
- 5.Goutte, C., Tsunozaki, M., Hale, V. A. & Priess, J. R. (2002) Proc. Natl. Acad. Sci. USA 99, 775-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Francis, R., McGrath, G., Zhang, J., Ruddy, D. A., Sym, M., Apfeld, J., Nicoll, M., Maxwell, M., Hai, B., Ellis, M. C., et al. (2002) Dev. Cell 3, 85-97. [DOI] [PubMed] [Google Scholar]
- 7.Takasugi, N., Tomita, T., Hayashi, I., Tsuruoka, M., Niimura, M., Takahashi, Y., Thinakaran, G. & Iwatsubo, T. (2003) Nature 422, 438-441. [DOI] [PubMed] [Google Scholar]
- 8.Kimberly, W. T., LaVoie, M. J., Ostaszewski, B. L., Ye, W., Wolfe, M. S. & Selkoe, D. J. (2003) Proc. Natl. Acad. Sci. USA 100, 6382-6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edbauer, D., Winkler, E., Regula, J. T., Pesold, B., Steiner, H. & Haass, C. (2003) Nat. Cell Biol. 5, 486-488. [DOI] [PubMed] [Google Scholar]
- 10.Esler, W. P., Kimberly, W. T., Ostaszewski, B. L., Ye, W., Diehl, T. S., Selkoe, D. J. & Wolfe, M. S. (2002) Proc. Natl. Acad. Sci. USA 99, 2720-2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolfe, M. S., Xia, W., Moore, C. L., Leatherwood, D. D., Ostaszewski, B., Donkor, I. O. & Selkoe, D. J. (1999) Biochemistry 38, 4720-4727. [DOI] [PubMed] [Google Scholar]
- 12.Shearman, M. S., Beher, D., Clarke, E. E., Lewis, H. D., Harrison, T., Hunt, P., Nadin, A., Smith, A. L., Stevenson, G. & Castro, J. L. (2000) Biochemistry 39, 8698-8704. [DOI] [PubMed] [Google Scholar]
- 13.Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T. & Selkoe, D. J. (1999) Nature 398, 513-517. [DOI] [PubMed] [Google Scholar]
- 14.Wolfe, M. S. & Haass, C. (2001) J. Biol. Chem. 276, 5413-5416. [DOI] [PubMed] [Google Scholar]
- 15.Li, Y. M., Xu, M., Lai, M. T., Huang, Q., Castro, J. L., DiMuzio-Mower, J., Harrison, T., Lellis, C., Nadin, A., Neduvelil, J. G., et al. (2000) Nature 405, 689-694. [DOI] [PubMed] [Google Scholar]
- 16.Esler, W. P., Kimberly, W. T., Ostaszewski, B. L., Diehl, T. S., Moore, C. L., Tsai, J.-Y., Rahmati, T., Xia, W., Selkoe, D. J. & Wolfe, M. S. (2000) Nat. Cell Biol. 2, 428-434. [DOI] [PubMed] [Google Scholar]
- 17.Wolfe, M. S., De Los Angeles, J., Miller, D. D., Xia, W. & Selkoe, D. J. (1999) Biochemistry 38, 11223-11230. [DOI] [PubMed] [Google Scholar]
- 18.Beher, D., Fricker, M., Nadin, A., Clarke, E. E., Wrigley, J. D., Li, Y. M., Culvenor, J. G., Masters, C. L., Harrison, T. & Shearman, M. S. (2003) Biochemistry 42, 8133-8142. [DOI] [PubMed] [Google Scholar]
- 19.Roberge, M., Santell, L., Dennis, M. S., Eigenbrot, C., Dwyer, M. A. & Lazarus, R. A. (2001) Biochemistry 40, 9522-9531. [DOI] [PubMed] [Google Scholar]
- 20.Das, C., Berezovska, O., Diehl, T. S., Genet, C., Buldyrev, I., Tsai, J. Y., Hyman, B. T. & Wolfe, M. S. (2003) J. Am. Chem. Soc. 125, 11794-11795. [DOI] [PubMed] [Google Scholar]
- 21.Lichtenthaler, S. F., Wang, R., Grimm, H., Uljon, S. N., Masters, C. L. & Beyreuther, K. (1999) Proc. Natl. Acad. Sci. USA 96, 3053-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kornilova, A. Y., Das, C. & Wolfe, M. S. (2003) J. Biol. Chem. 278, 16470-16473. [DOI] [PubMed] [Google Scholar]
- 23.Dorman, G. & Prestwich, G. D. (1994) Biochemistry 33, 5661-5673. [DOI] [PubMed] [Google Scholar]
- 24.Bihel, F., Das, C., Bowman, M. J. & Wolfe, M. S. (2004) J. Med. Chem. 47, 3931-3933. [DOI] [PubMed] [Google Scholar]
- 25.Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., et al. (1995) Nature 375, 754-760. [DOI] [PubMed] [Google Scholar]
- 26.Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D. M., Oshima, J., Pettingell, W. H., Yu, C. E., Jondro, P. D., Schmidt, S. D., Wang, K., et al. (1995) Science 269, 973-977. [DOI] [PubMed] [Google Scholar]
- 27.Moehlmann, T., Winkler, E., Xia, X., Edbauer, D., Murrell, J., Capell, A., Kaether, C., Zheng, H., Ghetti, B., Haass, C. & Steiner, H. (2002) Proc. Natl. Acad. Sci. USA 99, 8025-8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steiner, H., Kostka, M., Romig, H., Basset, G., Pesold, B., Hardy, J., Capell, A., Meyn, L., Grim, M. L., Baumeister, R., et al. (2000) Nat. Cell Biol. 2, 848-851. [DOI] [PubMed] [Google Scholar]
- 29.Song, W., Nadeau, P., Yuan, M., Yang, X., Shen, J. & Yankner, B. A. (1999) Proc. Natl. Acad. Sci. USA 96, 6959-6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maun, H. R., Eigenbrot, C. & Lazarus, R. A. (2003) J. Biol. Chem. 278, 21823-21830. [DOI] [PubMed] [Google Scholar]
- 31.Weihofen, A., Binns, K., Lemberg, M. K., Ashman, K. & Martoglio, B. (2002) Science 296, 2215-2218. [DOI] [PubMed] [Google Scholar]