

Abstract
Purpose of review
Up to twenty percent of patients with sarcoidosis develop pulmonary fibrosis, transforming an often benign disease into a highly morbid and potentially fatal one. We highlight the fibrotic pulmonary sarcoidosis phenotype as an area of intense clinical and translational investigation, review recent developments in treatment, and provide a roadmap for future research in sarcoidosis associated pulmonary fibrosis.
Recent findings
Granulomatous inflammation in a lymphatic distribution is the hallmark finding of pulmonary sarcoidosis and the nidus for fibrosis. Recent research demonstrates that fibrotic sarcoidosis begins in the setting of persistent, uncontrolled inflammation, and is aided by pro-fibrotic genetic features and immune responses. Comparison to other fibrotic lung diseases also reveals key features that inform our understanding of common pathways in fibrosis.
Summary
Understanding the mechanisms of fibrotic transformation in sarcoidosis enhances clinical care and facilitates development of novel therapeutic options. The impact of these findings in fibrotic sarcoidosis may be amplified through application to other interstitial lung diseases marked by inflammatory to fibrotic transformation. Important aspects of clinical management of fibrotic sarcoidosis include surveillance for co-morbidities, such as pulmonary hypertension, airway disease, and infection, and assessment for pulmonary disease activity that may benefit from immunosuppression.
Keywords: Sarcoidosis, Granulomatous inflammation, Pulmonary fibrosis, Bronchiectasis
Introduction
Granulomas are the key histopathologic features that establish the diagnosis and unify the multisystem signs and symptoms of sarcoidosis. Sarcoid granulomas are sterile, typically non-caseating and often found in a distinctly lymphatic distribution along the bronchovascular bundles of the pulmonary tree (Figure 1). The microarchitecture of the granuloma results from CD4 T cell mediated recruitment and activation of macrophages, which undergo epithelioid transformation and can also form multinucleated giant cells(1). Restriction of the T cell receptor repertoire (2) supports the hypothesis that sarcoidosis is an antigen driven process, although the antigen remains unknown. At least in acute disease, T cell polarization is Th1 predominant, and a host of pro-inflammatory cytokines are up-regulated, including interferon-gamma (IFN-ƴ) and tumor necrosis factor-alpha (TNF-α).
Figure 1.
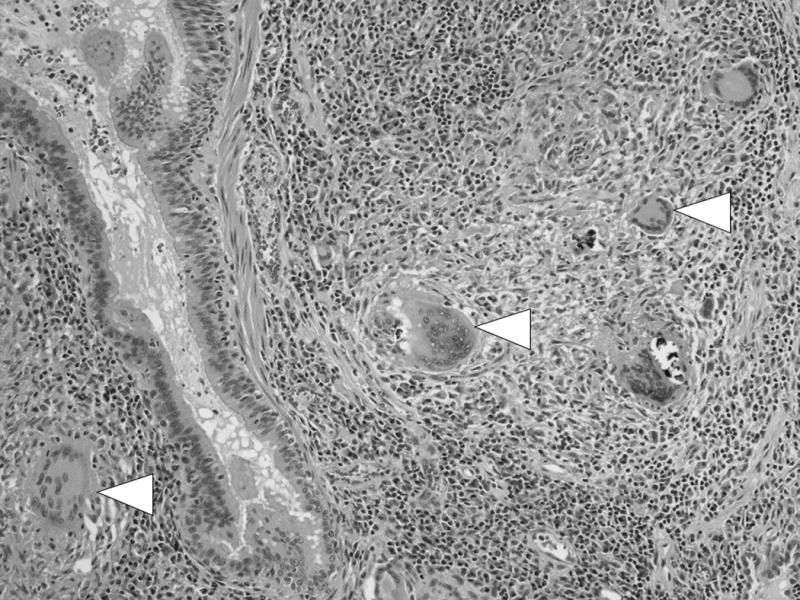
Multiple non-caseating granulomas distributed along the airway (arrowheads). Image courtesy of Aliya Husain MD.
Approximately two-thirds of patients enter clinical remission within years of diagnosis. Their long-term outcomes are generally excellent. However, a third of patients experience persistent or recurrent active disease. This is variably referred to as chronic, severe, advanced, refractory or progressive sarcoidosis (3–6). Regardless of the nomenclature, patients with long-standing disease are most at risk of developing fibrosis. Though the pathophysiology and natural history of fibrotic sarcoidosis are incompletely understood, research on this phenotype has increased and already revealed important immunologic and clinical features. While sarcoidosis can affect a variety of organs, the lung is the most common site of disease with the highest risk of developing fibrosis. As such, this review focuses on fibrotic pulmonary sarcoidosis. In part one, we summarize recent studies on the pathophysiology of fibrotic pulmonary sarcoidosis. We then survey data on clinical features, including imaging, histopathology, and outcomes. We end with a discussion of clinical management. Throughout, we suggest high impact pathways for future research.
Part I: Pathophysiology of fibrotic sarcoidosis
Inflammation both precedes fibrosis and anatomically centers it at the granuloma (Figure 2). The mechanisms driving the transition from inflammation to fibrosis are poorly understood, although two phases are evident. The first is development of chronic inflammation, and the second is fibrotic transformation.
Figure 2.
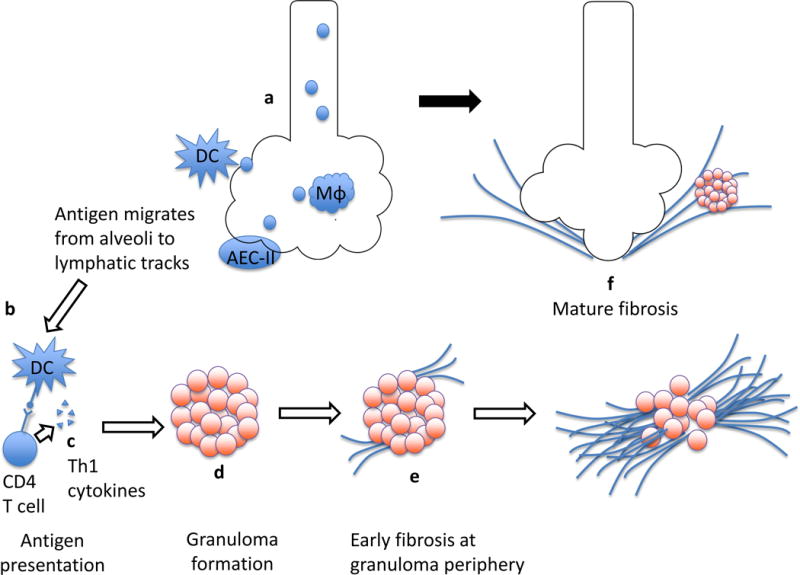
Granulomatous inflammation transitions to fibrosis. The pathophysiology of sarcoidosis begins with exposure to putative inhaled antigen(s) that in the appropriate genetic and epigenetic context initiate an immune cascade via activation of interstitial dendritic cells (DC), alveolar macrophages (Mφ), and type 2 alveolar epithelial cells (AEC-II) (a). Antigen is presented by cells such as DC and recognized by CD4+ T cells (b). The engagement of CD4+ cells leads to their activation and proliferation, and release of an array of pro-inflammatory cytokines (c). Antigen features, Th1 polarization of CD4+ lymphocytes, and the cytokine milieu all likely contribute to the recruitment and organization of macrophages into granulomas within lymphatic tracks (d). In some cases, fibrosis begins at the periphery of sarcoid granulomas (e), which serves to contain if not break up active granulomas. As it extends over time, this deposition of collagen results in mature fibrosis which obliterates parenchymal tissue, resulting in “end-stage” sarcoidosis (f).
Step one: Chronic sarcoidosis
Studies reveal that chronic sarcoidosis is not simply the persistence of acute sarcoidosis. Rather, it is fundamentally distinct from remitting disease, perhaps from the beginning.
Cytokine responses early in disease are distinct for those who experience remission versus chronic disease. TNF-α levels from alveolar macrophages are higher and transforming growth factor-beta (TGF-β) levels are lower for persons with progressive versus stable sarcoidosis at follow-up (7–9). Alveolar macrophages with higher TGF-β release on ex-vivo stimulation are associated with higher rates of spontaneous remission of sarcoidosis (9) These studies have piqued interest in TGF-β as an anti-inflammatory cytokine, with opposite roles in disease resolution and fibrosis. Pabst and colleagues demonstrated TGF-β single nucleotide polymorphism (SNP) frequencies differ for patients with chronic versus acute or limited sarcoidosis (10). These early cytokine responses may set the stage for resolution, versus persistent inflammation and fibrotic change via shifts in Th1 to Th2 mediated T helper cell responses(11).
Chronically activated T effector cells (Teff cells) often display markers of exhaustion, such as up-regulation of programmed death-1 (PD-1). In patients with chronic sarcoidosis, PD-1 was up-regulated on Teff cells. Moreover, PD-1 blockade restored their proliferative capacity (12). One hypothesis is that chronic inflammation results from insufficient, exhausted Teff cell activity with failure to clear antigen. Why antigen would not be cleared early in disease, when Teff cells are presumably potent and fully functional, is not clear. Studies of early CD4+ T cell function and long-term outcomes will be helpful.
In contrast to Teff cells, T regulatory cells (Treg cells) down-regulate immune responses. In patients with stage 2 sarcoidosis, higher serum Treg cell counts correlated with clinical remission after discontinuation of treatment, although the follow-up time of 3 months was short (13). In another recent study, highly activated circulating Treg cells, with up-regulation of inducible co-stimulator (ICOS), were observed in patients with Lofgren’s syndrome, who have a low risk of chronic disease(14). Finally, the ratio of Treg to Teff cells in bronchoalveolar lavage (BAL) was significantly higher in patients who, at follow-up, had inactive or remitted compared to chronic disease (15). This well-designed study was one of the first to characterize lung Treg cells in sarcoidosis and correlate Treg cells with long-term outcomes. However, sarcoidosis was not newly diagnosed, and studies of lung Treg cell activity early in disease will complement this work. Taken together, there seems to be an association between increased Treg cell activity and better long-term outcomes suggesting deficient Treg cell activity likely contributes to chronic sarcoidosis.
Step two: pulmonary fibrosis
Recent results support the hypothesis that fibrosis results from unbridled inflammation. Gene expression profiles of patients with progressive sarcoidosis, most of whom had evidence of fibrosis on imaging, were similar to those with inflammatory hypersensitivity pneumonitis(6). In contrast, expression profiles in progressive sarcoidosis did not resemble those of idiopathic pulmonary fibrosis (IPF). However, fibrosis-promoting gene polymorphisms may also mediate fibrotic transformation. Building on earlier work by Kruit and colleagues(16), a recent study evaluated TGF-β2 and TGF-β3 SNPs among various phenotypes of sarcoidosis, where TGF-β is also a key cytokine in fibrosis(10). TGF-β3 SNP frequencies differed for those with fibrotic and chronic sarcoidosis, defined by persistence of symptoms for at least two years, or two or more flares of disease.
Functional assays complement genetic studies. In a recent investigation, TGF-β levels in BAL did not correlate with Scadding stages (17). However, the cohort with fibrotic sarcoidosis (stage IV) was small, and larger studies are needed. Other cytokines may also be important, including IL-5, which was significantly higher in sera of patients with fibrotic compared to non-fibrotic disease (18) The same study demonstrated a trend towards higher IL-7 levels among fibrotic patients. Further studies examining early versus late cytokine levels will clarify whether these alterations contribute to the pathophysiology, or are merely a marker of fibrotic sarcoidosis.
Comparing sarcoidosis and other fibrotic ILDs
Several recent studies have directly compared fibrotic sarcoidosis to other fibrotic ILDs, with illuminating results. These studies have focused on alveolar epithelial cell repair and plasticity. MRP14, also called S100A9, was significantly elevated in BAL from both sarcoidosis and IPF patients, and correlated with chest radiograph stage in sarcoidosis (19). Xu and colleagues recently examined the effect of S100A9 in human embryo lung fibroblasts, and found that it promoted fibroblast proliferation and type III collagen type deposition (20). Matrix metalloproteinases (MMPs) modify the lung microarchitecture via fibroblast expansion, myofibroblast differentiation, and extracellular matrix accumulation. In contrast to findings of MRP14/S100A9 elevation, serum MMP1 and MMP7 levels were significantly higher in IPF versus fibrotic sarcoidosis (21). Divergent MMP signatures may influence lung remodelling, particularly in relation to development of usual interstitial pneumonia (UIP) in IPF, versus other patterns of fibrotic change seen in non-IPF ILD, including sarcoidosis.
Part II: Clinical features of fibrotic sarcoidosis
Recent efforts to better characterize radiographic and pathologic features of fibrotic pulmonary sarcoidosis both refine our understanding and impact prognosis.
Radiographic features of fibrotic sarcoidosis
Radiographic findings of fibrotic sarcoidosis include thin linear bands, dense fibrotic masses, and cystic lung disease. The degree of parenchymal destruction varies from minimal to extensive (Figure 3). Cysts develop from bronchiectasis or, less commonly, represent typical UIP honeycombing. Abehsera and colleagues were one of the first groups to systematically catalogue radiographic findings in fibrotic sarcoidosis(22). In a more recent review, Nishino and colleagues further highlight that radiographic fibrosis, following the distribution of inflammatory sarcoidosis, is middle and upper lobe predominant, as are honeycomb cysts when they are observed (23).
Figure 3.
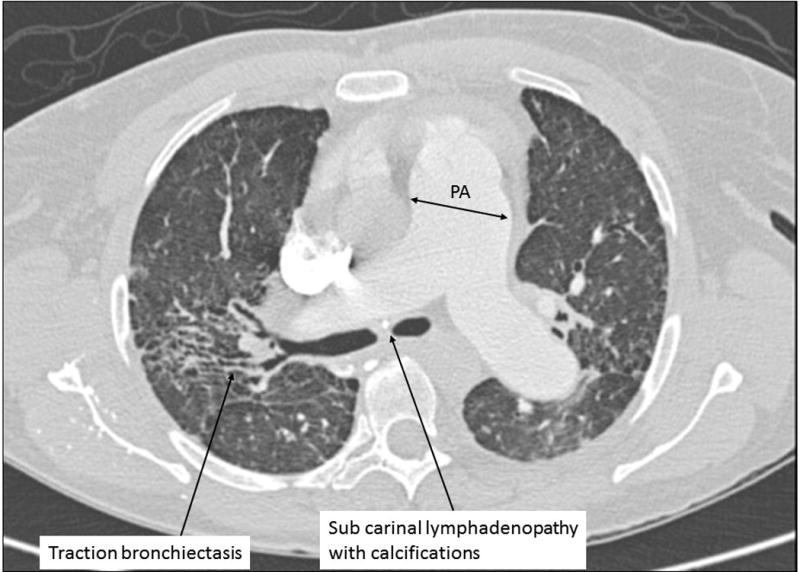
Chest CT of a patient with fibrotic pulmonary sarcoidosis and pulmonary hypertension (axial view at level of carina).
An important question is whether radiographic features in early sarcoidosis can predict eventual development of a pulmonary fibrosis. Scadding chest radiograph stages correlate with long-term disease activity (24). Whether specific parenchymal features, best discerned on CT imaging, can predict development of fibrosis in particular is not known. Polverosi’s group recently assessed the relationship between early radiographic features and chronic disease, a prerequisite to fibrosis (25). Patients with typical sarcoid findings, including nodular lesions in a lymphatic distribution, were more likely to have clinical improvement at follow-up compared to patients with atypical findings. While fibrosis was noted in at least one patient with atypical findings, the outcome of pulmonary fibrosis was not specifically measured. Further longitudinal imaging studies will be instructive.
Histopathology features of fibrotic sarcoidosis
The key histopathologic finding is fibrotic destruction at sites of prior granulomatous inflammation. This process may so extensive that often only a few to no granulomas are observed in end-stage disease. Three recent pathology studies, with somewhat contradictory findings, raise interesting questions about the lung injury response in sarcoidosis. Shigemitsu and colleagues reviewed the explants of seven patients with sarcoidosis(26). Four patients had interstitial lymphocytic infiltrates, an unexpected finding in chronic sarcoidosis. Those with such infiltrates progressed more quickly to end-stage disease (4.8 versus 23.3 years), raising the possibility that a subset of patients with sarcoidosis may be rapid progressors. In addition, two had an overall pattern “indistinguishable from UIP”, defined as the presence of patchy infiltrates with fibroblast foci. Zhang and colleagues also found 3 of 12 patients who underwent lung transplantation for sarcoidosis had either overlapping features of sarcoidosis and UIP, or UIP alone (27) In a review of nine explants of patients with sarcoidosis, honeycombing in sarcoidosis represented bronchiectasis, where central airway dilation was present in all patients(28). The question of whether a UIP injury response is a potential manifestation of sarcoidosis, as it is in other granulomatous diseases such as hypersensitivity pneumonitis, is important to consider, as wound healing responses impact natural history. Further pathology studies will continue to inform our understanding of the pathophysiology, particularly when histopathology features are correlated with imaging and with important clinical features, such as length of disease.
Outcomes in fibrotic sarcoidosis
The development of pulmonary fibrosis in sarcoidosis converts a relatively benign disease into a highly morbid and potentially fatal one (29). Patients who develop fibrotic pulmonary sarcoidosis have increased rates of death, with 75% of mortality due to respiratory causes (30). In one study highlighting racial differences in outcomes among the United States population, African Americans died at an earlier age and were more likely to have pulmonary hypertension, while Caucasian decedents had higher rates of pulmonary fibrosis (31). Along with pulmonary hypertension, pulmonary fibrosis is a key harbinger of worse prognosis and is heavily weighted in the recently reported clinical algorithm for sarcoid outcome prediction (32).
Part III: Clinical management of fibrotic sarcoidosis
Fibrotic sarcoidosis results in numerous cardiopulmonary sequelae that impair quality of life and increase mortality (33). Fibrocavitary parenchymal lung disease results in chronic pulmonary symptoms and exercise limitation and may cause respiratory insufficiency associated with airflow obstruction, bronchial stenosis, and bronchiectasis. Airflow obstruction may be caused by granulomatous airway inflammation or, in patients with advanced sarcoidosis, by parenchymal fibrosis (3). Other potential complications of fibrotic sarcoidosis include pulmonary hypertension (PH), pulmonary aspergillus syndromes including aspergilloma and chronic aspergillus infection, and opportunistic infection. These complications and their treatment approaches are summarized in Table 1. Pulmonary hypertension results from a variety of mechanisms including obliteration of the pulmonary capillary bed by fibrosis, extrinsic compression of pulmonary arteries from mediastinal adenopathy or fibrosis, direct granulomatous involvement of the pulmonary vasculature, pulmonary veno-occlusive disease, chronic hypoxemia, and left heart dysfunction (34–36). Immune dysregulation and impairments in airway clearance foster development of aspergillus infection in some patients(33).
Table 1.
Serious cardiopulmonary complications of fibrotic pulmonary sarcoidosis
| Specific Diagnosis | Clues to Diagnosis | Treatment | |
|---|---|---|---|
|
|
|||
| Airway Disease | Airflow obstruction Bronchiectasis Endobronchial stenosis due to intraluminal narrowing or extrinsic compression |
Wheezing, reduced FEV1/FVC Cough, sputum, positive cultures, bronchial distortion or bronchiectasis on imaging Cough, fixed wheeze, localized end expiratory rhonchi, positional dyspnea, obstructive atelectasis on imaging |
Bronchodilators, inhaled corticosteroids Airway clearance measures (Nebulized) antibiotics as indicated Systemic corticosteroids and/or steroid sparing agents, bronchoscopy with balloon dilatation and/or stenting for refractory lesions |
| Aspergillus Syndromes | Simple aspergilloma Chronic pulmonary aspergillosis |
CT imaging Massive hemoptysis Fever, weight loss, Elevated IgG aspergillus |
Observation Bronchial artery embolization Anti-fungal therapy |
| Pulmonary vascular disease | Pulmonary arterial hypertension* Left heart disease |
Disproportionate reduction in DLCO, dyspnea out of proportion to lung disease Elevated BNP, echocardiogram |
Oxygen, Vasodilators Treat diastolic and systolic dysfunction as indicated |
Requires right heart catheterization for diagnosis
Assessing for Active Inflammation
The complications listed in Table 1 should be considered for all patients with fibrotic disease and pulmonary symptoms or limitations. In addition, some patients require immunosuppressive therapy. While patients with fibrotic pulmonary sarcoidosis may have “burnt out” disease, a subset has active, potentially treatment-responsive inflammation. Unfortunately, determining which patients are in this group is not always easy or straight-forward. A high resolution CT (HRCT) scan can be useful. Micronodular lesions and ground glass abnormalities are associated with improvement on therapy (37). There is also increasing interest in fluorodeoxyglucose-PET/computed tomography (FDG-PET CT) scans to detect metabolic activity in granulomas. Mostard et al retrospectively assessed 95 patients with sarcoidosis for whole body PET scan and HRCT findings (38). Overall, pulmonary PET positivity was detected in 56 patients (59%), and the vast majority of patients with fibrotic changes on HRCT had positive PET findings. A semi-quantitative HRCT score for typical parenchymal sarcoidosis was higher in patients with positive compared to negative PET scans. These results contrast with those of a larger retrospective study, in which fibrotic sarcoidosis had a lower rate of PET positivity (39). Pulmonary PET scans were positive in 16 of 22 patients with Scadding radiographic stage II and III sarcoidosis, but only 2 of 22 patients with stage IV pulmonary disease. Notably, eleven patients with repeat scans after corticosteroid therapy demonstrated decreased PET positivity. Along with the correlation of PET-CT findings observed in Mostard’s study, this supports the validity of PET scans to detect disease activity in sarcoidosis. In addition to CT and PET findings, assessment of symptom progression and pulmonary function aid in treatment decisions(40). A decrease in FVC > 10% or DLCO > 20% in one year is suggestive of active, progressive lung disease.
Treating Fibrotic Pulmonary Sarcoidosis
The treatment approach for fibrotic sarcoidosis with co-existing inflammation is similar to that of non-fibrotic sarcoidosis. Corticosteroids, begun at the dose equivalent of 20 to 40 mg of prednisone daily and tapered to the lowest possible effective dose, are first-line therapy. Additional immunosuppressive agents should be considered for patients intolerant of corticosteroids, requiring high long-term doses (prednisone > 10 mg daily), or whose lung disease continues to progress. Methotrexate is a common second-line agent (5). The TNF-α inhibitors infliximab and adalimumab have reasonable safety profiles in appropriately selected patients. Infliximab is the only immunosuppressive agent with a randomized-controlled study to include patients with fibrotic sarcoidosis (41, 42). Rituximab has been used in refractory pulmonary disease with descriptions of stabilization in lung function and radiographic imaging in a recent case report (43). Other sarcoidosis immunosuppressive therapies may also be effective in patients with co-existing fibrosis and inflammation.
Similar to other forms of severe parenchymal lung disease, acute exacerbations are not infrequent in sarcoidosis and may require augmented therapy. A recent retrospective study of fibrotic pulmonary sarcoidosis demonstrated a median of 3 acute worsening events per year (44). A review of the literature suggests that risk factors for acute exacerbations include African-American race, longer disease duration and previous treatment with corticosteroids (45). Acute exacerbations may respond to augmented corticosteroid therapy after infection and other etiologies of respiratory worsening such as pulmonary embolism and cardiac dysfunction are ruled out.
Clinical management of fibrotic sarcoidosis is not limited to immunosuppression. A recent study demonstrated > 10% improvement in six-minute walk distance after a 12-week physical training program in 6 patients with end-stage fibrotic pulmonary sarcoidosis (46). Sarcoidosis associated PH may be responsive to vasodilator therapies used for idiopathic pulmonary arterial hypertension (36). Aspergillosis usually stabilizes or improves on long-term anti-fungal treatment. Finally, appropriate referral for lung transplantation is important (47). Despite being uncommon, lung transplantation for pulmonary sarcoidosis is successful with outcomes comparable to other lung transplant recipients (47).
Conclusion
Not all patients with long standing inflammation develop end-stage disease, and predicting who will progress is difficult. Once pulmonary fibrosis occurs, the greatest challenge in clinical management is determining if active inflammation is also present. When it is, a trial of immunosuppressive therapy is often warranted. Ongoing surveillance for disease complications is also essential.
Looking ahead, more research is needed to better understand fibrotic sarcoidosis, the most under-studied phenotype of pulmonary sarcoidosis. Fibrotic sarcoidosis comprises a heterogeneous group, where pulmonary impairments range from minimal to life threatening. Sub-phenotyping patients is helpful, perhaps by radiographic features (22), or by severity of pulmonary impairment (48). Important topics for future studies are reviewed above. We highlight future studies of key cytokines such as TNF-α, and examination of T cell subsets and function throughout the course of sarcoidosis as particularly important. Mechanisms of fibrotic transformation in other chronic inflammatory diseases may ultimately inform our understanding of fibrotic sarcoidosis(49).
Key points.
Fibrotic sarcoidosis results from progression to chronic inflammatory disease and fibrotic transformation.
Methodologies for studying fibrotic sarcoidosis are (1) correlating findings early in disease with long-term outcomes, and (2) studying immune and genetic features of patients who progress to fibrotic disease.
Recent studies highlight deficiencies in immuno-regulation and inflammatory and pro-fibrotic genetic signatures as important contributors to the pathophysiology.
Fibrotic pulmonary sarcoidosis is associated with increased morbidity and mortality due to underlying disease and the sequelae of airway disease, pulmonary hypertension and infection.
Fibrotic sarcoidosis is clinically heterogeneous: the natural history and pulmonary and functional impairments should be considered in the assessment of disease severity for each patient.
Footnotes
Supplementary material
None
Disclosure of funding received for this work: Dr. Bonham received funding from NIH K12 HL119995.
The authors have no relevant conflicts of interest or sources of funding.
References
- 1.Rosen Y. Pathology of sarcoidosis. Semin Respir Crit Care Med. 2007;28(1):36–52. doi: 10.1055/s-2007-970332. [DOI] [PubMed] [Google Scholar]
- 2.Grunewald J, Kaiser Y, Ostadkarampour M, Rivera NV, Vezzi F, Lotstedt B, et al. T-cell receptor-HLA-DRB1 associations suggest specific antigens in pulmonary sarcoidosis. Eur Respir J. 2016;47(3):898–909. doi: 10.1183/13993003.01209-2015. [DOI] [PubMed] [Google Scholar]
- 3.Kouranos V, Jacob J, Wells AU. Severe Sarcoidosis. Clin Chest Med. 2015;36(4):715–26. doi: 10.1016/j.ccm.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet P-Y, Müller-Quernheim J. Sarcoidosis. The Lancet. 2014;383(9923):1155–67. doi: 10.1016/S0140-6736(13)60680-7. [DOI] [PubMed] [Google Scholar]
- 5.Korsten P, Strohmayer K, Baughman RP, Sweiss NJ. Refractory pulmonary sarcoidosis - proposal of a definition and recommendations for the diagnostic and therapeutic approach. Clin Pulm Med. 2016;23(2):67–75. doi: 10.1097/CPM.0000000000000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lockstone HE, Sanderson S, Kulakova N, Baban D, Leonard A, Kok WL, et al. Gene set analysis of lung samples provides insight into pathogenesis of progressive, fibrotic pulmonary sarcoidosis. Am J Respir Crit Care Med. 2010;181(12):1367–75. doi: 10.1164/rccm.200912-1855OC. **This study examines the disparate gene expression profiles of self-limiting versus progressive pulmonary sarcoidosis including patients with pulmonary fibrosis, and compares gene expression pathways in progressive sarcoidosis to that of other interstitial lung diseases. [DOI] [PubMed] [Google Scholar]
- 7.Ziegenhagen MW, Benner UK, Zissel G, Zabel P, Schlaak M, Muller-Quernheim J. Sarcoidosis: TNF-alpha release from alveolar macrophages and serum level of sIL-2R are prognostic markers. Am J Respir Crit Care Med. 1997;156(5):1586–92. doi: 10.1164/ajrccm.156.5.97-02050. [DOI] [PubMed] [Google Scholar]
- 8.Ziegenhagen MW, Rothe ME, Zissel G, Muller-Quernheim J. Exaggerated TNFalpha release of alveolar macrophages in corticosteroid resistant sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19(3):185–90. [PubMed] [Google Scholar]
- 9.Zissel G, Homolka J, Schlaak J, Schlaak M, Muller-Quernheim J. Anti-inflammatory cytokine release by alveolar macrophages in pulmonary sarcoidosis. Am J Respir Crit Care Med. 1996;154(3 Pt 1):713–9. doi: 10.1164/ajrccm.154.3.8810610. [DOI] [PubMed] [Google Scholar]
- 10.Pabst S, Franken T, Schonau J, Stier S, Nickenig G, Meyer R, et al. Transforming growth factor-{beta} gene polymorphisms in different phenotypes of sarcoidosis. Eur Respir J. 2011;38(1):169–75. doi: 10.1183/09031936.00120410. [DOI] [PubMed] [Google Scholar]
- 11.Patterson KC, Hogarth K, Husain AN, Sperling AI, Niewold TB. The clinical and immunologic features of pulmonary fibrosis in sarcoidosis. Transl Res. 2012;160(5):321–31. doi: 10.1016/j.trsl.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braun NA, Celada LJ, Herazo-Maya JD, Abraham S, Shaginurova G, Sevin CM, et al. Blockade of the programmed death-1 pathway restores sarcoidosis CD4(+) T-cell proliferative capacity. Am J Respir Crit Care Med. 2014;190(5):560–71. doi: 10.1164/rccm.201401-0188OC. * Braun’s ex vivo investigation of sarcoidosis CD4(+) T-cell proliferative capacity delineates PD-1 blockade as an actionable target for immunotherapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Qiu L, Wang Y, Aimurola H, Zhao Y, Li S, et al. The Circulating Treg/Th17 Cell Ratio Is Correlated with Relapse and Treatment Response in Pulmonary Sarcoidosis Patients after Corticosteroid Withdrawal. PLoS One. 2016;11(2):e0148207. doi: 10.1371/journal.pone.0148207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakthivel P, Grunewald J, Eklund A, Bruder D, Wahlstrom J. Pulmonary sarcoidosis is associated with high-level inducible co-stimulator (ICOS) expression on lung regulatory T cells - possible implications for the ICOS/ICOS-ligand axis in disease course and resolution. Clin Exp Immunol. 2016;183(2):294–306. doi: 10.1111/cei.12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prasse A, Zissel G, Lutzen N, Schupp J, Schmiedlin R, Gonzalez-Rey E, et al. Inhaled vasoactive intestinal peptide exerts immunoregulatory effects in sarcoidosis. Am J Respir Crit Care Med. 2010;182(4):540–8. doi: 10.1164/rccm.200909-1451OC. ** This study is the first to correlate T regulatory cell counts in the lung and long-term disease activity, which is the most significant risk factor for pulmonary fibrosis in sarcoidosis. [DOI] [PubMed] [Google Scholar]
- 16.Kruit A, Grutters JC, Ruven HJ, van Moorsel CH, Weiskirchen R, Mengsteab S, et al. Transforming growth factor-beta gene polymorphisms in sarcoidosis patients with and without fibrosis. Chest. 2006;129(6):1584–91. doi: 10.1378/chest.129.6.1584. [DOI] [PubMed] [Google Scholar]
- 17.Ahmadzai H, Cameron B, Chui J, Lloyd A, Wakefield D, Thomas PS. Measurement of neopterin, TGF-beta1 and ACE in the exhaled breath condensate of patients with sarcoidosis. J Breath Res. 2013;7(4):046003. doi: 10.1088/1752-7155/7/4/046003. [DOI] [PubMed] [Google Scholar]
- 18.Patterson KC, Franek BS, Muller-Quernheim J, Sperling AI, Sweiss NJ, Niewold TB. Circulating cytokines in sarcoidosis: phenotype-specific alterations for fibrotic and non-fibrotic pulmonary disease. Cytokine. 2013;61(3):906–11. doi: 10.1016/j.cyto.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korthagen NM, Nagtegaal MM, van Moorsel CH, Kazemier KM, van den Bosch JM, Grutters JC. MRP14 is elevated in the bronchoalveolar lavage fluid of fibrosing interstitial lung diseases. Clin Exp Immunol. 2010;161(2):342–7. doi: 10.1111/j.1365-2249.2010.04181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu X, Chen H, Zhu X, Ma Y, Liu Q, Xue Y, et al. S100A9 promotes human lung fibroblast cells activation through receptor for advanced glycation end-product-mediated extracellular-regulated kinase 1/2, mitogen-activated protein-kinase and nuclear factor-kappaB-dependent pathways. Clin Exp Immunol. 2013;173(3):523–35. doi: 10.1111/cei.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morais A, Beltrao M, Sokhatska O, Costa D, Melo N, Mota P, et al. Serum metalloproteinases 1 and 7 in the diagnosis of idiopathic pulmonary fibrosis and other interstitial pneumonias. Respir Med. 2015;109(8):1063–8. doi: 10.1016/j.rmed.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Abehsera M, Valeyre D, Grenier P, Jaillet H, Battesti JP, Brauner MW. Sarcoidosis with pulmonary fibrosis: CT patterns and correlation with pulmonary function. AJR Am J Roentgenol. 2000;174(6):1751–7. doi: 10.2214/ajr.174.6.1741751. **This was a hallmark study which systematically characterized CT findings in sarcoidosis and correlated CT patterns with clinical outcomes. [DOI] [PubMed] [Google Scholar]
- 23.Nishino M, Lee KS, Itoh H, Hatabu H. The spectrum of pulmonary sarcoidosis: variations of high-resolution CT findings and clues for specific diagnosis. Eur J Radiol. 2010;73(1):66–73. doi: 10.1016/j.ejrad.2008.09.038. [DOI] [PubMed] [Google Scholar]
- 24.Scadding JG. Prognosis of intrathoracic sarcoidosis in England. A review of 136 cases after five years’ observation. Br Med J. 1961;2(5261):1165–72. doi: 10.1136/bmj.2.5261.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polverosi R, Russo R, Coran A, Battista A, Agostini C, Pomerri F, et al. Typical and atypical pattern of pulmonary sarcoidosis at high-resolution CT: relation to clinical evolution and therapeutic procedures. Radiol Med. 2014;119(6):384–92. doi: 10.1007/s11547-013-0356-x. [DOI] [PubMed] [Google Scholar]
- 26.Shigemitsu H, Oblad JM, Sharma OP, Koss MN. Chronic interstitial pneumonitis in end-stage sarcoidosis. Eur Respir J. 2010;35(3):695–7. doi: 10.1183/09031936.00150609. [DOI] [PubMed] [Google Scholar]
- 27.Zhang C, Chan KM, Schmidt LA, Myers JL. Histopathology of Explanted Lungs From Patients With a Diagnosis of Pulmonary Sarcoidosis. Chest. 2016;149(2):499–507. doi: 10.1378/chest.15-0615. [DOI] [PubMed] [Google Scholar]
- 28.Xu L, Kligerman S, Burke A. End-stage sarcoid lung disease is distinct from usual interstitial pneumonia. Am J Surg Pathol. 2013;37(4):593–600. doi: 10.1097/PAS.0b013e3182785a2d. * This study further defines the histopathology of fibrotic pulmonary sarcoidosis, and addresses an evolving controversy on whether the usual interstitial pneumonia pattern is part of the spectrum of this phenotype. [DOI] [PubMed] [Google Scholar]
- 29.Gerke AK. Morbidity and mortality in sarcoidosis. Curr Opin Pulm Med. 2014;20(5):472–8. doi: 10.1097/MCP.0000000000000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nardi A, Brillet PY, Letoumelin P, Girard F, Brauner M, Uzunhan Y, et al. Stage IV sarcoidosis: comparison of survival with the general population and causes of death. Eur Respir J. 2011;38(6):1368–73. doi: 10.1183/09031936.00187410. [DOI] [PubMed] [Google Scholar]
- 31.Mirsaeidi M, Machado RF, Schraufnagel D, Sweiss NJ, Baughman RP. Racial difference in sarcoidosis mortality in the United States. Chest. 2015;147(2):438–49. doi: 10.1378/chest.14-1120. * This investigation highlights important differences in clinical features and mortality outcomes among United States citizens with self-identified racial backgrounds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walsh SLF, Wells AU, Sverzellati N, Keir GJ, Calandriello L, Antoniou KM, et al. An integrated clinicoradiological staging system for pulmonary sarcoidosis: a case-cohort study. The Lancet Respiratory Medicine. 2014;2(2):123–30. doi: 10.1016/S2213-2600(13)70276-5. **This study outlines and tests a clinically relevant prognostic tool for sarcoidosis using widely available clinical parameters. [DOI] [PubMed] [Google Scholar]
- 33.Patterson KC, Strek ME. Pulmonary fibrosis in sarcoidosis. Clinical features and outcomes. Ann Am Thorac Soc. 2013;10(4):362–70. doi: 10.1513/AnnalsATS.201303-069FR. [DOI] [PubMed] [Google Scholar]
- 34.Baughman RP, Engel PJ, Nathan S. Pulmonary Hypertension in Sarcoidosis. Clin Chest Med. 2015;36(4):703–14. doi: 10.1016/j.ccm.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 35.Maimon N, Salz L, Shershevsky Y, Matveychuk A, Guber A, Shitrit D. Sarcoidosis-associated pulmonary hypertension in patients with near-normal lung function. Int J Tuberc Lung Dis. 2013;17(3):406–11. doi: 10.5588/ijtld.12.0428. [DOI] [PubMed] [Google Scholar]
- 36.Bonham CA, Oldham JM, Gomberg-Maitland M, Vij R. Prostacyclin and oral vasodilator therapy in sarcoidosis-associated pulmonary hypertension: a retrospective case series. Chest. 2015;148(4):1055–62. doi: 10.1378/chest.14-2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Criado E, Sanchez M, Ramirez J, Arguis P, de Caralt TM, Perea RJ, et al. Pulmonary sarcoidosis: typical and atypical manifestations at high-resolution CT with pathologic correlation. Radiographics. 2010;30(6):1567–86. doi: 10.1148/rg.306105512. [DOI] [PubMed] [Google Scholar]
- 38.Mostard RL, Verschakelen JA, van Kroonenburgh MJ, Nelemans PJ, Wijnen PA, Voo S, et al. Severity of pulmonary involvement and (18)F-FDG PET activity in sarcoidosis. Respir Med. 2013;107(3):439–47. doi: 10.1016/j.rmed.2012.11.011. *This study demonstrates that many patients with fibrotic pulmonary sarcoidosis show signs of ongoing inflammation as measured by (18)F-FDG PET activity. [DOI] [PubMed] [Google Scholar]
- 39.Teirstein AS, Machac J, Almeida O, Lu P, Padilla ML, Iannuzzi MC. Results of 188 whole-body fluorodeoxyglucose positron emission tomography scans in 137 patients with sarcoidosis. Chest. 2007;132(6):1949–53. doi: 10.1378/chest.07-1178. [DOI] [PubMed] [Google Scholar]
- 40.Amin EN, Closser DR, Crouser ED. Current best practice in the management of pulmonary and systemic sarcoidosis. Ther Adv Respir Dis. 2014;8(4):111–32. doi: 10.1177/1753465814537367. [DOI] [PubMed] [Google Scholar]
- 41.Russell E, Luk F, Manocha S, Ho T, O’Connor C, Hussain H. Long term follow-up of infliximab efficacy in pulmonary and extra-pulmonary sarcoidosis refractory to conventional therapy. Semin Arthritis Rheum. 2013;43(1):119–24. doi: 10.1016/j.semarthrit.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 42.Rossman MD, Newman LS, Baughman RP, Teirstein A, Weinberger SE, Miller W, Jr, et al. A double-blinded, randomized, placebo-controlled trial of infliximab in subjects with active pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2006;23(3):201–8. [PubMed] [Google Scholar]
- 43.Cinetto F, Compagno N, Scarpa R, Malipiero G, Agostini C. Rituximab in refractory sarcoidosis: a single centre experience. Clin Mol Allergy. 2015;13(1):19. doi: 10.1186/s12948-015-0025-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baughman RP, Lower EE. Frequency of acute worsening events in fibrotic pulmonary sarcoidosis patients. Respir Med. 2013;107(12):2009–13. doi: 10.1016/j.rmed.2013.10.014. [DOI] [PubMed] [Google Scholar]
- 45.Panselinas E, Judson MA. Acute pulmonary exacerbations of sarcoidosis. Chest. 2012;142(4):827–36. doi: 10.1378/chest.12-1060. [DOI] [PubMed] [Google Scholar]
- 46.Strookappe B, Swigris J, De Vries J, Elfferich M, Knevel T, Drent M. Benefits of Physical Training in Sarcoidosis. Lung. 2015;193(5):701–8. doi: 10.1007/s00408-015-9784-9. * This study demontrates the utility of physical training and, in so doing, highlights the role of non-pharmacologic intervention in patients with chronic fatigue and exercise limitations. [DOI] [PubMed] [Google Scholar]
- 47.Shlobin OA, Nathan SD. Management of end-stage sarcoidosis: pulmonary hypertension and lung transplantation. Eur Respir J. 2012;39(6):1520–33. doi: 10.1183/09031936.00175511. [DOI] [PubMed] [Google Scholar]
- 48.Baughman RP, Drent M, Kavuru M, Judson MA, Costabel U, du Bois R, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med. 2006;174(7):795–802. doi: 10.1164/rccm.200603-402OC. [DOI] [PubMed] [Google Scholar]
- 49.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–40. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]