

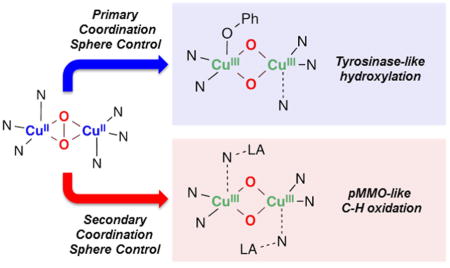
Abstract
Copper-dependent metalloenzymes are widespread throughout metabolic pathways, coupling the reduction of O2 with the oxidation of organic substrates. Small-molecule synthetic analogs are useful platforms to generate L/Cu/O2 species that reproduce the structural, spectroscopic, and reactive properties of some copper-/O2-dependent enzymes. Landmark studies have shown that the conversion between dicopper(II)-peroxo species (L2CuII2(O22−) either side-on peroxo, SP, or end-on trans-peroxo, TP) and dicopper(III)-bis(μ-oxo) (L2CuIII2(O2−)2:O) can be controlled through ligand design, reaction conditions (temperature, solvent, and counteranion), or substrate coordination. We recently published (J. Am. Chem. Soc. 2012, 134, 8513, DOI: 10.1021/ja300674m) the crystal structure of an unusual SP species [(MeAN)2CuII2(O22−)]2+ (SPMeAN, MeAN: N-methyl-N,N-bis[3-(dimethylamino)propyl]amine) that featured an elongated O–O bond but did not lead to O–O cleavage or reactivity toward external substrates. Herein, we report that SPMeAN can be activated to generate OMeAN and perform the oxidation of external substrates by two complementary strategies: (i) coordination of substituted sodium phenolates to form the substrate-bound OMeAN-RPhO− species that leads to ortho-hydroxylation in a tyrosinase-like fashion and (ii) addition of stoichiometric amounts (1 or 2 equiv) of Lewis acids (LA’s) to form an unprecedented series of O-type species (OMeAN-LA) able to oxidize C–H and O–H bonds. Spectroscopic, computational, and mechanistic studies emphasize the unique plasticity of the SPMeAN core, which combines the assembly of exogenous reagents in the primary (phenolates) and secondary (Lewis acids association to the MeAN ligand) coordination spheres with O–O cleavage. These findings are reminiscent of the strategy followed by several metalloproteins and highlight the possible implication of O-type species in copper-/dioxygen-dependent enzymes such as tyrosinase (Ty) and particulate methane monooxygenase (pMMO).
Graphical Abstract
INTRODUCTION
Dicopper centers are a common motif found in the active sites of several O2-activating metalloproteins that perform an array of physiological functions.1 Tyrosinase (Ty) is a paradigmatic example of a metalloenzyme with a coupled dicopper active center (Figure 1A, left), which combines the reduction of dioxygen with the ortho-hydroxylation of tyrosine for melanin biosynthesis. Although its catalytic activity has been widely studied during the last several decades, there is still some debate on the identity of the reactive Cu2/O2 intermediate that is responsible for substrate oxidation. There is agreement on the first reaction step: Reduced Ty (dicopper(I)) reacts with dioxygen to form a dicopper(II) species with a side-on peroxide moiety (SP). It is proposed that coordination of tyrosinate (phenolate form, Figure 1A center) to one of the copper(II) centers in oxy-Ty is followed by electrophilic attack of the peroxide to the aromatic ring.1–3 Alternatively, some authors propose that prior to the electrophilic attack O–O cleavage occurs thus generating a dicopper(III) bis(μ-oxo) species (O) as the active oxidant (Figure 1A, right).4,5
Figure 1.

Active site and proposed Cu/O2 reactive species of (A) tyrosinase (Ty) and (B) particulate methane monooxygenase (pMMO).
Another ambiguity exists in copper-dependent particulate methane monooxygenase (pMMO). Detailed structural and spectroscopic studies have led to the proposal that the active site is a dicopper center with only two residues coordinating each Cu, with one Cu intriguingly bound to a N-terminal histidine (Figure 1B, left). Due to the technological implications of methane oxidation,6,7 elucidation of the identity of the Cu/O2 active species is of considerable general interest. However, the little information available has led to speculation that oxidation could be carried out by a CuII(O22−)CuII (SP), a CuIII(O2−)2CuIII (O), or a mixed-valent CuII(O2−)2CuIII (and possibly protonated) species (Figure 1B, right).5,8–11
Several research groups, including ours, have extensively explored the O2 chemistry of Cu(I) model complexes, which has led to the characterization of many distinct types of mononuclear and dinuclear Cun/O2 species (Figure 2, top).12–16 Especially relevant has been the study of the equilibrium between peroxodicopper(II) (SP or TP) and bis(μ-oxo) dicopper(III) (O) species, which can be controlled by ligand design (including ligand donation to Cu), varying reaction conditions (i.e., temperature, solvent, and/or counter-anion), and substrate coordination (Figure 2, bottom).12–21 From these studies, several general trends have emerged for controlling this equilibrium. It has been generally established that (i) tridentate ligation (i.e., N3) favors SP formation while use of bidentate ligands (i.e., N2) usually leads to O species; (ii) weakly coordinating counteranions and polar solvents favor O assemblies; and (iii) coordination of anionic substrates such as phenolates can lead to O–O cleavage prior to substrate oxidation.4
Figure 2.

Top: Common LCu/O2 species derived from the oxygenation of copper(I) complexes. Bottom: Control of the SP/O equilibrium by different means.
Recently, we published the crystal structure of the SP system [(MeAN)2CuII2(O22−)]2+ (SPMeAN) (MeAN: N-methyl-N,N-bis[3-(dimethylamino)propyl]amine, Figure 3 top).22 Strikingly, despite the unusually long O–O distance (1.54 Å) in this complex, there was no observable SPMeAN/OMeAN equilibrium or reactivity with external substrates. We had attributed this to the weak backbonding from the Cu 3dxy orbital to the peroxide σ* orbital (Figure 3, middle), which is crucial for O–O cleavage.
Figure 3.

Top: SPMeAN X-ray structure and its geometric and spectroscopic features. Middle: Molecular orbital diagrams of the SPMeAN core that lead to lengthening of the O–O bond without concomitant O–O cleavage. Bottom: Subjects of this report: coordination of phenolates and Lewis acids to SPMeAN imparts tyrosinase-like hydroxylation and pMMO-like C–H oxidation reactivity through OMeAN.
Herein, we report that the oxidative properties of SPMeAN can be activated by two different approaches (Figure 3, bottom). As previously reported in other systems, coordination of a series of substituted sodium phenolates leads to O–O cleavage to generate observable OMeAN-RPhO− species that decay to generate the ortho-hydroxylated product.4,23 A surprising and very new finding is that the SPMeAN/OMeAN equilibrium can also be controlled by addition of various Lewis acids (LA) which are able to trigger the O–O cleavage of SPMeAN species to generate distinctive OMeAN-LA species with enhanced and tunable oxidative reactivity toward C–H and O–H bonds.
EXPERIMENTAL SECTION
General Considerations
All reagents and solvents were of commercially available quality except as noted. Acetone was distilled from Drierite under argon atmosphere. [(MeAN)CuI](BArF) (BArF: B(C6F5)4−) was synthesized as previously described.24 Sodium phenolates were obtained using a synthetic method similar to previous results.23 All UV–vis measurements were carried out using a Hewlett-Packard 8453 diode array spectrophotometer with a 10 mm quartz cell. The spectrometer was equipped with HP Chemstation software and a Unisoku cryostat for low temperature experiments. 1H NMR spectra were recorded on a Bruker 400 instrument. Resonance Raman (rR) samples were excited using a Coherent I90C–K Kr+ ion laser at 413.1 or 568.2 nm while the sample was immersed in a liquid-nitrogen-cooled (77 K) EPR finger dewar (Wilmad). Power was ~20 mW at the sample for the 413.1 nm line and ~130 mW at 568.2 nm. Data were recorded while rotating the sample to minimize photodecomposition. The spectra were recorded using a Spex 1877 CP triple monochromator with 600, 1200, or 2400 grooves/mm holographic spectrograph grating and detected by an Andor Newton CCD cooled to −80 °C (413.1 nm) or an Andor IDus CCD cooled to −80 °C (568.2 nm). Spectra were calibrated on the energy axis to toluene at room temperature.
[(MeAN)2CuII2(O22−)]2+ (SPMeAN) Generation and Reaction with Sodium Phenolates
A 3 mL aliquot of a [(MeAN)CuI](BArF) solution (0.2 mM) in acetone was placed in a 10 mm path quartz cell equipped with a stir bar and capped with a rubber septum. After cooling down the cell to −90 °C (acetone), dioxygen was added to generate [(MeAN)2CuII2(O22−)]2+ (SPMeAN) (λmax: 365 nm ε = 22 mM−1 cm−1). After complete formation, 100 μL of an acetone solution containing excess of the corresponding substrate (substituted phenols, PPh3, DHA, thioanisole, Me2Fc, or Fc) was added. No spectral change was observed within 60 min after substrate addition. In contrast, addition of 100 μL of an acetone solution containing the corresponding 4-substituted sodium phenolate (to give a final concentration of 0.2 mM) to SPMeAN led to the immediate formation of the putative [(MeAN)2CuIII2(O2−)2(R-PhO−)]+, followed by its decay (see Supporting Information). Kinetic analysis was performed by fitting the exponential decay of [(MeAN)2CuIII2(O2−)2(R-PhO−)]+ at 400 nm. The kobs obtained are summarized in the Supporting Information. Quantification of the corresponding catechols was performed using a method previously described (see Supporting Information).25,26
OMeAN-LA Generation
A 0.2 mM solution of SPMeAN in acetone was prepared as described above at −90 °C. Then, 100 μL of an acetone solution containing the corresponding equivalents of Lewis acid (0.2 mM DMF·CF3SO3H, 0.1 mM Sc(CF3SO3)3, or 0.1 mM B(C6F5)3) was added to generate the corresponding OMeAN-LA species. See Supporting Information for titration experiments.
SPMeAN/OMeAN-LA Reversibility
Formation of OMeAN-2H+ could be reversed by addition of proton sponge (1,8-bis(dimethylamino)-naphthalene) in excess (0.6 mM) leading to full regeneration of SPMeAN (see Supporting Information). OMeAN-2H+ could be formed again by readdition of DMF·CF3SO3H (0.6 mM was required for full formation, due to the presence of proton sponge excess in solution). Similarly, formation of OMeAN-Sc3+ could be reversed by addition of 1,10-phenanthroline in excess (0.8 mM), leading only to partial formation (75%) of SPMeAN (see Supporting Information) due to the ability of 1,10-phenanthroline to trap Cu(I) (430 and 455 nm bands, see Figure S9). OMeAN-Sc3+ could be partially formed again by readdition of Sc(CF3SO3)3 (0.2 mM was required for full formation, due to the presence of excess 1,10-phenanthroline in solution). We could also reverse the formation of OMeAN-2BR3 by addition of TASF (tris(dimethylamino)sulfonium difluorotrimethylsilicate, a F− source) in excess (0.6 mM), leading to partial formation (60%) of SPMeAN (see Supporting Information). OMeAN-2BR3 could be partially formed again by readdition of B(C6F5)3.
OMeAN-LA Reactivity toward Weak C–H Bonds and Substituted Phenols
After generation of the corresponding OMeAN-LA as described above, 100 μL of an acetone solution containing various amounts of the corresponding substrate (weak C–H bonds or substituted phenols) was added, causing the decay of the OMeAN-LA spectral features. The absorbance changes (λ = 410 nm) were fitted to single exponential decays. In all cases, a linear correlation between the reaction rate (kobs) and the substrate concentration was found, from which the second-order rate constants were obtained. Quantification of the oxidation products derived from the C–H (BNAH) and O–H oxidation (2,4-tBu-PhOH) was performed using methods previously described (see Supporting Information for details).27
Resonance Raman Experiments
In a typical experiment, 0.57 mL of a [(MeAN)CuI]+ (1.0 mM) solution in acetone were placed in a 5 mm NMR tube capped with a septum. After cooling the tube to −90 °C (acetone/N2(liq) bath), dioxygen was bubbled through the solution mixture to generate [(MeAN)2Cu2(O22−)]2+ (SPMeAN). To generate the [(MeAN)2CuIII2(O2−)2(2,6-F2–PhO−)]+, 50 μL of an acetone solution containing sodium 2,6-F2-phenolate (3 mM) was added. To generate OMeAN-2H+, 50 μL of an acetone solution containing 2 equiv of DMF·CF3SO3H (1 mM) was added. To generate the OMeAN-Sc3+, 50μL of an acetone solution containing 1 equiv of Sc(CF3SO3)3 (0.5 mM) was added. To generate the OMeAN-2BR3, 50 μL of an acetone solution containing 2 equiv of B(C6F5)3 (0.5 mM) was added. Samples were frozen either 10 or 300 s after phenolate addition.
DFT Calculations
All calculations were performed with Gaussian 09 (revision D.01)28 using the spin-unrestricted hybrid density functional B3LYP29–31 and a split basis set (6-311G(d) on Cu, N, and O; 6-31G on all other atoms) on an ultrafine integration grid. All calculations included solvation correction (PCM, acetone solvent). Analytical frequencies were calculated on all geometry-optimized species to verify that the species were true local minima without negative frequencies. Calculations involving 2,6-difluorophenolate included Grimme’s D3 dispersion correction.32 For broken-symmetry singlet states of the side-on peroxos, the Yamaguchi formalism33 was used to eliminate triplet contamination and obtain a corrected SCF energy (eq 1, where 1E is the corrected singlet energy, BSE and 3E are energies of the broken-symmetry and triplet states, respectively, and 〈S2〉BS is the calculated spin expectation value obtained from the broken-symmetry wave function).
| (1) |
For time-dependent DFT calculations, the calculated electronic absorption spectra were generated using SWizard34 with 3000 cm−1 line widths and Gaussian bandshapes.
RESULTS AND DISCUSSION
Ortho Hydroxylation of Phenolates
We previously reported22 that SPMeAN was unreactive toward external substrates including 9,10-dihydroanthracene (DHA, BDFE(C–H) = 76 kcal/mol), 2,4-di-tert-butylphenol (2,4-DTB-PhOH, BDFE(O–H) = 82 kcal/mol), and thioanisole. We expanded the substrate scope to other substrates even more prone to oxidation by electrophilic metal-peroxo species including 1-benzyl-1,4-dihydronicotinamide (BNAH, BDFE(C–H) = 71 kcal/mol),27 2,6-di-tert-butyl-4-methoxyphenol (BDFE(O–H) = 80 kcal/mol),35 and triphenylphosphine, but no decay of the SPMeAN UV–vis features were observed, even 1 h after addition of the substrate. We speculated that the unreactive nature of SPMeAN was due to the inability of the complex to form an OMeAN species, which typically oxidizes these types of substrates.14,36
This has led us to attempt oxidation of other types of substrates. SPMeAN reacts with a series of 4-substituted sodium phenolates (4-MeO, 4-Me, 4-H, 4-Cl, and 4-CN), which generates the corresponding ortho-hydroxylated products in a tyrosinase-like fashion in moderate yields (30–40%). Kinetic studies using the series of 4-R-PhO− were conducted in order to gather mechanistic details for this oxidative process. Addition of the substrate to SPMeAN caused an immediate change of the UV–vis spectrum, followed by a fast first-order decay (see Supporting Information for details). The disappearance of these fleeting species was followed by UV–vis and fit to an exponential decay. When plotting the reaction rates obtained (kobs) for the varying substrates against the Hammett parameter (σ+), a linear correlation was found (ρ = −2.2), suggesting an electrophilic aromatic substitution mechanism (see Figure S2). These findings are analogous to the results found by Stack and co-workers and other research groups,4,23 pointing toward a mechanism where substrate coordination to SPMeAN triggers O–O cleavage to form a [(MeAN)2CuIII2(O2−)2(R-PhO−)]+ (OMeAN-RPhO−) intermediate prior to substrate oxidation (Figure 4, top).
Figure 4.

Top: Postulated reaction mechanism of SPMeAN toward sodium phenolates to generate the corresponding ortho-hydroxylated products through OMeAN-RPhO−. Bottom left: Core structures of DFT-optimized SPMeAN and OMeAN-RPhO− with relevant metrical parameters, calculated Cu–O normal modes, TD-DFT-calculated LMCT energies, and energy of peroxo isomers relative to the corresponding bis(μ-oxo) isomers. Bottom right: Absorption spectra of SPMeAN (brown) and intermediate OMeAN−F2PhO− (blue). Inset: Resonance Raman spectra of SPMeAN (λ = 413.1 nm) and OMeAN−F2PhO− (λ = 568.2 nm) with 16O2 (blue), 18O2 (brown), and the 16O2-18O2 difference spectra (green).
In a recent contribution, Serrano-Plana et al. reported that the MeAN-inspired [(m-XYLMeAN)2CuIII2(O2−)2] 2+ complex was also able to perform the ortho-hydroxylation/defluorination of 2-fluoro-substituted phenolates in a selective fashion.26 Using similar reaction conditions, we found that the SPMeAN oxidized 2,6-F2-phenolate to the corresponding 3-F-catechol in moderate yield (30%). Interestingly, coordination of the substrate to SPMeAN (Figure 4 bottom right, brown spectrum) was found to form a phenolate-bound O-type species [(MeAN)2CuIII2-(O2−)2(F2PhO−)]+ (OMeAN-F2PhO−, Figure 4, blue spectrum) with spectroscopic characteristics similar to other phenolate-coordinated O species.23
Due to the slower reaction rate found for the fluorinated substrate, we were able to cold-trap and characterize OMeAN-F2PhO− by rR spectroscopy (Figure 4, bottom right inset). Upon laser excitation at 568.2 nm, three main isotope-sensitive modes were observed at 663 (Δ (18O2) = −23 cm−1), 604 (Δ (18O2) = −13 cm−1), and 590 cm−1 (Δ (18O2) = −31 cm−1), which are assigned as νCu–O vibrations of the Cu2O2 core of OMeAN-F2PhO−.37 The characteristic νO–O = 722 cm−1 mode of SPMeAN (Figure 4, bottom left inset) was not observed after phenolate addition, demonstrating full conversion to OMeAN-F2PhO−. One additional mode with very small 16/18O2 sensitivity (513 cm−1, Δ (18O2) = −3 cm−1) is also observed in OMeAN-F2PhO−, which is likely a predominantly Cu–N or Cu–Ophenolate mode that gains minor 16/18O2 isotope sensitivity through mixing of a core Cu–O mode. The number of observed Cu–O modes in rR can be a useful reporter of the local symmetry of the Cu2O2 core, since only symmetric modes (i.e., with A symmetry) are rR active.38 A maximally symmetric Cu2O2 bis(μ-oxo) of D2h symmetry has only one of four Cu–O stretching modes symmetry allowed (A1), while descent in symmetry to C2h or C2v results in two allowed modes (Ag), and further descent to Cs results in all four modes being symmetry allowed (A′). The observation of three Cu–O modes in OMeAN-F2PhO− thus suggests local pseudo-Cs symmetry, which is consistent with coordination of phenolate to one Cu.
DFT calculations were used to support the assignment of OMeAN-F2PhO− and investigate the role of phenolate in modulating the SP/O equilibrium. Two different isomers of the resulting OMeAN-F2PhO− adduct were optimized (Figure 4, bottom left): one with the phenolate coordinated to one of the copper(III) centers in an axial position (OMeAN-F2PhO− (ax)), which was 10 kcal/mol higher in energy than the other with phenolate coordinated in an equatorial position (OMeAN-F2PhO− (eq)). Only the equatorial phenolate model correctly reproduced the observed direction of the SP/O equilibrium, with the bis(μ-oxo) favored over the side-on peroxo isomer by 7.7 kcal/mol (for the axial phenolate, SP was favored by 3.1 kcal/mol). Furthermore, calculated normal modes for OMeAN-F2PhO− (eq) better matched the experimental rR data than OMeAN-F2PhO− (ax) (See Supporting Information for details); thus, the observed species is most consistent with possessing a OMeAN-F2PhO− (eq) structure.
TD-DFT calculations of OMeAN-F2PhO− (eq) also reproduced the absorption spectrum of the observed bis(μ-oxo) species. While both OMeAN-F2PhO− isomers showed typical spectra for a O-type species (two intense absorptions centered around 300 and 400 nm), only the spectrum calculated for the OMeAN-F2PhO− (eq) isomer had the intense lower energy bands (450–550 nm), which are assigned as phenolate-to-copper charge transfer transitions based on orbital analysis. This arises from the orientation of the phenolate in the Cu2O2 plane, which provides good overlap between the donor (phenolate π) and acceptor (Cu dx2–y2) orbitals and thus intense LMCT character.
As previously reported by Stack and co-workers,37 we propose that initial coordination of the phenolate occurs at one of the available axial positions (OMeAN-F2PhO− (ax)) and then rapidly isomerizes to the spectroscopically observed equatorial form (OMeAN-F2PhO− (eq)). The substrate is then electrophilically attacked by the CuIII2(O2−)2 core. In the axial form, DFT calculations suggest there is a weak CuIII···F interaction (d(Cu···F) = 3.02 Å) worth ~2 kcal/mol as compared to that of the phenolate-bound complex without the ortho F (calculated using 2,4-F2-phenolate, see Supporting Information). However, this weak Cu–F interaction is not sufficiently strong to prevent isomerization to the equatorially bound species (OMeAN-F2PhO− (eq), 10 kcal/mol more stable) which would then lead to ortho-hydroxylation and defluorination.
O–O Cleavage Promoted by Lewis Acids
Due to the unreactive character of SPMeAN core, we wondered if addition of Lewis acids could boost its oxidative power, as observed with iron or manganese oxo (or peroxo) complexes.39–42 Strikingly, addition of Sc(CF3SO3)3 to SPMeAN (λmax = 365 nm; ε = 22 mM−1 cm−1) led to the formation of a new species with UV–vis features (Figure 5) that are characteristic of O-type complexes (λmax = 412 nm; ε = 24 mM−1 cm−1). Titration experiments established that only 1 equiv of Sc3+ was required for full formation of the adduct, thus formulated as OMeAN-Sc3+ (Figure 5, top). Surprisingly, this reactivity is not exclusive for Sc3+: Addition of nonmetallic Lewis acids such as B(C6F5)3 or DMF·CF3SO3H (Brønsted acid) also led to O–O cleavage and formation of O-type species. In these latter cases, titration experiments suggested that 2 equiv of B(C6F5)3 or 2 equiv of DMF·CF3SO3H were necessary to form the corresponding adducts, thus formulating the products as the species OMeAN-2BR3 (λmax = 414 nm; ε = 24 mM−1 cm−1) and OMeAN-2H+ (λmax = 410 nm; ε = 21 mM−1 cm−1), respectively.
Figure 5.

Top: Generation of the different OMeAN-LA complexes. Middle: Spectroscopic characterization of the OMeAN-LA series by UV–vis (inset shows titration experiments) and rR at 413.1 nm excitation (16O2 in blue, 18O2 in red, and Cu(I) background in black). Mode assignments are indicated, and instrument artifacts are marked with (*). Bottom: Structures of DFT-optimized OMeAN-LA species with relevant metrical parameters, calculated Cu–O normal modes, TD-DFT-calculated LMCT energies, and energy of peroxo isomers relative to the corresponding bis(μ-oxo) isomers.
We made use of rR spectroscopy to characterize this family of OMeAN-LA species. Laser excitation (λexc. = 413 nm) of samples of OMeAN-Sc3+, OMeAN-2BR3 and OMeAN-2H+ prepared with 16O2 and 18O2 enhanced three isotope-sensitive modes at 514–602 cm−1 (ν2, ν3, and ν4 in Figure 5 middle) characteristic of Cu–O core modes of O-type species, and similar to OMeAN-F2PhO−. Combination bands (ν2 + ν4 at ~1120 cm−1 and ν1 + ν4 at ~740 cm−1, where ν1 is a low-energy isotope-insensitive mode at ~140 cm−1) and an overtone (2ν4 at ~1210 cm−1) are also observed.43,44 Surprisingly, the rR spectra of the different OMeAN-LA were almost superimposable, suggesting that the bis(μ-oxo) dicopper(III) cores had very similar structures. This indicates that the Lewis acids are not interacting directly with the oxygen atoms of the Cu2O2 core but rather coordinating to the axial N donors of the ligand. DFT calculations support this hypothesis: We found that in the computationally optimized structures for all three OMeAN-LA,43,45 the Lewis acid was bound to the N ligands of MeAN (Figure 5, bottom). Moreover, our calculations were able to reproduce the stoichiometry found in our titration experiments: While the Sc3+ ion was able to coordinate one N from each of the MeAN ligands, two molecules of borane or acid were required for O–O cleavage. The stabilization energy calculated upon binding of the Lewis acid was found to be very similar for all three OMeAN-LA complexes (between 3 and 6 kcal/mol, see Supporting Information). The optimized geometries for the OMeAN-LA species are very similar in all the cases, where the copper(III) ions are bound to two of the N donors of the MeAN ligand and two oxygen atoms. Similarly, the calculated UV–vis and rR features were also nearly identical for all adducts, corroborating our experimental findings (see Supporting Information).
To investigate the role of Lewis acids in stabilizing the OMeAN isomer relative to SPMeAN, we optimized a structure of SPMeAN with both axial N ligand arms uncoordinated (Figure 6A, right). Although removing the axial ligands is calculated to cost ΔG = +8 kcal/mol, the “arm off” isomer favors an O configuration by ΔG = −3.5 kcal/mol, which is similar to the preference for O in the Lewis acid adducts (–3.5 to −6.7 kcal/mol stabilization of O relative to that of SP, Figure S23), and inverse from that of the “arm on” isomer, which favors the SP configuration by ΔG = −2.2 kcal/mol. The “arm off” peroxo model featured a longer O–O bond (1.504 vs 1.488 Å) and shorter Cu–O (average 1.93 vs 1.98 Å) and Cu–N bonds (1.99 vs 2.03 Å) compared to the “arm on” structure (Figure 6A, top). The “arm off” structure thus allows better orbital overlap with the equatorial N ligands, which is also consistent with the less pyramidalized Cu (3° deviation from the Cu2O2 plane vs 27°, Figure 6A, center). Therefore, by sequestering the axial N, addition of Lewis acid allows stronger equatorial ligand donation, which in turn leads to a greater degree of O2 activation (Figure S26) that results in formation of a low energy OMeAN species via O–O cleavage.43
Figure 6.

(A) DFT-optimized structures of SPMeAN with the axial amine ligand arm in an (left) “on” binding position as observed experimentally by crystallography, and (right) “off” binding position. Top: core geometry and bond lengths (Å). Bottom: core structure viewed perpendicular to the Cu2O2 plane, showing the smaller deviation of the equatorial N ligands out of the Cu2O2 plane in the “arm off” isomer. (B) Role of Lewis acids in O–O bond formation at the active center of Photosystem II (left), the reversible O–O cleavage shown in the SPMeAN/OMeAN− LA series (middle) and O–O cleavage in nonheme iron(III)-peroxo reduction/oxidation (right).
Surprisingly, the O–O bond cleavage could be reversed by addition of Lewis acid chelating reagents. For example, 1,10-phenanthroline was able to capture Sc3+ from OMeAN-Sc3+ to recover 75% of SPMeAN, and 1,8-bis(dimethylamino)-naphthalene (proton sponge) was added to OMeAN-2H+ to fully regenerate SPMeAN (see Supporting Information for details).46 Recently, Que and co-workers have studied the effect of Sc3+ addition to the mononuclear nonheme iron(II) complex, which when combined with dioxygen led to the formation of a metastable iron(III)-peroxo-Sc3+ adduct that then irreversibly transformed to a corresponding high-valent iron(IV)-oxo complex (Figure 6, right).47 Nam and co-workers expanded that reactivity to other Lewis acids, where depending on the nature of the Lewis acid identity, the iron(III)-peroxo-LA adduct could be reduced to liberate dioxygen (LA: Ca2+, Sr2+) or led to O–O cleavage (LA: Zn2+, Lu3+, Y3+, Sc3+).48 To the best of our knowledge, the reactivity reported herein is the first example of Lewis acid mediated reversible O–O bond cleavage/formation, which is reminiscent of the chemistry occurring in water oxidation process catalyzed by the Mn4Ca2+ cluster in photosystem II (Figure 6, left).
Reactivity of OMeAN-LA toward C–H Bonds
At this point, we decided to test the reactivity of OMeAN-LA species toward external substrates, alongside the analogous parent SPMeAN complex. Surprisingly, we found that both SPMeAN and OMeAN-LA were unable to oxidize substrates that are prone to accept O atoms, such as triphenylphosphine and thioanisole.14 The first difference in reactivity was found when substrate one-electron (1e−) reductants were added. While both SPMeAN and OMeAN-LA reacted with strong 1e− reducing reagents (Me10Fc or Me8Fc, E0 < – 0.4 V vs Fc+/0), only the OMeAN-LA series was found to be able to oxidize the more difficult substrate, Me2Fc (E0 = −0.12 V vs Fc+/0). Based on this bracketing analysis, we can conclude that the OMeAN-LA species possess higher reduction potentials than SPMeAN, that is, they are stronger one-electron oxidants.
Another main difference in reactivity was found in reactions with substrates with weak C–H bonds. As previously reported, SPMeAN was not able to oxidize dihydroanthracene (DHA; BDE: 78 kcal/mol).22 Here, we find that the OMeAN-LA species are also not able to abstract a H atom (H•) from DHA. However, when 1-benzyl-1,4-dihydronicotinamide (BNAH, BDE: 71 kcal/mol) was used, the SPMeAN core was unreactive, but the series of OMeAN-LA complexes reacted to generate the corresponding BNA+ product in 75–99% yield (Figure 7A, see Supporting Information for product quantification). Kinetic interrogation of this reaction showed pseudo-first-order decay behavior with respect to [BNAH] (see Supporting Information). Second-order rate constants were obtained (Table 1), with the OMeAN-2BR3 reacting slightly faster than the OMeAN-2H+ and OMeAN-Sc3+ cores (Table 1). The same set of experiments was carried out using the deuterated analogue BNAD, which led to the determination of primary kinetic isotope effects of 10.3 for OMeAN-2BR3, 8.5 for OMeAN-2H+, and 6.1 for OMeAN-Sc3+ (temperature: −90 °C). These values are consistent with C–H bond cleavage during the rate-determining step (r.d.s.).
Figure 7.

C–H oxidations performed by OMeAN-LA cores: (A) kinetic analysis of the reaction (see Supporting Information for further details); (B) substrates used and proposed mechanism for HAT + e− transfer.
Table 1.
Kinetic Parameters of C–H Oxidation Reactions by OMeAN-LA
| substrate | BDE (H•) (kcal/mol) | BDE (H−) (kcal/mol) | kOMeAN32H+ (M−1 s−1) | kOMeAN3Sc3+ (M−1 s−1) | kOMeAN32BR3 (M−1 s−1) | notes |
|---|---|---|---|---|---|---|
| BNAH | 70.7 | 64.2 | 66 ± 1 | 58 ± 1 | 138 ± 2 | yield: 80–99% |
| BNAD | 7.8 ± 0.1 (KIE: 8.5) | 5.7 ± 0.1 (KIE: 10.3) | 22.7 ± 0.3 (KIE: 6.1) | |||
| BzImH | 73.4 | 49.5 | 12.1 ± 0.2a | 6.0 ± 0.1b | 19.1 ± 0.2c | kBNAH/BzImH: 5.5, 9.7, 7.2a,b,c |
| AcrH2 | 73.0 | 81.1 | too slow | too slow | too slow | less than 5% decay after 1 h |
| DHA | 76 | no reaction | no reaction | no reaction | no decay (t = 1 h, −90 °C) |
kOMeAN−2H+.
kOMeAN−Sc3+.
kOMeAN−2BR3.
Two mechanistic scenarios are possible: (i) stepwise H• transfer (HAT) during the r.d.s. followed by e− transfer or (ii) a one-step hydride (H−) transfer during the r.d.s. We previously reported that a copper(I)-dioxygen adduct, formally a cupric-superoxide species, also preferentially effected the energetically more difficult H•-abstraction in preference to hydride abstraction chemistry.27 In order to distinguish between these two possibilities, the reaction between 1,3-dimethyl-2,3-dihydrobenzimidazole (BzImH) and the OMeAN-LA series was tested. BzImH is a better hydride donor (49.5 kcal/mol heterolytic C–H BDE, vs 64.2 kcal/mol for BNAH) but a poorer H• donor (73.4 kcal/mol homolytic C–H BDE vs 70.7 kcal/mol for BNAH). For all the OMeAN-LA complexes, slower reaction rates for BzImH were observed (5–10 times slower, see Table 1), consistent with HAT during the r.d.s. (Figure 7) rather than concerted hydride transfer.
The reactivity described herein is consistent with previous reports that suggest that O-type species rather than SP carry out the intra- or intermolecular oxidation of C–H bonds.14,20,21 Although the difference in the reaction rates found for the different OMeAN-LA species was modest (the OMeAN-2BR3 adduct was 2–3 times faster for all the substrates tested), it is worth mentioning that subtle changes in the stereoelectronic properties of the OMeAN-LA cores, tuned by the use of different Lewis acids, could lead to differential and useful oxidation kinetics/chemistries. Our findings are in accordance with the data reported by Goldberg and co-workers, where the MnIV(O):B(C6F5)3 adduct ([(TBP8Cz•+)MnIV(O):B(C6F5)3]) (TBP8Cz = octakis(p-tert-butylphenyl)corrolazinato) was found to perform the oxidation of C–H and O–H bonds via HAT faster than the corresponding MnIV(O):Zn2+ adduct.40,46
Reactivity of OMeAN-LA toward O–H Bonds
We have also explored the reactivity of the SPMeAN and OMeAN-LA toward phenols. As previously reported, SPMeAN was found to be unreactive toward 2,4-tBu2-phenol. As shown in Table 2, we tested the reactivity of SPMeAN/OMeAN-LA with a series of 4-substituted phenols with different O–H bond strengths and varying one-electron redox potentials (E0). The SPMeAN complex was unreactive toward all of the phenols tested. However, the family of OMeAN-LA cores was found to be reactive in the 1e−/1H+ oxidation of phenols with BDEs lower than 84.6 kcal/mol and E0 values lower than 1.12 V (Table 2, see Supporting Information for product quantification). The oxidation of 2,4-di-tert-butylphenol (2,4-tBu-PhOH) by the OMeAN-LA cores led to the formation of the C–C coupling product (see Supporting Information for details on its detection and quantification).
Table 2.
Kinetic Parameters of O–H Oxidation Reactions by OMeAN-LA
| substrate | BDE (H•) (kcal/mol) | E0 (V vs Fc+/0) | kOMeAN32H+ (M−1 s−1) | kOMeAN3Sc3+ (M−1 s−1) | kOMeAN32BR3 (M−1 s−1) | notes |
|---|---|---|---|---|---|---|
| 4-MeO-PhOH | 80.9 | 1.01 | 64 ± 1 | 53 ± 1 | 2.9 ± 0.2 | a |
| 4-MeO-PhOD | 1.01 | 52 ± 1 (KIE: 1.25) | 42 ± 1 (KIE: 1.19) | 2.4 ± 0.1 (KIE: 1.17) | a | |
| 2,4-tBu2-PhOH | 81.9 | 1.04 | 1.8 ± 0.2 | 1.1 ± 0.1 | 0.12 ± 0.01 | yield: 60–70% |
| 4-PhO-PhOH | 1.07 | 0.51 ± 0.02 | 0.44 ± 0.02 | 0.046 ± 0.001 | a | |
| 4-tBu-PhOH | 1.10 | 0.025 ± 0.001 | 0.023 ± 0.001 | 0.0027 ± 0.0003 | a | |
| 4-Me-PhOH | 84.6 | 1.12 | 0.035 ± 0.002 | 0.027 ± 0.001 | 0.0024 ± 0.0002 | a |
| 4-Cl-PhOH | 85.7 | 1.21 | no reaction | no reaction | no reaction | no decay (t = 1 h) |
Product not quantified (see text and Supporting Information).
Kinetic analyses of these reactions (Figure 8A) led to pseudo-first-order behavior with varying phenol concentrations and gave linear second-order plots allowing for the calculation of second-order rate constants, which are listed in Table 2. Overall, the trends found in phenol (O–H) oxidation are reversed from BNAH/BzImH (C–H) oxidation; the OMeAN-2H+ and OMeAN-Sc3+ complexes exhibit higher reaction rates (10–20 times) than the corresponding OMeAN-2BR3 complex. When the reaction rates found were plotted against the 1e−reduction potentials of the phenols ((RT/F)·ln(k) vs E0), a linear correlation was obtained (Figure 8A, right).35,49,50
Figure 8.

O–H oxidation performed by OMeAN-LA cores: (A) kinetic analysis; (B) possible reaction mechanisms and comparison of KIEs (see Figure S18) and reduction potential effects to literature examples.
According to Marcus theory, the slope value obtained in this correlation can be used to distinguish between pure H• transfer (HAT: slope = 0.0) from electron transfer (ET) or proton-coupled electron transfer (PCET: slope = −0.5 if ΔG0et < 0; slope = −1.0 if ΔG0et > 0).49 The low KIE’s observed (1.1–1.2, see Figure S18) along with the slope values (around −1.0) suggest a mechanistic scenario where endergonic electron transfer (ET) is followed by fast proton transfer (PT). This proposal is in agreement with the 1e− reactivity observed with substituted Fc (see Supporting Information), where the OMeAN-LA complexes were only reduced by donors with E0 equal or lower than −0.12 V (e.g., Me2Fc), a value substantially lower (by 1.0–1.5 V) than the phenol 1e− oxidation potential. (The OMeAN-LA complexes were not reduced by Fc, E0 = 0.0 V vs Fc+/0.) An alternative mechanism with rate-limiting PT followed by 1e− oxidation of the phenolate is not consistent with the large negative slope of the rate/potential correlation or the small primary H/D isotope effect.
The mechanistic study of the 1e−/1H+ oxidation of phenols by LCu/O2 derived species was first reported by Itoh and coworkers, where SP and O species were found to oxidize 4-substituted phenols via proton-coupled electron transfer (PCET) with somewhat smaller slopes (−0.72 and −0.71, respectively) and similar KIEs (1.2–1.6 and 1.2–1.5, respectively), where the smaller slope indicates less endergonic ET such that ET and PT are kinetically similar.49 Similarly, Costas and co-workers reported that end-on peroxo species are also able to oxidize phenols via PCET (slope = −0.49, KIE: 1.5–2.0), where the slope of ~ −0.5 is consistent with exergonic ET.51 Very recently, we reported that a mononuclear end-on copper(II)-superoxide intermediate (ES) oxidizes phenols via HAT (slope: −0.29, KIE: 9.0).35 The results reported herein (stepwise ET-PT, slope = −1.0, KIE: 1.1–1.3) are unprecedented for the reactivity shown by Cu/O2-derived species, in particular the ability to perform substrate oxidation that involves endergonic ET.52 Furthermore, compared to SPMeAN, which is unreactive toward phenols, the enhanced oxidative reactivity of OMeAN-LA suggests that one important consequence of Lewis acid binding is to raise the reduction potential of the Cu2O2 core to allow heightened oxidative reactivity. Thus, Lewis acid coordination is a strategy that can not only effect the core structure (SP/O equilibrium) but also may tune the reduction potential of the Cu2O2 core to facilitate substrate oxidation.
CONCLUSIONS
In this paper, we showed that the unreactive Cu2O2 core of SPMeAN could be activated by substrate binding (i.e., sodium 4-R-phenolates) and by Lewis acid addition triggering O–O cleavage to form OMeAN cores. First, phenolate binding to the copper ion (primary coordination sphere) led to formation of a fleeting phenolate-bound dicopper(III) species (OMeAN-RPhO−) which was characterized by UV–vis and rR spectroscopies. TD-DFT calculations provided evidence that sequential phenolate binding as an axial ligand and isomerization to the equatorial form occurs before the r.d.s., where the O core electrophilically attacks the phenyl ring.
Second, we demonstrated that addition of Lewis acids to SPMeAN also led to O–O cleavage (OMeAN-LA formation) with coordination of the different Lewis acids occurring at the secondary coordination sphere via binding to what was the axial N of the MeAN ligand. Importantly, the oxidative properties of the OMeAN-LA species toward external substrates were influenced by the Lewis acid identity. Mechanistic studies provided evidence that C–H oxidation occurred via hydrogen atom transfer, with the borane adduct OMeAN-2BR3 reacting slightly faster than did the OMeAN-2H+and OMeAN-Sc3+ cores. Conversely, phenol oxidation was found to happen via stepwise electron-transfer proton-transfer (ET-PT), with the OMeAN-2H+ and OMeAN-Sc3+ cores reacting much faster than the OMeAN-2BR3 analogue.
These findings can inspire new routes to develop oxidation systems based on first row metals and dioxygen by utilizing Lewis acids to fine-tune the stereoelectronic properties of the reactive intermediates, leading potentially to novel selectivity patterns. Moreover, these results emphasize the importance of understanding the influence of the secondary as well as the primary coordination spheres in order to explain the speciation (type of Cu/O2 species) and reactivity of metalloenzymes such as pMMO.
Supplementary Material
Acknowledgments
This research was supported by the U.S. NIH (GM60353 to K.D.K., DK31450 to E.I.S., and NRSA postdoctoral fellowship F32-GM105288 to R.E.C.). I.G.-B. thanks the Robert A. Welch Foundation for financial support (grant N-1900).
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b12990.
UV–vis experimental details and spectra, product analyses, rR experiments, DFT calculations, and xyz coordinates for the DFT structures (PDF)
References
- 1.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Chem Rev. 2014;114:3659. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solem E, Tuczek F, Decker H. Angew Chem, Int Ed. 2016;55:2884. doi: 10.1002/anie.201508534. [DOI] [PubMed] [Google Scholar]
- 3.Decker H, Schweikardt T, Tuczek F. Angew Chem, Int Ed. 2006;45:4546. doi: 10.1002/anie.200601255. [DOI] [PubMed] [Google Scholar]
- 4.Mirica LM, Vance M, Rudd DJ, Hedman B, Hodgson KO, Solomon EI, Stack TDP. Science. 2005;308:1890. doi: 10.1126/science.1112081. [DOI] [PubMed] [Google Scholar]
- 5.Citek C, Herres-Pawlis S, Stack TDP. Acc Chem Res. 2015;48:2424. doi: 10.1021/acs.accounts.5b00220. [DOI] [PubMed] [Google Scholar]
- 6.Rosenzweig AC. Nature. 2015;518:309. doi: 10.1038/nature14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lawton TJ, Rosenzweig AC. J Am Chem Soc. 2016;138:9327. doi: 10.1021/jacs.6b04568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balasubramanian R, Rosenzweig A. Acc Chem Res. 2007;40:573. doi: 10.1021/ar700004s. [DOI] [PubMed] [Google Scholar]
- 9.Culpepper MA, Cutsail GE, Hoffman BM, Rosenzweig AC. J Am Chem Soc. 2012;134:7640. doi: 10.1021/ja302195p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Citek C, Lin BL, Phelps TE, Wasinger EC, Stack TDP. J Am Chem Soc. 2014;136:14405. doi: 10.1021/ja508630d. [DOI] [PubMed] [Google Scholar]
- 11.Yoshizawa K, Shiota Y. J Am Chem Soc. 2006;128:9873. doi: 10.1021/ja061604r. [DOI] [PubMed] [Google Scholar]
- 12.Mirica LM, Ottenwaelder X, Stack TDP. Chem Rev. 2004;104:1013. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 13.Himes RA, Karlin KD. Curr Opin Chem Biol. 2009;13:119. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis EA, Tolman WB. Chem Rev. 2004;104:1047. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 15.Itoh S. Curr Opin Chem Biol. 2006;10:115. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 16.Serrano-Plana J, Garcia-Bosch I, Company A, Costas M. Acc Chem Res. 2015;48:2397. doi: 10.1021/acs.accounts.5b00187. [DOI] [PubMed] [Google Scholar]
- 17.Halfen JA, Mahapatra S, Wilkinson EC, Kaderli S, Young VG, Jr, Que L, Jr, Zuberbühler AD, Tolman WB. Science. 1996;271:1397. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- 18.Kieber-Emmons MT, Ginsbach JW, Wick PK, Lucas HR, Helton ME, Lucchese B, Suzuki M, Zuberbühler AD, Karlin KD, Solomon EI. Angew Chem, Int Ed. 2014;53:4935. doi: 10.1002/anie.201402166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim S, Ginsbach JW, Billah AI, Siegler MA, Moore CD, Solomon EI, Karlin KD. J Am Chem Soc. 2014;136:8063. doi: 10.1021/ja502974c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatcher LQ, Karlin KD. JBIC, J Biol Inorg Chem. 2004;9:669. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 21.Hatcher LQ, Karlin KD. Adv Inorg Chem. 2006;58:131. [Google Scholar]
- 22.Park GY, Qayyum MF, Woertink J, Hodgson KO, Hedman B, Narducci Sarjeant AA, Solomon EI, Karlin KD. J Am Chem Soc. 2012;134:8513. doi: 10.1021/ja300674m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Company A, Palavicini S, Garcia-Bosch I, Mas-Balleste R, Que L, Rybak-Akimova EV, Casella L, Ribas X, Costas M. Chem - Eur J. 2008;14:3535. doi: 10.1002/chem.200800229. [DOI] [PubMed] [Google Scholar]
- 24.Liang HC, Zhang CX, Henson MJ, Sommer RD, Hatwell KR, Kaderli S, Zuberbuehler AD, Rheingold AL, Solomon EI, Karlin KD. J Am Chem Soc. 2002;124:4170. doi: 10.1021/ja0125265. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Bosch I, Company A, Frisch JR, Torrent-Sucarrat M, Cardellach M, Gamba I, Gúell M, Casella L, Que L, Jr, Ribas X, Luis JM, Costas M. Angew Chem, Int Ed. 2010;49:2406. doi: 10.1002/anie.200906749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serrano-Plana J, Garcia-Bosch I, Miyake R, Costas M, Company A. Angew Chem, Int Ed. 2014;53:9608. doi: 10.1002/anie.201405060. [DOI] [PubMed] [Google Scholar]
- 27.Peterson RL, Himes RA, Kotani H, Suenobu T, Tian L, Siegler MA, Solomon EI, Fukuzumi S, Karlin KD. J Am Chem Soc. 2011;133:1702. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09 revision D.01. Gaussian Inc; Wallingford, CT: 2009. [Google Scholar]
- 29.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]
- 30.Lee C, Yang W, Parr RG. Phys Rev B: Condens Matter Mater Phys. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 31.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623. [Google Scholar]
- 32.Grimme S, Antony J, Ehrlich S, Krieg H. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi K, Jensen F, Dorigo A, Houk KN. Chem Phys Lett. 1988;149:537. [Google Scholar]
- 34.Gorelsky SI. SWizard, version 4.2. University of Ottawa; Ottawa, Ontario, Canada: 2013. http://www.sg-chem.net/swizard/ [Google Scholar]
- 35.Lee JY, Peterson RL, Ohkubo K, Garcia-Bosch I, Himes RA, Woertink J, Moore CD, Solomon EI, Fukuzumi S, Karlin KD. J Am Chem Soc. 2014;136:9925. doi: 10.1021/ja503105b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsumoto T, Ohkubo K, Honda K, Yazawa A, Furutachi H, Fujinami S, Fukuzumi S, Suzuki M. J Am Chem Soc. 2009;131:9258. doi: 10.1021/ja809822c. [DOI] [PubMed] [Google Scholar]
- 37.Op’t Holt BT, Vance MA, Mirica LM, Heppner DE, Stack TDP, Solomon EI. J Am Chem Soc. 2009;131:6421. doi: 10.1021/ja807898h.(b) The hydroxylation must occur through the equatorially-coordinated phenolate since the substrate ring must be attacked by the oxo in the Cu2O2 plane. Axial coordination of the phenolate places the substrate ring above the Cu2O2 plane; thus, the axial isomer must isomerize to the equatorial isomer to effect hydroxylation.
- 38.(a) Holland PL, Cramer CJ, Wilkinson EC, Mahapatra S, Rodgers KR, Itoh S, Taki M, Fukuzumi S, Que L, Tolman WB. J Am Chem Soc. 2000;122:792. [Google Scholar]; (b) Tang J, Albrecht AC. In: In Raman Spectroscopy. Szymanski HA, editor. Vol. 2 Plenum; New York: 1970. [Google Scholar]
- 39.Chen J, Lee YM, Davis KM, Wu X, Seo MS, Cho KB, Yoon H, Park YJ, Fukuzumi S, Pushkar YN, Nam W. J Am Chem Soc. 2013;135:6388. doi: 10.1021/ja312113p. [DOI] [PubMed] [Google Scholar]
- 40.(a) Baglia RA, Dürr M, Ivanović-Burmazović I, Goldberg DP. Inorg Chem. 2014;53:5893. doi: 10.1021/ic500901y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baglia RA, Krest CM, Yang T, Leeladee P, Goldberg DP. Inorg Chem. 2016;55:10800. doi: 10.1021/acs.inorgchem.6b02109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoon H, Lee YM, Wu X, Cho KB, Sarangi R, Nam W, Fukuzumi S. J Am Chem Soc. 2013;135:9186. doi: 10.1021/ja403965h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park J, Lee YM, Nam W, Fukuzumi S. J Am Chem Soc. 2013;135:5052. doi: 10.1021/ja311662w. [DOI] [PubMed] [Google Scholar]
- 43.Henson MJ, Mukherjee P, Root DE, Stack TDP, Solomon EI. J Am Chem Soc. 1999;121:10332. [Google Scholar]
- 44.The presence of this ~140 cm−1 mode was not directly observed due to significant elastic scattering of the Raman samples below 200 cm−1. This mode has been directly observed along with its combination bands in a similar Cu2(μ-O)2 complex; see ref 43.
- 45.(a) Several isomers of the OMeAN-LA complexes were surveyed, including cis/trans and syn/anti orientation of the two uncoordinated ligand arms. The lowest energy structures are given in Figure 5. (b) An O-protonated OMeAN-2H+ species was also evaluated computationally and was found to be energetically similar to the N-protonated species in Figure 5 (+0.5 kcal/mol); however, the calculated normal modes were inconsistent with the rR data (see Figure S25). Attempts to optimize OMeAN-2BMe3 or OMeAN-Sc(OTf)2 with O-bound Lewis acids invariably led to cleavage of a Cu–O bond and opening of the diamond core. Since these structures would not be consistent with the spectroscopy indicating bis(μ-oxo) cores, they were not evaluated further.
- 46.Leeladee P, Baglia RA, Prokop KA, Latifi R, de Visser SP, Goldberg DP. J Am Chem Soc. 2012;134:10397. doi: 10.1021/ja304609n. [DOI] [PubMed] [Google Scholar]
- 47.Li F, Van Heuvelen KM, Meier KK, Münck E, Que L. J Am Chem Soc. 2013;135:10198. doi: 10.1021/ja402645y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bang S, Lee YM, Hong S, Cho KB, Nishida Y, Seo MS, Sarangi R, Fukuzumi S, Nam W. Nat Chem. 2014;6:934. doi: 10.1038/nchem.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Osako T, Ohkubo K, Taki M, Tachi Y, Fukuzumi S, Itoh S. J Am Chem Soc. 2003;125:11027. doi: 10.1021/ja029380+. [DOI] [PubMed] [Google Scholar]
- 50.A linear relationship was also found between the reaction rates and the BDFE of the phenols (only three values available), with the BDFE directly proportional to E0 and the pKa.
- 51.Garcia-Bosch I, Ribas X, Costas M. Chem - Eur J. 2012;18:2113. doi: 10.1002/chem.201102372. [DOI] [PubMed] [Google Scholar]
- 52.Dhar D, Yee GM, Markle TF, Mayer JM, Tolman WB. Chem Sci. 2017;8:1075. doi: 10.1039/c6sc03039d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.