

Abstract
Recent investigations have shed much light on the nuclear and electronic factors that control the rates of long-range electron tunneling through molecules in aqueous and organic glasses as well as through bonds in donor–bridge–acceptor complexes. Couplings through covalent and hydrogen bonds are much stronger than those across van der Waals gaps, and these differences in coupling between bonded and nonbonded atoms account for the dependence of tunneling rates on the structure of the media between redox sites in Ru-modified proteins and protein–protein complexes.
Keywords: electron tunneling, hopping, glass, protein
Independent efforts by Gamow (1) in late 1928 and Gurney and Condon (2) in early 1929 to explain radioactive decay using the “new quantum mechanics” produced an expression for the probability that a particle will tunnel through a square potential energy barrier. The transmission coefficient (κ) decays exponentially with increasing barrier width (r) and the decay constant varies as the square root of the product of the barrier height (ΔE) times the mass (m) of the particle (Eq. 1).
 |
[1] |
This theory rationalized the microsecond tunneling of an α-particle through a 3 × 10-4-Å, 6-MeV barrier (2) and predicted that in the same time period an electron could tunnel 19 Å through a 1-eV barrier. In the intervening years, electron tunneling has been found to play pivotal roles in solid-state physics (3), chemistry (4, 5), and biology (5–7).
A Landau-Zener treatment of the reactant-product transition probability produces the familiar semiclassical expression for the rate of nonadiabatic electron transfer (ET) between a donor (D) and acceptor (A) held at fixed distance (Eq. 2) (5).
 |
[2] |
The electronic coupling matrix element (HAB) reflects the strength of the interaction between reactants and products at the nuclear configuration of the transition state. McConnell (8), building on prior work by Halpern and Orgel (9), argued that the interaction energy (2HAB) between two redox centers separated by a covalent bridge composed of n identical repeat units depends on the coupling strength between the redox sites and the bridge (hAb, hbB), the coupling between adjacent bridge elements (hbb), and the tunneling energy gap (Δε), defined as the vertical energy required to remove an electron from the donor, or a hole from the acceptor, and place it on the bridge. If the donor-acceptor separation (d) is a linear function of n (d = do + nδ), then
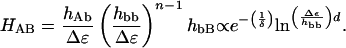 |
[3] |
HAB will decay exponentially with distance (hbb ≪ Δε; Eq. 3). Using the result from semiclassical theory that ET rates are proportional to 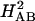 (Eq. 2), it is possible to define effective tunneling barriers (ΔEeff) in terms of superexchange parameters, as well as the exponential decay constant (β) describing the variation of rates with distance (Eq. 4).
(Eq. 2), it is possible to define effective tunneling barriers (ΔEeff) in terms of superexchange parameters, as well as the exponential decay constant (β) describing the variation of rates with distance (Eq. 4).
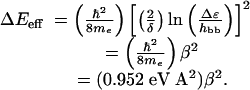 |
[4] |
Saturated hydrocarbon spacers typically exhibit β ≈1.0 Å-1 (ΔEeff = 0.95 eV) (10–12). The decay constant for phenylene bridges depends on the dihedral angle between adjacent aromatic rings [0.4–0.8 Å-1 (13, 14); ΔEeff = 0.15–0.61 eV]. Polyene and phenylenevinylene bridges exhibit remarkably efficient ET over very long distances: β-values as low as 0.04 Å-1 (ΔEeff = 0.002 eV) have been reported (15).
Aqueous and Organic Glasses
Several experimental investigations have demonstrated that solvent hole and electron states can mediate long-range electron tunneling (16–21). In fluid solution, when the positions of D and A are not constrained by a covalent bridge, diffusion places an upper limit on the time scale (<10-9 s) and, therefore, the tunneling distance range (<9 Å for β = 1.0 Å-1). Longer tunneling distances can be examined if D and A are immobilized. In a typical experiment, a small concentration of electron or hole donors is embedded in a glassed solvent amid a higher concentration of randomly distributed acceptors. The donor is a photoexcited chromophore or a radiolytically generated radical. The time-dependent survival probability of the donor depends on the concentration of acceptors, the rate constant for electron/hole transfer when D and A are in van der Waals contact (ko), and the distance decay factor β. Extracting reliable values for ko and β from time-resolved spectroscopic measurements, however, can be rather difficult because the two parameters are highly correlated (19, 22). In the case of photoinitiated ET in glasses, measurements of luminescence decay kinetics and luminescence quantum yields at several different quencher concentrations provide enough information to decouple ko and β, permitting reliable values to be determined for each parameter (19).
Our experimental investigation of  luminescence quenching by
luminescence quenching by 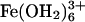 in aqueous acidic glasses placed rigorous limits on the distance decay constant for tunneling through water (23). The luminescence lifetime of
in aqueous acidic glasses placed rigorous limits on the distance decay constant for tunneling through water (23). The luminescence lifetime of  in aqueous glasses is long enough to allow a significant distance range (≈25 Å) to be probed. A distance decay constant of 1.58(5) Å-1 was obtained for H2SO4/H2O, HSO3F/H2O, and D2SO4/D2O glasses (ΔEeff = 2.4 eV) (14).
in aqueous glasses is long enough to allow a significant distance range (≈25 Å) to be probed. A distance decay constant of 1.58(5) Å-1 was obtained for H2SO4/H2O, HSO3F/H2O, and D2SO4/D2O glasses (ΔEeff = 2.4 eV) (14).
We also have determined β and ΔEeff values for electron tunneling from electronically excited [Ir(μ-pyrazolyl)(1,5-cyclooctadiene)]2 to 2,6-dichloro-1,4-benzoquinone in 2-methyltetrahydrofuran (MTHF) and toluene glasses at 77 K (14). The effective barrier height for electron tunneling through toluene (ΔEeff = 1.4 eV) is substantially lower than the barrier in MTHF (ΔEeff = 2.6 eV); and the barrier for tunneling in aqueous sulfuric acid glasses (ΔEeff = 2.4 eV) is very near that for MTHF. Distance decay parameters are 1.62 (MTHF), 1.58 (H2O), and 1.23 Å-1 (toluene) (14). In toluene and MTHF, coupling between bridge units is mediated by van der Waals contacts, whereas the aqueous glass is interlaced with strong hydrogen bonds. It is possible that the hydrogen bonds between molecules in the aqueous glass compensate for the large molecular orbital (H2O) energy gap to produce a tunneling barrier on par with that of MTHF.
The 1.62-Å-1 distance decay constant for MTHF confirms that there is a significant coupling penalty associated with tunneling across the van der Waals gaps between solvent molecules. Taking 20 Å as a reference distance, we find that tunneling across an alkane bridge (β ≈ 1.0 Å-1) is ≈40,000 times faster than tunneling through MTHF; and, to underscore the point, recent experiments on D-oligoxylene-A complexes have shown that 20-Å tunneling across covalently linked xylenes is almost 3,000 times faster than tunneling through a toluene glass (Fig. 1) (14).
Fig. 1.

Timetable for activationless electron tunneling through various media: vacuum (black, β = 2.9–4.0 Å-1), MTHF glass (violet, β = 1.57–1.67 Å-1), aqueous glass (cyan, β = 1.55–1.65 Å-1), and toluene glass (green, β = 1.18–1.28 Å-1). Investigations of ET rates in D-(bridge)-A complexes have produced exponential distance dependences: xylyl bridges, β = 0.76 Å-1 (red) (12); alkane bridges, β = 1.0 Å-1 (orange) (10); and β-strand bridges in ruthenium-modified azurin, β = 1.1 Å-1 (yellow).
Ru-Proteins
The protein fold plays a central role in lowering the reorganization energy of a biological ET reaction (24, 25). Continuum models suggest that embedding a redox center inside a low dielectric cavity can lower the outer-sphere λ by as much as 50% (26). Moreover, by constraining the coordination environment around metal centers, the inner-sphere λ can be reduced as well (25). Copper is a case in point. The reorganization energy for electron self-exchange in 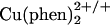 is ≈2.4 eV; the value for Cu(II/I) in Pseudomonas aeruginosa azurin is 0.7 eV (24). The 1.7-eV reduction in λ reflects the transition-state stabilization imposed by the azurin fold.
is ≈2.4 eV; the value for Cu(II/I) in Pseudomonas aeruginosa azurin is 0.7 eV (24). The 1.7-eV reduction in λ reflects the transition-state stabilization imposed by the azurin fold.
In 1982, we demonstrated long-range electron tunneling through Ru-modified cytochrome (cyt) c (27). Subsequent work in our laboratory has focused on the elucidation of distant electronic couplings between redox sites in several Ru-proteins (7, 28–32). In particular, work on Ruazurin has provided a reference point for electron tunneling through folded polypeptide structures (7, 31, 32). The copper center in azurin is situated at one end of an eight-stranded β-barrel, ligated in a trigonal plane by two imidazoles (His-46 and His-117) and a thiolate (Cys-112); in addition, there are weak axial interactions (Met-121 thioether sulfur and Gly-45 carbonyl oxygen) (33, 34). The azurin from P. aeruginosa has two additional His residues, one of which (His-83) reacts readily with Ru-labeling reagents. A H83Q base mutant was prepared and individual mutant His residues were introduced at five locations on β-strands extending from the Cys-112 and Met-121 ligands (K122H, T124H, T126H, Q107H, and M109H) (31, 32). Tunneling distances (Ru-Cu) in these five Ru(bpy)2(im)(HisX)2+-azurins and Ru(bpy)2(im)(His-83)2+-azurin are shown in Fig. 2.
Fig. 2.

Tunneling distances and backbone structure models showing locations of the Cu active site (blue) and the Ru(bpy)2(im)(HisX)2+ label (orange) in Ru-azurins: Ru-Cu (Å), HisX (X = 122, 15.9; 83, 16.9; 109, 17.9; 124, 20.6; 107, 25.7; 126, 26.0).
Measurements of Cu(I) → Ru(III) ET (-ΔG° = 0.7 eV) in the set of Ru-azurins established the distance dependence of ET along β-strands (7, 31, 32). The driving force-optimized azurin tunneling timetable reveals a nearly perfect exponential distance dependence, with a decay constant (β) of 1.1 Å-1, and an intercept at close contact (ro = 3 Å) of 1013 s-1. This decay constant is quite similar to that found for superexchange-mediated tunneling across saturated alkane bridges (β ≈1.0 Å-1) (12, 35), strongly indicating that a similar coupling mechanism is operative in the polypeptide (Fig. 1). Importantly, kinetics data obtained by Farver and Pecht (36) in their studies of long-range ET from radiolytically generated disulfide radical anion to the blue copper center in azurin also have been interpreted successfully in terms of this coupling model.
Our studies have shown that Cu(I) to Ru(III) or Os(III) ET rates in labeled azurin crystals are nearly identical with solution values for each donor–acceptor pair (34). The energy gap between the donor–acceptor redox levels and those of oxidized or reduced intermediate states is the primary criterion in determining when hole or electron hopping becomes important. In Ru-azurin, photogenerated Ru(bpy)2(im)(His)3+ (E° = 1.0 V vs. NHE) (37) potentially could oxidize Trp or Tyr residues (7). If the Cu(I) center is replaced by redox-inert Zn(II) in the protein, however, we find that photogenerated holes in Ru(bpy)2(im)(HisX)3+ complexes remain localized on the metal center. The energy gap between the Ru(III) hole and oxidized bridge states therefore must be >75 meV (3 kBT at 295 K). Our finding that the Cu(I) → Ru(III) ET rate in Ru(bpy)2(im)(HisX)-azurin does not decrease in going from 300 to 240 K and actually increases slightly at 160 K demonstrates that hopping does not occur in this case, as a reaction with an endergonic step would be highly disfavored at low temperature. We conclude that the Ru-azurin timetable (Fig. 1) provides a benchmark for superexchange-mediated electron tunneling through proteins.
The rates of high driving-force ET reactions have been measured for >30 Ru(dii-mine)-labeled metalloproteins (7, 29, 30, 38). Driving-force-optimized values are scattered around the Ru-azurin 1.1-Å-1 exponential distance decay. Rates at a single distance can differ by as much as a factor of 103, and D/A distances that differ by as much as 5 Å can produce identical rates (Fig. 3). In seminal work, Beratan, Onuchic, and coworkers (39–41) developed a generalization of the McConnell superexchange coupling model that accounts for rate scatter attributable to protein structural complexity. In this tunneling-pathway model, the medium between D and A is decomposed into smaller subunits linked by covalent bonds, hydrogen bonds, or through-space jumps. More elaborate computational protocols also have shed light on the factors that determine distant couplings in proteins (42–46). Indeed, in this issue of PNAS, Beratan and coworkers (47) present a detailed analysis, including the effects of protein dynamics on the distant couplings of six Ru-azurins (Fig. 2).
Fig. 3.

Tunneling timetable for intraprotein ET in Ru-modified azurin (blue circles), cyt c (red circles), myoglobin (yellow triangles), cyt b562 (green squares), HiPIP (orange diamonds), and for interprotein ET Fe:Zn-cyt c crystals (fuchsia triangles). The solid lines illustrate the tunneling-pathway predictions for coupling along β-strands (β = 1.0 Å-1) and α-helices (β = 1.3 Å-1); the dashed line illustrates a 1.1-Å-1 distance decay. Distance decay for electron tunneling through water is shown as a cyan wedge. Estimated distance dependence for tunneling through vacuum is shown as the black wedge.
Medium Dynamics
The nonadiabatic ET model embodied in Eq. 2 rests on the assumption that the electronic transition from the reactant potential energy surface (D + A) to the product surface (D+ + A-) is much slower than the frequency of nuclear motion on these surfaces. Both theoreticians and experimentalists have long been interested in charge-transfer processes that are not well described by this model (48–54).
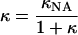 |
[5] |
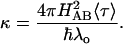 |
[6] |
Theory suggests that, under certain circumstances, the time scales for reorientation of solvent molecules can be slower than the reactant-product transition frequency. In this solvent-controlled adiabatic limit, reactions are limited by the dynamics of solvent relaxation. Bixon and Jortner (53, 54) developed a generalized expression for ET rates that explicitly accounts for solvent relaxation dynamics (Eqs. 5 and 6). The factor κ is a solvent adiabaticity factor and kNA is the nonadiabatic ET rate given by Eq. 2. Small values of κ correspond to the nonadiabatic limit; large values result in solvent-controlled adiabatic processes. Typical solvent relaxation times (〈τ〉) are ≤10-11 s, so that solvent relaxation dynamics are expected to become important only in relatively strongly coupled systems. An adiabaticity factor of 1 in a solvent with a 1-ps relaxation time, for example, corresponds to a coupling matrix element of ≈50 cm-1 (λo = 0.5 eV).
Our studies of ET in Ru-proteins indicate that a large part of the contribution to λo comes from reorientation of the polypeptide matrix (34). The dynamics of large-scale nuclear motions in polypeptides are expected to be substantially slower than those of most solvents. Relaxation times ranging from picoseconds to microseconds have been reported for the heme pocket of myoglobin (55–58). Indeed, electrochemical measurements by Waldeck and coworkers (59) using cyt c adsorbed onto self-assembled monolayers suggest that the characteristic relaxation time for protein ET (〈τ〉) is on the order of 200 ns. We emphasize, however, that eight tunneling times measured for four different Ru-proteins are <200 ns (Fig. 3).
Bixon and Jortner (53, 54) have noted that there are several examples of ET rates exceeding the solvent-controlled adiabatic limit; model calculations suggest a possible explanation. Reactions at low driving force (-ΔG° < λ) require substantial reorganization along solvent coordinates and rates are predicted to exhibit a pronounced dependence on relaxation dynamics. The calculations suggest, however, that the rates of activationless (-ΔG° ≈ λ) and inverted (-ΔG° > λ) reactions will be nearly independent of κ and, hence, the dynamics of medium relaxation, as we have found in the case of Ru(diimine)-protein ET reactions (7).
Protein–Protein Reactions
At least three elementary steps are required to complete a redox reaction between soluble proteins: (i) formation of an active donor–acceptor complex; (ii) electron tunneling within the donor–acceptor complex; and (iii) dissociation of the oxidized and reduced products. Because the dynamics of the first and third steps obscure the electron tunneling reaction, experimental studies often focus on ET reactions within protein–protein complexes that form at low ionic strength. It has been difficult to interpret the results, however, as neither the donor–acceptor docking geometries nor the conformations of these complexes are known with certainty. With the aid of rapid triggering methods, it has been possible to measure rates of long-range ET between redox sites in protein–protein complexes. In many complexes, there are multiple binding sites and it is not uncommon to find that the ET kinetics often are regulated by the dynamics of conformational changes in the complex (60, 61). The usual interpretation is that surface diffusion of the two proteins produces a transient complex with enhanced electronic coupling and faster ET. Consequently, rates depend strongly on solvent viscosity rather than intrinsic ET parameters. A further complication associated with studies of protein–protein ET in solution is that binding sites and, hence, locations of redox cofactors, often are unknown.
Crystals containing photoactivatable donors and acceptors at specific lattice sites are ideal media for investigating tunneling between proteins. In crystal lattices of tuna cyt c, chains of protein molecules form helices with a 24.1-Å separation between neighboring metal centers (62). By doping Zn-cyt c into this lattice, interprotein ET between triplet-excited Znporphyrin and a neighboring Fe(III)-cyt c could be investigated; the rate constant was found to be 4(1) × 102 s-1, and charge recombination was about four times faster [2.0(5) × 103 s-1] (62).
Rapid relay of electrons involving at least one soluble redox enzyme requires the formation of short-lived, weakly bound protein–protein complexes. The recognition sites between proteins in such complexes tend to be smaller (<1,200 Å2) and include more water molecules than the interfaces between subunits in oligomeric proteins (63). The interprotein interactions in crystals of tuna cyt c involve relatively few contacts: 760 Å2 of surface area is buried in an interface with 31 van der Waals contacts (3.2 ≤ d ≤ 3.9 Å) and 16 water molecules (3 of which form bridging hydrogen bonds across the interface) but only one direct hydrogen bond bridging the two proteins. Indeed, the cyt c–cyt c interface is reminiscent of that between natural redox partners, e.g., cyt c and cyt c peroxidase (ccp) (770 Å2) (64), or cyt c2 and the photosynthetic reaction center, discussed in this issue of PNAS by Onuchic and coworkers (65). Our finding that ET rates in Zn-doped tuna cyt c crystals fall well within the protein range in the Ru-protein tunneling timetable (Fig. 3) (7, 62) demonstrates that small interaction zones of low density are quite effective in mediating interprotein redox reactions.
Kinetics measurements on crystallographically characterized metal-substituted hemoglobin (Hb) hybrids provided some of the earliest insights into interprotein ET rates (66, 67). Because Hb is a very strongly bound complex of four polypeptide subunits, ET measurements are not complicated by the dynamical problems that plague interpretation of rates in more weakly bound assemblies. Replacement of the native Fe center in the β-subunits of Hb with Zn or Mg creates the opportunity for photoinitiated ET reactions. The reacting metal centers in the Hb hybrids are separated by 25 Å so that rates are relatively slow even at high driving forces. The time constant for ET from a triplet-excited Zn-porphyrin in the β-subunit to an Fe(III) center in the α-subunit is ≈16 ms (66). Extensive studies of temperature dependence of hybrid Hb ET rates led to the conclusion that the reorganization energy for these reactions (λ ≈1 eV) is dominated by outer-sphere contributions (68, 69). Measurements of ET rates in cryogenic glasses suggest that the polypeptide is the primary outer-sphere medium for the reaction and that bulk solvent reorganization does not play an important role. Moreover, it was suggested that even at room temperature, the protein medium in Hb acts like a frozen glass. Results from measurements on Ru-azurin crystals also indicate that bulk solvent makes only a minor contribution to protein ET reorganization energies (34).
The ET reaction between cyt c and cyt b5 has been the subject of experimental and theoretical investigations for >40 years (70, 71). The structural model proposed by Salemme in 1976 (72) for the precursor complex of this protein pair stimulated a great deal of experimental work. Careful spectroscopic studies revealed that these oppositely charged proteins form a stable 1:1 complex at low ionic strength [KA = 8(3) × 104 M-1, pH 7, μ 10 mM; KA = 4(3) × 106 M-1, pH 7, μ 1 mM] (73).
McLendon and Miller (74) used a combination of photochemical and pulse-radiolytic methods to probe the driving-force dependence of heme-heme ET in the 1:1 complex. The ET rates exhibited a near-Gaussian free energy dependence, in excellent agreement with a 0.8-eV reorganization energy. Evidence for more complex ET processes came from studies in which photochemically generated reductants injected electrons into preformed Fe-cyt b5/Fe-cyt c complexes. In one study, the rate of b5 → c ET (1.7 × 103 s-1) was reported to depend on viscosity and surface mutations (75). Later laser flash photolysis experiments revealed a rate-limiting second-order reduction of Fe-cyt b5/Fe-cyt c and no sign of saturation, suggesting that the intracomplex ET rate was >104 s-1 (76).
Durham, Millett, and coworkers (71) and Meyer et al. (76) used Ru-modified cyt b5 and photochemical triggering methods to examine the kinetics of ET in cyt b5/c complexes. Rapid intraprotein reduction (<100 ns) of Fe(III)-cyt b5 by electronically excited 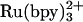 made it possible to probe b5 → c ET kinetics. Two concentration-independent ET rates (4 × 105 s-1 and 3.4 × 104 s-1) were observed, suggesting that two cyt b5/c species are present in solution. Studies of ionic strength dependences and the effects of mutations indicated that the slower Fe(III)-cyt c reduction phase may be limited by conformational changes within one of the complexes (71).
made it possible to probe b5 → c ET kinetics. Two concentration-independent ET rates (4 × 105 s-1 and 3.4 × 104 s-1) were observed, suggesting that two cyt b5/c species are present in solution. Studies of ionic strength dependences and the effects of mutations indicated that the slower Fe(III)-cyt c reduction phase may be limited by conformational changes within one of the complexes (71).
Ccp catalyzes the two-electron reduction of H2O2 by ferrocyt c. Peroxide reacts rapidly with the resting ferric form of ccp to produce a species referred to as compound I, which contains a ferryl [Fe(IV)O2+] heme and a protein radical located on Trp-191. The ET reactions involving these physiological redox partners have been studied in great detail (77). At low ionic strength, acidic ccp and basic cyt c form a stable complex. A model of a 1:1 complex, based on the crystal structures of the two independent proteins, was proposed by Poulos and Kraut (78) in 1980. Twelve years later, Pelletier and Kraut (64) reported the crystal structure of a 1:1 complex of the two yeast proteins. Interestingly, the complex between yeast ccp and horse cyt c exhibited a slightly different structure. Analysis of the yeast/yeast complex suggested an electronic coupling pathway from the cyt c heme to the ccp heme via Trp-191. On the basis of these crystallographic results, Pelletier and Kraut argued that ccp and cyt c form a highly specific 1:1 ET complex.
Hoffman and coworkers (77) have used metal-substituted ccp and cyt c to explore the ET kinetics between these two proteins. Results from quenching studies, temperature and ionic strength dependences, species variations, and electrostatic modeling provide compelling evidence for two distinct cyt c binding sites on ccp. The higher-affinity binding site is the locus for Trp-191 radical reduction by cyt c. Heme (ccp) reduction by cyt c can occur from either the high- or low-affinity binding site but, when exchange between the two is rapid, reduction from the low-affinity site dominates (77). These studies of ccp/cyt c and cyt b5/c ET, including an important contribution by Hoffman and coworkers (79) in this issue of PNAS, have shed much light on the mechanisms of protein–protein ET processes.
In the terminal reaction of the respiratory chain, cyt c oxidase (CcO) removes electrons from cyt c and passes them on to O2 (80). CcO is a multisubunit membrane-bound enzyme with four redox cofactors (CuA, cyt a, cyt a3, and CuB). The locations of these metal complexes in CcO were revealed in the 1990s by the x-ray crystal structures of bacterial (81) and bovine enzymes (82, 83). CuA, a binuclear site with bridging S(Cys) atoms, is the primary electron acceptor from cyt c. Studies with Ru-modified cyt c reveal rapid (6 × 104 s-1) (84) electron injection from Fe(II) into CuA at low driving force (ΔG° = -0.03 eV) (85). Modeling suggests that cyt c binds to the enzyme at an acidic patch on subunit II (86, 87). The cyt c heme is very near the Trp-104 (subunit II) indole ring, a residue that appears from mutagenesis experiments to be critical for rapid cyt c → CuA ET (88–91). Solomon and coworkers (92) have identified a possible electron tunneling path from this cyt c binding site through Trp-104 to the bridging S(Cys-200) ligand on CuA.
The 19.6-Å ET from CuA to cyt a proceeds rapidly at low driving force (≈104 s-1; ΔG° ≈-0.05 eV) (84, 93). Ramirez et al. (80), Regan et al. (94), and Solomon and coworkers (92, 95) have identified a coupling route that proceeds from CuA ligand His-204 (subunit II) across one hydrogen bond to Arg-438 (subunit I) [H204(Nε)-R438(O), 3.36 Å], and another H-bond (2.95 Å) from the Arg-438 N-amide to the cyt a heme propionate. Based on a tunneling currents analysis, Stuchebrukhov and colleagues (96) suggested a slightly different CuA-to-cyt a coupling route through His-204. It is likely that, owing to strong Cu-S(Cys) electronic interactions, pathways involving the bridging Cys residues are important for mediating coupling even though they involve more bonds than the His-204 route. The current view is that the sequence Cys-200/Ile-199/Arg-439/heme-propionate (cyt a) is the dominant CuA to cyt a electron tunneling pathway (92, 96).
Both Regan et al. (94) and Stuchebrukhov and colleagues (96) have identified pathways between cyt a and cyt a3. Included among these routes is a direct covalent pathway from the heme-a axial ligand His-378 through Phe-377 to the a3 His-376. Importantly, although the CuA-cyt a3 distance (22.4 Å) is similar to that of CuA to cyt a, neither Regan et al. (94) nor Stuchebrukhov and colleagues (96) found a coupling pathway that would facilitate electron flow to a3 in a single step from the binuclear copper center.
Hopping
Electron tunneling times must be in the millisecond to microsecond range for biological redox machines to function properly. As a result, the maximum center-to-center distance for single-step tunneling through proteins can be no more than ≈20 Å (Fig. 4). The structures of several redox enzyme assemblies, however, suggest that charge transport may occur over distances that far exceed this single-step limit (7, 97, 98). How can charge transport in proteins cover distances well over 20 Å? One possibility is by hopping, as it can be shown that coupled tunneling reactions, particularly with endergonic steps, can in favorable cases deliver electrons or holes rapidly to very distant sites (7, 30, 99). Requirements for functional hopping include optimal positioning of redox centers and fine-tuning of reaction driving forces.
Fig. 4.

Tunneling-time (1/kobs) contours as functions of donor-acceptor distance (β = 1.1 Å-1) and driving force [in units of λ; kBT/λ = kB(295 K)/(0.8 eV) = 0.318].
Modeling the kinetics of electron hopping is a straightforward problem that can be solved analytically without using simplifying approximations (7).
 |
[7] |
Using the well defined properties of ET reactions (Eq. 2), and the average distance dependence defined by Ru-protein tunneling timetables, we can predict hopping rates for any set of driving-force, temperature, and distance parameters. Consider the two-step tunneling reaction defined in Eq. 7 (reactants, R = D-I-A; redox intermediate, H = D+-I--A or D-I+-A-; products, P = D+-I-A-) The general solution to the rate law for this process calls for biexponential production of P, although under some circumstances the appearance of P can be approximated by a single exponential function. Taking a value of λ = 0.8 eV for both tunneling reactions (i.e., R → H and H → P) and a distance decay constant of 1.1 Å-1, we can calculate the time dependence of the populations of all three reacting species for various values of ΔGRH °, ΔGHP °, rRH, and rHP (7). Results for the particular case in which ΔGRH ° = -ΔG°HP and rRH = rHP are illustrated in Fig. 5. This model approximates a biological electron-transport chain (ΔGRP ° = 0) reaction with a single endergonic step. Transport across 20 Å is 104 times faster than a single tunneling step at this distance and submillisecond transfers can be realized. For D-A separations <20 Å, endergonic intermediate steps as large as 0.4–0.5 eV will afford submillisecond transport times. An important conclusion is that hopping can facilitate electron flow over distances >20 Å in cases where the free-energy changes for endergonic intermediate steps are no more than 0.2 eV.
Fig. 5.

Distance dependences of the rates of single-step and two-step electron tunneling reactions. Solid line indicates theoretical distance dependence for a single-step, ergoneutral (ΔGRP = 0) tunneling process (β = 1.1 Å-1). Dashed lines indicate distance dependence calculated for two-step ergoneutral tunneling (R ;HP) with the indicated free-energy changes for the RH step.
;HP) with the indicated free-energy changes for the RH step.
Concluding Remarks
More than 75 years have passed since the Gamow (1) and Gurney-Condon (2) papers appeared. Activity in the electron tunneling field over the last 20 years has been intense, most especially on the experimental side, where investigators have elucidated many of the factors that control reaction rates through bonded as well as nonbonded atoms in molecules and molecular assemblies, including the important case of folded polypeptide structures.
In 2005, the field is booming, as tunneling-related solar cell, sensor, and other device technologies are being developed at a rapid pace. A critical issue here is understanding bridge energy effects on charge transport through molecular materials, as discussed in this issue of PNAS by Ratner, Wasielewski, and coworkers (100). The role of dynamics in protein ET is another hot topic these days; and two articles in this issue of PNAS report exciting results in this area (47, 79). Distant (>20 Å) charge transport in DNA is still another area of great interest. Much work in this area has been done by Barton and coworkers; and, in this issue of PNAS, a report from her laboratory, in collaboration with the David group at Utah, suggests that guanine radicals, which facilitate iron-sulfur cluster oxidation in a DNA/MutY complex, may stimulate DNA repair (101).
Controlled electron flow is an absolute requirement for efficient storage and conversion of all forms of energy. It also is essential for successful operation of molecular-scale electronic devices. We have laid a firm foundation for these applications, but we must greatly ramp up both theoretical and experimental investigations of multielectron and other coupled redox processes if we are to realize the full potential of this simplest of chemical reactions.
Acknowledgments
Our work is supported by the National Institutes of Health, the National Science Foundation, BP, and the Arnold and Mabel Beckman Foundation.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ET, electron transfer; cyt, cytochrome; ccp, cyt c peroxidase; MTHF, 2-methyltetrahydrofuran; Hb, hemoglobin.
References
- 1.Gamow, G. (1928) Z. Phys. 51, 204-212. [Google Scholar]
- 2.Gurney, R. W. & Condon, E. U. (1929) Phys. Rev. 33, 127-140. [Google Scholar]
- 3.Esaki, L. (1974) Science 183, 1149-1155. [DOI] [PubMed] [Google Scholar]
- 4.Miller, J. R. (1975) Science 189, 221-222. [DOI] [PubMed] [Google Scholar]
- 5.Marcus, R. A. & Sutin, N. (1985) Biochim. Biophys. Acta 811, 265-322. [Google Scholar]
- 6.De Vault, D., Parkes, J. H. & Chance, B. (1967) Nature 215, 642-644. [DOI] [PubMed] [Google Scholar]
- 7.Gray, H. B. & Winkler, J. R. (2003) Q. Rev. Biophys. 36, 341-372. [DOI] [PubMed] [Google Scholar]
- 8.McConnell, H. M. (1961) J. Chem. Phys. 35, 508-515. [Google Scholar]
- 9.Halpern, J. & Orgel, L. E. (1960) Disc. Faraday Soc. 32-41.
- 10.Oevering, H., Paddon-Row, M. N., Heppener, M., Oliver, A. M., Cotsaris, E., Verhoeven, J. W. & Hush, N. S. (1987) J. Am. Chem. Soc. 109, 3258-3269. [Google Scholar]
- 11.Johnson, M. D., Miller, J. R., Green, N. S. & Closs, G. L. (1989) J. Phys. Chem. 93, 1173-1176. [Google Scholar]
- 12.Smalley, J. F., Finklea, H. O., Chidsey, C. E. D., Linford, M. R., Creager, S. E., Ferraris, J. P., Chalfant, K., Zawodzinsk, T., Feldberg, S. W. & Newton, M. D. (2003) J. Am. Chem. Soc. 125, 2004-2013. [DOI] [PubMed] [Google Scholar]
- 13.Helms, A., Heiler, D. & McLendon, G. (1992) J. Am. Chem. Soc. 114, 6227-6238. [Google Scholar]
- 14.Wenger, O. S., Leigh, B. S., Villahermosa, R. M., Gray, H. B. & Winkler, J. R. (2005) Science 307, 99-102. [DOI] [PubMed] [Google Scholar]
- 15.Davis, W. B., Svec, W. A., Ratner, M. A. & Wasielewski, M. R. (1998) Nature 396, 60-63. [Google Scholar]
- 16.Sakata, Y., Tsue, H., O'Neil, M. P., Wiederrecht, G. P. & Wasielewski, M. R. (1994) J. Am. Chem. Soc. 116, 6904-6909. [Google Scholar]
- 17.Newton, M. D. (1997) J. Electroanal. Chem. 438, 3-10. [Google Scholar]
- 18.Hayashi, S. & Kato, S. (1998) J. Phys. Chem. A 102, 3333-3342. [Google Scholar]
- 19.Weidemaier, K., Tavernier, H. L., Swallen, S. F. & Fayer, M. D. (1997) J. Phys. Chem. A 101, 1887-1902. [Google Scholar]
- 20.Miller, J. R. (1975) J. Phys. Chem. 79, 1070-1078. [Google Scholar]
- 21.Miller, J. R., Beitz, J. V. & Huddleston, R. K. (1984) J. Am. Chem. Soc. 106, 5057-5068. [Google Scholar]
- 22.Swallen, S. W., Weidemaier, K., Tavernier, H. L. & Fayer, M. D. (1996) J. Phys. Chem. 100, 8106-8117. [Google Scholar]
- 23.Ponce, A., Gray, H. B. & Winkler, J. R. (2000) J. Am. Chem. Soc. 122, 8187-8191. [Google Scholar]
- 24.Winkler, J. R., Wittung-Stafshede, P., Leckner, J., Malmström, B. G. & Gray, H. B. (1997) Proc. Natl. Acad. Sci. USA 94, 4246-4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray, H. B., Malmström, B. G. & Williams, R. J. P. (2000) J. Biol. Inorg. Chem. 5, 551-559. [DOI] [PubMed] [Google Scholar]
- 26.Simonson, T. (2002) Proc. Natl. Acad. Sci. USA 99, 6544-6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winkler, J. R., Nocera, D. G., Yocom, K. M., Bordignon, E. & Gray, H. B. (1982) J. Am. Chem. Soc. 104, 5798-5800. [Google Scholar]
- 28.Winkler, J. R. & Gray, H. B. (1992) Chem. Rev. 92, 369-379. [Google Scholar]
- 29.Gray, H. B. & Winkler, J. R. (1996) Annu. Rev. Biochem. 65, 537-561. [DOI] [PubMed] [Google Scholar]
- 30.Winkler, J. R., Di Bilio, A., Farrow, N. A., Richards, J. H. & Gray, H. B. (1999) Pure Appl. Chem. 71, 1753-1764. [Google Scholar]
- 31.Langen, R., Chang, I.-J., Germanas, J. P., Richards, J. H., Winkler, J. R. & Gray, H. B. (1995) Science 268, 1733-1735. [DOI] [PubMed] [Google Scholar]
- 32.Regan, J. J., Di Bilio, A. J., Langen, R., Skov, L. K., Winkler, J. R., Gray, H. B. & Onuchic, J. N. (1995) Chem. Biol. 2, 489-496. [DOI] [PubMed] [Google Scholar]
- 33.Adman, E. T. (1991) Adv. Protein Chem. 42, 145-197. [DOI] [PubMed] [Google Scholar]
- 34.Crane, B. R., Di Bilio, A. J., Winkler, J. R. & Gray, H. B. (2001) J. Am. Chem. Soc. 123, 11623-11631. [DOI] [PubMed] [Google Scholar]
- 35.Smalley, J. F., Feldberg, S. W., Chidsey, C. E. D., Linford, M. R., Newton, M. D. & Liu, Y.-P. (1995) J. Phys. Chem. 99, 13141-13149. [Google Scholar]
- 36.Farver, O. & Pecht, I. (1999) Adv. Chem. Phys. 107, 555-589. [Google Scholar]
- 37.Chang, I.-J., Gray, H. B. & Winkler, J. R. (1991) J. Am. Chem. Soc. 113, 7056-7057. [Google Scholar]
- 38.Babini, E., Bertini, I., Borsari, M., Capozzi, F., Luchinat, C., Zhang, X., Moura, G. L. C., Kurnikov, I. V., Beratan, D. N., Ponce, A., et al. (2000) J. Am. Chem. Soc. 122, 4532-4533. [Google Scholar]
- 39.Beratan, D. N. & Onuchic, J. N. (1989) Photosynth. Res. 22, 173-186. [DOI] [PubMed] [Google Scholar]
- 40.Onuchic, J. N., Beratan, D. N., Winkler, J. R. & Gray, H. B. (1992) Annu. Rev. Biophys. Biomol. Struct. 21, 349-377. [DOI] [PubMed] [Google Scholar]
- 41.Beratan, D. N., Betts, J. N. & Onuchic, J. N. (1991) Science 252, 1285-1288. [DOI] [PubMed] [Google Scholar]
- 42.Regan, J. J. & Onuchic, J. N. (1999) Adv. Chem. Phys. 107, 497-553. [Google Scholar]
- 43.Balabin, I. A. & Onuchic, J. N. (1998) J. Phys. Chem. B 102, 7497-7505. [Google Scholar]
- 44.Skourtis, S. S. & Beratan, D. N. (1997) J. Biol. Inorg. Chem. 2, 378-386. [Google Scholar]
- 45.Kumar, K., Kurnikov, I. V., Beratan, D. N., Waldeck, D. H. & Zimmt, M. B. (1998) J. Phys. Chem. A 102, 5529-5541. [Google Scholar]
- 46.Stuchebrukhov, A. A. (1996) J. Chem. Phys. 105, 10819-10829. [Google Scholar]
- 47.Skourtis, S. S., Balabin, I. A., Kawatsu, T. & Beratan, D. N. (2005) Proc. Natl. Acad. Sci. USA 102, 3552-3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calef, D. F. & Wolynes, P. G. (1983) J. Phys. Chem. 87, 3387-3400. [Google Scholar]
- 49.Calef, D. F. & Wolynes, P. G. (1983) J. Chem. Phys. 78, 470-482. [Google Scholar]
- 50.Maroncelli, M., MacInnis, J. & Fleming, G. (1989) Science 243, 1674-1681. [DOI] [PubMed] [Google Scholar]
- 51.Hynes, J. T. (1986) J. Phys. Chem. 90, 3701-3706. [Google Scholar]
- 52.Sumi, H. & Marcus, R. A. (1986) J. Chem. Phys. 84, 4894-4914. [Google Scholar]
- 53.Bixon, M. & Jortner, J. (1993) Chem. Phys. 176, 467-481. [Google Scholar]
- 54.Bixon, M. & Jortner, J. (1999) Adv. Chem. Phys. 106, 35-202. [Google Scholar]
- 55.Genberg, L., Richard, L., McLendon, G. & Miller, R. J. D. (1991) Science 251, 1051-1054. [DOI] [PubMed] [Google Scholar]
- 56.Bashkin, J. S., McLendon, G., Mukamel, S. & Marohn, J. (1990) J. Phys. Chem. 94, 4757-4761. [Google Scholar]
- 57.Pierce, D. W. & Boxer, S. G. (1992) J. Phys. Chem. 96, 5560-5566. [Google Scholar]
- 58.Hagen, S. J. & Eaton, W. A. (1996) J. Chem. Phys. 104, 3395-3398. [Google Scholar]
- 59.Khoshtariya, D. E., Wei, J., Liu, H., Yue, H. & Waldeck, D. H. (2003) J. Am. Chem. Soc. 125, 7704-7714. [DOI] [PubMed] [Google Scholar]
- 60.Stemp, E. D. A. & Hoffman, B. M. (1993) Biochemistry 32, 10848-10865. [DOI] [PubMed] [Google Scholar]
- 61.Pletneva, E. V., Fulton, D. B., Kohzuma, T. & Kostic, N. M. (2000) J. Am. Chem. Soc. 122, 1034-1046. [Google Scholar]
- 62.Tezcan, F. A., Crane, B. R., Winkler, J. R. & Gray, H. B. (2001) Proc. Natl. Acad. Sci. USA 98, 5002-5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lo Conte, L., Chothia, C. & Janin, J. (1999) J. Mol. Biol. 285, 2177-2198. [DOI] [PubMed] [Google Scholar]
- 64.Pelletier, H. & Kraut, J. (1992) Science 258, 1748-1755. [DOI] [PubMed] [Google Scholar]
- 65.Miyashita, O., Okamura, M. Y. & Onuchic, J. N. (2005) Proc. Natl. Acad. Sci. USA 102, 3558-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McGourty, J. L., Blough, N. V. & Hoffman, B. M. (1983) J. Am. Chem. Soc. 105, 4470-4472. [Google Scholar]
- 67.Kuila, D., Baxter, W. W., Natan, M. J. & Hoffman, B. M. (1991) J. Phys. Chem. 95, 1-3. [Google Scholar]
- 68.Peterson-Kennedy, S. E., McGourty, J. L., Kalweit, J. A. & Hoffman, B. M. (1986) J. Am. Chem. Soc. 108, 1739-1746. [Google Scholar]
- 69.Dick, L. A., Malfant, I., Kuila, D., Nebolsky, S., Nocek, J. M., Hoffman, B. M. & Ratner, M. A. (1998) J. Am. Chem. Soc. 120, 11401-11407. [Google Scholar]
- 70.Mauk, A. G., Mauk, M. R., Moore, G. R. & Northrup, S. H. (1995) J. Bioenerg. Biomemb. 27, 311-330. [DOI] [PubMed] [Google Scholar]
- 71.Durham, B., Fairris, J. L., McLean, M., Millett, F., Scott, J. R., Sligar, S. G. & Willie, A. (1995) J. Bioenerg. Biomemb. 27, 331-340. [DOI] [PubMed] [Google Scholar]
- 72.Salemme, F. R. (1976) J. Mol. Biol. 102, 563-568. [DOI] [PubMed] [Google Scholar]
- 73.Mauk, M. R., Reid, L. S. & Mauk, A. G. (1982) Biochemistry 21, 1843-1846. [DOI] [PubMed] [Google Scholar]
- 74.McLendon, G. & Miller, J. R. (1985) J. Am. Chem. Soc. 107, 7811-7816. [Google Scholar]
- 75.Qin, L., Rodgers, K. K. & Sligar, S. G. (1991) Mol. Cryst. Liq. Cryst. 194, 311-316. [Google Scholar]
- 76.Meyer, T. E., Rivera, M., Walker, F. A., Mauk, M. R., Mauk, A. G., Cusanovich, M. A. & Tollin, G. (1993) Biochemistry 32, 622-627. [DOI] [PubMed] [Google Scholar]
- 77.Nocek, J. M., Zhou, J. S., DeForest, S., Priyadarshy, S., Beratan, D. N., Onuchic, J. N. & Hoffman, B. M. (1996) Chem. Rev. 96, 2459-2489. [DOI] [PubMed] [Google Scholar]
- 78.Poulos, T. L. & Kraut, J. (1980) J. Biol. Chem. 255, 10322-10330. [PubMed] [Google Scholar]
- 79.Hoffman, B. M., Celis, L. M., Cull, D. A., Patel, A. D., Seifert, J. L., Wheeler, K. E., Wang, J., Yao, J., Kurnikov, I. V. & Nocek, J. M. (2005) Proc. Natl. Acad. Sci. USA 102, 3564-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ramirez, B. E., Malmström, B. G., Winkler, J. R. & Gray, H. B. (1995) Proc. Natl. Acad. Sci. USA 92, 11949-11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Iwata, S., Ostermeier, C., Ludwig, B. & Michel, H. (1995) Nature 376, 660-669. [DOI] [PubMed] [Google Scholar]
- 82.Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki, T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono, R. & Yoshikawa, S. (1995) Science 269, 1071-1074. [DOI] [PubMed] [Google Scholar]
- 83.Yoshikawa, S., Shinzawa-Itoh, K., Nakashima, R., Yaono, R., Yamashita, E., Inoue, N., Yao, M., Fei, J. M., Libeu, C. P., Mizushima, T., et al. (1998) Science 280, 1723-1729. [DOI] [PubMed] [Google Scholar]
- 84.Geren, L. M., Beasley, J. R., Fine, B. R., Saunders, A. J., Hibdon, S., Pielak, G. J., Durham, B. & Millett, F. (1995) J. Biol. Chem. 270, 2466-2472. [DOI] [PubMed] [Google Scholar]
- 85.Pan, L. P., Hibdon, S., Liu, R.-Q., Durham, B. & Millett, F. (1993) Biochemistry 32, 8492-8498. [DOI] [PubMed] [Google Scholar]
- 86.Roberts, V. A. & Pique, M. E. (1999) J. Biol. Chem. 274, 38051-38060. [DOI] [PubMed] [Google Scholar]
- 87.Flöck, D. & Helms, V. (2002) Proteins Struct. Funct. Genet. 47, 75-85. [DOI] [PubMed] [Google Scholar]
- 88.Witt, H., Malatesta, F., Nicoletti, F., Brunori, M. & Ludwig, B. (1998) J. Biol. Chem. 273, 5132-5136. [DOI] [PubMed] [Google Scholar]
- 89.Zhen, Y. J., Hoganson, C. W., Babcock, G. T. & Ferguson-Miller, S. (1999) J. Biol. Chem. 274, 38032-38041. [DOI] [PubMed] [Google Scholar]
- 90.Wang, K. F., Zhen, Y. J., Sadoski, R., Grinnell, S., Geren, L., Ferguson-Miller, S., Durham, B. & Millett, F. (1999) J. Biol. Chem. 274, 38042-38050. [DOI] [PubMed] [Google Scholar]
- 91.Drosou, V., Malatesta, F. & Ludwig, B. (2002) Eur. J. Biochem. 269, 2980-2988. [DOI] [PubMed] [Google Scholar]
- 92.George, S. D., Metz, M., Szilagyi, R. K., Wang, H., Cramer, S. P., Lu, Y., Tolman, W. B., Hedman, B., Hodgson, K. O. & Solomon, E. I. (2001) J. Am. Chem. Soc. 123, 5757-5767. [DOI] [PubMed] [Google Scholar]
- 93.Farver, O., Einarsdóttir, O. & Pecht, I. (2000) Eur. J. Biochem. 267, 950-954. [DOI] [PubMed] [Google Scholar]
- 94.Regan, J. J., Ramirez, B. E., Winkler, J. R., Gray, H. B. & Malmström, B. G. (1998) J. Bioenerg. Biomembr. 30, 35-39. [DOI] [PubMed] [Google Scholar]
- 95.Gamelin, D. R., Randall, D. W., Hay, M. T., Houser, R. T., Mulder, T. C., Canters, G. W., de Vries, S., Tolman, W. B., Lu, Y. & Solomon, E. I. (1998) J. Am. Chem. Soc. 120, 5246-5263. [Google Scholar]
- 96.Medvedev, D. M., Daizadeh, I. & Stuchebrukhov, A. A. (2000) J. Am. Chem. Soc. 122, 6571-6582. [Google Scholar]
- 97.Sjöberg, B. M. (1997) Struct. Bonding 88, 139-173. [Google Scholar]
- 98.Stubbe, J., Nocera, D. G., Yee, C. S. & Chang, M. C. Y. (2003) Chem. Rev. 103, 2167-2201. [DOI] [PubMed] [Google Scholar]
- 99.Page, C. C., Moser, C. C., Chen, X. & Dutton, P. L. (1999) Nature 402, 47-52. [DOI] [PubMed] [Google Scholar]
- 100.Goldsmith, R. H., Sinks, L. E., Kelley, R. F., Betzen, L. J., Liu, W., Weiss, E. A., Ratner, M. A. & Wasielewski, M. R. (2005) Proc. Natl. Acad. Sci. USA 102, 3540-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yavin, E., Boal, A. K., Stemp, E. D. A., Boon, E. M., Livingston, A. L., O'Shea, V. L., David, S. S. & Barton, J. K. (2005) Proc. Natl. Acad. Sci. USA 102, 3546-3551. [DOI] [PMC free article] [PubMed] [Google Scholar]