

Abstract
Prolyl endopeptidases (PEPs) are a unique class of serine proteases with considerable therapeutic potential for the treatment of celiac sprue. The crystal structures of two didomain PEPs have been solved in alternative configurations, thereby providing insights into the mode of action of these enzymes. The structure of the Sphingomonas capsulata PEP, solved and refined to 1.8-Å resolution, revealed an open configuration of the active site. In contrast, the inhibitor-bound PEP from Myxococcus xanthus was crystallized (1.5-Å resolution) in a closed form. Comparative analysis of the two structures highlights a critical role for the domain interface in regulating interdomain dynamics and substrate specificity. Structure-based mutagenesis of the M. xanthus PEP confirms an important role for several interfacial residues. A salt bridge between Arg-572 and Asp-196/Glu-197 appears to act as a latch for opening or closing the didomain enzyme, and Arg-572 and Ile-575 may also help secure the incoming peptide substrate to the open form of the enzyme. Arg-618 and Asp-145 are responsible for anchoring the invariant proline residue in the active site of this postproline-cleaving enzyme. A model is proposed for the docking of a representative substrate PQPQLPYPQPQLP in the active site, where the N-terminal substrate residues interact extensively with the catalytic domain, and the C-terminal residues stretch into the propeller domain. Given the promise of the M. xanthus PEP as an oral therapeutic enzyme for treating celiac sprue, our results provide a strong foundation for further optimization of the PEP's clinically useful features.
Keywords: celiac sprue, gluten, prolyl oligopeptidase, serine protease
Prolyl endopeptidases (PEPs), also known as prolyl oligopeptidases or postproline cleaving enzymes, are a family of serine proteases that cleave after proline residues in peptides. These endoproteolytic enzymes are widely distributed in bacteria, fungi, animals, and plants (1–5). The human PEP is a cytosolic enzyme involved in physiological processes such as the degradation of certain peptide hormones and neuropeptides. It has been implicated in the regulation of blood pressure and in neurological disorders and is, therefore, an attractive pharmacological target (6, 7). Recently, PEPs have also been evaluated as oral therapeutic agents for celiac sprue because of a unique ability to accelerate the breakdown of proline-rich gluten in the gut lumen (8, 9).
From a structural and mechanistic standpoint, PEPs are relatively unusual serine proteases. As peptidases, their activity is restricted to substrates that are shorter than 30 amino acid residues, with the possible exception of the Flavobacterium meningosepticum and Pyrococcus furiosus PEPs, which can break down longer peptides (10, 11). PEPs are also significantly larger than typical serine proteases (75 kDa vs. 30 kDa). The x-ray crystal structure of a prototypical PEP from porcine muscle was solved by Polgar and coworkers (12–14) and revealed a distinctive two-domain structure, a smaller N-terminal catalytic domain, and an unusual β-propeller domain. The catalytic domain resembles canonical serine proteases such as trypsin and chymotrypsin, whereas the unique propeller domain forms a tight barrel-shaped lid over the active site and is postulated to regulate substrate size.
Mechanisms have been proposed to account for the observed bias of PEPs for shorter substrates. It had long been speculated that some conformational change might be involved in substrate binding (15). One mechanism suggested that the oscillating β-propeller blades act as a “gating filter” during catalysis to let only short peptide substrates into the active site via the central tunnel of the propeller (12, 13, 16, 17). This proposal was supported by experiments that connected the first and seventh blades of the propeller domain with disulfide bonds, which resulted in reduced enzymatic activity. A more recent study suggests that relative movement of the two domains might be required to allow substrates into the active site (18). The nature of the substrate-binding mode is pertinent from both fundamental mechanistic and practical therapeutic points of view. Control of chain-length specificity is of particular importance for the therapeutic use of PEPs in celiac sprue, because many of the immunotoxic peptides released by the gastrointestinal metabolism of dietary gluten are relatively long (8, 19).
Here, we have solved the x-ray crystal structures of two therapeutically promising bacterial PEPs from Myxococcus xanthus (MX) and Sphingomonas capsulata (SC) (10). Comparison of the architecture of the two enzymes, one crystallizing in an open (i.e., unoccupied) configuration and the other in a closed (i.e., inhibitor-bound) configuration, provided direct insight into the mechanism by which substrate access to the active site is controlled. A docking model of MX PEP with peptide PQPQLPYPQPQLP further revealed an extended binding pocket that could accommodate a larger substrate. Supporting the structural analysis, mutagenesis of a salt bridge that hinges the two domains yielded enzymes with unaffected catalytic capability but altered chain-length specificity. Alteration of residues responsible for anchoring the invariant proline side chain in the substrate led to greatly reduced catalytic activity, illustrating the delicate interplay between specificity and catalysis in this class of enzymes.
Materials and Methods
Expression and Purification of SC and MX PEP. Both PEPs were expressed as recombinant proteins with C-terminal His6-tags in Escherichia coli and purified according the protocols described in ref. 10. Site-directed mutagenesis was performed by using the PCR QuikChange method (Stratagene).
Crystallization and Diffraction Data Collection. Initial crystallization conditions for native SC PEP were determined by using Wizard I and Wizard II crystal screens (Emerald Biostructures, Bainbridge Island, WA) with 19.4 mg/ml protein in 20 mM Hepes (pH 7.0)/2 mM DTT/5% glycerol. Crystals were grown at 22°C by using the hanging-drop vapor-diffusion technique. The optimized crystallization conditions involved mixing 1.2 μl of protein solution with 1.2 μl of reservoir solution containing 23% PEG 8K and 0.1 M Tris (pH 8.6). Diffraction-quality crystals appeared in 2–3 days. Crystals were cryoprotected by 2-min stepwise soaks in solutions of the well solution plus 2%, 5%, 8%, 12%, and, finally, 17% glycerol and were then flash-frozen in liquid nitrogen.
The complex of MX PEP–Z-Ala-prolinal was prepared by mixing the protein sample with a 5 M excess of the ligand. Crystals of the complex were grown at 4°C by using the hanging-drop vapor-diffusion technique. The optimized crystallization condition consisted of 26% methoxy PEG 5K and 0.1 M Mes (pH 6.01). A cryoprotectant solution of 2.5% glycerol in 32% methoxy PEG 5K in 0.1 M Mes was used for flash-freezing the crystals in liquid nitrogen.
Data for the SC and MX PEP crystals were collected at the Advanced Light Source (Lawrence Berkeley National Laboratory, Berkeley, CA) on beamlines 8.2.1 and 8.2.2, respectively. The data for SC PEP was measured in 1.0°-oscillation steps with 8-s exposure times by using a Quantum 210 CCD detector (Area Detector Systems, Poway, CA) with a crystal-to-detector distance of 200 mm. This was followed by a higher-resolution data set with 25-s exposure times and a crystal-to-detector distance of 150 mm. The data for MX PEP was measured in 1.0°-oscillation steps with 5-s exposure times by using a Quantum 315 CCD detector (Area Detector Systems) with a crystal-to-detector distance of 190 mm. Both data sets were processed and scaled by using hkl2000 (HKL Research, Charlottesville, VA) (20, 21). Final data-processing statistics are shown in Table 1.
Table 1. Data collection and refinement statistics.
| SC | MX+bocNFP | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P21 |
| Unit cell, Å | 53.34, 91.22, 79.79 | 65.69, 114.72, 99.28 |
| β = 91.0° | β = 103.6° | |
| Wavelength, Å | 1.00 | 1.00 |
| Resolution, Å | 50.0–1.80 (1.86–1.80) | 50.0–1.50 (1.55–1.50) |
| No. of reflections | 353,562 | 918,495 |
| No. of unique reflections | 68,648 | 225,366 |
| Redundancy | 5.2 (2.1) | 4.1 (4.0) |
| Completeness | 98.9 (92.2) | 98.9 (97.7) |
| Rsym, % | 7.1 (50.2) | 6.8 (47.6) |
| I/σ | 13.0 (1.7) | 12.8 (2.8) |
| Refinement | ||
| Resolution, Å | 30.0–1.80 (1.85–1.80) | 30.0–1.50 (1.54–1.50) |
| Reflections (working) | 62,633 (4,393) | 202,805 (15,555) |
| Reflections (test) | 3,538 (237) | 11,252 (812) |
| Rwork,* % | 16.1 (25.0) | 16.0 (22.4) |
| Rfree,† % | 18.6 (26.3) | 18.2 (25.6) |
| No. of protein atoms | 5,252 | 10,717 |
| No. of ligand atoms | 0 | 67 |
| No. of buffer/glycerol/ion atoms | 12 | 41 |
| No. of water atoms | 660 | 1,371 |
| rms deviations | ||
| Bonds, Å | 0.017 | 0.015 |
| Angles, ° | 1.56 | 1.49 |
| Average B factor, Å2 | ||
| Protein atoms | 16.7 | 11.0 |
| Ligand atoms | N/A | 19.6 |
| Buffer/glycerol/ions | 47.4 | 28.5 |
| Solvent atoms | 34.7 | 25.0 |
Values for the outer shell are given in parentheses.
Rwork = Σhkl ‖Fo| – |Fc‖Σhkl |Fo|, where Fo and Fc are observed and calculated structure factors, respectively
For Rfree, the above summation is extended over a subset of reflections (5%) that were excluded from all stages of refinement
Structure Determination and Refinement. The structure of the SC PEP protein was determined by molecular replacement by using the program molrep from the ccp4 suite (30). Because the initial attempts with reported structures (closed forms) of the PEPs were unsuccessful, individual searches for the catalytic domain and the β-barrel domain were performed. This procedure enabled the structure solution and revealed the open conformation of the SC PEP structure.
The MX PEP structure was determined by molecular replacement by using the program cns (22, 23). The successful solutions were obtained by using only the closed forms of the reported oligopeptidase structures. The best solution resulted from using the structure of the porcine prolyl oligopeptidase (purified directly from porcine muscle) bound to Z-Pro-prolinal (13) as the search model.
The initial partial tracings of both structures were performed by using the program resolve (24). The atomic model was then built manually with the program o (25) by using maps at various resolution ranges generated from the resolve phases. Alternate rounds of model building in o by using maps with Fourier coefficients (2 Fo - Fc) and (Fo - Fc), and refinements with cns (22) or refmac5 as implemented in the ccp4 suite resulted in the final models summarized in Table 1. The final model for the SC PEP includes a protein monomer, two glycerol molecules, and 660 water molecules in the asymmetric unit. No electron density was observed for residues 1–37 (signal peptide was cleaved), 158–161, 231–238, 689–697, and the last three histidines of the His6-tag. The final model for the MX PEP includes a protein dimer, two covalently bound ligands and a free ligand, three Mes buffer molecules, one 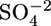 ion, and 1,371 water molecules in the asymmetric unit (Table 1). No electron density was observed for residue 1 and residues 678–689. The C-terminal His6-tag is disordered in the structure. procheck (26) indicates that 89.2% of the residues in the SC PEP structure and 91.4% of the residues in the MX PEP structure are located in the most favorable regions of the Ramachandran plot (27). There are two outliers in both PEP structures. One outlier in both structures is the active-site residue (Ser-575 in SC PEP and Ser-533 in MX PEP). The corresponding residue in the porcine structure (13) is also located in the disallowed region. The second disallowed residue in the MX PEP structure (Gln-295) is also in the disallowed region in the SC PEP structure (Lys-341) and the porcine structure. All outlier residues are well ordered in both structures and show good electron density (see Fig. 5, which is published as supporting information on the PNAS web site).
ion, and 1,371 water molecules in the asymmetric unit (Table 1). No electron density was observed for residue 1 and residues 678–689. The C-terminal His6-tag is disordered in the structure. procheck (26) indicates that 89.2% of the residues in the SC PEP structure and 91.4% of the residues in the MX PEP structure are located in the most favorable regions of the Ramachandran plot (27). There are two outliers in both PEP structures. One outlier in both structures is the active-site residue (Ser-575 in SC PEP and Ser-533 in MX PEP). The corresponding residue in the porcine structure (13) is also located in the disallowed region. The second disallowed residue in the MX PEP structure (Gln-295) is also in the disallowed region in the SC PEP structure (Lys-341) and the porcine structure. All outlier residues are well ordered in both structures and show good electron density (see Fig. 5, which is published as supporting information on the PNAS web site).
Accession Numbers. The coordinates for the SC and MX–Z-Ala-Pro complex have been deposited in the Protein Data Bank (PDB ID codes 1YR2 and 2BKL, respectively).
Kinetic Analysis. Hydrolysis of Suc-Ala-Pro-p-nitroanilide (pNA) by SC and MX PEPs was monitored at 25°C in a 300-μl mixture with a final concentration of 0.01–0.02 μM enzyme and Suc-Ala-Pro-pNA at final concentrations of between 100 μM and 4 mM in PBS. The release of p-nitroaniline was spectrophotometrically detected at a 410-nm wavelength, which was used to calculate KM and kcat according to the Michaelis–Menten relationship. PEP specificity toward a 13-mer substrate, PQPQLPYPQPQLP, was assessed in a competitive assay in which 100 μM peptide and 100 μM Suc-Ala-Pro-pNA were mixed and reacted with 0.02–0.2 μM PEP. The initial rate of Suc-Ala-Pro-pNA cleavage was measured at 410 nm, whereas the initial velocity of PQPQLPYPQPQLP hydrolysis was determined by HPLC. The relative specificity, kcat/KM, for the hydrolysis of PQPQLPYPQPQLP could be determined based on the known kcat/KM of the enzyme for Suc-Ala-Pro-pNA and the observed reaction rates of the two substrates, as follows: (kcat/KM)A = (kcat/KM)B([B] vA)/([A] vB), where A is PQPQLPYPQPQLP and B is Suc-Ala-Pro-pNA. This alternative method for measuring kcat/KM values allowed us to quantify the specificity of wild-type and mutant PEPs for the peptide with considerably fewer HPLC runs as compared with standard 4 × 4 plots. RP-HPLC was performed on a system consisting of a Beckman LC125, a 340 UV detector (Varian, Palo Alto, CA) set at 215 nm. Solvent A was H2O with 0.1% TFA, and solvent B was acetonitrile with 0.1% TFA (gradient 10–55% B for 30 min; flow 1 ml/min). Separation was performed on a reverse-phase C18 column (Vydac, Hesperia, CA).
Results and Discussion
Architecture of MX PEP. The MX PEP has a similar structure to that from porcine muscle or brain (13, 14, 17). The enzyme consists of a catalytic domain with a typical α/β-hydrolase fold, which is covalently connected to a cylindrical barrel-shaped propeller domain (Fig. 1A). The catalytic domain is made up of N-terminal residues 1–67 and C-terminal residues 410–678; the propeller domain includes residues 71–406. Two linear strands formed by residues 67–70 and 407–409 covalently connect the two domains.
Fig. 1.

Tertiary structures of MX and SC PEPs [drawn and rendered with pymol (Delano Scientific, San Carlos, CA)]. (A) Side view of inhibitor-bound MX PEP, represented by ribbon diagrams. The bound inhibitor Z-Ala-prolinal is colored in magenta. The inhibitor interacts only with the front half of the propeller domain. The two domains are covalently connected by two linkers consisting of residues 67–70 (marked as 1) and 407–409 (marked as 2). (B) Side view of the open SC structure. The two connecting strands between the domains are numbered as 1 (Ile-106 ∼ Glu-109, in blue) and 2 (Thr-453 ∼ Pro-449, in yellow). The C-terminal His6-tag is fitted into an α-helix trailing on the right, in red.
The catalytic domain, also called the peptidase domain, is ≈56 Å long × 50 Å wide, and the height is ≈30 Å. The catalytic triad (Ser-533, Asp-616, and His-651) is located at the bottom center. There are a total of 10 α-helices throughout the domain (see Fig. 6, which is published as supporting information on the PNAS website). Two parallel, long helices consisting of Pro-33–Ala-51 (α1*) and Asp-500–Gln-518 (αB) line up the sides. Other helices form vertical columns between the two long helices. The α2*-helix (Arg-56–Phe-67) is at the N terminus and connects to the propeller domain by a short strand (residues 67–70). The helix consisting of Gln-656–Val-674 (αF) is at the C terminus of the enzyme. The active-site serine is at the beginning of the helix αC (Ser-533–Thr-544). Aside from these α-helices, 10 β-strands contribute to the scaffold of the catalytic domain. A central, eight-stranded β-sheet (β1–8) partially wraps around the central four α-helices. The bend in the β-sheet topology is distributed fairly evenly across the strands, with each strand at an angle of ≈20° to the next one.
The β-propeller domain in the MX PEP is an unusual domain that is conserved throughout this subfamily of serine proteases. Its structure suggests an important role for this propeller domain in limiting substrate accessibility. It is shaped like a hollow, circular barrel ≈30 Å high (Fig. 1A). The central tunnel inside the propeller domain decreases in diameter from ≈40 Å to ≈5 Å as it goes deeper. This interesting structure is comprised of seven blades of β-sheets. The β-strands are connected by loops that form the top and bottom surfaces of the propeller. These loops appear similar, with the exception of the loops corresponding to residues Leu-187–Thr-202 and Lys-161–Pro-173. The former forms a short α-helix that strengthens contacts with the catalytic domain; the latter loop is exceptionally short and leaves a small gap at the otherwise tight seam of the domain interface. Residues situated at the interdomain loops participate in the interactions between the propeller and catalytic domains; for example, Asp-196 and Glu-197 (propeller side) form hydrogen bonds with Arg-572 (catalytic side), and Asp-145 (propeller side) forms a salt bridge with Arg-618 (catalytic side).
The active site of the MX PEP binds the inhibitor, Z-Ala-prolinal, which forms a hemiacetal with the active-site Ser-533. The substrate residues are anchored by extensive noncovalent interactions in the P1, P2, and P3 pockets of the protein (Fig. 2). In the P1 pocket, the S1 carbonyl forms a hydrogen bond with the backbone NH1 of Asn-534 and the phenol hydrogen of Tyr-453. Water molecules also play an important role in stabilizing the substrate in the binding pocket. The phenol hydrogen of Tyr-453 is stabilized by a water molecule. In the P2 pocket, the S2 carbonyl oxygen forms a hydrogen bond with Arg-618, which closely interacts with Asp-145 through a salt bridge and hydrogen bonds. The S2 backbone amide is hydrogen-bonded with another water molecule. Notably, residues Asn-534, Tyr-453, and Arg-618 are conserved in the PEP family, and they might be crucial for controlling the PEP specificity. The S3 site in the inhibitor is the benzyloxycarbonyl group, whose carbonyl oxygen is stabilized by two hydrogen bonds with the side chain of Trp-574, and a water molecule, which is further involved in a hydrogen-bond network with Tyr-170, Thr-573, and another water molecule.
Fig. 2.

MX interactions mapping. Residues (Y453, N534, S533, H651, W574, Y578, R618, D145, Y170, and T573) involved in the inhibitor-binding pocket are labeled in black, and their corresponding hydrogen-bond network is indicated by blue dashed lines. Z-Ala-prolinal is colored in orange. Additional residues (V458, G532, R572, I575, D196, and E197) and interactions of interest are indicated in the map labeled in gray and are further examined through mutagenesis. Notably, two sets of interface residues, D145 (from the propeller domain) with R618 (from the catalytic domain) and D196, E197 (propeller) with R572 (catalytic), contribute to the domain–domain interactions by forming salt bridges and hydrogen bonds.
Architecture of SC PEP. The structure of the SC PEP offers a structural observation of an open configuration in the class of propeller-containing serine proteases (Fig. 1B). The opening of the two domains causes shifts of both the global and local features of the structure, although each domain retains characteristics similar to those in the closed form. The only covalent connections between the domains are by means of two strands consisting of residues Ile-106–Glu-109 and Thr-453–Pro-449. Interestingly, the domain opening is asymmetric, with one side of the domains separated by ≈30 Å and the other side of the two domains still interacting with each other. Specifically, the β1 strand (Ala-457–Ser-468) and the loop region between β4 and β5 (Asn-499–Ser-507) in the catalytic domain are still in close contact.
Despite the overall similarity of the seven-blade propeller domains in the two proteins (the rms deviation between the two homologous domains is 2.0 Å for 324 fitted Cα atoms), the flexible-loop regions in the SC PEP propeller domain are significantly distorted (see Fig. 7, which is published as supporting information on the PNAS web site). For example, residues Phe-428, Gly-407, and Ala-384, situated in loops at the interface of the catalytic domain, have been pushed outward from the radial center of the propeller domain. Their neighboring loops are pushed down and protrude at the bottom. On the other hand, the catalytic domains of the two enzymes align quite well as a result of the highly stable secondary structures in the domain. The β-strands in the catalytic domain of SC PEP superimpose well with those in MX PEP. Most of the α-helices are unchanged, except for αF, αA, and α2*, which are situated next to the C-terminal His6-tag of the enzyme.
A Well Arranged Domain Interface. The open structure of the SC PEP illustrates the extensive motions of the enzyme. Interestingly, the open conformation of the SC enzyme is stabilized by the C-terminal His6-tag of a neighboring molecule in the crystal lattice, which extends into the active-site region of the enzyme (Fig. 3). Comparisons between the MX and SC PEP crystal structures provide important insights into the substrate-binding mechanism of PEPs by means of domain opening. Three stretches of residues, Trp-157∼Gly-161, Thr-689∼Lys-697, and Lys-231∼Ala-238, all from the domain interface, have become unstructured in the open structure. Their involvement in domain interaction can be visualized by examining the corresponding residues in the closed MX PEP structure, Trp-115∼Gly-119, Ala-647∼Val-657, and Asp-190∼Glu-197. The loop consisting of Trp-115∼Gly-119 is stabilized by hydrogen bonding with the glutamine in the loop of Ala-647∼Val-657. Most interestingly, residues Asp-190∼Glu-197, situated in the uniquely stabilized helical loop from the propeller, interacts extensively with a loop from the catalytic domain, with D196 and E197 forming a salt bridge with R572 (Figs. 2 and 4). D196 also contributes to a hydrogen-bond network of R572 and I575. Another salt bridge is found at the interface between Asp-145 from the propeller domain and R618 from the catalytic domain. The extensive domain–domain interaction is presumably the driving force for the closed structure. It is proposed that the electrostatic points of interaction provide the “latches” for domain opening and closing.
Fig. 3.

Open structure of the SC PEP stabilized by the neighboring molecule's His6-tag through its interaction with the catalytic domain. The α-helix at the mouth region of the catalytic domain interacts extensively with the incoming peptide (cyan).
Fig. 4.

Docking model of MX PEP. MX is docked with PQPQLPYPQPQLP. The peptide is in a green/red/blue color configuration. The active-site serine is labeled. Residues D196 and R572, which form a salt bridge at the mouth of the didomain enzyme, are also shown.
An Extended Binding Site. To investigate the extended binding site in PEP that might be required for the binding of longer peptides, the crystal structure of MX PEP was docked against a model of a 13-mer peptide PQPQLPYPQPQLP. Earlier work has already shown that MX and SC PEPs have a higher specificity toward PQPQLPYPQPQLP compared with succinyl-Ala-Pro-pNA, and MX preferentially cleaves at the P|Y motif (10). Manual peptide docking was carried out by using the graphics program o, based on the known peptide cleavage site, and resulted in a docking model that achieved both good geometry and extensive hydrophobic interactions and hydrogen bonding between the peptide and the binding site. The predicted 13-mer binding pocket has an additional buried surface area of ≈250 Å2 in comparison with that of the observed Z-Ala-prolinal inhibitor. The model reveals a plausible binding mode of the 13-mer peptide in the active-site pocket of MX PEP (Fig. 4), based on the following observations. The S1′∼S6′ residues (PQPQLP) interact extensively with the binding pocket, much more so than the bound inhibitor. The N terminus of the peptide points toward the domain–domain interface, and, as in the case of Z-Ala-prolinal, it tilts toward the short loop consisting of Lys-161–Pro-173, which could possibly act as an initial opening site. The S1 and S2 residues (Tyr and Pro) are in close contact with the catalytic pocket, whereas the C-terminal residues fit into the propeller central cavity. As outlined below, mutagenesis of the binding-site residues supported this model for substrate binding.
Mutagenesis Confirms Critical Domain-Interface Regions. A panel of site-directed MX PEP mutants was generated with selected residues within 10 Å of the bound substrate based on the MX PEP crystal structure and sequence alignments of MX, SC, and F. meningosepticum PEPs (10) (see Fig. 8, which is published as supporting information on the PNAS web site). These mutants were assayed with substrates of varying lengths [the short chromogenic substrate Suc-Ala-Pro-pNA and a longer 13-mer peptide (PQPQLPYPQPQLP)] (8, 10). These mutants display an interesting range of catalytic properties and chain-length specificity (Table 2 and Fig. 2).
Table 2. Kinetics analysis of MX PEP and mutants.
| Suc-Ala-Pro-pNA
|
PQPQLPYPQPQLP
|
|||
|---|---|---|---|---|
| PED | kcat, s-1 | Km, μM | kcat/Km, s-1·mM-1 | kcat/Km |
| Wild type | 46 ± 5 | 400 ± 30 | 120 ± 23 | 480 ± 60 |
| V458I | 37 ± 3 | 282 ± 80 | 131 ± 31 | 270 ± 35 |
| G532R | 36 ± 2 | 372 ± 61 | 98 ± 27 | 255 ± 50 |
| R572A | 41 ± 7 | 2028 ± 610 | 21 ± 11 | 228 ± 15 |
| R572Q | 49 ± 7 | 1620 ± 310 | 28 ± 13 | 338 ± 38 |
| I575A | 45 ± 3 | 779 ± 112 | 58 ± 22 | 776 ± 83 |
| F229Y | 40 ± 2 | 1633 ± 194 | 23 ± 9 | 589 ± 169 |
| D196S | 39 ± 4 | 387 ± 64 | 106 ± 28 | 662 ± 175 |
| E197Q | 27 ± 2 | 270 ± 54 | 100 ± 31 | 232 ± 65 |
| D145N | 0.06 ± 0.02 | |||
| D145E | 0.37 ± 0.04 | |||
| R618Q | 0.016 ± 0.002 | |||
| R618K | 0.04 ± 0.02 | |||
The first set of amino acids, V458 and G532, are close to the catalytic Ser-533 in the binding pocket. Their mutants retain wild-type activity toward Suc-Ala-Pro-pNA but show reduced activity toward the 13-mer peptide. Their ability to discriminate between the chromogenic substrate and the longer peptides suggests that these residues may play a role in binding to the C-terminal side of the scissile bond.
As discussed above, residues R572 and D196/E197 stood out from the structural analysis as potential key points of interaction between the two domains. Mutagenesis of these and neighboring residues not only confirmed their importance at the domain interface but also provided greater insight into the substrate-binding mode. Whereas the wild-type MX PEP exhibits a 4-fold-higher specificity toward PQPQPLPYPQPQLP as compared with the short (chromogenic) substrate, mutants R572A/Q, I575A, and F229Y have a >10-fold-higher specificity for the longer substrate (Table 2). In most cases, this difference can be accounted for by a markedly higher KM for Suc-Ala-Pro-pNA. In contrast, mutation of the propeller residues, D196 and, to a lesser degree, E197, does not significantly affect activity or specificity. Together with the two structures described above, these results suggest that, not only do longer substrates pry open the two domains, but they also interact extensively with the catalytic domain (R572 is 18 Å away from the active-site serine). Indeed, in the open structure of the SC PEP, the C-terminal His6 peptide (≈10 residues long and presumably acting as a surrogate substrate) is observed to interact closely with a helical region (Fig. 3) in the catalytic domain that includes residues corresponding to R572 and I575. Mutation of R572 in MX PEP results in reduced Suc-Ala-Pro-pNA binding affinity, presumably because of the absence of extended interactions between this position and either the short substrate or the propeller domain. In contrast, for the longer substrate PQPQLPYPQPQLP, secondary interactions between the substrate and the domains help compensate for the absence of the salt-bridge latch that ordinarily allows the two domains to envelope the substrate. Similarly, the affinity of Suc-Ala-Pro-pNA for the F229Y mutant is reduced, because Phe-229 directly contributes to the substrate stabilization (Fig. 2). On the other hand, the specificity of the mutant for the 13-mer substrate is minimally affected.
Mutagenesis has also shed light on another set of critical residues for PEP activity. Asp-145 and Arg-618 (from MX PEP) are conserved throughout the PEP family. Their key roles in catalysis are appreciated from the observation that mutations at these positions leads to a loss of >99% of activity (Table 2). As seen in the MX structure, Arg-618 is involved in stabilizing the bound inhibitor by contributing a hydrogen bond to the S2 carbonyl oxygen. Asp-145, which hydrogen bonds to Arg-618, plays a role in the accurate positioning of the latter residue. Thus, the invariant S1-proline residue in the substrate is tightly anchored by this network of hydrogen bonds, which presumably contributes to the catalytic strength as well as unique substrate specificity of this class of proteases.
Mechanism and Implications. The observation of an open structure of the SC PEP has unveiled a unique substrate-binding mode in the serine-protease family. On the basis of the x-ray crystal structures of the open, unoccupied form of SC PEP and the closed MX PEP-inhibitor complex, as well as on mutagenesis and modeling studies, the following mechanism is proposed for the substrate access and binding of PEPs. The incoming peptide substrate induces a conformational change of the enzyme and causes the domain interface to open. As a comparison, in the case of prolyl-specific exopeptidase, dipeptidyl peptidase IV, the substrate enters through a large side cavity at the domain interface leading to the active site (28, 29). The open PEP structure is temporarily stabilized by interactions between the substrate and the catalytic domain of the enzyme. Residues Arg-618 and Asp-145 have been found to contribute to the unique substrate specificity and activity of the enzyme. The extent of the substrate–catalytic-domain interaction depends on the size of the peptide. Residues from the catalytic-domain mouth region serve as anchors for longer substrates, whereas key residues such as Arg-572 and Ile-575 help control the opening and closing of the enzyme structure. In contrast, Asp-196 and Glu-197 in the propeller domain are important in stabilizing the closed (unbound) structure but do not appear to interact with the bound substrate.
Whereas the previously established closed-structure complexes of a mammalian PEP provided a high-resolution view of the binding state of the substrate, they could not reflect a dynamic substrate-binding mechanism. In fact, the mammalian PEP enzyme was previously thought to have a rigid structure, allowing only local oscillations between propeller blades to filter substrates through the propeller tunnel (17). Kinetic studies on the mammalian enzyme have suggested the possibility of interdomain movement (15, 18), but until now, the visualization of domain opening upon substrate binding has not been possible. This mechanism has significant physiological and therapeutic implications for this unique family of peptidases. The limited surface area created by domain opening efficiently selects the size of the oligopeptides and protects larger structured peptides and proteins from proteolysis. The open form also offers a limited P′ pocket and thus controls the “bite size” on peptide substrates, as observed previously in MX PEP, where approximately four to six residues are cleaved at a time (10). Given the promising roles of bacterial PEPs for detoxification of immunotoxic proline-rich peptides from gluten in celiac sprue patients, the findings here provide a strong basis for further engineering of this class of enzymes. For example, the acid stability, gastrointestinal-protease resistance, and substrate specificity of these peptidases could be optimized to suit pharmaceutical needs.
Supplementary Material
Acknowledgments
We thank Shiou-Chuan Tsai for assistance with crystallization. This work was supported by National Institutes of Health Grant DK063158 (to C.K.). L.S. is a recipient of a Stanford Graduate Fellowship. I.I.M. is associated with the Structural Molecular Biology Program at the Stanford Synchrotron Radiation Laboratory, which is supported by the U.S. Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and National Institute of General Medical Sciences. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences Division, of the U.S. Department of Energy under Contract DE-AC03-76SF00098 with the Lawrence Berkeley National Laboratory.
Author contributions: L.S., I.I.M., and C.K. designed research; L.S. and I.I.M. performed research; L.S., I.I.M., and C.K. analyzed data; and L.S., I.I.M., and C.K. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MX, Myxococcus xanthus; PEP, prolyl endopeptidase; pNA, p-nitroanilide; SC, Sphingomonas capsulata.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2BKL and 1YR2).
References
- 1.Yoshimoto, T., Walter, R. & Tsuru, D. (1980) J. Biol. Chem. 255, 4786-4792. [PubMed] [Google Scholar]
- 2.Yoshimoto, T., Kanatani, A., Shimoda, T., Inaoka, T., Kokubo, T. & Tsuru, D. (1991) J. Biochem. (Tokyo) 110, 873-878. [DOI] [PubMed] [Google Scholar]
- 3.Yoshimoto, T., Miyazaki, K., Haraguchi, N., Kitazono, A., Kabashima, T. & Ito, K. (1997) Biol. Pharm. Bull. 20, 1047-1050. [DOI] [PubMed] [Google Scholar]
- 4.Kabashima, T., Fujii, M., Meng, Y., Ito, K. & Yoshimoto, T. (1998) Arch. Biochem. Biophys. 358, 141-148. [DOI] [PubMed] [Google Scholar]
- 5.Rennex, D., Hemmings, B. A., Hofsteenge, J. & Stone, S. R. (1991) Biochemistry 30, 2195-2203. [DOI] [PubMed] [Google Scholar]
- 6.Mentlein, R. (1988) FEBS Lett. 234, 251-256. [DOI] [PubMed] [Google Scholar]
- 7.Atack, J. R., Suman-Chauhan, N., Dawson, G. & Kulagowski, J. J. (1991) Eur. J. Pharmacol. 205, 157-163. [DOI] [PubMed] [Google Scholar]
- 8.Shan, L., Molberg, O., Parrot, I., Hausch, F., Filiz, F., Gray, G. M., Sollid, L. M. & Khosla, C. (2002) Science 297, 2275-2279. [DOI] [PubMed] [Google Scholar]
- 9.Hausch, F., Shan, L., Santiago, N. A., Gray, G. M. & Khosla, C. (2002) Am. J. Physiol. 283, G996-G1003. [DOI] [PubMed] [Google Scholar]
- 10.Shan, L., Marti, T., Sollid, L. M., Gray, G. M. & Khosla, C. (2004) Biochem. J. 383, 311-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris, M. N., Madura, J. D., Ming, L. J. & Harwood, V. J. (2001) J. Biol. Chem. 276, 19310-19317. [DOI] [PubMed] [Google Scholar]
- 12.Polgar, L. (2002) Cell. Mol. Life Sci. 59, 349-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fulop, V., Bocskei, Z. & Polgar, L. (1998) Cell 94, 161-170. [DOI] [PubMed] [Google Scholar]
- 14.Fulop, V., Szeltner, Z., Renner, V. & Polgar, L. (2001) J. Biol. Chem. 276, 1262-1266. [DOI] [PubMed] [Google Scholar]
- 15.Polgar, L. (1992) Biochem. J. 283, 647-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polgar, L. & Patthy, A. (1992) Biochemistry 31, 10769-10773. [DOI] [PubMed] [Google Scholar]
- 17.Fulop, V., Szeltner, Z. & Polgar, L. (2000) EMBO Rep. 1, 277-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szeltner, Z., Rea, D., Juhasz, T., Renner, V., Fulop, V. & Polgar, L. (2004) J. Mol. Biol. 340, 627-637. [DOI] [PubMed] [Google Scholar]
- 19.Marti, T., Molberg, O., Li, Q., Gray, G. M., Khosla, C. & Sollid, L. M. (2005) J. Pharmacol. Exp. Ther. 312, 19-26. [DOI] [PubMed] [Google Scholar]
- 20.Minor, W., Tomchick, D. & Otwinowski, Z. (2000) Structure Fold. Des. 8, R105-R110. [DOI] [PubMed] [Google Scholar]
- 21.Otwinowski, Z., Borek, D., Majewski, W. & Minor, W. (2003) Acta Crystallogr. A 59, 228-234. [DOI] [PubMed] [Google Scholar]
- 22.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 23.Brunger, A. T., Adams, P. D. & Rice, L. M. (1998) Curr. Opin. Struct. Biol. 8, 606-611. [DOI] [PubMed] [Google Scholar]
- 24.Terwilliger, T. C. & Berendzen, J. (1999) Acta Crystallogr. D 55, 849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard (1991) Acta Crystallogr. A 47, 110-119. [DOI] [PubMed] [Google Scholar]
- 26.Laskowski, R. A., Moss, D. S. & Thornton, J. M. (1993) J. Mol. Biol. 231, 1049-1067. [DOI] [PubMed] [Google Scholar]
- 27.Ramachandran, G. N. & Sasisekharan, V. (1968) Adv. Protein Chem. 23, 283-438. [DOI] [PubMed] [Google Scholar]
- 28.Thoma, R., Loffler, B., Stihle, M., Huber, W., Ruf, A. & Hennig, M. (2003) Structure (London) 11, 947-959. [DOI] [PubMed] [Google Scholar]
- 29.Aertgeerts, K., Ye, S., Tennant, M. G., Kraus, M. L., Rogers, J., Sang, B. C., Skene, R. J., Webb, D. R. & Prasad, G. S. (2004) Protein Sci. 13, 412-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collaborative Computational Project 4 (1994) Acta Crystallogr. D 50, 760-763.15299374 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.