

ABSTRACT
Seneca Valley virus (SVV) is an oncolytic RNA virus belonging to the Picornaviridae family. Its nucleotide sequence is highly similar to those of members of the Cardiovirus genus. SVV is also a neuroendocrine cancer-selective oncolytic picornavirus that can be used for anticancer therapy. However, the interaction between SVV and its host is yet to be fully characterized. In this study, SVV inhibited antiviral type I interferon (IFN) responses by targeting different host adaptors, including mitochondrial antiviral signaling (MAVS), Toll/interleukin 1 (IL-1) receptor domain-containing adaptor inducing IFN-β (TRIF), and TRAF family member-associated NF-κB activator (TANK), via viral 3C protease (3Cpro). SVV 3Cpro mediated the cleavage of MAVS, TRIF, and TANK at specific sites, which required its protease activity. The cleaved MAVS, TRIF, and TANK lost the ability to regulate pattern recognition receptor (PRR)-mediated IFN production. The cleavage of TANK also facilitated TRAF6-induced NF-κB activation. SVV was also found to be sensitive to IFN-β. Therefore, SVV suppressed antiviral IFN production to escape host antiviral innate immune responses by cleaving host adaptor molecules.
IMPORTANCE Host cells have developed various defenses against microbial pathogen infection. The production of IFN is the first line of defense against microbial infection. However, viruses have evolved many strategies to disrupt this host defense. SVV, a member of the Picornavirus genus, is an oncolytic virus that shows potential functions in anticancer therapy. It has been demonstrated that IFN can be used in anticancer therapy for certain tumors. However, the relationship between oncolytic virus and innate immune response in anticancer therapy is still not well known. In this study, we showed that SVV has evolved as an effective mechanism to inhibit host type I IFN production by using its 3Cpro to cleave the molecules MAVS, TRIF, and TANK directly. These molecules are crucial for the Toll-like receptor 3 (TLR3)-mediated and retinoic acid-inducible gene I (RIG-I)-like receptor (RLR)-mediated signaling pathway. We also found that SVV is sensitive to IFN-β. These findings increase our understanding of the interaction between SVV and host innate immunity.
KEYWORDS: Seneca Valley virus, innate immunity, 3C-like protease, MAVS, TRIF, TANK, RNA virus, cell signaling, interferons
INTRODUCTION
Seneca Valley virus (SVV) is a positive single-stranded RNA virus that belongs to the Picornaviridae (1). Analysis of the full-length genome of SVV-001 suggests that SVV is closely related to Cardiovirus (1). The SVV genome contains a single open reading frame (ORF) that encodes a polyprotein. This polyprotein is processed by viral 2A protein, 3C protease (3Cpro), and host proteases to produce three structural proteins and eight nonstructural ones. The structural proteins, namely, VP0, VP3, and VP1, form the viral capsid, while the nonstructural ones play an important role in viral replication and virus-host interactions (1). SVV also exhibits a putative oncolytic property which selectively targets neuroendocrine cancers (2–5). It has been identified as a potent oncolytic virus against several human cancers (3, 5–7). However, basic knowledge regarding the etiology and immunology of SVV is limited because it is a newly emerged virus, so detailed information on it is yet to be obtained.
Innate immunity is the first line of host defense against invading pathogens. During viral infection, host pattern recognition receptors (PRRs) capture pathogen ligands and subsequently induce interferon (IFN) production mediated by two transcription regulators, IFN regulatory factors (IRFs) and nuclear factor κB (NF-κB). IFN molecules bind to IFN receptors and activate the Janus kinase (JAK) signal transducer and activator of transcription (STAT) pathway. Thus, numerous IFN-stimulated genes (ISGs) become transcribed. These ISGs exert numerous antiviral functions directly or indirectly (8, 9). Several PRRs have been identified to recognize viral RNA and induce the production of IFN, including Toll-like receptors (TLR3, TLR7, TLR8, and TLR9) as well as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) (10–12). Mitochondrial antiviral-signaling protein (MAVS), also known as IPS-1, Cardif, and VISA, participates in the regulation of antiviral responses mediated by RIG-I-like receptors (RLRs) (13). Once a viral RNA is recognized by RIG-I and MDA5, they form homo-oligomers and interact with MAVS through their cascade recruitment domain (CARD). Subsequently, MAVS transduces signals to a complex of TANK-binding kinase 1 (TBK1) and inducible IκB kinases (IKKs) and thus activates IRF3 and NF-κB. In the TLR3 signaling pathway, Toll/interleukin 1 (IL-1) receptor domain-containing adaptor inducing IFN-β (TRIF) regulates TLR3-mediated IRF3 and NF-κB activation (14). In addition, tumor necrosis factor receptor-associated factor (TRAF) family member-associated NF-κB activator (TANK) plays a critical role in the regulation of RLR- and TLR-mediated interferon production (15). TANK interacts with several signal molecules, such as MAVS, TRIF, TBK1, and IRF3, to modulate the TBK1-IKK-mediated IFN antiviral response (15).
Although hosts have developed highly efficient strategies to detect and control invading viruses, most viruses have evolved various strategies to evade host defenses and thus effectively infect and replicate in host cells. For instance, picornaviruses evade RLR recognition, undergo cleavage, or degrade crucial innate immune molecules, including adaptors, kinases, and transcriptional factors (16). Poliovirus, echovirus, and encephalomyocarditis virus (EMCV) infections were shown to induce the cleavage of RIG-I (17). Enteroviruses (EVs) target various innate immune molecules for cleavage by viral nonstructural proteins (18). EV proteins, including 2A, 2C, and 3C, have been identified as certain antiviral antagonists, which can target MDA5, RIG-I, MAVS, TRIF, TAK1, IRF3, IRF7, and IRF9 for cleavage. As a result, IFN production and its effects are prevented (18–24).
SVV, an emerging novel picornavirus, has been identified as an oncolytic virus to treat various human neuroendocrine cancers. However, basic information regarding SVV, particularly its interaction with hosts, is limited. In this study, the association between SVV and host IFN antiviral responses was thus investigated. Our results revealed that this virus has developed many strategies to antagonize type I IFN signaling. SVV infection did not significantly induce host type I IFN responses. SVV also targeted the MAVS, TRIF, and TANK adaptors to induce cleavage at specific sites on the basis of its 3Cpro properties. The cleaved MAVS and TANK lose the ability to modulate RLR-mediated IFN production. Therefore, SVV has evolved a mechanism to cleave host innate immune adaptors by 3Cpro and consequently escape host antiviral innate immunity.
RESULTS
SVV infection does not trigger host type I interferon production.
It is well known that type I IFN plays an important role in host innate immune response to defend against viral infection. In this study, the potential interaction between SVV and host type I IFN production was investigated by conducting a luciferase activity assay for the IFN-β reporter gene. We found that Sendai virus (Sev) infection of 293T cells significantly induced IFN-β promoter activity in a time-dependent manner. Meanwhile, IFN-β promoter-driven luciferase activity was scarcely detectable in SVV-infected cells at any time points (Fig. 1A). To confirm these results, a quantitative real-time reverse transcription-PCR (qRT-PCR) assay was performed to investigate whether SVV infection induced the expression of IFN-β mRNA. As shown in Fig. 1B, SVV failed to induce the endogenous transcription of IFN-β at any time point examined. These results suggested that SVV infection was barely able to trigger host IFN-β expression.
FIG 1.

SVV infection does not trigger host type I interferon production. (A) Luciferase activity assay from 293T cells infected with Sev (HA titer, 32) or SVV (MOI, 1) for the indicated times. (B) qRT-PCR assay performed with 293T cells infected with Sev (HA titer, 32) and SVV (MOI, 1) for the indicated times. (C) Immunoblot analysis for IRF3 activation in 293T cells infected with Sev (HA titer, 32) or SVV (MOI, 1) for the indicated times. Quantification of the extent of the increase in phosphorylated IRF3 was normalized to total IRF3 (right panel). (D) Immunofluorescence microscopy analysis for the nuclear translocation of IRF3 in 293T cells infected with Sev (HA titer, 32) or SVV (MOI, 1) for 12 h. IRF3 is green and the nucleus is blue. (E) 293T cells were pretreated with IFN-β at the indicated concentration for 12 h and then infected with VSV-green fluorescent protein (GFP) or SVV. CPE was observed at 24 h postinfection. (F) The virus progeny from panel E were determined by plaque assay. Data are shown as means ± SD. ***, P < 0.001. Data are representative of those from at least three independent experiments.
IRF3 is a crucial transcriptional regulator upstream of type I IFN transcription and production. Therefore, we investigated whether SVV infection impacts IRF3 activation. Specifically, we analyzed the phosphorylation and nuclear translocation of IRF3 in SVV-infected 293T cells. We found that the SVV infection of 293T cells barely induced IRF3 activation at any time point as determined by Western blotting of phosphorylated IRF3 (Fig. 1C). In addition, immunofluorescence microscopy analysis showed that the proportions of IRF3 in the nucleus in Sev- and SVV-infected 293T cells were 72% and 4%, respectively. This implies that SVV infection cannot significantly induce IRF3 transport into the nucleus. Meanwhile, IRF3 activation was significantly induced in Sev-infected 293T cells (Fig. 1C and D). Subsequently, we were interested in the effect of IFN-β in SVV infection. We evaluated the effect of IFN-β pretreatment on infection by vesicular stomatitis virus (VSV), an IFN-β-sensitive virus, and SVV in 293T cells. We found that IFN-β pretreatment significantly inhibited SVV infection-induced cytopathic effect (CPEs) in a dose-dependent manner (Fig. 1E, bottom). Likewise, the progeny SVV in culture medium were reduced in the presence of IFN-β (Fig. 1F). Together, these results indicated that SVV exerts certain mechanisms to inhibit host type I IFN production upstream of IRF3 activation, which resulted in facilitating its replication.
SVV 3Cpro inhibits Sev-induced type I IFN production.
Upon investigating the role of SVV infection in the IFN-β production pathway, we found that SVV infection significantly inhibited Sev-induced IFN-β production (Fig. 2A). Based on this finding, we became interested in whether SVV was directly involved in this inhibition. We thus performed a luciferase reporter assay to identify potential viral proteins that participate in the suppression of Sev-induced IFN-β expression. We found that viral proteins VP2, 3Cpro, and 3D showed different levels of inhibitory function in Sev-induced IFN-β promoter activity (Fig. 2B).
FIG 2.

SVV 3Cpro antagonizes Sev-induced type I IFN production. (A) IFN-β and ISG56 mRNA expression as measured by qRT-PCR from 293T cells infected with SVV (MOI, 5) for 3 h and then infected or not with Sev (HA titer, 32) for another 6 and 9 h. (B) Luciferase activity assay of 293T cells transfected with plasmids expressing the indicated SVV protein for 20 h and then infected or not with Sev (HA titer, 32) for another 9 h. Protein expression was detected by Western blotting (bottom). GST, recombinant protein glutathione S-transferase. (C) Luciferase activity assay of 293T cells transfected with increasing levels of SVV 3Cpro-expressing plasmids (50, 100, and 250 ng) for 20 h and then infected or not with Sev (HA titer, 32) for another 9 h. (D) IFN-β and ISG56 mRNA expression as assessed by qRT-PCR from 293T cells transfected with SVV 3Cpro-expressing plasmids or empty vector for 20 h and then infected or not with Sev (HA titer, 32) for another 9 h. (E) Immunoblot analysis of 293T cells transfected with increasing levels of SVV 3Cpro-expressing plasmids (0, 200, and 800 ng) for 20 h and then infected or not with Sev (HA titer, 32) for another 9 h. Data are shown as means ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data are representative of those from at least three independent experiments.
Previous studies have reported that picornaviruses exert various strategies to evade host innate immune responses through certain viral proteins, such as Lpro, VP2, VP3, 2Apro, 2Cpro, 3Apro, and 3Cpro (17, 18, 25–31). In this study, we found that the overexpression of SVV proteins VP2, 3Cpro, and 3D reduced Sev-induced IFN-β production (Fig. 2B). Moreover, 3Cpro showed an extremely inhibitory effect. It is known that 3Cpro plays a crucial role in the viral life cycle and virus-host interaction (32). This implies that SVV 3Cpro serves as an antagonist factor for SVV to inhibit type I IFN production. Confirming this hypothesis, we found that SVV 3Cpro inhibited Sev-induced IFN-β promoter activity in a dose-dependent manner (Fig. 2C). In addition, Sev-induced endogenous transcripts of IFN-β and ISG56 were obviously decreased in cells expressing SVV 3Cpro (Fig. 2D). Moreover, we also found that Sev-induced IRF3 activation was inhibited by SVV 3Cpro overexpression. As shown in Fig. 2E, the generation of phosphorylated IRF3 was significantly restricted by SVV 3Cpro in a dose-dependent manner. Collectively, these results suggest that SVV 3Cpro contributes to the ability of SVV to inhibit type I IFN production.
SVV 3Cpro antagonizes Sev-induced type I IFN production by targeting MAVS, TRIF, and TANK.
It has been reported that RIG-I, MDA5, and TLR3 serve as RNA sensors for some picornaviruses (32). However, the sensor for SVV is still unknown. In this study, we investigated the functions of several PRRs and their positive adaptor molecules in SVV-infected cells. We found that putative MDA5-, RIG-I-(N)-, MAVS-, TRIF-, TBK1-, TANK-, and IKKε-mediated IFN-β promoter activity was reduced in the presence of SVV infection (Fig. 3A). In contrast, SVV infection had no influence on IRF3/5D-mediated IFN-β promoter activity (Fig. 3A). Subsequently, we investigated the potential role of SVV 3Cpro in the IFN-β promoter activity mediated by these molecules. A similar observation was made, except in the case of IRF3/5D, namely, that the mediation of IFN-β promoter activity by these molecules was inhibited in the presence of SVV 3Cpro (Fig. 3B). These results indicate that SVV 3Cpro exerts an inhibitory function upstream of IRF3. Again, we found that SVV 3Cpro significantly inhibited the endogenous transcription of IFN-β and ISG56 that was mediated by the indicted adaptors but had no effect on IRF3/5D (Fig. 3C). Taken together, these results demonstrate that SVV 3Cpro has an inhibitory function upstream of IRF3.
FIG 3.

SVV 3Cpro mediates the cleavage of MAVS, TRIF, and TANK. (A) Luciferase activity assay from 293T cells transfected with the indicated adaptor-expressing plasmids or empty vector for 20 h and then infected or not with SVV (MOI, 5) for another 9 h. (B) Luciferase activity assay from 293T cells cotransfected with the indicated adaptors plus SVV 3Cpro or empty vector for 24 h. (C) IFN-β and ISG56 mRNA expression as assessed by qRT-PCR from 293T cells cotransfected with the indicated adaptors plus SVV 3Cpro or empty vector for 24 h. (D) Immunoblot analysis of 293T cells cotransfected with the indicated adaptors (N-terminally Flag tagged) plus SVV 3Cpro or empty vector for 24 h. (E to G) Plasmids expressing Flag-MAVS (E), Flag-TRIF (F), and Flag-TANK (G) were cotransfected with plasmids expressing HA-tagged 3Cpro derived from EMCV (JQ864080) and EV71 (JN230523) into 293T cells. The protein patterns were detected by immunoblotting. (H) Immunoblot analysis of 293T cells infected with SVV (MOI, 5) for the indicated times. Antibodies against MAVS, TANK, and SVV VP1 were used. (I) Immunoblot analysis of 293T cells transfected with Flag-TRIF for 20 h and then infected with SVV (5 MOI) for the indicated times. Antibodies against TRIF and SVV VP1 were used. Data are shown as means ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data are representative of at least three independent experiments.
Previous studies have shown that some picornaviruses evade host innate immune responses through viral 3Cpro-mediated cleavage of several innate immune adaptors. Foot-and-mouth disease virus (FMDV) 3Cpro and hepatitis A virus (HAV) 3Cpro cleave NEMO to impair type I IFN production (30, 31). Enterovirus 3Cpro can target RIG-I, MAVS, TRIF, and IRF7 for cleavage to evade host antiviral innate immunity (18). To investigate whether SVV 3Cpro targets certain adaptors in order to cleave them to inhibit IRF3 activation, we assessed the pattern of expression of these adaptor molecules in SVV 3Cpro-overexpressing 293T cells by Western blotting. N-terminal Flag-tagged adaptor expression plasmids were used in this study. We found that the expression of SVV 3Cpro induced the cleavage of MAVS, TRIF, and TANK (Fig. 3D). A single small pattern, corresponding to a molecular mass of approximately 15 to 25 kDa, was observed in cells cotransfected with MAVS and TRIF plus SVV 3Cpro (Fig. 3D, lanes 6 and 8). Two close patterns, corresponding to a molecular mass of approximately 35 to 40 kDa, were observed in cells cotransfected with TANK and SVV 3Cpro (Fig. 3D, lane 12). These findings demonstrated that SVV 3Cpro can mediate the cleavage of MAVS, TRIF, and TANK. To investigate whether SVV 3Cpro cleaves MAVS, TRIF, and TANK in a specific manner, we assessed the expression pattern of these molecules in 293T cells cotransfected with 3Cpro from another two picornaviruses, EMCV and EV71. As shown in Fig. 3E, a pattern of similar molecule size was observed in 293T cells cotransfected with MAVS and EMCV 3Cpro (lane 4), while no cleavage product was detected in 293T cells cotransfected with EV71 3Cpro (lane 4). No specific cleavage products were detected in 293T cells cotransfected with TRIF with EMCV 3Cpro or with EV71 3Cpro (Fig. 3F, lanes 3 and 4). A single protein band was observed in 293T cells cotransfected with each of TANK, EMCV, and EV71 3Cpro (Fig. 3G, lanes 3 and 4).
To investigate the endogenous cleavage of MAVS and TANK, 293T cells were infected with SVV at a multiplicity of infection (MOI) of 5. By Western blotting, no similar cleavage products of MAVS and TANK in SVV-infected 293T cells were detected, but the protein abundances of endogenous MAVA and TANK were significantly reduced (Fig. 3H). Similarly, we did not observe cleavage fragments of TRIF during SVV infection but found a reduction in TRIF protein abundance (Fig. 3I). Collectively, our results showed that SVV 3Cpro targets host adaptor molecules, including MAVS, TRIF, and TANK, for cleavage in a specific manner.
SVV 3Cpro-mediated cleavage of MAVS, TRIF, and TANK depends on its protease activity.
A previous study reported that both MAVS and TRIF can be cleaved by caspases (33). To investigate whether the SVV 3Cpro-mediated cleavage is also caspase dependent, the broad-spectrum caspase inhibitor Z-VAD-FMK, the proteasome inhibitor MG132, and the lysosome inhibitor NH4Cl were used in this study. Z-VAD-FMK and NH4Cl had little effect on the SVV 3Cpro-mediated cleavage of MAVS (Fig. 4A), TRIF (Fig. 4B), and TANK (Fig. 4C). Interestingly, we found that the abundance of the full-length MAVS and cleaved MAVS fragment was significantly reduced in the presence of proteasome inhibitor MG132 (Fig. 4A, lane 4). Previous studies have reported that the cleavage of MAVS was induced during cellular apoptosis and that both MG132 and Z-VAD-FMK can limit apoptosis-induced cleavage of MAVS (33, 34). We found that Z-VAD-FMK can inhibit SVV 3C-mediated cleavage of MAVS (Fig. 4A, lane 6). In addition, we also found SVV 3C overexpression can significantly induce cellular apoptosis. These results suggest that the cellular apoptosis and degradation process has hardly any effect on the SVV 3Cpro-induced cleavage of TRIF and TANK but impairs SVV 3Cpro-induced cleavage of MAVS.
FIG 4.

Protease activity is required for SVV 3Cpro-mediated cleavage of MAVS, TRIF, and TANK. Plasmids expressing Flag-MAVS (A), Flag-TRIF (B), and Flag-TANK (C) were cotransfected with plasmids expressing SVV 3Cpro or empty vector into 293T cells. The inhibitors MG132 (10 μM), NH4CL (10 mM), and z-VAD-FMK (50 μM) were added as indicated at 12 h posttransfection and for another 12 h. The protein patterns were assessed by immunoblotting. (D) Amino acid sequence alignment of 3Cpro derived from SVV (APA28975), FMDV (NP_74046), EMCV (NP_740410), HAV (AKI05745), and EV71 (AFP66570). (E to G) Plasmids expressing Flag-MAVS (E), Flag-TRIF (F), and Flag-TANK (G) were cotransfected with plasmids expressing wild-type SVV 3Cpro and indicated 3Cpro mutations for 24 h. The protein patterns were assessed by immunoblotting.
Similar to 3Cpro from other picornaviruses, SVV 3Cpro contains the conserved catalytic box with histidine (His) and cysteine (Cys) residues (Fig. 4D). Therefore, we next constructed three mutants with the abolition of protease activity, with single-site mutations H48A and C160A and the double-site mutation H48A-C160A (3Cdm). Compared to the overexpression of wild-type SVV 3Cpro (3Cwt), all 3Cpro molecules without protease activity lost the ability to mediate the cleavage of MAVS (Fig. 4E, lanes 3 to 5), TRIF (Fig. 4F, lanes 3 to 5), and TANK (Fig. 4G, lanes 3 to 5). These findings together indicate that the SVV 3Cpro-mediated cleavage of MAVS, TRIF, and TANK is strictly dependent on its protease activity.
SVV 3Cpro interacts with MAVS, TRIF, and TANK.
We found that SVV 3Cpro induces the cleavage of MAVS, TRIF, and TANK, which indicates that 3Cpro interacts with these adaptors. Supporting this notion, coimmunoprecipitation and immunofluorescence microscopy analysis of wild-type and double-site-mutant 3Cpro and the desired adaptors were conducted. As shown in Fig. 5A, Flag-MAVS can precipitate both HA-3Cwt and HA-3Cdm. In this study, we found that the SVV 3Cpro-mediated cleavage of TRIF is rapid and of high efficiency. We observed Flag-TRIF precipitated HA-3Cdm, while expression of HA-3Cwt induced the complete degradation of Flag-TRIF as assessed by coimmunoprecipitation (Fig. 5B). A previous study reported that overexpressed TRIF forms speckled structures in the cytosol (35). In this study, we found it difficult to detect the interaction between HA-3Cwt and Flag-TANK by immunoprecipitation (IP) assay, while Flag-TANK can precipitate HA-3Cdm (Fig. 5C). These findings demonstrated that SVV 3Cpro interacts with MAVS, TRIF, and TANK, and this interaction provides the basic conditions for the 3Cpro-mediated direct cleavage of these adaptors.
FIG 5.
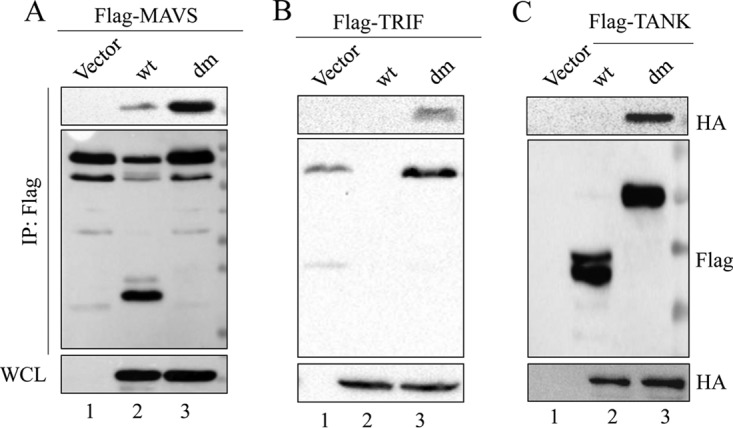
SVV 3Cpro interacts with MAVS, TRIF, and TANK. 293T cells were transfected with Flag-MAVS and empty vector, HA-3Cwt, or HA-3Cdm for 24 h. Subsequently, the interaction between MAVS and 3Cpro was measured by anti-Flag immunoprecipitation (A). (B) Anti-Flag immunoprecipitation for the interaction between Flag-TRIF, HA-3Cwt, and HA-3Cdm. (C) Anti-Flag immunoprecipitation for the interaction between Flag-TANK, HA-3Cwt, and HA-3Cdm. WCL, whole-cell lysate.
SVV 3Cpro cleaves MAVS, TRIF, and TANK at specific sites.
Picornavirus 3C protease preferentially recognizes glutamine-glycine (Q-G) or glutamic acid-glutamine (E-Q) pairs in the viral polyprotein and host factors (36). The putative SVV 3Cpro cleavage sites were proposed based on sequence homology in a previous study (Fig. 6A) (1). As shown in Fig. 3D, the cleavage of N-terminally Flag-tagged MAVS and TRIF by SVV 3Cpro generated a single 15- to 25-kDa band (lanes 6 and 8), and two protein bands between 35 and 40 kDa were observed in TANK (lane 12).
FIG 6.

SVV 3Cpro cleaves MAVS, TRIF, and TANK at specific sites. (A) SVV polyprotein organization and the proposed SVV 3Cpro cleavage sites (red). The indicated viral proteins were colored. (B) (Top) Schematic representation of Flag-MAVS and its mutants. (Bottom) 293T cells transfected with the indicated Flag-MAVS construct and empty vector or HA-3Cwt were lysed and subjected to immunoblot analysis. CARD, caspase recruitment domain; TM, transmembrane. (C) (Top) Schematic representation of Flag-TRIF and its mutants. (Bottom) 293T cells transfected with the indicated Flag-TRIF construct and empty vector or HA-3Cwt for were lysed and subjected to immunoblot analysis. NTD, N-terminal domain; TIR, Toll/interleukin-1 receptor domain; SIA, sufficient to induce apoptosis. (D) (Top) Schematic representation of Flag-TANK and its mutants. (Bottom) 293T cells transfected with the indicated Flag-TANK construct and empty vector or HA-3Cwt for were lysed and subjected to immunoblot analysis. TBD, TRAF binding domain; ZF, zinc finger.
Based on these results, we created a series of site-directed mutations within MAVS, TRIF, and TANK at the Q residues, which may act as SVV 3Cpro cleavage sites. Five Q residues (Q148, Q159, Q162, Q196, and Q198) in the MAVS N terminus were changed to alanine (A) (Fig. 6B, top) and cotransfected into 293T cells with HA-3Cwt or empty vector. As shown in Fig. 6B, the cleavage pattern entirely disappeared in the Q148A mutant (bottom, lane 8). Two Q residues (Q159 and Q190) were replaced with A (Q159A and Q190A) within TRIF (Fig. 6C, top). The Q159A mutation was resistant to SVV 3Cpro-mediated cleavage (Fig. 6C, bottom, lane 5). To identify the residues within TANK, we constructed a series of site-directed mutations in TANK with the replacement of potential Q and E residues with alanine residues. We found that the two cleavage fragments separately disappeared in E272A and Q291A TANK. Next, a double-site mutant of TANK (E272A-Q291A; TANKdm) was cotransfected into 293T cells with HA-3Cwt. The results showed that E272A-Q291A TANK was resistant to SVV 3Cpro-mediated cleavage (Fig. 6D, bottom, lane 8). Collectively, these results demonstrated that SVV 3Cpro mediated the cleavage of MAVS, TRIF, and TANK at specific sites.
The cleavage fragments of MAVS, TRIF, and TANK lost their functions to induce type I IFN production.
MAVS contains a CARD and a proline-rich region in the N terminus and a mitochondrial transmembrane region in the C terminus, both of which are essential for MAVS-mediated signaling (37). In this study, we found that SVV 3Cpro cleaves MAVS at a specific site, Q148, within the proline-rich region. To investigate whether the 3Cpro-induced cleavage disrupts MAVS-mediated type I production, a luciferase activity assay was performed. Wild-type MAVS and Q148A MAVS were cotransfected into 293T cells with HA-3Cwt, HA-3Cdm, or empty vector. The results showed that wild-type MAVS-mediated IFN-β promoter activity was reduced in the presence of HA-3Cwt but not HA-3Cdm. Meanwhile, Q148A MAVS-mediated IFN-β promoter activity was resistant to both HA-3Cwt and HA-3Cdm (Fig. 7A). To confirm this effect, qRT-PCR analysis was conducted to determine the MAVS-mediated endogenous transcripts of IFN-β and ISG56. Again, Q148A MAVS remained functionally active (Fig. 7B). To assess whether the cleavage fragments of MAVS remain active, wild-type MAVS and the N-terminal (residues 1 to 148; N148) and the C-terminal (residues 149 to 540; C149) fragments of MAVS were cotransfected into 293T cells with HA-3Cwt, HA-3Cdm, or empty vector. Subsequently, the effects were assessed by luciferase activity and qRT-PCR assay. The results showed that both N148 and C149 MAVS abolished IFN-β production (Fig. 7C and D).
FIG 7.

Cleaved MAVS, TRIF, and TANK lose their functions to trigger type I IFN production. (A) Luciferase activity assay of 293T cells transfected with wild-type and Q148A mutant MAVS plus empty vector, HA-3Cwt, or HA-3Cdm. (B) IFN-β expression as determined by qRT-PCR from 293T cells transfected with wild-type and Q148A mutant MAVS plus empty vector, HA-3Cwt, or HA-3Cdm. (C and D) The effects of cleavage fragments of MAVS by SVV 3Cpro on MAVS-mediated IFN-β production were assessed by IFN-β reporter assay (C) and qRT-PCR assay (D). (E) Luciferase activity assay of 293T cells transfected with wild-type and Q159A mutant TRIF plus empty vector, HA-3Cwt, or HA-3Cdm. (F) IFN-β expression as determined by qRT-PCR of 293T cells transfected with wild-type and Q159A mutant TRIF plus empty vector, HA-3Cwt, or HA-3Cdm. (G and H) The effects of cleavage fragments of TRIF by SVV 3Cpro on TRIF-mediated IFN-β production were assessed by IFN-β reporter assay (G) and qRT-PCR assay (H). (I) Luciferase activity assay of 293T cells transfected with wild-type and E272A-Q291A mutant TANK plus empty vector, HA-3Cwt, or HA-3Cdm. (J) IFN-β expression as determined by qRT-PCR of 293T cells transfected with wild-type and E272A-Q291A mutant TANK plus empty vector, HA-3Cwt, or HA-3Cdm. (K and L) The effects of cleavage fragments of TANK by SVV 3Cpro on TANK-mediated IFN-β production were assessed by IFN-β reporter assay (K) and qRT-PCR assay (L). Data are shown as means ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data are representative of those from at least three independent experiments.
To assess the effect of TRIF cleavage by SVV 3Cpro on TRIF-mediated type I IFN production, wild-type TRIF and Q159A TRIF were cotransfected into 293T cells with HA-3Cwt, HA-3Cdm, or empty vector. Subsequently, IFN-β promoter activity assay and qRT-PCR assay for the endogenous transcripts of IFN-β and ISG56 were conducted. Wild-type TRIF-mediated IFN-β production was restricted in HA-3Cwt-expressing cells. Meanwhile, Q159A TRIF remained functionally active (Fig. 7E and F). Furthermore, we explored whether possible SVV 3Cpro-cleaved N-terminal (residues 1 to 159; N159) and C-terminal (residues 160 to 712; C160) fragments of TRIF could remain active in the induction of IFN-β. We found that the N159 fragment lost the ability to induce IFN-β production, while the C160 fragment still was functionally active (Fig. 7G and H). This result is consistent with those of previous studies (14, 23, 33, 38, 39).
We found that IFN-β production mediated by E272A-Q291A double-site mutant TANK was resistant to 3Cpro-mediated inhibitory function as assessed by luciferase activity assay and qRT-PCR assay (Fig. 7I and J). We also found that the N-terminal (residues 1 to 291; N291) cleavage fragment of TANK is null for induction of IFN-β production (Fig. 7K and L). Similar to the case with a previous study (40), we found that the C-terminal (residues 272 to 425) fragment of TANK could not be detected in transfected cells. Thus, we did not assess its effect on IFN-β production. Taken together, these results demonstrate that SVV 3Cpro targets MAVS, TRIF, and TANK at specific sites for cleavage, resulting in the suppression of type I IFN production.
3Cpro-mediated cleavage of TANK facilitates TRAF6-triggered NF-κB activation.
It has been reported that TANK associates with the monocyte chemoattractant protein-induced protein 1 (MCPIP1)–ubiquitin-specific peptidase 10 (USP10) complex to decrease TRAF6 ubiquitination, resulting in the inhibition of TRAF6-mediated NF-κB signaling (41). In this study, we investigated the effect of the cleavage of TANK by SVV 3Cpro on its suppression in TRAF6-mediated NF-κB activation. The results showed that TANK expression significantly inhibits TRAF6-mediated NF-κB promoter activity; this inhibitory activity was negated in the presence of wild-type SVV 3Cpro but not 3Cpro lacking protease activity (Fig. 8A). We also found that TRAF6-mediated NF-κB promoter activity was enhanced under the expression of wild-type SVV 3Cpro (Fig. 8A). In addition, the qRT-PCR results demonstrated that wild-type SVV 3Cpro expression promoted TRAF6-mediated endogenous transcription of tumor necrosis factor alpha (TNF-α) (Fig. 8B). To confirm these results, the degradation of IκBα was analyzed. As shown in Fig. 8C, only slight degradation of IκBα occurred when only Flag-TRAF6 was expressed (Fig. 8C, lane 2). However, IκBα was significantly degraded when Flag-TRAF6 and HA-3Cwt were coexpressed (Fig. 8C, lanes 3 and 4) but not when HA-3Cdm was coexpressed with Flag-TRAF6. Moreover, Flag-TANK expression restricted TRAF6-mediated IκBα degradation (Fig. 8C, lane 6). Meanwhile, HA-3Cwt expression significantly abrogated TANK-mediated inhibition (Fig. 8C, lanes 7 and 8). Next, our data showed that TANK with the double-cleavage-site mutation retained its activity to inhibit TRAF6-mediated NF-κB activation (Fig. 8D).
FIG 8.

SVV 3Cpro-mediated TANK cleavage facilitates the TRAF6-NF-κB signaling pathway. (A) NF-κB reporter assay of 293T cells transfected with Flag-TRAF6 and Flag-TANK, HA-3Cdm, or HA-3Cwt for 24 h. (B) Endogenous transcription of TNF-α as determined by qRT-PCR from 293T cells transfected with Flag-TRAF6 and either Flag-TANK, HA-3Cdm, or HA-3Cwt. (C) Immunoblot analysis for IκBα from 293T cells transfected with Flag-TRAF and empty vector, Flag-TANK, HA-3Cdm, or HA-3Cwt for 24 h. (D) NF-κB reporter assay from 293T cells transfected with Flag-TRAF6 and empty vector, Flag-TANKwt, Flag-TANKdm, or HA-3Cwt for 24 h. (E) NF-κB reporter assay from 293T cells transfected with Flag-TRAF6 and empty vector, Flag-tagged wild-type TANK, and the N291 fragment of TANK for 24 h. (F) 293T cells transfected with empty vector or amounts increased wild-type SVV 3Cpro for 20 h and then infected or not with Sev (HA titer, 32) for another 9 h. The cells were lysed and subjected to immunoblot analysis. (G) Endogenous transcription of TNF-α and IL-6 as measured by qRT-PCR from 293T cells infected with SVV (MOI, 5) at the indicated time points. IκBα and VP1 protein expression was detected by Western blotting (bottom). Data are shown as means ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data are representative of those from at least three independent experiments.
To investigate whether the cleavage fragments of TANK retained their activity, wild-type TANK, the N-terminal (residues 1 to 291; N291) fragment of TANK, or empty vector was cotransfected with TRAF6 into 293T cells. The NF-κB reporter assay showed that N291 TANK lost its activity (Fig. 8E). In this study, we also found that SVV 3Cpro promoted Sev-induced IκBα degradation (Fig. 8F). Collectively, these findings suggest that SVV 3Cpro cleaves TANK to negate the suppression of TANK in TRAF6-mediated NF-κB activation. Next, we wished to determine whether the NF-κB is activated during SVV infection. For this, 293T cells were infected with SVV at an MOI of 5, and then the endogenous transcription of NF-κB activation-dependent proinflammatory cytokines TNF-α and IL-6 was determined by qRT-PCR. As shown in the top portion of Fig. 8G, the mRNA expression of TNF-α and IL-6 was significantly increased along with SVV infection. Meanwhile, we also observed significant degradation of IκBα during SVV infection (Fig. 8G, bottom). These results indicate that SVV 3Cpro-mediated cleavage of TANK contributes to SVV-induced NF-κB activation.
DISCUSSION
RLR- and TLR-mediated type I IFN production is implicated in mechanisms of host defense against invading picornaviruses (16, 18). However, picornaviruses have evolved several strategies, such as suppressing type I IFN production and blocking IFN signal transduction, to evade host antiviral innate immunity (16, 18). Enterovirus infection induces host factors, including RIG-I, MDA5, MAVS, TRIF, IRF7/9, and other molecular cleavage or degradation substances. As a result, IFN production and signal transduction are suppressed. FMDV infection restricts the expression of RIG-I, DMA5, and MAVS to attenuate host innate immune responses (18, 27, 29). FMDV can also target NEMO to cleave it to inhibit NF-κB- and IRF3-mediated IFN production (30). HAV ablates the RLR- and TLR-mediated type I IFN response by cleaving MAVS and NEMO (31, 42).
In this study, we found that infection with SVV, a member of the Picornavirus genus, does not trigger a host innate immune response. 293T cells prior to infection with SVV significantly inhibited the Sev-induced type I IFN response (Fig. 2). Our further data showed that SVV 3Cpro targets the host adaptor molecules MAVS, TRIF, and TANK for cleavage to abrogate host innate immune responses (Fig. 3). Compared to 3Cpro derived from EMCV and EV71, SVV 3Cpro cleaves MAVS, TRIF, and TANK to product-specific protein bands (Fig. 3E to G). Meanwhile, we found that EMCV 3Cpro cleaves MAVS to produce a single protein band like that for SVV 3Cpro. In addition, EV71 3Cpro targets TANK for cleavage to produce only one band corresponding to a molecular mass of 35 to 40 kDa. Unfortunately, we failed to detect the endogenous cleavage fragments of MAVS, TRIF, and TANK in SVV-infected cells, but we observed a reduction of the protein abundance (Fig. 3H and I). Studies have reported that picornaviral proteases, including Lpro, 2Apro, and 3Cpro, shut off host protein translation by cleaving eIF4G (43–45). Thus, we have to question whether SVV infection induced host protein translation shutoff. But it is very difficult to demonstrate this, because SVV 3C protease activity is crucial for the cleavage of viral polyprotein and host proteins. In a further study, we demonstrated that this cleavage is not associated with cellular apoptosis-, proteasome-, or lysosome-mediated protein degradation (Fig. 4A to C). Further data confirmed that the SVV 3Cpro-mediated cleavage of these molecules depends on its protease activity (Fig. 4E to G). Because the protease activity of 3C is required for proteolytic cleavage of SVV polyprotein, we failed to rescue a 3Cpro-dead SVV to confirm host cleavage. Unfortunately, we also failed to detect endogenous cleavage of MAVS, TRIF, and TANK during SVV infection. Previous studies have reported that picornaviruses utilize different strategies to inhibit host proteins translation, particularly through Lpro- and 3Cpro-mediated the cleavage of eIF4G (43–46). A recent study identified FMDV 2B protein as a novel antagonism factor to counteract RIG-I-mediated antiviral response by reducing protein abundance of RIG-I (27). Less is known about SVV and host cellular translation. We cannot confirm whether SVV infection induced host translation shutoff and resulted in lower protein abundance of these molecules. We trust that SVV-induced host translation shutoff may also contribute to its combat with host innate immune responses. We were able to confirm that SVV 3C not only mediated the cleavage of these molecules in a protease activity-dependent manner but also interacted with MAVS, TRIF, and TANK (Fig. 5).
Based on the consensus Q↓G cleavage sequence in the SVV polyprotein cleaved by SVV 3Cpro, we constructed a series of point mutants of MAVS, TRIF, and TANK in which potential Q residues were replaced with A residues. We found that the Q148A mutant of MAVS and the Q159A mutant of TRIF were resistant to 3C-mediated cleavage (Fig. 6B and C). For TANK, two amino acid residues, E272 and Q291, were identified as the cleavage sites for SVV 3Cpro (Fig. 6D). Similar to the case with a previously reported study (40), we found that the N-terminal (residues 292 to 425) cleavage fragment of TANK is unstable in cells. Moreover, our data demonstrated that the fragments of MAVS, TRIF, and TANK cleaved by SVV 3Cpro lost the ability to induce the production of IFN-β (Fig. 7).
MAVS and TRIF induce apoptosis independently of their function in PRR-mediated type I IFN production (38, 39, 47, 48). Early apoptosis is a host strategy to suppress viral replication and inhibit virus spreading (49). Lei et al. demonstrated that the transmembrane (TM) domain and CARD are essential for MAVS-induced apoptosis (48). In this study, SVV 3C mediated the cleavage of MAVS at Q148, and this cleavage separated CARD and the TM domain from MAVS (Fig. 6B). As a result, MAVS became nonfunctional in PRR-mediated IFN production and MAVS-induced apoptosis. For TRIF, the N- and C-terminal domains perform different functions in the regulation of innate immunity and apoptosis. The N-terminal domain of TRIF is required for the initiation of IFN-β promoter activity, while both the N- and C-terminal domains are involved in the activation of NF-κB (38). Furthermore, the RIP homotypic interaction motif in the C terminus is essential for TRIF-induced apoptosis (23, 39). In this study, SVV 3C targeted TRIF at Q159 to process two fragments, namely, residues 1 to 159 and 160 to 712 (Fig. 6C). Although the N-terminal domain is separated from TRIF, the C terminus is still implicated in the initiation of IFN signaling, NF-κB activation, and apoptosis (38, 39, 47). Interestingly, we found that SVV infection induced extremely cellular apoptosis responses starting from 6 h postinfection. A broad-spectrum caspase inhibitor, Z-VAD-FMK, can inhibit SVV infection-induced cellular apoptosis and virus replication (data not shown). These results suggested that SVV inhibited host antiviral IFN production but induced apoptosis to facilitate viral replication and viral spreading during a productive infection.
In previous studies, TANK was shown to form a complex with MAVS, TRIF, TRAF3, and TBK1 to modulate TBK1/IKKi-mediated IFN responses (15). TANK also suppressed TRAF6-mediated NF-κB activation by downregulating TRAF6 ubiquitination (41, 50, 51). In this study, we found that wild-type 3Cpro promoted TRAF6-mediated NF-κB activation even when TANK was overexpressed. Conversely, 3C with abolished protease activity could not disrupt the inhibitory function of TANK (Fig. 8A and B). TANK with mutations at two cleavage sites was also associated with resistance to the suppression of wild-type SVV 3Cpro (Fig. 8D). Moreover, we found that SVV 3Cpro expression significantly promoted TRAF6- and Sev-induced IκBα degradation (Fig. 8D and F). These results are consistent with previous findings (40, 41, 52). These findings, to a certain extent, probably can answer why different degrees of inflammation were observed in SVV-infected animals.
Host innate immune responses play an important role in defense against virus invasion in the early stages of infection (53–55). Meanwhile, viruses also developed some ingenious mechanisms to disrupt host innate immune responses (56). In this study, we demonstrated that SVV, a picornavirus, blocks host antiviral innate immunity by cleaving various adaptor proteins in the antiviral innate immune signaling pathway, including MAVS, TRIF, and TANK. These findings are informative to our understanding of the interaction between SVV and the host. Moreover, it also implies that SVV 3C protease could be a target to restrict SVV replication.
MATERIALS AND METHODS
Cells, viruses, antibodies, and reagents.
Baby hamster kidney BHK-21 cells (ATCC; CCL-10) and human embryonic kidney 293T cells (HEK293T; ATCC; CRL-11268) were grown in Dulbecco's modified essential medium (DMEM; Invitrogen, USA) containing 10% fetal bovine serum (Gibco, USA), 100 U/ml of penicillin (Genview, USA), and 10 μg/ml of streptomycin sulfate (Genview) at 37°C in a humidified 5% CO2 incubator. SVV and VSV were used in this study, and virus titers were determined using a PFU assay in BHK-21 cells. Sev was amplified in chicken eggs and viral titer was determined using the hemagglutination (HA) test. Specific-pathogen-free embryonated chicken eggs (7 to 9 days old) were purchased from Beijing Merial Vital Laboratory Animal Technology Co., Ltd.
Mouse monoclonal antibodies against Flag tag (M185-3L) and HA tag (M180-3) were obtained from Medical and Biological Laboratories (Japan). Alexa Fluor 555 goat anti-mouse (A21424) and Alexa Fluor 488 goat anti-rabbit (A11034) were obtained from Life Technologies, USA. Mouse anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) monoclonal antibody (60004-1-Ig) and rabbit anti-IκBα polyclonal antibody (10268-1-AP) were obtained from ProteinTech Group, China. Rabbit polyclonal antibodies against IRF3 (A2172), phosphor-IRF3-S396 (AP0623), MAVS (A5764), and TANK (A6763) were obtained from ABclonal (Boston, MA).
Dimethyl sulfoxide (DMSO; ST038), NH4Cl, caspase inhibitor Z-VAD-FMK (C1202), and proteasome inhibitor MG132 (S1748) were obtained from Beyotime Biotechnology, China. Recombinant human IFN-β (300-02BC) was obtained from PerproTech, USA. Mouse anti-Flag M2 affinity gel (A2220) was obtained from Sigma (St. Louis, MO). Protein A/G Plus agarose (sc-2003) was from Santa Cruz Biotechnology (Dallas, TX).
Constructs.
The full-length coding sequences (CDs) of individual SVV proteins were amplified from SVV (KX377924) (57) cDNA. VP1, VP2, VP3, 2C, 3C, and 3D were cloned into vector pcDNA3.0-HA. Lpro, 2B, and 3A were cloned into pEBG-GST. Flag-TANK was a kind gift from Changjiang Weng (Harbin Veterinary Research Institute of Chinese Academy of Agricultural Sciences). Flag-MAVS and Flag-TRIF were kindly provided by Meilin Jin (Huazhong Agricultural University). HA-EV71-3C was kindly provided by Mingzhou Chen (Wuhan University). Mutagenesis of SVV 3C, MAVS, TRIF, and TANK constructs was performed using overlap PCR or site-directed mutagenesis (C214-01/02; Vazyme, China). All constructs were identified through DNA sequencing. For primer pairs used in this study, see in Table S1 in the supplemental material.
Immunoblotting and IP.
Cells were collected and treated with lysis buffer containing 1.19% HEPES, 0.88% NaCl, 0.04% EDTA, 1% NP-40, and a protease inhibitor (Roche, UK). The protein concentration of whole-cell lysates was determined using the bicinchoninic acid protein assay kit (Thermo Scientific) to evaluate protein expression. Equal amounts of protein were separated using 12% sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (Roche, UK). These membranes were blocked with 5% nonfat milk in 1×Tris-buffered saline (TBS) with 5% Tween 20 (DGBio, Beijing, China) for 4 h at room temperature. They were subsequently incubated with diluted primary antibodies at room temperature or at 4°C for 2 or 16 h. Anti-rabbit or anti-mouse IgG antibodies conjugated to horseradish peroxidase (HRP) were used as secondary antibodies. An enhanced chemiluminescence substrate was used in detection using an HRP kit (Thermo Scientific, USA). All immunoblot imaging was performed using a Bio-Rad ChemiDoc XRS+ instrument and imaging software. For IP, the cells were lysed with lysis buffer containing protease inhibitors. Samples were subjected to centrifugation for 10 min at 4°C to remove cellular debris. Cell lysates were incubated with anti-Flag M2 affinity gel in a rolling incubator at 4°C overnight. Lysates were discarded after centrifugation at 3,000 × g for 5 min at 4°C. The beads were washed five times with cold lysis buffer prior to elution by incubation at 95°C in 1× sample buffer (62.5 mM Tris [pH 6.8], 10% glycerol, 15 mM EDTA, 4% 2-mercaptoethanol 2% SDS, and bromophenol blue).
Reporter gene assay.
293T cells (1 × 105) were seeded in 24-well plates 24 h prior to transfection. Cells were cotransfected with a reporter plasmid encoding NF-κB and IFN-β-Luc plus pTK-Renilla and the desired expression plasmids. The empty vector was used as a negative control to adjust for the total amount of transfected DNA. The firefly and Renilla luciferase activities were determined using a dual-luciferase reporter assay system (E1910; Promega, Madison, WI), in accordance with the manufacturer's instructions. All reporter assays were repeated three times. Data are presented as means ± standard deviations (SD).
Real-time qRT-PCR.
Total RNA was extracted with the TRIzol reagent (Invitrogen, Grand Island, NY) in accordance with the manufacturer's instructions. Total RNA (1.0 μg) was reverse transcribed using a First Strand cDNA synthesis kit (Toyobo, Japan) also in accordance with the manufacturer's instructions. The mRNA levels of the desired targeted genes were determined through relative quantitative real-time reverse transcription-PCR (qRT-PCR) using SYBR green real-time PCR master mix (Toyobo) with specific primer pairs (see Table S1 in the supplemental material). All reactions were performed in triplicate, and the mRNA level of the housekeeping GAPDH gene was used as an endogenous reference control.
Immunofluorescence assay.
Cells were fixed in 4% paraformaldehyde for 20 min and permeabilized with 0.1% Triton X-100 at room temperature for 10 min. The cells were then washed three times with phosphate-buffered saline (PBS) and incubated with the desired primary antibodies for 2 h. After three washes with PBS, the cells were incubated with the desired secondary antibodies for 1 h. The cells were washed again three times with PBS and then incubated with 4′,6-diamidino-2-phenyl-indole (DAPI; Sigma) for 5 min. Fluorescent images were obtained using a confocal laser scanning microscope (LSM 510 Meta; Carl Zeiss).
Statistical analysis.
All experiments were performed at least three times. Data are shown as the means ± SD. All data were analyzed using GraphPad Prism software, version 5 (GraphPad Software, La Jolla, CA). The various treatments were compared using an unpaired, two-tailed Student t test with an assumption of unequal variance. A P value of ≤0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Program on Key Research Project of China (2016YFD0501505 to Ping Qian), the National Natural Science Foundation of China (31572495 to Xiangmin Li), and Fundamental Research Funds for the Central Universities (2662017PY108 to Ping Qian).
We thank Changjiang Weng (Harbin Veterinary Research Institute of Chinese Academy of Agricultural Sciences), Meilin Jin (Huazhong Agricultural University), and Mingzhou Chen (Wuhan University) for kindly providing plasmids. We are grateful to Ren-Jye Lin (Taibei Medical University), Li Lin (Huazhong Agricultural University), and Bo Zhong (Wuhan University) for giving constructive suggestions for this study.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00823-17.
REFERENCES
- 1.Hales LM, Knowles NJ, Reddy PS, Xu L, Hay C, Hallenbeck PL. 2008. Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J Gen Virol 89:1265–1275. doi: 10.1099/vir.0.83570-0. [DOI] [PubMed] [Google Scholar]
- 2.Poirier JT, Dobromilskaya I, Moriarty WF, Peacock CD, Hann CL, Rudin CM. 2013. Selective tropism of Seneca Valley virus for variant subtype small cell lung cancer. J Natl Cancer Inst 105:1059–1065. doi: 10.1093/jnci/djt130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudin CM, Poirier JT, Senzer NN, Stephenson J, Loesch D, Burroughs KD, Reddy PS, Hann CL, Hallenbeck PL. 2011. Phase I clinical study of Seneca Valley virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin Cancer Res 17:888–895. doi: 10.1158/1078-0432.CCR-10-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morton CL, Houghton PJ, Kolb EA, Gorlick R, Reynolds CP, Kang MH, Maris JM, Keir ST, Wu J, Smith MA. 2010. Initial testing of the replication competent Seneca Valley virus (NTX-010) by the pediatric preclinical testing program. Pediatr Blood Cancer 55:295–303. doi: 10.1002/pbc.22535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z, Zhao X, Mao H, Baxter PA, Huang Y, Yu L, Wadhwa L, Su JM, Adesina A, Perlaky L, Hurwitz M, Idamakanti N, Police SR, Hallenbeck PL, Hurwitz RL, Lau CC, Chintagumpala M, Blaney SM, Li XN. 2013. Intravenous injection of oncolytic picornavirus SVV-001 prolongs animal survival in a panel of primary tumor-based orthotopic xenograft mouse models of pediatric glioma. Neuro Oncol 15:1173–1185. doi: 10.1093/neuonc/not065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reddy PS, Burroughs KD, Hales LM, Ganesh S, Jones BH, Idamakanti N, Hay C, Li SS, Skele KL, Vasko A, Yang J, Watkins DN, Rudin CM, Hallenbeck PL. 2007. Seneca Valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J Natl Cancer Inst 99:1623–1633. doi: 10.1093/jnci/djm198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burke MJ. 2016. Oncolytic Seneca Valley virus: past perspectives and future directions. Oncolytic Virother 5:81–89. doi: 10.2147/OV.S96915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. 2008. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe 3:352–363. doi: 10.1016/j.chom.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Kumar H, Kawai T, Akira S. 2009. Pathogen recognition in the innate immune response. Biochem J 420:1–16. doi: 10.1042/BJ20090272. [DOI] [PubMed] [Google Scholar]
- 12.Roers A, Hiller B, Hornung V. 2016. Recognition of endogenous nucleic acids by the innate immune system. Immunity 44:739–754. doi: 10.1016/j.immuni.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, Yamamoto M, Uematsu S, Ishii KJ, Takeuchi O, Akira S. 2006. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med 203:1795–1803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. 2003. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol 4:161–167. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 15.Guo B, Cheng G. 2007. Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J Biol Chem 282:11817–11826. doi: 10.1074/jbc.M700017200. [DOI] [PubMed] [Google Scholar]
- 16.Feng Q, Langereis MA, van Kuppeveld FJM. 2014. Induction and suppression of innate antiviral responses by picornaviruses. Cytokine Growth Factor Rev 25:577–585. doi: 10.1016/j.cytogfr.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barral PM, Sarkar D, Fisher PB, Racaniello VR. 2009. RIG-I is cleaved during picornavirus infection. Virology 391:171–176. doi: 10.1016/j.virol.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lei X, Xiao X, Wang J. 2016. Innate immunity evasion by enteroviruses: insights into virus-host interaction. Viruses 8:1–13. doi: 10.3390/v8010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, Jin Q, Zhao Z. 2013. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog 9(3):e1003231. doi: 10.1371/journal.ppat.1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lei X, Liu X, Ma Y, Sun Z, Yang Y, Jin Q, He B, Wang J. 2010. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J Virol 84:8051–8061. doi: 10.1128/JVI.02491-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiang Z, Liu L, Lei X, Zhou Z, He B, Wang J. 25 November 2015. The 3C protease of enterovirus D68 inhibits the cellular defense mediated by interferon regulatory factor 7. J Virol doi: 10.1128/JVI.02395-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. 2011. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J Virol 85:8811–8818. doi: 10.1128/JVI.00447-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang T, Coyne CB. 2011. The coxsackievirus B 3Cpro protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog 7(3):e1001311. doi: 10.1371/journal.ppat.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lei X, Han N, Xiao X, Jin Q, He B, Wang J. 2014. Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB1/TAB2/TAB3 complex. J Virol 88:9830–9841. doi: 10.1128/JVI.01425-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knolle PA. 2017. Hitting the right button: MAVS-mediated defense against HAV infection. Cell Res 27:7–8. doi: 10.1038/cr.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drahos J, Racaniello VR. 2009. Cleavage of IPS-1 in cells infected with human rhinovirus. J Virol 83:11581–11587. doi: 10.1128/JVI.01490-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu Z, Wang G, Yang F, Cao W, Mao R, Du X, Zhang X, Li C, Li D, Zhang K, Shu H, Liu X, Zheng H. 2016. Foot-and-mouth disease virus viroporin 2B antagonizes RIG-I mediated antiviral effects by inhibition of its protein expression. J Virol 90:11106–11121. doi: 10.1128/JVI.01310-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li D, Yang W, Yang F, Liu H, Zhu Z, Lian K, Lei C, Li S, Liu X, Zheng H. 2016. The VP3 structural protein of foot-and-mouth disease virus inhibits the IFN-beta signaling pathway. FASEB J 30:1757–1766. doi: 10.1096/fj.15-281410. [DOI] [PubMed] [Google Scholar]
- 29.Li D, Lei C, Xu Z, Yang F, Liu H, Zhu Z, Li S. 2016. Foot-and-mouth disease virus non-structural protein 3A inhibits the interferon-β signaling pathway. Sci Rep 6:21888. doi: 10.1038/srep21888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang D, Fang L, Li K, Zhong H, Fan J, Ouyang C, Zhang H, Duan E, Luo R, Zhang Z, Liu X, Chen H, Xiao S. 2012. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J Virol 86:9311–9322. doi: 10.1128/JVI.00722-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang D, Fang L, Wei D, Zhang H, Luo R, Chen H, Li K, Xiao S. 2014. Hepatitis A virus 3C protease cleaves NEMO to impair induction of beta interferon. J Virol 88:10252–10258. doi: 10.1128/JVI.00869-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun D, Chen S, Cheng A, Wang M. 2016. Roles of the picornaviral 3C proteinase in the viral life cycle and host cells. Viruses 8:1–22. doi: 10.3390/v8030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rebsamen M, Meylan E, Curran J, Tschopp J. 2008. The antiviral adaptor proteins Cardif and Trif are processed and inactivated by caspases. Cell Death Differ 15:1804–1811. doi: 10.1038/cdd.2008.119. [DOI] [PubMed] [Google Scholar]
- 34.Scott I, Norris KL. 2008. The mitochondrial antiviral signaling protein, MAVS, is cleaved during apoptosis. Biochem Biophys Res Commun 375:101–106. doi: 10.1016/j.bbrc.2008.07.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Funami K, Sasai M, Ohba Y, Seya T, Matsumoto M. 2007. Spatiotemporal mobilization of Toll/IL-1 receptor domain-containing adaptor molecule-1 in response to dsRNA. J Immunol 179:6867–6872. doi: 10.4049/jimmunol.179.10.6867. [DOI] [PubMed] [Google Scholar]
- 36.Blom N, Hansen J, Blaas D, Brunak S. 1996. Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Sci 5:2203–2216. doi: 10.1002/pro.5560051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seth RB, Sun L, Ea C, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. 2002. A novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-β promoter in the Toll-Like receptor signaling. J Immunol 169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 39.Han KJ, Su X, Xu LG, Bin LH, Zhang J, Shu HB. 2004. Mechanisms of the TRIF-induced interferon-stimulated response element and NF-κB activation and apoptosis pathways. J Biol Chem 279:15652–15661. doi: 10.1074/jbc.M311629200. [DOI] [PubMed] [Google Scholar]
- 40.Huang L, Liu Q, Zhang L, Zhang Q, Hu L, Li C, Wang S, Li J, Zhang Y, Yu H, Wang Y, Zhong Z, Xiong T, Xia X, Wang X, Yu L, Deng G, Cai X, Cui S, Weng C. 2015. Encephalomyocarditis virus 3C protease relieves TRAF family member-associated NF-κB activator (TANK) inhibitory effect on TRAF6-mediated NF-κB signaling through cleavage of TANK. J Biol Chem 290:27618–27632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W, Huang X, Xin HB, Fu M, Xue A, Wu ZH. 2015. TRAF family member-associated NF-κB activator (TANK) inhibits genotoxic nuclear factor κB activation by facilitating deubiquitinase USP10-dependent deubiquitination of TRAF6 ligase. J Biol Chem 290:13372–13385. doi: 10.1074/jbc.M115.643767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, Lemon SM. 2007. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci U S A 104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belsham GJ, Inerney GMMC, Ross-smith N. 2000. Foot-and-mouth disease virus 3C protease induces cleavage of translation initiation factors eIF4A and eIF4G within infected cells. J Virol 74:272–280. doi: 10.1128/JVI.74.1.272-280.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glaser W, Skern T. 2000. Extremely efficient cleavage of eIF4G by picornaviral proteinases L and 2A in vitro. FEBS Lett 480:151–155. doi: 10.1016/S0014-5793(00)01928-1. [DOI] [PubMed] [Google Scholar]
- 45.de Breyne S, Bonderoff JM, Chumakov KM, Lloyd RE, Hellen CUT. 2008. Cleavage of eukaryotic initiation factor eIF5B by enterovirus 3C proteases. Virology 378:118–122. doi: 10.1016/j.virol.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aminev AG, Amineva SP, Palmenberg AC. 2003. Encephalomyocarditis viral protein 2A localizes to nucleoli and inhibits cap-dependent mRNA translation. Virus Res 95:45–57. doi: 10.1016/S0168-1702(03)00162-X. [DOI] [PubMed] [Google Scholar]
- 47.Kaiser WJ, Offermann MK. 2005. Apoptosis induced by the Toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol 174:4942–4952. doi: 10.4049/jimmunol.174.8.4942. [DOI] [PubMed] [Google Scholar]
- 48.Lei Y, Moore CB, Liesman RM, O'Connor BP, Bergstralh DT, Chen ZJ, Pickles RJ, Ting JP-Y. 2009. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS One 4:e5466. doi: 10.1371/journal.pone.0005466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cuconati A, White E. 2002. Viral homologs of Bcl-1: role of apoptosis in the regulation of virus infection. Genes Dev 16:2465–2478. doi: 10.1101/gad.1012702. [DOI] [PubMed] [Google Scholar]
- 50.Maruyama K, Kawagoe T, Kondo T, Akira S, Takeuchi O. 2012. TRAF family member-associated NF-κB activator (TANK) is a negative regulator of osteoclastogenesis and bone formation. J Biol Chem 287:29114–29124. doi: 10.1074/jbc.M112.347799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawagoe T, Takeuchi O, Takabatake Y, Kato H, Isaka Y, Tsujimura T, Akira S. 2009. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat Immunol 10:965–972. doi: 10.1038/ni.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen ZJ. 2012. Ubiquitination in signaling to and activation of IKK. Immunol Rev 246:95–106. doi: 10.1111/j.1600-065X.2012.01108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takeuchi O, Akira S. 2007. Recognition of viruses by innate immunity. Immunol Rev 220:214–224. doi: 10.1111/j.1600-065X.2007.00562.x. [DOI] [PubMed] [Google Scholar]
- 54.Takeuchi O, Akira S. 2009. Innate immunity to virus infection. Immunol Rev 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seth RB, Sun L, Chen ZJ. 2006. Antiviral innate immunity pathways. Cell Res 16:141–147. doi: 10.1038/sj.cr.7310019. [DOI] [PubMed] [Google Scholar]
- 56.Beachboard DC, Horner SM. 2016. Innate immune evasion strategies of DNA and RNA viruses. Curr Opin Microbiol 32:113–119. doi: 10.1016/j.mib.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qian S, Fan W, Qian P, Chen H, Li X. 2016. Isolation and full-genome sequencing of Seneca Valley virus in piglets from China, 2016. Virol J 13:173. doi: 10.1186/s12985-016-0631-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.