

Abstract
The human dopamine (DA) transporter (hDAT) is a key regulator of neurotransmission and a target for antidepressants and addictive drugs. Despite the recent resolution of dDAT structures from Drosophila melanogaster, complete understanding of its mechanism of function and even information on its biological assembly is lacking. The resolved dDAT structures are monomeric, but growing evidence suggests that hDAT might function as a multimer, and its oligomerization may be relevant to addictive drug effects. Here, using structure-based computations, we examined the possible mechanisms of hDAT dimerization and its dynamics in a lipid bilayer. Using a combination of site-directed mutagenesis, DA-uptake, and cross-linking experiments that exploited the capacity of Cys-306 to form intermonomeric disulfide bridges in the presence of an oxidizing agent, we tested the effects of mutations at transmembrane segment (TM) 6 and 12 helices in HEK293 cells. The most probable structural model for hDAT dimer suggested by computations and experiments differed from the dimeric structure resolved for the bacterial homolog, LeuT, presumably because of a kink at TM12 preventing favorable monomer packing. Instead, TM2, TM6, and TM11 line the dimer interface. Molecular dynamics simulations of the dimeric hDAT indicated that the two subunits tend to undergo cooperative structural changes, both on local (extracellular gate opening/closure) and global (transition between outward-facing and inward-facing states) scales. These observations suggest that hDAT transport properties may be allosterically modulated under conditions promoting dimerization. Our study provides critical insights into approaches for examining the oligomerization of neurotransmitter transporters and sheds light on their drug modulation.
Keywords: docking, dopamine transporter, molecular dynamics, mutagenesis, protein assembly
Introduction
Dopamine (DA) 3 transporter (DAT) regulates dopaminergic signaling by reuptake of DA into presynaptic terminals, thus preventing excess DA levels at the synapse. The process is assisted by co-transport of two sodium ions and the permeation of a chloride ion. The human DAT, hDAT, is a major target for therapeutic antidepressants and addictive drugs such as amphetamine (AMPH) and cocaine (1–3). Dysfunction of hDAT has been implicated in many neurological and psychiatric disorders (2, 3).
DAT belongs to the SLC6A3 family of neurotransmitter sodium symporters (NSSs). NSSs comprise structural and functional homologs such as norepinephrine transporter (NET), serotonin transporter (SERT), and GABA transporter (GAT). It is generally accepted that NSS members transport their substrate via the classic alternating access mechanism (4), i.e. the transporter alternates between outward-facing open (OFo) conformation for extracellular (EC) binding of DA and Na+/Cl− ions and inward-facing open (IFo) conformation for their intracellular (IC) release. The passage between these states takes place via intermediates occluded to both EC and IC regions either in substrate/Na+-loaded form or in the apo state after the release of the cargo. In addition, eukaryotic NSS members such as DAT and SERT allow for the efflux of their substrate to the cell exterior, which has been attributed to the modulating effects of addictive psychostimulants such as AMPH (5, 6).
NSS members generally share the topology of the bacterial leucine transporter LeuT. The LeuT fold is composed of 12 transmembrane (TM) helices (TM1–TM12) organized in two pseudo-symmetric inverted repeats (7). LeuT has long served as a prototype for understanding the mechanism of function of transporters that share its fold, as the first structurally characterized member of the LeuT fold family. The Gouaux laboratory has resolved the X-ray structures of LeuT in four different states: OFo, stabilized by inhibitors (8), substrate-free OFo (9), substrate-bound OFc where the EC gate is closed (10), and apo-IFo (9). Molecular modeling and simulations based on known LeuT structures have helped draw useful inferences on the structure and dynamics of hDAT (11–15). On a broader scale, computational studies performed for transporters that share the LeuT fold provided insights into the mechanistic aspects of substrate transport favored by NSSs (16–23).
The elucidation of the first DAT structure (antidepressant-bound dDAT, from Drosophila melanogaster) in the OFo state (24) and subsequent resolution of DA-, AMPH-, or cocaine-bound dDAT (25), have led the way to structure-based explorations of DAT mechanism of function (26–28). dDAT has >50% sequence identity with hDAT as opposed to 22% with LeuT. As such, it served as a reliable model for modeling its human counterpart. Using these structural data, we studied the mechanism of function of hDAT monomer (27), with dual-boost accelerated MD (aMD) (29, 30) and conventional MD (cMD) simulations. Our simulations revealed the structure of hDAT at various stages of the transport cycle, including DA-bound OFc state, which shows close resemblance to Leu-bound LeuT (10), and the IFo state, leading to the release of DA and Na+ ions. The transport events revealed for hDAT monomer (27) showed striking similarities to those disclosed in our recent investigation of LeuT transport cycle (16, 17). Yet there are notable differences in the sequence and structure of DAT and LeuT, which evidently underlie their specific functionalities.
A prominent structural difference is the kink in the TM12 helix of dDAT (24) which is also retained in hDAT (Fig. 1). This kink occurs at the position of two serines (Ser-567–Ser-568 in hDAT) that are conserved among human monoamine transporters hDAT, hSERT, and hNET as well as inhibitory transporters like hGAT1 and glycine transporters (hGly1). The recently resolved crystal structure of hSERT in an antidepressant-bound OFo also shows a kink at TM12 (31).
Figure 1.
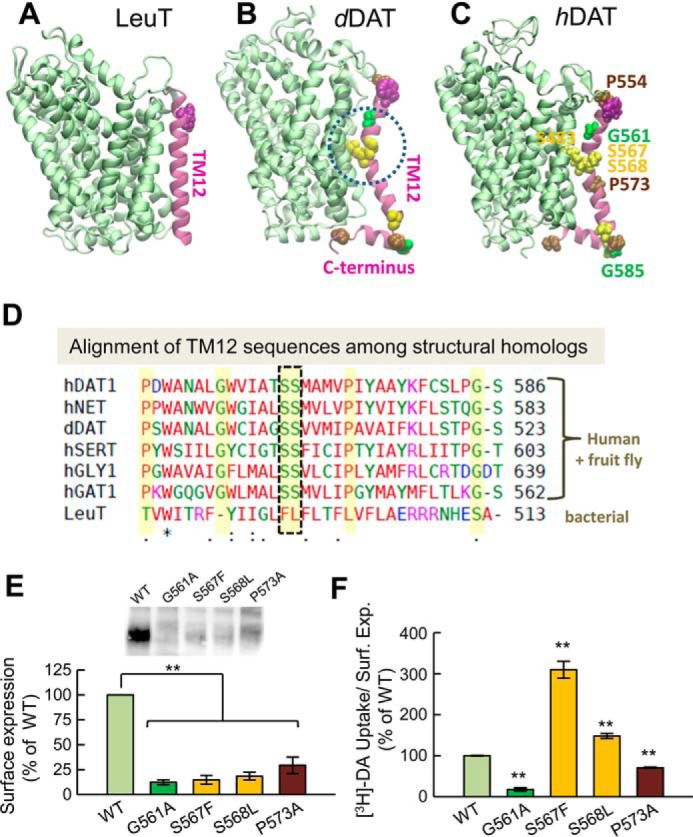
The kink in the TM12 segment of DAT and its functional significance. Structural and sequence differences between LeuT from Aquifex aeolicus (A), dDAT from D. melanogaster (B), and hDAT (human) (C). There is a kink (dashed circle in B) in the TM12 segment of eukaryotic DATs near two serines conserved among the eukaryotic transporters. DAT has an additional C-terminal helix, with a glycine (G585 in hDAT) conserved among human paralogs. Notably, Ser-568 forms hydrogen bonding with Ser-483 in the TM10 segment. D, sequence alignment of TM12 in different members of transporters that share the LeuT fold. The sequences belong to human paralogs hDAT, hNET, hSERT, hGLY1, and hGAT1 and the fruit fly DAT (dDAT) shown in B. The last sequence is that of bacterial LeuT (shown in A). Selected residues conserved among all human paralogs are highlighted in yellow (also labeled in panel C). The kink-forming serines on TM12 are highlighted and enclosed in a dashed box. E and F, effect of mutations at TM12 kink-forming residues on DAT expression and DA uptake. E, surface expression of WT, G561A, S567F, S568L, and P573A hDAT. Transfected HEK293 cells were incubated with sulfo-NHS-SS-biotin and isolated with Neutravidin-agarose beads after cell lysis. For detection of DAT protein an anti-DAT (rat, Millipore MAB369) antibody was used. Densitometry analysis was performed with Image Lab (Bio-Rad) and SigmaPlot 12.5 (Systat Software). Bars represent the percentage of the mean value of the WT group (mean ± S.E.). F, transport activity. Uptake of [3H]DA was performed in HEK293 cells. DA accumulation was normalized with respect to the percentage of cell-surface expression (Surf. Exp.) for each mutant and expressed as the percentage of WT. The statistical analysis was performed with a two-tailed Student's t test versus WT group with an accepted significance level of p < 0.05 (**).
At present, there is no consensus on whether the preferred functional state of NSS members is monomeric or dimeric or even oligomeric. LeuT OFo and OFc crystal structures were resolved as dimers (8–10). Moreover, the Gouaux and co-workers (32) studied LeuT crystal structures in a range of environments and reported that a common feature was the occurrence of a parallel LeuT dimer. Even though dDAT (24, 25) and hSERT (31) have been resolved as monomeric constructs, there is growing evidence in support of the formation of multimeric structures for monoamine transporters such as hDAT or SERT, indicated by radiation inactivation (33), cross-linking (34), mutagenesis (35), co-immunoprecipitation (36, 37), and single molecule analysis (38).
Using fluorescence resonance energy transfer microscopy, Sitte and co-workers (39) provided the first visual evidence in support of the oligomerization of SERT and GAT1 in living cells. The same group subsequently showed the importance of a leucine heptad repeat in GAT1 oligomerization (40). They have also shown that GAT1 functional subunit is the monomer. Oligomerization of NSSs has been suggested to be a determinant of transporter trafficking to the plasma membrane in addition to efficient substrate transport (37, 40, 41). A cooperativity has been observed recently within DAT oligomers (42), and NSS oligomeric constructs have been proposed to be implicated in AMPH action via an oligomer-based counter-transport model (43), as reviewed by Sitte and Freissmuth (5, 44). An early experimental study by Javitch and co-workers (34) found that the dimer interface of hDAT formed intermonomeric cross-links between the Cys-306 residues belonging to the TM6 of the two subunits. Torres et al. (35) proposed that the TM2 segment of hDAT was involved in dimerization. On the other hand, the dimer interface observed in prokaryotic LeuT is mainly formed by TM9 and TM12 (8, 10).
No rigorous computational study of potential dimerization interface has been performed after the elucidation of the monomeric DAT structure. Given the neurobiological significance of the transporter and the availability of structures, we are now in a good position to examine the possible dimerization geometry and/or the most favorable dimerization interface as well as their implication on DAT functional dynamics using a combination of computations and experiments. To this aim, we take advantage of the wealth of conformational data generated in our hDAT monomer simulations (26, 27): mainly, we will explore the dimerization propensities of hDAT in different states in the presence of the membrane remodeling effect.
Strikingly, the most probable hDAT dimer model indicated by the present study differs from the LeuT dimeric structure with regard to its dimerization interface. Cys-306 residues are confirmed to form intermonomer cross-links in the presence of the oxidizing agent copper-phenanthroline (CuP), in support of the computationally predicted dimerization interface, and in consistency with the observations made by Javitch and co-workers (34). Our results indicated that residues in (spatial) proximity of Cys-306 are important for hDAT stabilization and dimer formation. Arg-304 and Glu-307 appear to be able to form intersubunit salt-bridges in our computed model. Substitutions at those sites (changes to alanine, R304A and E307A, or charge inversions, R304E and E307R) resulted in impairment of the formation of the CuP-induced dimer. Moreover, our study highlights the functional significance of the kink in the TM12 segment of hDAT. Substitution of helix-breaking residues (Gly-561, Ser-567, Ser-568, and Pro-573) at this kink by their (helix-favoring) orthologous counterparts in LeuT reduced the surface expression of DAT and altered its DA-reuptake. The importance of the TM12 residues revealed a new region relevant to function and/or expression.
Results
Validation of the computed LeuT dimer structure with the LeuT dimer structurally resolved
To assess the accuracy of ClusPro (45) for predicting the dimeric structure of a member of LeuT fold family, we first examined LeuT in different conformation states. LeuT dimer in the OFo state has been resolved by X-ray crystallography (9). We took one monomer (subunit A) from the OFo LeuT dimer crystal structure (PDB code 3TT1) and then generated structural models for a dimer composed of these monomers using ClusPro. Among the 23 computationally predicted models for LeuT dimer, only one satisfied the four criteria of acceptance (see “Experimental Procedures”). Supplemental Fig. S1A compares this computationally predicted LeuT OFo dimer with that structurally resolved. The root mean square deviation (r.m.s.d.) between the computed structural model (yellow) and experimentally resolved structure (orange) is 1.39 Å, indicating that the model predicted for the LeuT dimer in OFo state yields satisfactory agreement with the structure determined by X-ray crystallography.
The LeuT dimer in the IFo state has not yet been resolved. We generated a structural model for the IFo LeuT dimer using ClusPro, based on the resolved IFo LeuT monomer (PDB code 3TT3) using precisely the same protocol as adopted for OFo dimer (which will also be adopted for hDAT below). Interestingly, among a total of 27 LeuT IFo dimer models, again only one satisfied the four criteria set forth for selecting the optimal model. Supplemental Fig. S1B displays the structural alignment of the computationally predicted LeuT dimers in the OFo (yellow) and IFo (green) states. The r.m.s.d. between these two structural models is ∼6 Å. Similar protein orientation and interfacial contacts were preserved in different states. The interface predominantly involves contacts between the pair of TM12 helices belonging to the two subunits, consistent with experimental data (Fig. 2A).
Figure 2.
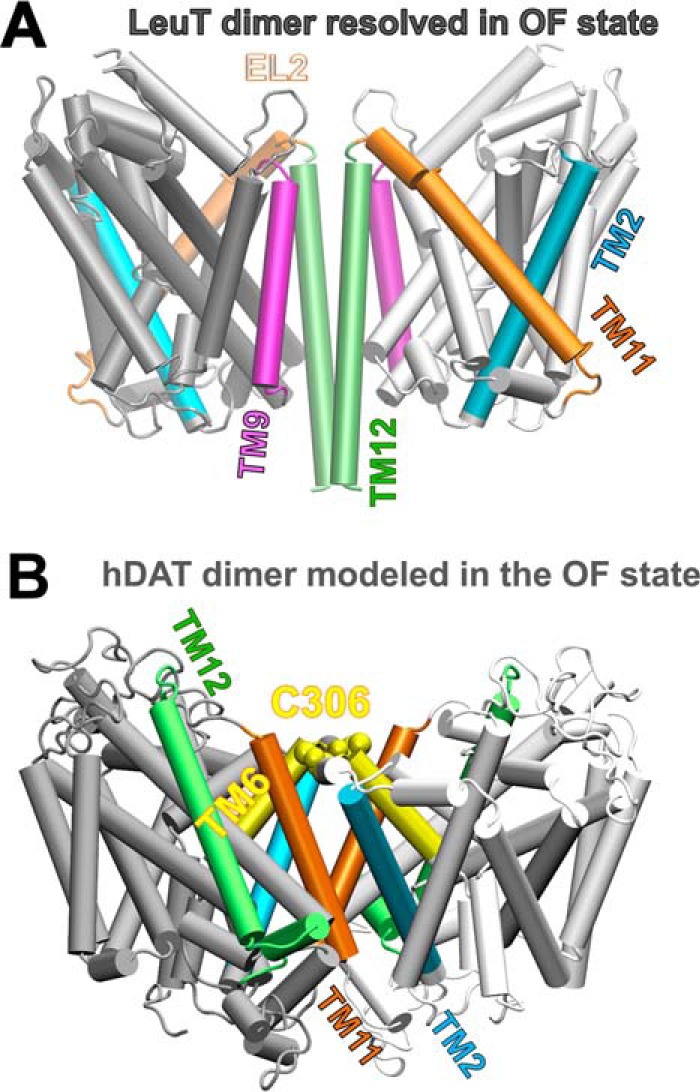
Difference in dimerization architecture between the known LeuT dimer and the computationally predicted hDAT dimer in the outward-facing conformation. A, LeuT dimer has interfacial TM segments TM9 (purple) and TM12 (lime). B, computationally predicted hDAT dimer makes interfacial contacts between TM6 (yellow), TM11 (orange), and TM2 (cyan) helices.
Taken together, ClusPro dimer docking tool is verified here to closely reproduce the dimerization geometry of the OFo monomers with an r.m.s.d. of 1.39 Å with respect to the crystal structure (9). It also helps us build a structural model for the dimer formed by two monomers in the IFo state, which satisfies the constraints inferred from previous experiments yet to be experimentally resolved. We now proceed to the evaluation of dimerization geometry for hDAT.
Discrepancy between experimental LeuT dimer contact and computational prediction for hDAT dimer
The LeuT dimers resolved in the OFo (9) and OFc states (10) make interfacial contacts between their transmembrane helices TM12, complemented by TM9, and a small segment from the EL2 loop (Ile-161 to Gys-163) (see Fig. 2A). In contrast, the same methodology applied to hDAT ended up yielding a significantly different dimerization interface (Fig. 2B). The best structural model computed hDAT dimer showed an association between TM2, TM6, and TM11 portions. Specifically, the predicted hDAT dimer interface includes Val-100–Pro-112 in TM2 and part of TM6a (Phe-302–Trp-311) and Ser-517–Phe-543 in TM11 (see Fig. 2B). Javitch and co-workers (34) show that the hDAT dimer was associated by potential cross-linking of Cys-306 at the TM6 segment. In the hDAT OFo and IFo dimer models predicted by ClusPro, the distance between the Cys-306 residues from the two subunits is within 10.0 ± 3.5 Å. The distance is close enough to allow for the formation of cross-links in the presence of oxidizing agents, facilitated by fluctuations in interresidue distances.
In our hDAT dimer models there is a large hydrophobic patch at the interface (supplemental Fig. S2A). Experimental work by Torres et al. (35) suggested that a leucine zipper (Leu-99, Met-106, Leu-113, and Leu-120) in TM2 of hDAT might contribute to the dimerization of hDAT. In our computed dimer model, the suggested leucine zipper was not in direct contact (supplemental Fig. S2B). Instead, residues Val-100, Val-107, and Pro-112 contributed to hydrophobic interfacial interactions. This shift by one residue notwithstanding, both computational modeling and experimental study suggest that the TM2 segment may contribute to stabilize the dimer interface (see Fig. 2B).
Significance of the kink in the TM12 segment of DAT
In summary, on the one hand, the structural model-predicted hDAT dimer showed good accord with cross-linking (34) and mutagenesis studies on hDAT (35). On the other, the predicted intersubunit interface in hDAT differed from that seen in the prokaryotic LeuT dimer crystal structures (see Fig. 2). This difference could be attributed to the sharp kink in the TM12 segment (Fig. 1), which prevents the registration of the two TM12 helices at the interface. Notably, the same kink has been observed in the crystal structures of dDAT (24, 25) and hSERT (31). Furthermore, the eukaryotic NSSs and bacterial LeuT show a significant structural difference at the C-terminal segment (Fig. 1). Eukaryotic transporters have a large C-terminal domain, which may further hinder the efficient packing of these regions against each other in the dimeric state.
To investigate how the kink in TM12 affects hDAT function, we generated a series of DAT mutants, expressed them into HEK293 cells, and measured their cell-surface expression and DA-uptake capacity. Substitution of the kink forming residues Gly-561, Ser-567, and Ser-568 by their orthologous counterparts in LeuT (i.e. G561A, S567F, and S568L) and the helix-breaking Pro-573 by alanine (P573A) resulted in a significant reduction in surface expression levels of the mutants compared with the wild type (WT) transporter (Fig. 1E). Because the protein expression for all mutants was different, we normalized the specific [3H]DA uptake properties of the mutants with respect to their surface expression so as to allow for the comparison of the DA transport activities of the mutants to that of WT-hDAT (Table 1). Fig. 1F shows that the transport activities of G561A and P573A were significantly reduced. In contrast, the mutants S567F and S568L, which displayed similar levels of expression, showed an increased DA transport activity compared with WT-hDAT (Fig. 1F).
Table 1.
Effects of hDAT mutations on transport activity and cell-surface expression
Transport activity and cell-surface biotinylation experiments were performed in HEK293 cells transfected with hDAT WT and mutants (see “Experimental Procedures”). Values for uptake and surface expression are expressed as the mean ± S.E. and calculated as the percentage of the mean value for the WT group in every independent experiment (n). Statistical values were obtained with Student's t tests.
| Construct | [3H]DA uptake | Surface expression | [3H]DA uptake/surface expression |
|---|---|---|---|
| % of WT | % of WT | ||
| WT-hDAT | 100.0 ± 0.8 (n = 8) | 100 | 100 |
| G561A | 2.2 ± 0.5 (n = 6) | 12.3 ± 2.5 (n = 3) | 17.9 ± 4.1a |
| S567F | 45.6 ± 3.0 (n = 6) | 14.7 ± 4.4 (n = 3) | 309.7 ± 20.6a |
| S568L | 27.4 ± 1.2 (n = 10) | 18.5 ± 4.0 (n = 3) | 148.2 ± 6.3a |
| P573A | 20.8 ± 0.6 (n = 6) | 29.4 ± 8.2 (n = 3) | 70.7 ± 2.0a |
| C306A | 122.8 ± 1.9 (n = 6) | 127.2 ± 15.1 (n = 4) | 96.5 ± 1.5b. |
| C523A | 90.4 ± 2.5 (n = 10) | 49.3 ± 5.6 (n = 6) | 183.3 ± 5.2c |
| R304A | 80.1 ± 1.9 (n = 6) | 22.3 ± 1.5 (n = 4) | 359.3 ± 8.3a |
| E307A | 82.7 ± 1.6 (n = 6) | 31.1 ± 4.3 (n = 4) | 265.5 ± 5.0a |
| R304E | 31.2 ± 1.3 (n = 6) | 10.0 ± 2.8 (n = 6) | 313.7 ± 12.7a |
| E307R | 70.1 ± 1.3 (n = 6) | 44.2 ± 9.2 (n = 5) | 158.6 ± 2.9c |
a p < 0.001.
b Not significant.
c p < 0.05.
Structural transitions of protomers in hDAT dimer resembled those observed in the monomer alone
To further investigate the structural stability and dynamics of the computationally predicted hDAT dimer in the OFo and IFo state, we performed four 100-ns MD simulations of hDAT dimers embedded in solvated POPC lipids. A typical MD simulation system constructed for simulating the dynamics of hDAT dimer is shown in Fig. 3A. In all four runs (two performed for each dimeric structure), the r.m.s.d. of the dimer reached an asymptotic value of 3.0 ± 0.4 Å after 10-ns simulations and fluctuated near this value in the remaining 90 ns, in support of the stability of the structural models embedded in the lipid bilayer. Fig. 3B illustrates the alignment of 100-ns MD equilibrated hDAT dimer in the OFo and IFo states. The corresponding r.m.s.d. is ∼3.5 Å. Notably, the two dimers show different tiltings at the EC-exposed and IC-exposed TM helices, e.g. TM1a (IC-exposed half of the broken helix TM1) is wide open in the IF state, whereas the helices near the EC vestibule are relatively more closed than their OF counterparts. Overall, the opening/closure of the EC and IC vestibule in the dimer (Fig. 3C) closely resembles those previously observed in the monomer dynamics (27).
Figure 3.
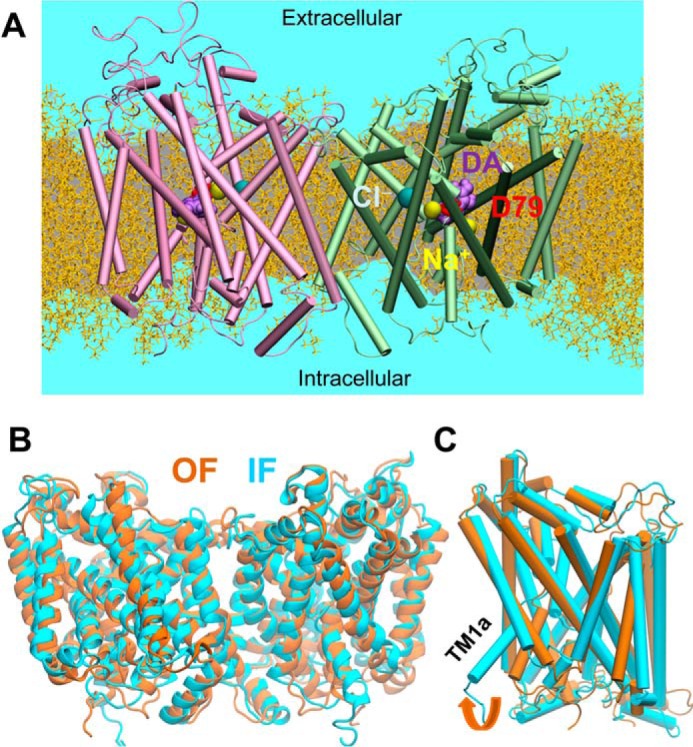
MD simulations of hDAT dimer dynamics, embedded into membrane lipids. A, typical MD set-up for hDAT dimer (colored in pink and green) in a lipid bilayer (yellow licorice format (55)) and solvated by 0.15 m NaCl (light blue). The bound substrate DA and the co-transported Na+ and Cl− are represented by purple, yellow, and cyan spheres. B, alignment of 100-ns MD equilibrated hDAT dimer in the outward-facing (OF; orange ribbon diagram) and inward-facing (IF; cyan) state. The r.m.s.d. between the OF and IF dimers levels off at 3.4 ± 0.3 Å. C, comparison of helical orientations in the OF and IF states. For clarity, one monomer (in schematic format) taken from the dimer is shown for each state.
For a quantitative analysis of the structural characteristics of the OF and IF dimers, we further examined the time evolution of the relative positions of selected pairs of helices, which provide a metric for probing the degree of exposure of the EC and IC vestibules. In line with previous work (26, 27), two pairs of helical segments were used as probes: TM1b and TM10 for exposure to the EC medium, and TM1a and TM6b for exposure to the IC medium. We performed four runs of 100 ns each (duplicate for each dimer, designated OF-1, OF-2, IF-1, and IF-2). Fig. 4 shows that the time evolution of the distance between these pairs of helices in the OF (panels A and B) and IF (panels C and D) dimers.
Figure 4.

Time evolution of the interhelical distances. The opening/closure of the EC and IC vestibules is characterized by the respective interhelical distances of the EC-exposed TM1b-TM10 and the IC-exposed TM1a-TM6b, shown for both subunits, as labeled (see Refs. 26 and 27). Results from four runs are presented: A, OF-1, in which the closure of the EC vestibule led to the OFc state in both subunits. B, OF-2, in which both subunits remained in the OFo state, as indicated by the separation between the EC-exposed TM1b and TM10 segments (light and dark blue) and the close proximity of the IC-exposed helical segments TM1a-TM6b (light and dark red). C, IF-1, in which the IC vestibule remained open in both subunits (see the red curves). D, IF-2, in which the initiation of a transition to the OF conformation could be observed, but the IC vestibule remained open in both subunits. The characteristic distances for TM1b-TM10 obtained in the hDAT dimer simulations are 20.0 ± 2.0 Å and 16.5 ± 0.5 Å in the OFo and OFc states, respectively. These values closely approximate those reported for hDAT monomer simulations (27). Distances are indicated by dotted and dashed lines in panels A and B. In the OF state the distance between the IC-exposed segments TM1a and TM6b remained 11.5 ± 1.0 Å, the same as that reported in the monomer simulations (27). In the IFo state, the IC vestibule is open for DA release when the distance between TM1a and TM6b is >15 Å in hDAT monomer (see Cheng and Bahar (27)).
The OF dimer in run OF-1 underwent a local structural transition from conformation OFo to OFc as can be seen from the time evolution of the TM1b-TM10 distance (light and dark blue curves, for respective subunits A and B) in Fig. 4A. Closer examination showed that the EC gates Phe-320–Tyr-156 and Arg-85–Asp-476 closed almost simultaneously within 20 ns in both subunits, leading to the OFc state (Fig. 5). Notably, the closure of EC gates was similarly observed in simulations of hDAT monomer (26, 27). In run OF-2, the subunits remained open (Fig. 4B); the TM1b-TM10 distance of 20 Å, typical of the OFo conformer, was maintained during the course of simulations, with fluctuations within the range ascribed (26, 27) to the OFo state. In both runs OF-1 and -2, the IC-exposed helical pairs TM1a and TM6b (dark/light red) remained tightly packed (Fig. 4, A and B), again consistent with the closed conformation of the IC gate in the OF state of the transporters.
Figure 5.
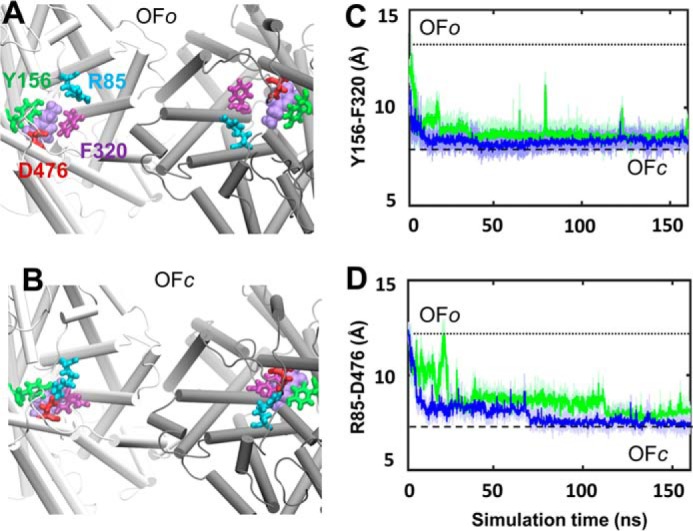
Structural transition observed from the OFo to OFc in the hDAT dimer. Top view of A, the initial OFo state with the opening of two EC gates: Arg-85–Asp-476 and Tyr-156–Phe-320. B, closure of the EC gates in the OFc state; a snapshot around 160 ns was used. Shown is time evolution of the center of the mass distances between the two EC gating residue pairs (C) aromatic residues Tyr-156 and Phe-320 and (D) salt-bridge-forming residue pairs Arg-85 and Asp-476. Note that the closure of the gates occurred almost simultaneously in the two subunits (colored in green and blue).
The relative distances of the two pairs of helices were inverted in the IF dimers (Fig. 4, C and D), consistent with the opening of the IC gate and closure of the EC gate in the IF state. In the run IF-1, the IC vestibule of subunit A remained open, whereas that of subunit B gradually closed, as can be seen from the time evolution of TM1a-TM6b helices. In run IF-2 (Fig. 4D), both helix pairs showed the same behavior, i.e. a transition toward the inward-facing closed (IFc) state. In three of four cases, the subunits in IFo dimer exhibited a natural tendency to rearrange themselves in favor of a potential transition into the OF state, a transition that is functionally required for completing the transport cycle.
Overall these data demonstrate that the OF and IF dimeric forms have distinctive interhelical packing characteristics (in Fig. 4, compare panels A and B for OF dimers to panels C and D for IF dimers), whereas the two subunits within the individual dimers exhibit similar dynamic patterns (compare the dark- and light-colored curves in each case). The equilibrium values observed for those interhelical distances and their fluctuation ranges were again typical of those sampled by the OF or IF monomers in isolation. Interestingly, the two subunits in a given dimer exhibited a tendency to undergo similar conformational transitions in most runs, i.e. both remained in the OFo state or both transitioned to OFc state in the respective runs OF-2 and OF-1, and both subunits exhibited a tendency to progress toward OF state in IF-2 (Fig. 4).
Oxidative cross-linking experiments showed that TM6 residues are important for hDAT stabilization and dimer formation
In the initial generation of the dimeric structure and the construction of the MD environment, we did not impose disulfide bond formation between Cys-306 residues from the neighboring subunits. During MD simulations, the average distance between the two Cys-306 decreased to 7.5 ± 2.5 Å and 8.0 ± 1.5 Å in the respective OF and IF dimers, in favor of a possible disulfide bridge formation. In addition to Cys-306, Cys-523 residues from the interfacial TM11 segments (Fig. 6A) came into proximity (11.0 ± 1.5 Å). Although a Cys at position 306 is unique to hDAT, Cys-523 is fully conserved among the eukaryotic NSSs and could not be (solely) responsible for the distinct dimerization behavior of LeuT and hDAT (Fig. 2). We also noted in the computed model a number of charged amino acids that are likely to make interfacial contacts and even form salt bridges in the IF conformation of the dimer, e.g. Glu-307(I)-Arg-304(II) and/or Glu-307(II)-Arg-304(I) (Fig. 6B). Acidic and basic residues at the respective sequence positions 307 and 304 are highly conserved in the eukaryotic NSSs (Fig. 6C). We hypothesized that mutations at these residues might affect the intersubunit disulfide cross-linking at Cys-306 and/or overall hDAT function. We introduced alanine substitutions at Arg-304 and Glu-307 and at the two native cysteines Cys-306 and Cys-523. Like the mutations in TM12, the levels of expression were different for all mutants (Table 1) so we normalized the [3H]DA uptake with respect to surface expression so as to enable a comparative analysis across the mutants and with WT-hDAT. C306A mutant showed DA-uptake capacity comparable with that of WT hDAT (Table 1). Unexpectedly the mutants C523A, R304A, and E307A displayed increased transport activity compared with WT hDAT.
Figure 6.

Interfacial interactions. A, two interfacial cysteines between subunits I and II (colored pink and green) are likely to form cross-links between the dimers: Cys-306(I)- Cys-306(II) (superposed, not seen separately in the panel) and Cys-523(I)–Cys-523(II). They are within close distances (<11 Å) in both the OF and IF conformers. B, an interfacial salt bridge is likely to form between Glu-307 and Arg-304 in the IF conformation of the dimer. C, sequence alignment of the putative interfacial regions.
To explore whether these residues participate in the stabilization of the hDAT dimer, we examined the propensity of Cys-306 in hDAT to dimerize with the neighboring subunit in the presence of CuP in intact HEK293 cells. In WT hDAT-expressing cells, CuP treatment resulted in the appearance of a distinct band at 150 kDa in addition to immunoreactive bands at 50, 75, and 100 kDa (Fig. 7, A, B, and E), which represent the immature and mature hDAT monomeric forms (50 and 75 kDa) and the immature dimer (∼100 kDa) (34, 46, 47). It has been shown previously that the alanine substitution C306A completely abolishes the formation of DAT dimers by CuP (34). As expected, C306A did not produce a cross-linked species after CuP treatment (Fig. 7, A, B, and E), confirming that Cys-306 is the reactive residue at the CuP-induced dimer interface. CuP treatment of cells expressing C523A, R304A, and E307A resulted in the formation of the 150-kDa DAT adduct corresponding to CuP-induced dimer (Fig. 7, A and B).
Figure 7.

Oxidative cross-linking of DAT Cys-306 in TM6 hDAT mutants. A, representative blot of CuP cross-linking in intact cells WT and DAT mutants R304A, E307A, C306A, and C523A. Cross-linking reaction with 0.1 μm CuSO4 and 0.4 μm phenanthroline was performed for 5 min at room temperature. Detection of DAT protein was detected by Western blot using an anti-DAT (rat, Millipore MAB369) antibody. Different immunodetected DAT bands were detected in CuP non-treated (−) and treated lanes (+). Different transporter forms are indicated as follows: immature monomer (yellow circle), mature monomer (red rhombus), immature dimer (yellow rhombus), mature dimer (double red rhombus). B, quantification of the CuP-induced 150-kDa DAT abduct. Bars represent the intensity of the band as % of the total bands per lane. Densitometry analysis was performed with ImageLab (Bio-Rad) and SigmaPlot 12.5 (Systat Software). C and D, CuP cross-linking of WT, R304E, and E307R. E, the intensity lane profile (obtained with Image Lab Software) shows the relative position of the different DAT-forms for WT, C306A, and R304E. The shadowed peaks represent the mature forms (monomeric 75 kDa) and CuP-induced dimer (150 kDa). F, quantification of the CuP-induced dimers relative to the 150-kDa DAT band in the control group (CuP(−)) for each DAT construct. Bars represent the mean values (mean ± S.E.). *, p < 0.05; **, p < 0.01 for Student's t test; p value versus control group. # represents p < 0.05 analysis of variance one way; n.s., not significant.
We also generated two additional hDAT mutants, R304E and E307R, to evaluate the effect of the proposed salt bridges on hDAT function and oligomerization. The E307R substitution increased transport activity (Table 1) and retained modest ability to form the CuP-induced dimers (Fig. 7, C and D). The most dramatic effect on dimer formation was observed with the R304E mutation. In cells expressing R304E, treatment with CuP resulted in no formation of the 150-kDa DAT abduct (Fig. 7, D and E). Finally, we estimated the relative amount of CuP-induced dimer in comparison with the amount of dimer present in the control group (no CuP) for each mutant (Fig. 7F). In this analysis we observed that WT and C523A hDAT produced comparable amounts of CuP-induced DAT dimer. Moreover, other mutations in Arg-304 (R304A) and Glu-307 (E307A and E307R) resulted in a reduction of CuP-induced dimer compared with the dimer formed in the WT, and the mutation R304E completely abolished the formation of 150-kDa DAT abduct, in support of the idea that these nearby residues are important for maintaining the protein in a conformation that permits cross-linking and likely favors dimerization.
Discussion
In the present study we constructed hDAT dimers composed of subunits in the OF and IF states using structural data for monomers generated in our recent MD simulations of hDAT (27). Although the LeuT dimer displays interfacial contacts at the TM9 and TM12 segments (Fig. 2A) (indicated by experiments and reproduced by the presently adopted computational protocol), subunits in the computed hDAT dimer are preferentially associated through portions of their TM2, TM6, and TM11 helices (Fig. 2B). Among these three helices, TM6 participates in the substrate/Na+/Cl− binding pocket, and TM2 participates in chloride binding. Closer examination showed that the TM6 portions that make interfacial contacts (near Cys-306, EC exposed end) differ from those involved in coordinating the substrate and ions (almost halfway into the lipid bilayer) such that the substrate/Na+/Cl−-binding site is not affected by dimerization. Likewise, comparison of isolated monomer chloride-binding site and that in the monomer belonging to the dimer shows that the chloride coordination geometry is maintained in the dimer. There is a conserved tyrosine (Tyr-102 in hDAT) that interacts with the chloride ion, whose site-chain reorientation (away from the interface) remains unchanged. Overall, the dimerization interface does not have any direct effect on the local interactions in the substrate/Na+/Cl−-binding site, and the mechanism of opening/closure of the EC and IC vestibules in the dimeric form of DAT (Fig. 3C) closely resembles that observed (27) in its monomeric form.
A central kink was observed to prevent the parallel alignment of the pair of TM12 helices in hDAT (Fig. 1, B and C). This irregularity of TM12 at a sequence region composed of two consecutive serines is not unique to hDAT. On the contrary, this is a common feature among eukaryotic neurotransmitter transporters that share the LeuT fold, which distinguish them from the bacterial transporter LeuT (Fig. 1D). Although current experiments do not provide direct evidence on the position of TM12 with respect to the dimer interface, they reveal the functional significance of a series of residues at the TM12 kink site. Substitutions by their LeuT counterparts (G561A, S567F, S568L, or P573A) led to a decrease in surface expression levels (∼70% to ∼90% reduction). Their low levels of expression prevented us from pursuing the oxidative cross-linking experiments. We normalized the observed DA uptakes by the levels of surface expression to analyze the intrinsic uptake capacity of each mutant. In this analysis, we observed that two TM12 mutations, G561A and P573A, resulted in a significant loss of uptake activity (∼80 and ∼30%, respectively) (Fig. 1, E and F; Table 1). In contrast, the two others (S567F and S568L) showed an increase (∼200 and ∼50%, respectively) in the apparent uptake capacity when compared with WT DAT. These results revealed that these residues might play a role in protein maturation, folding, or structural dynamics and thereby on transport activity. The observed differences between their effects reflect that each mutation confers specific changes to the carrier. We conclude that G561A and P573A result in non-functional carriers, whereas S567F and S568L cause an impairment in DAT trafficking. Yet the small number of S567F and S568L mutants that actually reach the cell surface are fully functional. Additionally, the computational analysis shows that Ser-568 forms a hydrogen bond with Ser-483 from TM10 helix (Fig. 1C). TM10 has been shown in our recent study to undergo a significant reconfiguration during the transport (27). It remains to be tested, for example, with the help of the mutant S483G (LeuT counterpart), if such hydrogen-bond formation contributes to gain of transport activity. It is striking to see that these TM12 residues that partly form the scaffolding domain of hDAT and are not in direct contact with the DA-translocation pathway resulted in non-functional carriers in G561A and P573A.
The present study shows that in the presence of an oxidizing agent that promotes disulfide-bridge formation, hDAT exhibits a tendency to increase dimerization due to cross-linking of Cys-306(I)–Cys-306(II) (Fig. 7). This observation lends strong support to the dimerization interface different from that of LeuT, predicted by our computational model for hDAT dimer. Other residues proposed here to play key roles are the charged residues (Glu-307 and Arg-304) (Figs. 6 and 7) that potentially form an interfacial salt bridge.
For each state we performed two sets of 100-ns MD simulations of hDAT dimer embedded into solvated membrane (Fig. 3A) to examine the stability of the structural models and the local changes in structure that may be functionally relevant. In all runs, the r.m.s.d. were stabilized at 3.0 ± 0.5 Å with respect to the initial models within the first 10 ns and remained unchanged in the remaining 90-ns portion of the trajectories. r.m.s.d. between MD-equilibrated hDAT OFo and IFo dimers (Fig. 3B) was ∼3.5 ± 0.8 Å. The hDAT OFo and IFo dimer exhibited distinctive characteristics in their EC-exposed and IC-exposed inter-helical distances, with values similar to those observed in the respective monomeric OFo and IFo states (27). Notably, the closure of EC gates (Arg-85–Asp-476 and Tyr-156–Phe-320) upon binding of Na+, Cl−, and DA was clearly seen in the dimer (Figs. 4 and 5), similar to that observed in the hDAT monomer (26, 27).
Interestingly, the two subunits in a given dimer generally exhibited coupled transitions during MD runs (Figs. 4 and 5). Although it is hard to generalize based on four runs, the fact that coupled behavior was observed between subunits in several independent runs suggests that there may be a correlation between the dynamics of the two subunits. The majority of earlier experimental studies did not directly indicate cooperativity among protomers. Kilic and Rudnick (36) provided the first experimental evidence showing a dimeric form of SERT with functional interactions between subunits. Furthermore, cooperativity between the NSS monomers has been observed in recent experiments on DAT (42). Our recent computational study (48) using the anisotropic network model (49) suggests that dimerization of LeuT or hDAT alters the collective motions intrinsically accessible to the individual monomers in favor of the functional transitions (OF ↔ IF). Whether the subunits in the dimer tend to undergo coupled changes is worth further investigating. It is not clear whether this coupling is due to the adoption of the same conformational state for both subunits as initial configuration; the initial conformers relaxed within the first 20 ns. Mixed conformations (containing one subunit in the IF state and two others in the OF or one in an intermediate OF) have been observed for another family of transporters (50, 51), and it remains to be seen if the LeuT fold also favors such mixed conformations. In the future it might be of interest to explore the possibility of mixed (IF and OF) dimeric structures and examine their relative stability (or interfacial surface area) and dynamics. The present study does not preclude the possible occurrence of higher oligomerization states or a distribution of multimers in different oligomerization states. Likewise, there may be alternative dimerization states with different probabilities. Other neurotransmitter transporters have been observed for form dimers or trimers while maintaining the monomeric fold, and oligomerization properties of hDAT and their functional implications may need further analyses. Finally, although a symmetric arrangement was used as a criterion for generating possible dimerization geometries, the final models exhibited only pseudosymmetric arrangements.
The function of NSSs is mediated by regulatory proteins (i.e. CaMKII), additive drugs (i.e. AMPH), and anionic lipids (i.e. PIP2) (see Refs. 3 and 5). Interestingly, anionic lipid palmitoyl-oleoylphosphatidylglycerol was found to bind at the interface of trimeric NSS ortholog betaine transporter (BetP) (52). We hypothesize that anionic lipids (i.e. PIP2) may potentially bind at the interface of hDAT dimer, as resolved in its NSS ortholog BetP (52) or reported to mediate stable oligomer constructs in hSERT (53); in addition, regulatory proteins or additive drugs may bind at the interface of hDAT dimer or higher-order constructs for cooperative modulation. Our study provides insights into methods for exploring the oligomerization of NSSs and shedding light on the functional cooperativity and drug modulation of NSSs. In the future it would be of interest to explore the potential binding of regulatory proteins or anionic lipids PIP2 to NSS oligomer and understand the cooperativity between the NSS monomers using combined experimental and computational studies.
Experimental procedures
Computational
Construction of hDAT dimer
We generated in silico hDAT monomer conformations at different stages of the transport cycle (27). We focused on the conformers in the OFo and IFo states to construct structural models for hDAT dimer. For each state three representative conformers were selected that were used for generating dimers using the dimer docking module in ClusPro (45). For the OFo state, we chose the conformers stabilized after 2–10-ns MD equilibration in the presence of DA, two Na+ and a Cl−, before proceeding to the OFc state; for the IFo state, hDAT IF conformers sampled after releasing DA and ions to the IC region were selected that were further subjected to 2-μs MD simulation using the Anton special-purpose computer (54).
For each simulation performed with a different monomer conformer, up to 30 dimeric models were predicted by ClusPro. These were rank-ordered based on the docking score from ClusPro together with four criteria specific to NSS membrane proteins: 1) symmetric or pseudo-symmetric arrangement of the monomers; 2) exposure of the N and C termini to the IC region; 3) proper orientation of the aromatic residues (i.e. Trp) on the EC and IC sides to enable suitable anchoring the protein to the membrane; 4) maintenance of similar interfacial contacts between the dimer formed by the OF monomers and that formed by IF monomers. Although up to 90 structural models (3 runs with ∼30 models) were generated for each state, these requirements were met by 5–6 models for each state. The r.m.s.d. between any pair of dimeric conformers selected by this screening procedure was observed smaller than 2.0 Å for OFo dimers and 4.5 Å for IFo dimers. A representative conformer (which has the lowest r.m.s.d. with respect to all others in each ensemble) was selected for each (OFo and IFo) state. More details on the methodology are presented in a recent study (48).
For testing our protocol, we reconstructed in silico the LeuT dimer in the OFo state (Fig. 2A) using the structural data on one monomer (subunit A) from the dimeric structure resolved for LeuT (PDB code 3TT1) (8, 10). The structural model computationally predicted to be the most favorable dimerization state was verified to closely agree with that resolved by X-ray, lending support to the applicability of our protocol for constructing dimeric models.
Construction of MD simulation environment for hDAT dimers
The systems were set up using VMD (55). For the OFo hDAT dimer, a DA, two Na+ ions, and one chloride ion were bound using coordinates from previous work (27). No substrate/ions were included in the IFo dimer. The TM portion of the hDAT dimer was then inserted into the center of a pre-equilibrated 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer following a previous protocol (27). Fully equilibrated TIP3 waters were added to form a rectangular simulation box of 140 Å × 140 Å × 100 Å3. Na+ and Cl− ions were further added to obtain a 0.15 m neutral solution. Overall, each simulation system contained ∼1.8 × 105 atoms, including the hDAT dimer, 400 lipids, and 35,000 water molecules.
MD simulations and trajectory analysis
All simulations were performed using NAMD2 (56). Simulation parameters and methods were adopted from our simulations of hDAT monomer (27). Briefly, we used the CHARMM force field with CMAP corrections (57, 58) for hDAT, water, and lipid molecules; the parameters for the substrate DA were evaluated following the protocol in CHARMM General Force Field (CGenFF) for drug-like molecules (59). Before productive runs, each system was first energy-minimized for 50,000 steps followed by 0.5-ns constant volume and temperature (310 K) (NVT) simulations and a subsequent 4-ns Nosé-Hoover constant pressure (1 bar) and temperature (310 K) (NPT) simulation, during which the protein was fixed and constraints on the POPC head groups were gradually released. Subsequently, the constraints on the protein backbone were reduced from 10 kcal/mol to 0 within 3 ns. Finally, the unconstrained protein was subjected to NPT simulations.
For each dimer, two independent MD runs of 100 ns were performed designated as OF-1 and OF-2 and as IF-1 and IF-2. VMD (55) with in-house scripts was used for visualization and analysis of MD trajectories. For calculating the r.m.s.d., hDAT residues Arg-58–Glu-598 from both subunits were used, except for the structurally unresolved EL2 loop segment (Ser-190–Pro-212). The opening of the EC gates was assessed by the changes in center of mass distances between the residues Tyr-156 and Phe-320 that serve as the inner gate and Arg-85 and Asp-476 that serve as the outer gate. Calculations of interhelical distances were based on residues Lys-66–Val-78 (TM1a), Leu-80–Gln-93 (TM1b), Ala-308–Leu-322 (TM6a), Phe-326–Tyr-335 (TM6b), and Gly-467–Gly-481 (EC-exposed TM10 segment).
Experimental
cDNA constructs, mutagenesis, and heterologous expression
Single point mutations were introduced into a cDNA construct of hDAT inserted in the vector pcDNA3.1(+) (Invitrogen) using the site-directed mutagenesis kit Q5® (New England BioLabs). To generate primers the NEBaseChanger web-based tool was used. All constructs were verified by sequencing. HEK293 cells were obtained from the American Type Culture Collection. HEK293 cells were cultured in MEM supplemented with 10% fetal bovine serum (FBS) and 50 μg/ml each penicillin and streptomycin at 37 °C in a humidified, 5% CO2 incubator. Cells were transfected with the corresponding cDNAs using Lipofectamine2000 (Life Science) and following the manufacturer's instructions.
Uptake assay
HEK293 cells were seeded (1–1.5 × 105 cells per well) in 24-well plates precoated with poly-d-lysine. The uptake assay was carried out 48 h after the transfection. Before the experiment, cells were washed once with uptake buffer containing 5 mm Tris base, 7.5 mm HEPES, 120 mm NaCl, 5.4 mm KCl, 1.2 mm CaCl2, 1.2 mm MgSO4, 0.1 mm ascorbic acid, 0.1 mm pargyline, 1 mm tropolone, and 5 mm glucose at pH 7.4. The uptake experiment was performed at 37 °C. The accumulation of radiolabeled dopamine (3,4-[7-3H]dihydroxyphenylethylamine, l46.0 Ci/mmol; PerkinElmer Life Sciences) started with the addition of labeled (0.02 μm) and non-labeled dopamine (10.0 μm) in a final volume of 0.4 ml. After 5 min, cells were washed with ice-cold NaCl-free uptake buffer (LiCl-based) to stop uptake activity and lysed with 0.4 ml of 1% SDS. The incorporated [3H]DA was measured by liquid scintillation counting with a LSC6000 counter (Beckman Coulter). Nonspecific uptake was determined with 0.01 mm GBR12935. Uptake data were analyzed with SigmaPlot 12.5 (Systat Software, Inc). All data are represented as the percentage of the mean value with respect to the WT group (mean ± S.E.). Every experimental point had a minimum of six replicates per plate. The statistical analysis was performed with two-tailed Student's t test with an accepted significance level at p < 0.05.
Surface biotinylation
HEK293 were seeded and transfected in a poly-d-Lysine 6-well plate. Forty-eight hours later, transfected cells were washed with PBS and incubated with gentle agitation for 30 min at 4 °C with 1 ml of 1.5 mg/ml sulfo-NHS-SS-biotin (Thermo Scientific) prepared in biotinylation buffer (150 mm NaCl, 2 mm CaCl2, 10 mm triethanolamine, pH 7.8). The reaction was quenched by incubating the cells for an additional 10 min with 50 mm glycine in PBS. Cells were then washed with PBS and incubated in radioimmune precipitation assay buffer (10 mm Tris, 150 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100, and 1% sodium deoxycholate, pH 7.4) at 4 °C for 1 h. Protein concentration was immediately quantified by BCA protein assay (Thermo Scientific). Equal amounts of protein were incubated overnight with Neutravidin beads (Thermo Scientific). Then the beads were washed 3 times with cold radioimmune precipitation assay buffer, and biotinylated membrane proteins were eluted in 50 μl of 2× Laemmli sample buffer (Bio-Rad). Samples were separated by SDS-PAGE (10% TGX minigels, Bio-Rad) and transferred to PVDF membranes using Trans-blot Turbo (Bio-Rad). For Western blot analysis the anti-DAT antibody (MAB369, Millipore) and an HRP-conjugated secondary antibody (Jackson ImmunoResearch Laboratories) were used. Densitometry analysis of bands was performed with Image Lab Software (Bio-Rad) and SigmaPlot 12.5. All data are represented as the percentage of the mean value of the WT group (mean ± S.E.). The statistical analysis was performed with two-tailed Student's t test with an accepted significance level at p < 0.05.
Oxidative cross-linking
Experiments were performed 48 h after transfection of adhered HEK293 cells in 6-well plates. Cells were washed twice with PBS followed by the addition of 0.1 mm CuSO4 (Sigma) and 0.4 mm o-phenanthroline (Sigma) in PBS. The cross-linking reaction was carried out for 5 min at room temperature. The reaction was stopped by removal of the reagent and incubation for 20 min with 10 mm N-ethylmaleimide to block free sulfhydryl groups. After quenching, we proceeded to the cell lysis and membrane-solubilization following the protocol detailed in Hastrup et al. (34). After the extraction, 50 μl of sample were mixed with 50 μl of 4×-Laemmli loading buffer (Bio-Rad) without reducing agent and held at room temperature for 30 min before proceeding to the analysis by Western blotting. Samples were separated by SDS-PAGE (Criterion 7.5% TGX, Bio-Rad) and transferred to PVDF membranes using Trans-blot Turbo (Bio-Rad). For immunodetection, the anti-DAT antibody MAB369 (rat, Millipore) and an HRP-conjugated secondary antibody (Jackson ImmunoResearch Laboratories) were used. Densitometry analysis of bands was performed with Image Lab Software and SigmaPlot 12.5 (Systat Software). All data are reported as the percentage of the mean value with respect to the WT group (mean ± S.E.). The statistical analysis was performed with a two-tailed Student's t test and an accepted significance level at p < 0.05.
Author contributions
I. B., M. H. C., and J. G.-O. designed the project. M. H. C. performed the modeling and simulations. J. G.-O., S. W., and J. D. performed the experiments. M. H. C., J. G.-O., and I. B. analyzed the data and wrote the manuscript.
Supplementary Material
Acknowledgments
We gratefully acknowledge several useful discussions with Drs. Susan G. Amara and Delany Torres Salazar.
* This work was supported by National Institutes of Health Grants R01-GM099738, P30DA035778, and P41GM103712 (to I. B.) and ZIA MH002946-04 (to S. G. Amara and J. G.-O.) and by the Anton machine (P41GM103712). This work was also supported by a computing award from the NSF TeraGrid (TG-MCB130006). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1 and S2.
- DA
- dopamine
- DAT
- dopamine transporter
- h-
- human
- hDAT
- human dopamine transporter
- NSS
- neurotransmitter: sodium symporter
- EC
- extracellular
- IC
- intracellular
- TM
- transmembrane
- OFo
- outward-facing open
- OFc
- outward-facing closed
- IFo
- inward-facing open
- SERT
- serotonin transporter
- BetP
- betamine transporter
- LeuT
- leucine transporter
- r.m.s.d.
- root-mean-squared deviation(s)
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- NET
- norepinephrine transporter
- GAT1
- GABA transporter 1
- GLY1
- glycine transporter 1
- CuP
- copper-phenanthroline
- PIP2
- phosphatidylinositol 4,5-bisphosphate.
References
- 1. Amara S. G., and Sonders M. S. (1998) Neurotransmitter transporters as molecular targets for addictive drugs. Drug Alcohol Depend. 51, 87–96 [DOI] [PubMed] [Google Scholar]
- 2. Reith M. (2002) Neurotransmitter transporters: Structure, Function, and Regulations. Humana, Totowa, NJ [Google Scholar]
- 3. Vaughan R. A., and Foster J. D. (2013) Mechanisms of dopamine transporter regulation in normal and disease states. Trends. Pharmacol. Sci. 34, 489–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jardetzky O. (1966) Simple allosteric model for membrane pumps. Nature 211, 969–970 [DOI] [PubMed] [Google Scholar]
- 5. Sitte H. H., and Freissmuth M. (2015) Amphetamines, new psychoactive drugs, and the monoamine transporter cycle. Trends Pharmacol. Sci. 36, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sitte H. H., Schütz G. J., and Freissmuth M. (2015) Cooperativity between individual transporter protomers: new data fuelling old complexes. J. Neurochem. 133, 163–166 [DOI] [PubMed] [Google Scholar]
- 7. Forrest L. R. (2013) Structural biology: (pseudo-)symmetrical transport. Science 339, 399–401 [DOI] [PubMed] [Google Scholar]
- 8. Singh S. K., Piscitelli C. L., Yamashita A., and Gouaux E. (2008) A competitive inhibitor traps LeuT in an open-to-out conformation. Science 322, 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krishnamurthy H., and Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamashita A., Singh S. K., Kawate T., Jin Y., and Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 11. Stockner T., Montgomery T. R., Kudlacek O., Weissensteiner R., Ecker G. F., Freissmuth M., and Sitte H. H. (2013) Mutational analysis of the high-affinity zinc binding site validates a refined human dopamine transporter homology model. PLoS Comput. Biol. 9, e1002909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beuming T., Shi L., Javitch J. A., and Weinstein H. (2006) A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 70, 1630–1642 [DOI] [PubMed] [Google Scholar]
- 13. Huang X., and Zhan C. G. (2007) How dopamine transporter interacts with dopamine: insights from molecular modeling and simulation. Biophys. J. 93, 3627–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Indarte M., Madura J. D., and Surratt C. K. (2008) Dopamine transporter comparative molecular modeling and binding site prediction using the LeuT(Aa) leucine transporter as a template. Proteins 70, 1033–1046 [DOI] [PubMed] [Google Scholar]
- 15. Kniazeff J., Shi L., Loland C. J., Javitch J. A., Weinstein H., and Gether U. (2008) An intracellular interaction network regulates conformational transitions in the dopamine transporter. J. Biol. Chem. 283, 17691–17701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheng M. H., and Bahar I. (2013) Coupled global and local changes direct substrate translocation by neurotransmitter-sodium symporter ortholog LeuT. Biophys. J. 105, 630–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheng M. H., and Bahar I. (2014) Complete mapping of substrate translocation highlights the significance of LeuT N-terminal segment in regulating transport cycle. PLoS Comput. Biol. 10, e1003879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khafizov K., Perez C., Koshy C., Quick M., Fendler K., Ziegler C., and Forrest L. R. (2012) Investigation of the sodium-binding sites in the sodium-coupled betaine transporter BetP. Proc. Natl. Acad. Sci. U.S.A. 109, E3035–E3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shaikh S. A., and Tajkhorshid E. (2010) Modeling and dynamics of the inward-facing state of a Na+/Cl− dependent neurotransmitter transporter homologue. PLoS Comput. Biol. 6, e1000905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thomas J. R., Gedeon P. C., Grant B. J., and Madura J. D. (2012) LeuT conformational sampling utilizing accelerated molecular dynamics and principal component analysis. Biophys. J. 103, L1–L3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zomot E., and Bahar I. (2012) A conformational switch in a partially unwound helix selectively determines the pathway for substrate release from the carnitine/γ-butyrobetaine antiporter CaiT. J. Biol. Chem. 287, 31823–31832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zomot E., Gur M., and Bahar I. (2015) Microseconds simulations reveal a new sodium-binding site and the mechanism of sodium-coupled substrate uptake by LeuT. J. Biol. Chem. 290, 544–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gur M., Zomot E., Cheng M. H., and Bahar I. (2015) Energy landscape of LeuT from molecular simulations. J. Chem. Phys. 143, 243134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Penmatsa A., Wang K. H., and Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang K. H., Penmatsa A., and Gouaux E. (2015) Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 521, 322–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheng M. H., Block E., Hu F., Cobanoglu M. C., Sorkin A., and Bahar I. (2015) Insights into the modulation of dopamine transporter function by amphetamine, orphenadrine, and cocaine binding. Front. Neurol. 6, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheng M. H., and Bahar I. (2015) Molecular mechanism of dopamine transport by human dopamine transporter. Structure 23, 2171–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khelashvili G., Stanley N., Sahai M. A., Medina J., LeVine M. V., Shi L., De Fabritiis G., and Weinstein H. (2015) Spontaneous inward opening of the dopamine transporter is triggered by PIP2-regulated dynamics of the N terminus. ACS Chem. Neurosci. 6, 1825–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamelberg D., de Oliveira C. A., and McCammon J. A. (2007) Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys. 127, 155102. [DOI] [PubMed] [Google Scholar]
- 30. Miao Y., Nichols S. E., Gasper P. M., Metzger V. T., and McCammon J. A. (2013) Activation and dynamic network of the M2 muscarinic receptor. Proc. Natl. Acad. Sci. U.S.A. 110, 10982–10987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Coleman J. A., Green E. M., and Gouaux E. (2016) X-ray structures and mechanism of the human serotonin transporter. Nature 532, 334–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang H., Elferich J., and Gouaux E. (2012) Structures of LeuT in bicelles define conformation and substrate binding in a membrane-like context. Nat. Struct. Mol. Biol. 19, 212–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Berger S. P., Farrell K., Conant D., Kempner E. S., and Paul S. M. (1994) Radiation inactivation studies of the dopamine reuptake transporter protein. Mol. Pharmacol. 46, 726–731 [PubMed] [Google Scholar]
- 34. Hastrup H., Karlin A., and Javitch J. A. (2001) Symmetrical dimer of the human dopamine transporter revealed by cross-linking Cys-306 at the extracellular end of the sixth transmembrane segment. Proc. Natl. Acad. Sci. U.S.A. 98, 10055–10060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Torres G. E., Carneiro A., Seamans K., Fiorentini C., Sweeney A., Yao W. D., and Caron M. G. (2003) Oligomerization and trafficking of the human dopamine transporter. Mutational analysis identifies critical domains important for the functional expression of the transporter. J. Biol. Chem. 278, 2731–2739 [DOI] [PubMed] [Google Scholar]
- 36. Kilic F., and Rudnick G. (2000) Oligomerization of serotonin transporter and its functional consequences. Proc. Natl. Acad. Sci. U.S.A. 97, 3106–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sorkina T., Doolen S., Galperin E., Zahniser N. R., and Sorkin A. (2003) Oligomerization of dopamine transporters visualized in living cells by fluorescence resonance energy transfer microscopy. J. Biol. Chem. 278, 28274–28283 [DOI] [PubMed] [Google Scholar]
- 38. Anderluh A., Klotzsch E., Reismann A. W., Brameshuber M., Kudlacek O., Newman A. H., Sitte H. H., and Schütz G. J. (2014) Single molecule analysis reveals coexistence of stable serotonin transporter monomers and oligomers in the live cell plasma membrane. J. Biol. Chem. 289, 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schmid J. A., Scholze P., Kudlacek O., Freissmuth M., Singer E. A., and Sitte H. H. (2001) Oligomerization of the human serotonin transporter and of the rat GABA transporter 1 visualized by fluorescence resonance energy transfer microscopy in living cells. J. Biol. Chem. 276, 3805–3810 [DOI] [PubMed] [Google Scholar]
- 40. Scholze P., Freissmuth M., and Sitte H. H. (2002) Mutations within an intramembrane leucine heptad repeat disrupt oligomer formation of the rat GABA transporter 1. J. Biol. Chem. 277, 43682–43690 [DOI] [PubMed] [Google Scholar]
- 41. Sitte H. H., Farhan H., and Javitch J. A. (2004) Sodium-dependent neurotransmitter transporters: oligomerization as a determinant of transporter function and trafficking. Mol. Interv. 4, 38–47 [DOI] [PubMed] [Google Scholar]
- 42. Zhen J., Antonio T., Cheng S. Y., Ali S., Jones K. T., and Reith M. E. (2015) Dopamine transporter oligomerization: impact of combining protomers with differential cocaine analog binding affinities. J. Neurochem. 133, 167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seidel S., Singer E. A., Just H., Farhan H., Scholze P., Kudlacek O., Holy M., Koppatz K., Krivanek P., Freissmuth M., Sitte H. H. (2005) Amphetamines take two to tango: an oligomer-based counter-transport model of neurotransmitter transport explores the amphetamine action. Mol. Pharmacol. 67, 140–151 [DOI] [PubMed] [Google Scholar]
- 44. Sitte H. H., and Freissmuth M. (2010) The reverse operation of Na+/Cl−-coupled neurotransmitter transporters: why amphetamines take two to tango. J. Neurochem. 112, 340–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Comeau S. R., Gatchell D. W., Vajda S., and Camacho C. J. (2004) ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20, 45–50 [DOI] [PubMed] [Google Scholar]
- 46. Bartholomäus I., Milan-Lobo L., Nicke A., Dutertre S., Hastrup H., Jha A., Gether U., Sitte H. H., Betz H., and Eulenburg V. (2008) Glycine transporter dimers: evidence for occurrence in the plasma membrane. J. Biol. Chem. 283, 10978–10991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen N., and Reith M. E. (2008) Substrates dissociate dopamine transporter oligomers. J. Neurochem. 105, 910–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gur M., Cheng M. H., Zomot E., and Bahar I. (2017) Effect of dimerization on the dynamics of neurotransmitter: sodium symporters. J. Phys. Chem. B 121, 3657–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Atilgan A. R., Durell S. R., Jernigan R. L., Demirel M. C., Keskin O., and Bahar I. (2001) Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80, 505–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jiang J., Shrivastava I. H., Watts S. D., Bahar I., and Amara S. G. (2011) Large collective motions regulate the functional properties of glutamate transporter trimers. Proc. Natl. Acad. Sci. U.S.A. 108, 15141–15146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Verdon G., and Boudker O. (2012) Crystal structure of an asymmetric trimer of a bacterial glutamate transporter homolog. Nat. Struct. Mol. Biol. 19, 355–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Koshy C., Schweikhard E. S., Gärtner R. M., Perez C., Yildiz O., and Ziegler C. (2013) Structural evidence for functional lipid interactions in the betaine transporter BetP. EMBO J. 32, 3096–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Anderluh A., Hofmaier T., Klotzsch E., Kudlacek O., Stockner T., Sitte H. H., and Schütz G. J. (2017) Direct PIP2 binding mediates stable oligomer formation of the serotonin transporter. Nat. Commun. 8, 14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shaw D. E., Deneroff M. M., Dror R. O., Kuskin J. S., Larson R. H., Salmon J. K., Young C., Batson B., Bowers K. J., Jack CChao; Eastwood M. P., Gagliardo J., Grossman J. P., Ho C. R., Ierardi D. J., et al. (2008) Anton, a special-purpose machine for molecular dynamics simulation. Commun. ACM 51, 91–97 [Google Scholar]
- 55. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 56. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D. Jr., and Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mackerell A. D. Jr., Feig M., and Brooks C. L. 3rd (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 [DOI] [PubMed] [Google Scholar]
- 59. Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., and Mackerell A. D. Jr. (2010) CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.