

Abstract
Cell proliferation and release from the bone marrow have been demonstrated to be controlled by circadian rhythms in both humans and mice. However, it is unclear whether local circadian clocks in the bone marrow influence physiological functions and life span of erythrocytes. Here, we report that loss of the clock gene Per2 significantly decreased erythrocyte life span. Mice deficient in Per2 were more susceptible to acute stresses in the erythrocytes, becoming severely anemic upon phenylhydrazine, osmotic, and H2O2 challenges. 1H NMR–based metabolomics analysis revealed that the Per2 depletion causes significant changes in metabolic profiles of erythrocytes, including increased lactate and decreased ATP levels compared with wild-type mice. The lower ATP levels were associated with hyperfunction of Na+/K+-ATPase activity in Per2-null erythrocytes, and inhibition of Na+/K+-ATPase activity by ouabain efficiently rescued ATP levels. Per2-null mice displayed increased levels of Na+/K+-ATPase α1 (ATP1A1) in the erythrocyte membrane, and transfection of Per2 cDNA into the erythroleukemic cell line TF-1 inhibited Atp1a1 expression. Furthermore, we observed that PER2 regulates Atp1a1 transcription through interacting with trans-acting transcription factor 1 (SP1). Our findings reveal that Per2 function in the bone marrow is required for the regulation of life span in circulating erythrocytes.
Keywords: circadian clock, clock gene, energy metabolism, erythrocyte, Na+/K+-ATPase
Introduction
The circadian rhythm of cell proliferation and release from the mammalian bone marrow has been observed in vitro and in vivo (1, 2). Multipotent stem cells undergo strong circadian variations in DNA synthesis, cell cycle, and mitotic activity (3, 4). Diurnal variation in the numbers of peripheral blood cells, such as red blood cells (RBCs), 3 platelets, white blood cells, lymphocytes, neutrophils, eosinophils, and basophils, has also been described (5–8). RBCs are released from the bone marrow and play an essential role in oxygen transport and metabolism (9, 10). The mature RBC lacks a nucleus, losing its mRNA transcription and protein translation systems. Daily circadian oscillation has been observed in the oxidant status, ion content, deformability, radiosensitivity, and adenine nucleotide content in erythrocytes (11–15). These phenomena have been attributed to diurnal rhythm in RBCs released from bone marrow (16, 17).
RBC life span in the circulation is influenced by several complex factors (18–20). Characteristic changes in the activity of ion transporters and cation content are related to RBC aging (21, 22). Lack of several structural membrane skeleton proteins in RBCs reduces life span and induces anemia (23, 24). Deficiency of antioxidant enzymes decreases RBC life span (25). Abnormal transporters and channels induce RBC damage and reduce life span (26, 27). The content of ATP in the erythrocytes is thought to be essential for erythrocyte survival (28, 29). ATP levels in erythrocytes are regulated by glycolysis for ATP synthesis and ATP hydrolysis. Impaired glycolysis or increased ATP hydrolysis could induce anemia and reduce RBC life span (30–35). In humans, increased ATPase activity and lowered ATP levels in RBCs contributes to anemia. Decreased RBC ATP content reduces GSH levels, resulting in increases in sensitivity to oxidant stress and osmotic fragility (36, 37).
Whereas clock genes Per1, Per2, Bmal1, Cry1, Clock, and Rev-erb α display circadian expression patterns both in bone marrow and hematopoietic stem cells over a 24-h period, it remains unclear whether local circadian clocks in the bone marrow directly control or influence physiological functions of blood cells (38–40). Here, we show that mice deficient in Per2, a core circadian gene, display a reduction of RBC life span and an increase in susceptibility to stress. We demonstrate that shortened RBC life span in Per2-null mice is associated with ATP depletion due to increased ATPase activity. Our findings reveal that Per2 function in the bone marrow is required for the regulation of life span in circulating erythrocytes.
Results
Genetic depletion of Per2 causes abnormal RBCs and impaired oxygen transport in mice
Our previous studies showed that genetic depletion of Per2 in mice caused a significant reduction in white blood cell and platelet counts (41). Here we further analyzed possible changes in blood parameters and RBC function in Per2-deficient mice. No significant differences in RBC counts or total hemoglobin were observed between WT and Per2-null mice; however, both mean corpuscular volume and the red cell distribution width in Per2-null mice are significantly elevated versus WT (Table 1). Oxygen consumption, measured over 24 h (light/dark, 12 h/12 h; zeitgeber time 0 (ZT0), beginning of the light period; ZT12, beginning of the dark phase), was severely decreased in Per2-null mice (Fig. 1A). Blood gas analysis revealed that both PO2 (left) and SaO2 (right) were decreased in Per2-null mice (Fig. 1B) compared with WT mice. Light and scanning-electron microscopy of peripheral blood smears of Per2-null mice showed more abnormal morphology of erythrocytes (surface changes of varying size and shape along with prominent echinocytes) than that of WT mice (Fig. 1C). These results suggest that abnormal RBCs could associate with impaired oxygen transport in Per2-null mice.
Table 1.
Hematological values in WT and Per2-null mice
All values are expressed as means ± S.D. for 10 male mice/genotype. **, p < 0.01. MCHC, mean corpuscular hemoglobin concentration; HGB, hemoglobin; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; RDW, red blood cell distribution width.
| Parameter | WT | Per2-null |
|---|---|---|
| RBC (1012/liter) | 9.35 ± 0.41 | 9.13 ± 0.35 |
| Hematocrit (%) | 43.2 ± 5.38 | 41.9 ± 6.64 |
| MCHC (g/liter) | 330.7 ± 28.78 | 317.8 ± 33.2 |
| HGB (g/liter) | 143.0 ± 6.64 | 145.7 ± 6.96 |
| MCH (pg) | 15.3 ± 0.32 | 16.0 ± 2.21 |
| MCV(fl) | 47.3 ± 1.58 | 51.2 ± 1.58** |
| RDW (%) | 35.7 ± 2.53 | 40.3 ± 1.58** |
Figure 1.

Impaired oxygen transport and the abnormal morphology of erythrocytes in Per2-null mice. A, oxygen consumption was determined at 15-s intervals for 24 h (n = 5 mice/group). B, PO2 (left) and SaO2 (right) were decreased in Per2-null mice. Results represent mean ± S.D. (error bars); n = 5 in each group. *, p < 0.05. C, morphology of peripheral red blood cells. Shown are Wright–Giemsa–stained (left) light microscopy and scanning-electron (right) microscopy of peripheral blood smears of WT and Per2-null mice. Arrows, abnormal morphology of erythrocytes in the Per2-null mice. Bar, 10 μm (left) and 5 μm (right).
Increased susceptibility of Per2-null erythrocytes to oxidative and osmotic stress
We examined responses to oxidant stress by treating with phenylhydrazine hydrochloride, an oxidant that induces destruction of erythrocytes (hemolysis). Susceptibility to a lethal dose of phenylhydrazine hydrochloride (0.23 mg/g) was significantly different in Per2-null mice compared with WT; 40% of WT mice died within a week after injection, whereas death occurred in >90% of Per2-null mice (Fig. 2A). Mortality occurred mostly in the first 5 days of phenylhydrazine hydrochloride treatment in both groups. Hemolysis was accompanied by a marked depression in hematocrit level. We next performed the experiment using lower amounts of phenylhydrazine hydrochloride (0.18 mg/g). Compared with WT mice, Per2-null mice exhibited more rapid and profound decreases in hematocrit levels (Fig. 2B). The greater decline in hematocrit within the first few days after phenylhydrazine treatment suggests increased RBC hemolysis in Per2-null mice. We also observed that Per2-null RBCs were highly sensitive to H2O2-induced hemolysis in vitro (Fig. 2, C and D), indicating that Per2-null RBCs are highly susceptible to oxidant stress. Hypotonic stress by low-ionic strength solution was used to measure the osmotic fragility of Per2-null RBCs. RBCs from Per2-null mice showed increased osmotic fragility when exposed to hypotonic NaCl solution (Fig. 2, E and F). Together, these results indicate that loss of Per2 in mice increased susceptibility of erythrocytes to oxidative and osmotic stress.
Figure 2.

Enhanced susceptibility to oxidant and osmotic stress in Per2-null erythrocytes. A, percent survival of WT and Per2-null mice subjected to PHZ-induced hemolysis. Mice were injected with a single dose of PHZ (0.23 mg/g body weight) and monitored for 7 days. Each group consisted of 20 mice. B, decreased hematocrit in Per2-null erythrocytes after exposure to the oxidant PHZ. WT and Per2-null mice were treated with a single dose of PHZ (0.20 mg/g). C, time course of H2O2-induced hemolysis of erythrocytes. RBCs were incubated in 2 mm H2O2 for the indicated time. The extent of hemolysis was measured as described under “Materials and methods.” D, dose course of H2O2-induced hemolysis of RBCs. RBCs were incubated for 2 h in the indicated concentrations of H2O2. E, erythrocytes from WT and Per2-null mice were subjected to an osmotic fragility test as described under “Materials and methods.” Results are expressed as the percentage of lysis in graded salt concentrations after a 30-min incubation. F, erythrocytes were incubated in 0.55% NaCl for the indicated time. Results represent mean ± S.D. (error bars); n = 5 in each group; *, p < 0.05; **, p < 0.01.
Decrease of erythrocyte life span and erythropoietin (EPO)-induced compensatory erythropoiesis in Per2-null mice
To determine whether the observed phenotypes in Per2 mice were due to increased destruction of RBCs in the circulation, we measured erythrocyte life span using direct biotin labeling in vivo. WT and Per2-null mice were intravenously injected with N-succinimidyl-6-(biotinamido) hexanoate to label RBCs, and cell life span was determined by a flow cytometer analysis of circulating and biotinylated RBCs. The time required for 50% of labeled RBCs to be lost from WT mice was 24 days, and the time for Per2-null mice was 14 days (Fig. 3A), indicating a decrease in erythrocyte life span in Per2-null mice. We therefore examined the percentage of erythroid cells among total bone marrow cells as well as the maturation stage of differentiating erythroblasts in the bone marrow by determination of the surface expression of both Ter119 and transferrin receptor protein 1 (CD71) (42). Combining Ter119 and CD71 expression distinguishes four subpopulations of erythroid cells, Ter119med CD71high, Ter119high CD71high, Ter119high CD71med, and Ter119high CD71low, corresponding to increasingly mature RBC precursors. Using this approach, we found that Per2-null mice had a significantly increased early erythroblast population (Ter119high CD71high) (Fig. 3B).
Figure 3.

Decreased erythrocyte life span in Per2-null mice. A, representative experiments showing decreased life span of RBCs in Per2-null mice. The dashed line indicates the time for the 50% clearance of N-succinimidyl-biotin-labeled red blood cells in mice. B, FCM analysis of live WT and Per2-null bone marrow erythroid precursors according to their expression of CD71 and Ter119. R2, proerythroblasts; R3, basophilic erythroblasts; R4, late basophilic and polychromatophilic erythroblasts; R5, orthochromatic erythroblasts. C, peripheral blood cells were stained with new methylene blue. Arrows, reticulocytes of WT and Per2-null mice. D, percentage of reticulocytes in the blood of WT and Per2-null mice. The percentage of reticulocytes was enumerated from microscopic examination of peripheral blood smears of WT and Per2-null mice and was calculated by counting 1000 cells on each slide. E, mRNA expression of Epo in WT and Per2-null liver at ZT1 and ZT13. F, spleens from WT and Per2-null mice were embedded in paraffin, and 5-μm sections were cut and stained with Prussian blue. Representative sections are shown. G, quantitation of iron content in spleens of WT and Per2-null mice. All data are shown as mean ± S.D. (error bars); n = 5–8 in each group. **, p < 0.01.
EPO is the prime regulator of RBC production. Increased destruction of RBCs would be expected to result in a compensatory acceleration of erythropoiesis. Next, using new methylene blue-stained blood smears, we determined the number of reticulocytes and found that the reticulocyte percentage in Per2-null mice was about 2–3-fold higher than that in WT mice (Fig. 3, C and D). Accordingly, a marked increase in Epo mRNA expression was observed in the liver of Per2-null mice at both ZT1 and ZT13 time points as compared with WT mice (Fig. 3E). Other markers of red cell destruction, namely the levels of iron in the serum, and the degree of iron overload in organs were also markedly abnormal. Table 2 shows an increased serum iron and transferring saturation in Per2-null mice. Meanwhile, increased RBC destruction resulted in iron accumulation in the Per2-null spleen, as demonstrated by Prussian blue staining (Fig. 3F). Indeed, total iron levels in the spleen were significantly increased in Per2-null mice compared with WT mice (Fig. 3G). These data suggest that the increased RBC destruction and shortened RBC life span in Per2-null mice is ameliorated, at least in part, by a compensatory increase in erythropoietic rate.
Table 2.
Serum iron parameter in WT and Per2-null mice
Results were obtained from (8-week-old) WT and Per2-null mice. All values are presented as means ± S.D. for three male mice per genotype. Total iron binding capacity = serum iron + unbound iron binding capacity. Transferrin saturation = (serum iron/total iron binding capacity) × 100. *, p < 0.05. TIBC, total iron binding capacity; UIBC, unbound iron binding capacity.
| Serum iron parameter | WT | Per2-null |
|---|---|---|
| Serum iron (μg/dl) | 196 ± 10.39 | 250 ± 24.25* |
| UIBC (μg/dl) | 114 ± 6.93 | 67.4 ± 20.78* |
| Transferrin saturation (%) | 63.2 ± 0.35 | 78.8 ± 8.83* |
| TIBC (μg/dl) | 310 ± 17.32 | 317 ± 15.59 |
RBC metabolome of Per2-null mice
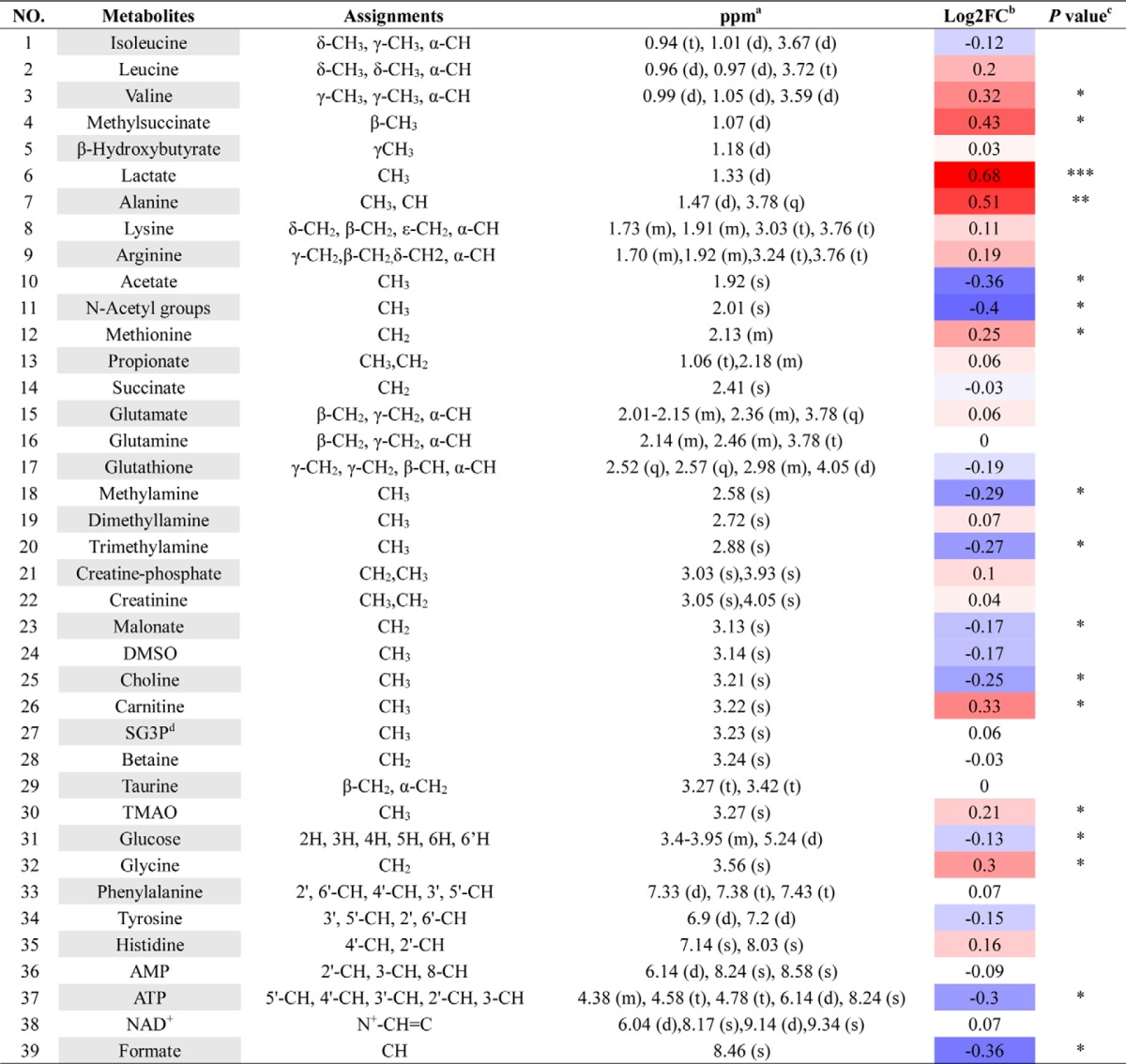
To understand the underlying mechanism of reduced RBC life span in Per2-null mice, we used the RBC metabolome to investigate metabolites produced by metabolic pathways related to vital functions in RBCs. In the OSC-PLS-DA score plot, RBCs were well separated, demonstrating that Per2 depletion causes obvious shifts in metabolic profiling (Fig. 4A). Typical 1H NMR spectra showed rich compositional information of 39 metabolites for the RBC extracts (Fig. 4C), and metabolites were identified with their 1H resonances assigned, and the detailed proton chemical shifts and their multiplicity are listed in Table 3. The S-plots (Fig. 4B) and color-coded loading plots (Fig. 4, D and E) show significant increases of lactate levels and clear decreases of glucose and ATP content in the Per2-null group as compared with the WT group (Table 3). The decreased ATP content in Per2-null erythrocytes could enhance glycolysis, resulting in progressive lactate accumulation.
Figure 4.

OSC-PLS-DA analyses based on 1H NMR data from erythrocyte extracts of WT and Per2-null group. Shown are a score plot (A), color-coded S-plot (B), and loading plots (D and E) of OSC-PLS-DA for RBC extracts obtained from eight WT and eight Per2-null mice. C, typical 500-MHz 1H NMR spectra of RBC extracts with metabolites assigned. 1, isoleucine; 2, leucine; 3, valine; 4, methylsuccinate; 5, β-hydroxybutyrate; 6, lactate; 7, alanine; 8, lysine; 9, arginine; 10, acetate; 11, N-acetyl groups; 12, methionine; 13, propionate; 14, succinate; 15, glutamate; 16, glutamine; 17, glutathione; 18, methylamine; 19, dimethyllamine; 20, trimethylamine; 21, creatine phosphate; 22, creatinine; 23, malonate; 24, DMSO; 25, choline; 26, carnitine; 27, sn-glycero-3-phosphocholine; 28, betaine; 29, taurine; 30, trimethylamine N-oxide; 31, glucose; 32, glycine; 33, phenylalanine; 34, tyrosine; 35, histidine; 36, AMP; 37, ATP; 38, NAD+; 39, formate. Metabolites that contributed to group separation were then visualized and color-coded according to the absolute correlation coefficient of each variable with each group. Color-coding is according to the -fold change in metabolites; red indicates a significant change.
Table 3.
Potential marker metabolites in mouse RBCs identified by 1HNMR and thier variations among groups and the associated p-values
Excessive activities of Na+/K+-ATPase caused decreased ATP level in Per2-null RBCs
To confirm the changes of ATP level in metabolic profiling of RBCs, HPLC analysis were performed to measure quantitatively ATP content. As shown in Fig. 5, A and B, Per2-null RBCs displayed a significant decrease in the level of ATP compared with WT RBCs. GSH is an antioxidant protein in RBCs, and its regeneration is dependent on ATP. Subsequent analysis showed the level of GSH was lower in Per2-null RBCs (Fig. 5C). GSH level in RBCs is negatively related to accumulation of malonic dialdehyde (MDA) in erythrocytes. As expected, Per2-null RBCs showed an increase in the level of MDA compared with WT RBCs (Fig. 5C). Moreover, Per2-null RBCs displayed an increase in methemoglobin formation after incubation with various concentrations of H2O2 (Fig. 5D), suggesting that hemoglobin in Per2-null RBCs is more easily oxidized due to the low level of GSH compared with WT RBCs. RBC ATP is primarily generated by glycolysis. Thus, we measured the activity of hexokinase (HK), phosphofructokinase (PFK), and pyruvate kinase (PK), three rate-limiting enzymes in glycolysis. There were no significant differences between two genotypes in the activity of HK, PFK, and PK at ZT1 and ZT13 (supplemental Fig. S1A). Also, the mRNA expression of Hk, Pfk, and Pk in Ter119+, Ter119−, and bone marrow was not reduced in Per2-null mice (supplemental Fig. S1, B–D). These results demonstrated that glycolysis in Per2-null RBCs is normal. It is well known that ATP depletion is highly associated with an increased Na+/K+-ATPase activity (33, 43). Then we measured Na+/K+-ATPase activities in isolated erythrocyte membranes. Fig. 5E shows that Per2-null erythrocyte membranes markedly increased Na+/K+-ATPase activities compared with WT. Consequently, the K+ concentration was elevated in Per2-null RBCs (Fig. 5F). Inhibition of Na+/K+-ATPase activity by digoxin (100 μg/ml) and ouabain (0.1 mm) efficiently rescued ATP level in Per2-null erythrocytes (Fig. 5G). Together, these results demonstrate that high Na+/K+-ATPase activity causes low ATP level in Per2-null erythrocytes.
Figure 5.

Enhanced Na+/K+-ATPase activities and decline of ATP level in Per2-null erythrocytes. A, HPLC profile of ATP and ADP in red blood cells. B, quantitation of ATP and ADP in RBCs of WT and Per2-null mice. C, analysis the content of GSH and MDA in RBCs of WT and Per2-null mice. D, elevated methemoglobin formation in Per2-null erythrocytes in response to various concentrations of H2O2 treatment. E, Na+/K+-ATPase activities were increased in the erythrocyte membrane of Per2-null mice. F, concentration of Na+ and K+ in the erythrocytes. G, quantitation of ATP in WT and Per2-null RBCs with or without digoxin (Dig) for 30 min and ouabain (Oua) treatment for 6 h. Results represent mean ± S.D. (error bars); n = 5–8 in each group; *, p < 0.05; **, p < 0.01.
PER2 represses Atp1a1 transcription through interaction with SP1
Next we examined the protein levels of ATP1A1 (Na+/K+-ATPase α1; one of the ATPase isoforms) between two genotypes. Western blot analysis showed a significant increase of ATP1A1 in Per2-null erythrocyte membranes compared with WT (Fig. 6A), suggesting that Per2 regulates Atp1a1 expression. To determine whether Per2 is expressed in mature erythrocytes or erythroid progenitors, Ter119+ cells were harvested from WT and Per2-null bone marrow using FACS. We found that Per2 mRNA expression was low at ZT1 and high at ZT13 in WT bone marrow, Ter119+ and Ter119− cells (Fig. 6B). Using an antibody specific for mouse PER2, we confirmed the rhythmic expression of PER2 in bone marrow and Ter119+ cells and the absence of PER2 in circulating mature RBCs (Fig. 6C). In Per2-null mice, Atp1a1 mRNA levels were elevated compared with WT mice at both ZT1 and ZT13 (Fig. 6D), and protein levels in bone marrow and RBC membranes showed a significant increase as well (Fig. 6E). Moreover, transfecting mouse Per2 cDNA into cultured TF-1 cells results in decreased Atp1a1 mRNA (Fig. 6F). SP1 (trans-acting transcription factor 1) is an important transcription factor that binds the Atp1a1 promoter. To investigate a potential association of PER2 with SP1, we performed co-immunoprecipitation assays using bone marrow cells. As shown in Fig. 6G, co-immunoprecipitation of SP1 using anti-PER2 antibody revealed that PER2 interacts with SP1 in bone marrow cells. As controls, Western blot analysis revealed the presence of PER2 and SP1 in the same immunoprecipitated cell lysate. ChIP-PCR with anti-SP1 antibody showed that PER2 protein had no effect on SP1 binding to Atp1a1 promoter (supplemental Fig. S2). These results suggested that PER2 may exert its inhibitory function by interacting with SP1 and repressing Atp1a1 transcription.
Figure 6.

PER2 interacts with SP1 to repress Atp1a1 transcription. A, ATP1A1 protein levels in WT and Per2-null RBC ghosts. Representative blotting shows increased ATPA1 protein level in RBC ghosts of Per2-null mice. B, bone marrow erythroblasts were fractionated according to the developmental marker Ter119. Ter119+, Ter119−, and total bone marrow (BM) cells from wild-type mice were analyzed for Per2 mRNA expression at ZT1 and ZT13. C, Western blot analysis showing PER2 protein expression in BM and Ter119+ cells at ZT1 and ZT13 in WT mice. D, increased mRNA expression of Atp1a1 in Per2-null Ter119+, Ter119−, and BM. E, quantitative real-time RT-PCR analysis reveals a decline in Atp1a1 mRNA in TF-1 cells transfected with Per2 cDNA plasmid for 12 and 24 h. All values are mean ± S.D. (error bars); n = 6 in each group;*, p < 0.05; **, p < 0.01. F, Western blot profiles of ATP1A1 in BM and the RBC ghosts of WT and Per2-null mice. G, PER2 was immunoprecipitated from mouse BM. Total lysates (INPUT) and immunoprecipitation samples were analyzed by immunoblotting (IB) with SP1 antibody. Shown are representative pictures of three independent experiments.
Discussion
It was well known that circadian genes are widely expressed in bone marrow and that blood cells are generated with a circadian rhythm (41, 44). Here, we provide molecular evidence that local circadian clocks in the bone marrow influence physiological functions of erythrocytes. Loss of Per2 in mice shortens erythrocyte life span. PER2 is a repressor that inhibits CLOCK/BMAL1-mediated transcription as a positive feedback loop in the clock system. Consistent with this, Per2-null mice display increased Clock mRNA expression (45). In Clock mutant mice, the spleen becomes enlarged as erythroid cells proliferate (44). Per2 expression is markedly reduced in Clock-null mice, suggesting that RBC effects in Clock-null mice arise secondarily through reduced Per2 expression (44, 45). ApcMin/+ Per2m/m double knock-out mice developed more severe anemia compared with ApcMin/+ mice (46). These observations imply that Per2 plays a functional role in erythrocytes.
In this study, we show that Per2-null mice have a much lower survival rate and hematocrit level than WT mice after phenylhydrazine treatment, and Per2-null RBCs were less resistant to hypo-osmotic and H2O2 stress compared with wild-type mice. In addition to shortened RBC life span, higher sensitivity to oxidant and osmotic stress could account at least in part for the accumulation of iron, EPO elevation, increased erythropoiesis, and increased reticulocytes that we observed in Per2-null mice. Increased reticulocytosis suggests that a mild, compensated anemia is present in the absence of Per2.
Decreased erythrocyte life span correlated with a decline of ATP levels in Per2-null erythrocytes. The erythrocyte depends solely on the anaerobic conversion of 90% glucose (the energy source) to pyruvate or lactate by glycolysis for the generation and storage of ATP (47). In erythrocytes, ATP represents the greatest reservoir of energy and supplies energy mainly to Na+/K+-ATPase, whose activity is responsible for maintaining cation gradients across the membrane, which is essential for their survival (48, 49). ATP regulates many aspects of RBC metabolism through participation in various biological processes, including GSH regeneration, oxygen affinity, glucose transport, deformation, and ion content (50, 51). ATP depletion in Per2-null RBCs could be the main reason that Per2-null RBCs are more sensitive to environment stressors. ATP depletion in RBCs contributed to inhibition of glycolysis or increased ATP consumption. Previous investigation had shown that increased levels of glycolytic enzymes in the muscle of Per2 mutant mice (52) and excessive ATP consumption lead to an accelerated anaerobic glycolysis in DMT1-mutant mice (53). In our study, 1H NMR–based metabolomics analysis revealed that there was an increased lactate level in Per2-null RBCs, suggesting that the glycolysis rate increased in these RBCs. We also found that the ATP content was significantly decreased in Per2-null erythrocytes, which could enhance glycolysis in a feedback way, resulting in progressive lactate accumulation. Increased activity of ATPase was thought to associate with increasing ATP consumption and lead to ATP depletion. In Fanconi's anemia, increased ATPase activity was considered as a cause for deceased ATP values (33). Congenital nonspherocytic hemolytic anemia was associated with increased activity of erythrocyte ATPase (54). Our results revealed that PER2 could bind with SP1 and repress the transcription of Atp1a1. Thus, loss of Per2 function increased ATP1A1 expression and Na+-K+-ATPase activities, consequently influencing ATP level in RBCs.
It remains to be investigated whether Per2 is essential for red blood cell-related diseases, but we have demonstrated that deleting this fundamental gene results in a physiological change in red blood cell. Our results suggest that Per2 is required for the regulation of RBC life span.
Materials and methods
Animals
The Per2-null mice used in this study were kindly provided by Dr. C. Lee (55). Per2-null mice on the 129SvEv background were bred onto the C57BL/6J (Jackson Laboratory, Bar Harbor, ME) background for 8–10 generations (N8–N10) according to standard genetic protocols. WT (C57BL/6J), Per2-null mice were housed in a standard animal maintenance facility under a 12-h/12-h light/dark cycle. All experiments were performed with sex- and age-matched mice under protocols approved by the institutional animal care committee of Nanjing University of Science and Technology (approval ACUC-NUST-20130016).
Erythrocyte life span analysis
In vivo biotinylation followed by flow cytometry (FCM) analysis were performed as described previously (56). Briefly, RBCs were labeled in vivo by intravenous injection of 30 mg/kg N-succinimidyl-6-(biotinamide) hexanoate (Thermo Fisher Scientific). Peripheral blood (5 μl) was collected from the tail vein 1 h after biotin labeling and stained using phycoerythrin (PE)-streptavidin. Staining was followed by FCM analysis to ensure that at least 95% of red cells were biotin-labeled. Blood samples were analyzed at 7-day intervals to quantify biotin-labeled cells remaining in the circulation. A flow cytometer assay was performed using a FACSCalibur (BD Biosciences), and FCM data were analyzed using Summit version 4.3 software.
Bone marrow erythropoiesis
Erythropoiesis was analyzed as described previously (42). Bone marrow cells from femora were collected in PBS with 0.5% BSA and passed through a 200-mesh filter. Single-cell suspensions were double-stained with antibodies against fluorescein isothiocyanate-conjugated Ter119 (Ter119-FITC; eBioscience) and PE-conjugated erythroid antigen CD71 (CD71-PE; eBioscience).
Hematological analysis and oxygen consumption assay
Whole blood (250 μl) was collected in potassium EDTA tubes. Complete blood counts were obtained using an ADVIA 120 automated hematological analyzer (Bayer, Tarrytown, NY) fitted with murine hematology software. The percentage of reticulocytes was determined by staining peripheral blood with new methylene blue (Sigma). The percentage of reticulocytes was calculated by counting 1000 cells on each slide.
Arterial blood gases and pH were measured using a Corning 178 blood gas/pH analyzer (Ciba Corning Diagnostics, Medford, MA). For oxygen consumption analysis, in vivo indirect open circuit calorimetry was performed in metabolic chambers at a controlled ambient temperature (23 ± 2 °C). A constant air flow (0.5 liters/min) was drawn through the chamber and monitored using a metabolic monitor (AccuScan Instruments, Columbus, OH). After 30 min allowed for adaptation to the metabolic chamber, VO2 was assessed at 15-s intervals for a 24-h period. Mice had free access to water and food during the 24-h period.
Light and electron microscopy
Blood smears were air-dried and viewed under a light microscope, either without staining by use of simple contrast enhancement closure of the iris and slight decentering of the condensator or with staining by Wright-Giemsa stain, and viewed in conventional bright field. For scanning-electron microscopy analysis, a drop of tail blood was suspended in 1 ml of 0.1 m phosphate buffer, pH 7.4, and centrifuged at 1500 rpm for 5 min at 4 °C. After centrifugation, the pelleted cells were resuspended in 1 ml of 2.5% glutaraldehyde and fixed for 1 h at room temperature on a rotating wheel, ensuring that the cells were continually spinning to allow a single-cell suspension. Once fixed, the cells were rinsed in three changes (15 min each) of phosphate buffer containing 5% sucrose, and 100 μl of the final cell suspension were placed on a Thermanox coverslip (Nunc, Thermo Fisher Scientific, Rochester, NY) and incubated at 4 °C until the cells adhered (30 min to 1 h). The cells attached to the coverslips were then post-fixed with 2.5% osmium tetroxide (OsO4) for 1 h at room temperature and rinsed extensively in distilled water (3 × 15 min changes). After rinsing, the cells were dehydrated through increasing concentrations of acetone (5-min changes in each of 70, 80, 90, 95, and 100%). Samples were then critical point-dried (Polaron critical point dryer, Quorum Technologies, East Sussex, UK), mounted on stubs with carbon dag (ProSciTech, Thuringowa, Australia), and sputter-coated with gold in an Edwards Sputter Coater (Edwards, West Sussex, UK). Gold-coated samples were observed using a 515 scanning-electron microscope (Phillips, Amsterdam, Netherlands) at 20 kV.
For transmission electron microscope analysis, washed red blood cells from WT and Per2-null mice were fixed in 2.5% glutaraldehyde and 2.0% paraformaldehyde in 0.1 m sodium cacodylate buffer, pH 7.4, overnight at 4 °C. The samples were then dehydrated in alcohol at progressively higher concentrations and embedded in Epon 812 resin (Polysciences, Warrington, PA). Consecutive thin and ultrathin sections were cut using a Reichert ultramicrotome. Ultrathin sections were collected on 100-mesh copper grids, counterstained with uranyl acetate and lead citrate, and observed with a JEOL 1011 transmission electron microscope (JEOL, Tokyo, Japan) at 120 kV.
Protein analysis
For PER2-SP1 co-immunoprecipitation assays, proteins from fresh mouse bone marrow nuclei was prepared according to the standard procedure (57). Protein G–agarose beads (Invitrogen) were added to the cleared lysates and incubated at 4 °C for 1 h. Beads were discarded, and lysates were incubated either alone or with mouse immunoglobulin G or mouse anti-PER2 antibodies overnight at 4 °C. Protein–antibody complexes were collected by incubating with protein G–agarose beads for 1 h at 4 °C, followed by centrifugation and three washes with radioimmune precipitation buffer (50 mm Tris–HCl,150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, pH 8.0). Immunoprecipitates were subjected to SDS-PAGE and immunoblotted with anti-PER2 or anti-SP1 antibody to detect PER2 or SP1, respectively.
For Western blot analysis, proteins were separated by standard SDS-PAGE and transferred to nitrocellulose membranes by a semidry transfer apparatus (Bio-Rad). Anti-ATP1A1 and anti-SP1 antibodies were from Bioworld Technology Inc. (Bioworld, St. Louis Park, MN). Anti-tubulin-α antibody was from Sigma. Anti-β-actin antibody was from KeyGEN BioTECH Inc. (Nanjing, China). Anti-PER2 antibody was a gift from Dr. Y. Xu (58).
Iron studies
Total serum iron and unsaturated iron-binding capacity were determined using assay kits from, Diagnostic Chemicals (Charlottetown, Canada), together with the calibrators and standards recommended by the manufacturer. To analyze iron content in the spleen, tissues were weighed wet and then dried overnight at 106 °C and weighed again. Dried samples were ashed at 500 °C for 17 h and then fully solubilized in 6 mol/liter HCl, and the final solution was adjusted with demineralized water to a final HCl concentration of 1.2 mol/liter. Iron concentration of the samples was determined by flame atomic absorption spectrometry (Varian SpectrAA 250 Plus, Varian, Mulgrave, Victoria, Australia). For histological analysis, spleens were fixed in 10% neutral buffered formalin for 24 h, processed to 70% alcohol, embedded in paraffin, sectioned (4 μm), and stained for iron content with Perls Prussian blue.
Phenylhydrazine treatment and in vitro hemolysis
Phenylhydrazine hydrochloride (Sigma) was prepared freshly in sterile PBS (20 mg/ml, pH 7.4). Age- and sex-matched 8-week-old mice were injected intraperitoneally (0.2–0.25 mg/g body weight), and 15–20 μl of blood was obtained serially from the tail vein for hematocrit determination using standard methods.
For in vitro hemolysis, EDTA-anticoagulated blood was centrifuged (600 × g, 5 min) to remove plasma. RBCs were washed in PBS, resuspended (PBS plus 20 mm glucose) at 5% packed cell volume, and incubated with the indicated concentration of H2O2 (37 °C, 20% O2, and 5% CO2). Erythrocytes were centrifuged (1000 × g, 10 min, 4 °C), and absorbance (540 nm) was measured from supernatant.
GSH and MDA levels
RBC GSH was measured by a spectrophotometric assay using DTNB and expressed as μg/mg protein. MDA in erythrocytes was estimated by measuring thiobarbituric acid reactivity. Values of product MDA in nmol/ml RBCs were determined using the extinction coefficient of MDA–thiobarbituric acid complex at 532 nm. The results were expressed as μg of MDA/mg of protein.
RBC metabolomics extraction and NMR spectroscopy
RBCs were obtained from WT and Per2-null mice at ZT13 and immediately extracted in ice-cold lysis/extraction buffer (methanol/acetonitrile/water, 5:3:2) at 1:10 dilutions. Samples were then agitated at 4 °C for 30 min and then centrifuged at 10,000 × g for 15 min at 4 °C. Protein and lipid pellets were discarded, and supernatants were frozen and lyophilized to dryness and stored at −80 °C until the NMR test. The NMR samples were prepared by dissolving dried extracts in 600 μl of D2O. After centrifugation at 12,000 × g for 10 min, the supernatant was transferred to a 5-mm NMR tube for 1H NMR analysis.
1H NMR spectra of the samples were recorded on a Bruker AV 500-MHz spectrometer at 298 K. For serum samples, a Carr-Purcell-Meibom-Gill spin-echo pulse sequence (90-(τ-180-τ) n-acquisition) with a total spin-echo delay (2 nτ) of 10 ms was adopted to attenuate the broad signals from macromolecules (i.e. proteins or lipoproteins), whereupon the signals of micromolecule metabolites were clearly observed. 1H NMR spectra were collected with 128 scans into 32,768 data points over a spectral width of 10,000 Hz. For tissue samples, modified nuclear Overhauser enhancement spectroscopy with a presaturation (NOESYPR) pulse sequence (relaxation delay-90°-μs-90°-tm-90°-acquire-FID) was used to suppress the residual water signal. Before the Fourier transformation, a line broadening of 0.3 Hz was applied to all spectra. An exponential weighting factor corresponding to a line broadening of 0.3 Hz was used to all acquired free induction decays before Fourier transformation and phase correction.
Spectral processing
1H NMR spectra of sample extracts were corrected for phase and baseline distortion and referenced manually to the TSP resonance at δ 0.00 using the TOPSPIN package (version 3.0; Bruker Biospin, Ettlingen, Germany). Spectral regions δ 0.50–9.50 were automatically binned using a dynamic adaptive binning approach with an equal width of 0.002 ppm. The noisy and residual water–affected regions (4.65–5.25 ppm) were removed. The remaining spectral data were normalized by probabilistic quotient normalization before pattern recognition analysis (59).
Methemoglobin formation and osmotic fragility assay
Methemoglobin concentration was determined by comparing the absorbance spectra at 630 nm before and after the addition of potassium cyanide (KCN) to hemolysates using a methemoglobin assay kit (Nanjing Jiancheng Bioengineering Institute) and was expressed as a percentage of total hemoglobin concentration. RBC osmotic fragility was measured on freshly collected heparized blood as described previously (26). Briefly, osmotic fragility of erythrocytes was measured on freshly collected blood in heparin from WT and Per2-null mice. RBCs were washed with the isotonic saline and suspended in varying concentrations of NaCl. NaCl solutions used were 0.300, 0.350, 0.400, 0.450, 0.475, 0.500, 0.525, 0.550, 0.575, 0.600, 0.625, 0.650, 0.700, and 0.800% (w/v). Samples were incubated at room temperature for 30 min and centrifuged at 1500 × g for 10 min at 4 °C. The supernatant was collected, and absorbance was measured at 540 nm with appropriate control. The percentage lysis of RBCs was calculated from the absorbance, and a fragility curve was generated by plotting varying salt concentration versus hemolysis. C50 values were determined by logarithmic linearization of the osmotic fragility curve.
HPLC analysis of ATP
RBCs were separated and were extracted from frozen samples using 0.4 n perchloric acid. Extracts were separated and quantified by using reversed-phase HPLC (Waters 1525 system, Millipore Corp., Bedford, MA) analysis on a Partisphere bonded phase C18 (reverse phase) cartridge column at a flow rate of 1.0 ml/min as described previously (60). For ouabain “rescue” experiments, WT and Per2-null RBCs were separated and washed three times in 10 mm PBS (pH 7.4). Then RBCs were cultured in RPMI 1640 (initially without potassium and sodium) containing 102.66 mm potassium chloride, 5.36 mm sodium chloride, 23.80 mm potassium hydrogen carbonate, and 5.63 mm dipotassium hydrogen phosphate with 20% fetal bovine serum. RBCs were incubated with or without ouabain (0.1 mm) for 6 h. After the incubation, erythrocytes were harvested and then measured by HPLC, whereas for digoxin rescue experiments, WT and Per2-null RBCs were separated and washed in the same way as described above. Then RBCs were cultured in RPMI 1640 with 20% fetal bovine serum. RBCs were incubated with or without digoxin (100 μg/ml) for 30 min (61). After the incubation, erythrocytes were harvested and then measured as described above.
Analysis of glycolysis
The activity of PK and HK in RBCs was detected using enzyme activity kits from the Nanjing Jiancheng Bioengineering Institute. The activity of PFK was determined by spectrophotometric analysis as described previously (62).
Na+/K+-ATPase activity assay
ATPase activity was measured in RBC membrane fragments as described (63). Briefly, freshly drawn blood samples containing heparin as an anticoagulant were centrifuged for 15 min at 950 × g in 4 °C. The erythrocytes were washed by ice-cold isotonic saline. 500 μl of packed RBCs were transferred into 5 ml of hypotonic medium (10 mm Tris-HCl, pH 7.4, containing 0.1 mm EGTA). The tubes were shaken vigorously and centrifuged at 4 °C for 20 min at 4.8 × 104 × g. After aspiration of the supernatant, the cells were resuspended in hypotonic medium and treated above two more times until the lysed membranes were free of hemoglobin. The final pellet of RBC membranes was resuspended in 200 μl of Tris-EGTA buffer. Total ATPase activity was determined for each subcellular fraction in the presence of 30 mm Tris-HCl–buffered medium (pH 7.4) containing 0.1 mm Tris-EDTA, 50 mm NaCl, 5 mm KC1, and 6 mm MgCl2. The Mg2+-ATPase was determined separately in 30 mm Tris-HCl buffered medium (pH 7.4) containing only MgCl2 (6 mm). The reaction was initiated by the addition of 1 mm ATP and stopped 10 min later with 5% trichloroacetic acid. The Pi released from ATP was determined by a molybdate colorimetric procedure. The (Na+K+)-ATPase activity present in each fraction was obtained by taking the difference between total ATPase activity and Mg2+-ATPase activity.
Measurement of RBC Na+ and K+
Na+ and K+ content in the RBCs was measured by atomic absorption spectrometry (Analyst 800, PerkinElmer Life Sciences) after washing the RBCs four times in cold (4 °C) solution with 110 mmol/liter MgCl2 (64).
Cell culture and plasmid transfections
Human erythroleukemic cell line TF-1 was grown in RPMI 1640 medium and 10% FCS (Gibco) supplemented with 5 ng/ml GM-CSF (PeproTech). Cells were grown at 37 °C in a humidified 5% CO2 atmosphere. The cells were transfected with mouse Per2 cDNA plasmids using Lipofectamine 2000 reagent (Invitrogen).
Ter119 sorting, RNA isolation, and quantitative real-time RT-PCR
Ter119+ and Ter119− cells from bone marrow were isolated using FACS. Bone marrow cell suspensions were stained with antibodies to Ter119-FITC before sorting on an Influx cell sorter. Total RNA was prepared from bone marrow, liver, TF-1 cells, Ter119+, and Ter119− using TRIzol reagent according to the manufacturer's instructions (Invitrogen), followed by DNase I treatment to remove potential containing genomic DNA. Total RNA (0.5 μg) was reverse-transcribed into cDNA in 20 μl of reverse transcription reaction mixture containing 2.5 μm random hexamers, 2 mm dNTP, 5 units RNase inhibitor, and 200 units Moloney murine leukemia virus reverse transcriptase. Real-time PCR was performed and analyzed using an ABI 7300 detection system in combination with SYBR Green dye. Relative expression in comparison with β-actin was calculated by the comparative CT method. The primer details can be seen in supplemental Table S1.
ChIP analysis
A ChIP assay was performed as described previously (65). Immunoprecipitated DNA was then used as a template for real-time PCR analysis. The sequences of primers used for ChIP-PCR are listed in supplemental Table S1. IgG control antibody was used as a negative control.
Multivariate data analysis
Multivariate data analysis was performed by a suite of scripts developed in-house running R software. Principle component analysis (PCA) is an exploratory unsupervised method to maximize the separation by providing model-free approaches for determining the latent or intrinsic information in the data set. However, no clustering was observed when variables were not selected. OSC-PLS-DA, which is a supervised method, was used to maximize covariance between the measured data (peak intensities in NMR spectra) and the response variable (predictive classifications) (66). All OSC-PLS-DA models were validated by a repeated 2-fold cross-validation method and permutation test. The parameters of R2 and Q2 represented the goodness of fit and the predictive ability of the models, respectively. The p value of the permutation test denoted the number of times that the permutated data yielded a better result than the one using the original labels. The -fold change values of metabolites among different groups were calculated. The p values obtained by permutation tests that were < 0.05 indicated the significance of the established OSC-PLS-DA model at a 95% confidence level. According to the integrated areas of metabolites, the increase or decrease factors were calculated with associated p values adjusted with the Benjamini and Hochberg method for multiple comparisons and visualized in colored tables (67, 68).
Statistical analysis
All values were expressed as mean ± S.D. Two-tailed p values were calculated by unpaired Student's t test. Differences were considered statistically significant at p values < 0.05.
Author contributions
Q. S. and Y. Z. performed the main research; Y. Y., M. L., and J. W. analyzed data; X. X., X. Y., and D. W. wrote part of paper; J. Z. designed the research and wrote the paper.
Supplementary Material
Acknowledgments
We thank Dr. Luanne L. Peters (Jackson Laboratory) for critical review of the paper. We thank Dr. Cheng Chi Lee (University of Texas, Houston, TX) for providing Per2-null mice and Per2 cDNA and Dr. Ying Xu (Nanjing University) for providing anti-PER2 antibody.
This work was supported by 973 Program of China Grant 2013CB945203 and National Science Foundation of China Grants 31671220 and 31471111. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Table S1 and Figs. S1 and S2.
- RBC
- red blood cell
- HK
- hexokinase
- MDA
- malonyl dialdehyde
- PFK
- phosphofructokinase
- PK
- pyruvate kinase
- OSC
- orthogonal signal correction
- PLS
- partial least squares
- DA
- discriminant analysis
- ZT
- zeitgeber time 0
- EPO
- erythropoietin
- FCM
- flow cytometry
- PE
- phycoerythrin
- BM
- bone marrow.
References
- 1. Smaaland R., Sothern R. B., Lote K., Sandberg S., Aakvaag A., and Laerum O. D. (1995) Circadian phase relationships between peripheral blood variables and bone marrow proliferative activity in clinical health. In Vivo 9, 379–389 [PubMed] [Google Scholar]
- 2. Méndez-Ferrer S., Lucas D., Battista M., and Frenette P. S. (2008) Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452, 442–447 [DOI] [PubMed] [Google Scholar]
- 3. Clark R. H., and Korst D. R. (1969) Circadian periodicity of bone marrow mitotic activity and reticulocyte counts in rats and mice. Science 166, 236–237 [DOI] [PubMed] [Google Scholar]
- 4. Laerum O. D., Sletvold O., and Riise T. (1988) Circadian and circannual variations of cell cycle distribution in the mouse bone marrow. Chronobiol. Int. 5, 19–35 [DOI] [PubMed] [Google Scholar]
- 5. Swoyer J., Haus E., and Sackett-Lundeen L. (1987) Circadian reference values for hematologic parameters in several strains of mice. Prog. Clin. Biol. Res. 227A, 281–296 [PubMed] [Google Scholar]
- 6. Cho C. S., Yoon H. J., Kim J. Y., Woo H. A., and Rhee S. G. (2014) Circadian rhythm of hyperoxidized peroxiredoxin II is determined by hemoglobin autoxidation and the 20S proteasome in red blood cells. Proc. Natl. Acad. Sci. U.S.A. 111, 12043–12048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pasqualetti P., Colantonio D., Casale R., Colangeli S., and Natali G. (1988) [Circadian rhythm of human lymphocyte subpopulations]. Quad. Sclavo Diagn. 24, 89–95 [PubMed] [Google Scholar]
- 8. Hartley P. S., Sheward J., Scholefield E., French K., Horn J. M., Holmes M. C., and Harmar A. J. (2009) Timed feeding of mice modulates light-entrained circadian rhythms of reticulated platelet abundance and plasma thrombopoietin and affects gene expression in megakaryocytes. Br. J. Haematol. 146, 185–192 [DOI] [PubMed] [Google Scholar]
- 9. Hamasaki N., and Yamamoto M. (2000) Red blood cell function and blood storage. Vox Sang. 79, 191–197 [DOI] [PubMed] [Google Scholar]
- 10. Wei H. S., Kang H., Rasheed I. Y., Zhou S., Lou N., Gershteyn A., McConnell E. D., Wang Y., Richardson K. E., Palmer A. F., Xu C., Wan J., and Nedergaard M. (2016) Erythrocytes are oxygen-sensing regulators of the cerebral microcirculation. Neuron 91, 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suplotov S. N., and Barkova E. N. (1986) [Diurnal and seasonal rhythms of lipid peroxides and superoxide dismutase in the erythrocytes of inhabitants of middle-latitude regions and the extreme north]. Lab. Delo 459–463 [PubMed] [Google Scholar]
- 12. Chakravarty S., and Rizvi S. I. (2011) Circadian modulation of sodium-potassium ATPase and sodium-proton exchanger in human erythrocytes: in vitro effect of melatonin. Cell Mol. Biol. 57, 80–86 [PubMed] [Google Scholar]
- 13. Yerer M. B., and Aydoğan S. (2006) The importance of circadan rhythm alterations in erythrocyte deformability. Clin. Hemorheol. Microcirc. 35, 143–147 [PubMed] [Google Scholar]
- 14. Newsome-Tabatabai R., and Rushton P. S. (1984) Daily variation in radiosensitivity of circulating blood cells and bone marrow cellularity of mice. Comp. Biochem. Physiol. A Comp. Physiol. 78, 779–783 [DOI] [PubMed] [Google Scholar]
- 15. Chagoya de Sánchez V., Hernández-Muñoz R., Díaz-Muñoz M., Villalobos R., Glender W., Vidrio S., Suárez J., and Yañez L. (1983) Circadian variations of adenosine level in blood and liver and its possible physiological significance. Life Sci. 33, 1057–1064 [DOI] [PubMed] [Google Scholar]
- 16. Antonelou M. H., Kriebardis A. G., and Papassideri I. S. (2010) Aging and death signalling in mature red cells: from basic science to transfusion practice. Blood Transfus. 8, s39–s47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Magnani M., Piatti E., Serafini N., Palma F., Dachà M., and Fornaini G. (1983) The age-dependent metabolic decline of the red blood cell. Mech. Ageing Dev. 22, 295–308 [DOI] [PubMed] [Google Scholar]
- 18. Ganzoni A. M., Oakes R., and Hillman R. S. (1971) Red cell aging in vivo. J. Clin. Invest. 50, 1373–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pretorius E., Swanepoel A. C., Buys A. V., Vermeulen N., Duim W., and Kell D. B. (2014) Eryptosis as a marker of Parkinson's disease. Aging 6, 788–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang Y. X., Wu Z. J., Mehrishi J., Huang B. T., Chen X. Y., Zheng X. J., Liu W. J., and Luo M. (2011) Human red blood cell aging: correlative changes in surface charge and cell properties. J. Cell. Mol. Med. 15, 2634–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mairbäurl H., Schulz S., and Hoffman J. F. (2000) Cation transport and cell volume changes in maturing rat reticulocytes. Am. J. Physiol. Cell Physiol. 279, C1621–C1630 [DOI] [PubMed] [Google Scholar]
- 22. Vokurková M., Rauchová H., Dobešová Z., Loukotová J., Nováková O., Kuneš J., and Zicha J. (2016) The influence of erythrocyte maturity on ion transport and membrane lipid composition in the rat. Physiol. Res. 65, 91–99 [DOI] [PubMed] [Google Scholar]
- 23. Peters L. L., and Barker J. E. (2001) Spontaneous and targeted mutations in erythrocyte membrane skeleton genes: mouse models of hereditary spherocytosis. In: Hematopoiesis: A Developmental Approach (Zon L. I., ed) pp. 582–608, Oxford University Press, New York [Google Scholar]
- 24. Robledo R. F., Ciciotte S. L., Gwynn B., Sahr K. E., Gilligan D. M., Mohandas N., and Peters L. L. (2008) Targeted depletion of α-adducin results in absent β- and γ-adducin, compensated hemolytic anemia, and lethal hydrocephalus in mice. Blood 112, 4298–4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee T. H., Kim S. U., Yu S. L., Kim S. H., Park D. S., Moon H. B., Dho S. H., Kwon K. S., Kwon H. J., Han Y. H., Jeong S., Kang S. W., Shin H. S., Lee K. K., Rhee S. G., and Yu D. Y. (2003) Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood 101, 5033–5038 [DOI] [PubMed] [Google Scholar]
- 26. De Franceschi L., Rivera A., Fleming M. D., Honczarenko M., Peters L. L., Gascard P., Mohandas N., and Brugnara C. (2005) Evidence for a protective role of the Gardos channel against hemolysis in murine spherocytosis. Blood 106, 1454–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weber Y. G., Storch A., Wuttke T. V., Brockmann K., Kempfle J., Maljevic S., Margari L., Kamm C., Schneider S. A., Huber S. M., Pekrun A., Roebling R., Seebohm G., Koka S., Lang C., et al. (2008) GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J. Clin. Invest. 118, 2157–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hortle E., Nijagal B., Bauer D. C., Jensen L. M., Ahn S. B., Cockburn I. A., Lampkin S., Tull D., McConville M. J., McMorran B. J., Foote S. J., and Burgio G. (2016) Adenosine monophosphate deaminase 3 activation shortens erythrocyte half-life and provides malaria resistance in mice. Blood 128, 1290–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cho J., Seo J., Lim C. H., Yang L., Shiratsuchi T., Lee M. H., Chowdhury R. R., Kasahara H., Kim J. S., Oh S. P., Lee Y. J., and Terada N. (2015) Mitochondrial ATP transporter Ant2 depletion impairs erythropoiesis and B lymphopoiesis. Cell Death Differ. 22, 1437–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keitt A. S. (1969) Hemolytic anemia with impaired hexokinase activity. J. Clin. Invest. 48, 1997–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Black J. A., Rittenberg M. B., Bigley R. H., and Koler R. D. (1979) Hemolytic anemia due to pyruvate kinase deficiency: characterization of the enzymatic activity from eight patients. Am. J. Hum. Genet. 31, 300–310 [PMC free article] [PubMed] [Google Scholar]
- 32. Etiemble J., Kahn A., Boivin P., Bernard J. F., and Goudemand M. (1976) Hereditary hemolytic anemia with erythrocyte phosphofructokinase deficiency: studies of some properties of erythrocyte and muscle enzyme. Hum. Genet. 31, 83–91 [DOI] [PubMed] [Google Scholar]
- 33. Syllm-Rapoport I., Gmyrek D., Altenbrunn H. J., Scheuch D., and Jacobasch G. (1965) [Studies on hemolysis in Fanconi's anemia: increase of ATP-ase as a possible cause for decreased ATP values]. Dtsch. Med. Wochenschr. 90, 290–296 [DOI] [PubMed] [Google Scholar]
- 34. Luthra M. G., and Sears D. A. (1982) Increased Ca++, Mg++, and Na+ + K+ ATPase activities in erythrocytes of sickle cell anemia. Blood 60, 1332–1336 [PubMed] [Google Scholar]
- 35. Eluwa E. O., Obidoa O., Obi G. O., and Onwubiko H. A. (1987) Variations in the relative activities of erythrocyte membrane ATPase with changes in severity of sickle cell anemia. Biochem. Med. Metab. Biol. 38, 142–148 [DOI] [PubMed] [Google Scholar]
- 36. Suzuki M., and Kurata M. (1992) Effects of ATP level on glutathione regeneration in rabbit and guinea-pig erythrocytes. Comp. Biochem. Physiol. B Comp. Biochem. 103, 859–862 [DOI] [PubMed] [Google Scholar]
- 37. Tozzi-Ciancarelli M. G., Di Massimo C., and Mascioli A. (1992) Aging of human erythrocytes: the role of membrane perturbations induced by in vitro ATP-depletion. Cell. Mol. Biol. 38, 303–310 [PubMed] [Google Scholar]
- 38. Chen Y. G., Mantalaris A., Bourne P., Keng P., and Wu J. H. (2000) Expression of mPer1 and mPer2, two mammalian clock genes, in murine bone marrow. Biochem. Biophys. Res. Commun. 276, 724–728 [DOI] [PubMed] [Google Scholar]
- 39. Tsinkalovsky O., Rosenlund B., Laerum O. D., and Eiken H. G. (2005) Clock gene expression in purified mouse hematopoietic stem cells. Exp. Hematol. 33, 100–107 [DOI] [PubMed] [Google Scholar]
- 40. Tsinkalovsky O., Smaaland R., Rosenlund B., Sothern R. B., Hirt A., Steine S., Badiee A., Abrahamsen J. F., Eiken H. G., and Laerum O. D. (2007) Circadian variations in clock gene expression of human bone marrow CD34+ cells. J. Biol. Rhythms 22, 140–150 [DOI] [PubMed] [Google Scholar]
- 41. Zhao Y., Zhang Y., Wang S., Hua Z., and Zhang J. (2011) The clock gene Per2 is required for normal platelet formation and function. Thromb. Res. 127, 122–130 [DOI] [PubMed] [Google Scholar]
- 42. Zhang J., and Lodish H. F. (2005) Identification of K-ras as the major regulator for cytokine-dependent Akt activation in erythroid progenitors in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 14605–14610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamasaki M., Nakamura K., Tamura N., Hwang S. J., Yoshikawa M., Sasaki N., Ohta H., Yamato O., Maede Y., and Takiguchi M. (2009) Effects and mechanisms of action of ionophorous antibiotics valinomycin and salinomycin-Na on Babesia gibsoni in vitro. J. Parasitol. 95, 1532–1538 [DOI] [PubMed] [Google Scholar]
- 44. Oishi K., Ohkura N., Kadota K., Kasamatsu M., Shibusawa K., Matsuda J., Machida K., Horie S., and Ishida N. (2006) Clock mutation affects circadian regulation of circulating blood cells. J. Circ. Rhythms 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhao Y., Zhang Y., Zhou M., Wang S., Hua Z., and Zhang J. (2012) Loss of mPer2 increases plasma insulin levels by enhanced glucose-stimulated insulin secretion and impaired insulin clearance in mice. FEBS Lett. 586, 1306–1311 [DOI] [PubMed] [Google Scholar]
- 46. Wood P. A., Yang X., Taber A., Oh E. Y., Ansell C., Ayers S. E., Al-Assaad Z., Carnevale K., Berger F. G., Peña M. M., and Hrushesky W. J. (2008) Period 2 mutation accelerates ApcMin/+ tumorigenesis. Mol. Cancer Res. 6, 1786–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Wijk R., and van Solinge W. W. (2005) The energy-less red blood cell is lost: erythrocyte enzyme abnormalities of glycolysis. Blood 106, 4034–4042 [DOI] [PubMed] [Google Scholar]
- 48. Desforges J. F. (1965) Erythrocyte metabolism in hemolysis. N. Engl. J. Med. 273, 1310–1321 [DOI] [PubMed] [Google Scholar]
- 49. Ataullakhanov F. I., and Vitvitsky V. M. (2002) What determines the intracellular ATP concentration. Biosci. Rep. 22, 501–511 [DOI] [PubMed] [Google Scholar]
- 50. Vorger P., and Ristori M. T. (1985) Effects of experimental anemia on the ATP content and the oxygen affinity of the blood in the rainbow trout (Salmo gairdnerii). Comp. Biochem. Physiol. A Comp. Physiol. 82, 221–224 [DOI] [PubMed] [Google Scholar]
- 51. Leitch J. M., and Carruthers A. (2007) ATP-dependent sugar transport complexity in human erythrocytes. Am. J. Physiol. Cell Physiol. 292, C974–C986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bae K., Lee K., Seo Y., Lee H., Kim D., and Choi I. (2006) Differential effects of two period genes on the physiology and proteomic profiles of mouse anterior tibialis muscles. Mol. Cells 22, 275–284 [PubMed] [Google Scholar]
- 53. Zidova Z., Kapralova K., Koralkova P., Mojzikova R., Dolezal D., Divoky V., and Horvathova M. (2014) DMT1-mutant erythrocytes have shortened life span, accelerated glycolysis and increased oxidative stress. Cell Physiol. Biochem. 34, 2221–2231 [DOI] [PubMed] [Google Scholar]
- 54. Yokoyama U., Numakura H., Takebe Y., Nagata N., and Nakamura S. (1971) [Congenital nonspherocytic hemolytic anemia with increased activity of erythrocyte ATP-ase]. Nihon Shonika Gakkai Zasshi 75, 341–347 [PubMed] [Google Scholar]
- 55. Fu L., Pelicano H., Liu J., Huang P., and Lee C. (2002) The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111, 41–50 [DOI] [PubMed] [Google Scholar]
- 56. Wang S., Dale G. L., Song P., Viollet B., and Zou M. H. (2010) AMPKα1 depletion shortens erythrocyte life span in mice: role of oxidative stress. J. Biol. Chem. 285, 19976–19985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sandoval S., Kraus C., Cho E. C., Cho M., Bies J., Manara E., Accordi B., Landaw E. M., Wolff L., Pigazzi M., and Sakamoto K. M. (2012) Sox4 cooperates with CREB in myeloid transformation. Blood 120, 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xu Y., Padiath Q. S., Shapiro R. E., Jones C. R., Wu S. C., Saigoh N., Saigoh K., Ptácek L. J., and Fu Y. H. (2005) Functional consequences of a CKIδ mutation causing familial advanced sleep phase syndrome. Nature 434, 640–644 [DOI] [PubMed] [Google Scholar]
- 59. Dieterle F., Ross A., Schlotterbeck G., and Senn H. (2006) Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 78, 4281–4290 [DOI] [PubMed] [Google Scholar]
- 60. Smolenski R. T., Lachno D. R., Ledingham S. J., and Yacoub M. H. (1990) Determination of sixteen nucleotides, nucleosides and bases using high-performance liquid chromatography and its application to the study of purine metabolism in hearts for transplantation. J. Chromatogr. 527, 414–420 [DOI] [PubMed] [Google Scholar]
- 61. Talansky B. E., Barg P. E., and Gordon J. W. (1987) Ion pump ATPase inhibitors block the fertilization of zona-free mouse oocytes by acrosome-reacted spermatozoa. J. Reprod. Fertil. 79, 447–455 [DOI] [PubMed] [Google Scholar]
- 62. Castaño J. G., Nieto A., and Felíu J. E. (1979) Inactivation of phosphofructokinase by glucagon in rat hepatocytes. J. Biol. Chem. 254, 5576–5579 [PubMed] [Google Scholar]
- 63. Reinila M., MacDonald E., Salem N. Jr, Linnoila M., and Trams E. G. (1982) Standardized method for the determination of human erythrocyte membrane adenosine triphosphatases. Anal. Biochem. 124, 19–26 [DOI] [PubMed] [Google Scholar]
- 64. Kedzierska K., Bober J., Ciechanowski K., Gołembiewska E., Kwiatkowska E., Noceń I., Dołegowska B., Dutkiewicz G., and Chlubek D. (2005) Copper modifies the activity of sodium-transporting systems in erythrocyte membrane in patients with essential hypertension. Biol. Trace Elem. Res. 107, 21–32 [DOI] [PubMed] [Google Scholar]
- 65. Spencer V. A., Sun J. M., Li L., and Davie J. R. (2003) Chromatin immunoprecipitation: a tool for studying histone acetylation and transcription factor binding. Methods 31, 67–75 [DOI] [PubMed] [Google Scholar]
- 66. Jung J. Y., Lee H. S., Kang D. G., Kim N. S., Cha M. H., Bang O. S., Ryu D. H., and Hwang G. S. (2011) 1H-NMR-based metabolomics study of cerebral infarction. Stroke 42, 1282–1288 [DOI] [PubMed] [Google Scholar]
- 67. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300 [Google Scholar]
- 68. Hochberg Y., and Benjamini Y. (1990) More powerful procedures for multiple significance testing. Stat. Med. 9, 811–818 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.