

Abstract
Alzheimer disease is associated with deposition of the amyloidogenic peptide Aβ in the brain. Passive immunization using Aβ-specific antibodies has been demonstrated to reduce amyloid deposition both in vitro and in vivo. Because N-terminally truncated pyroglutamate (pE)-modified Aβ species (AβpE3) exhibit enhanced aggregation potential and propensity to form toxic oligomers, they represent particularly attractive targets for antibody therapy. Here we present three separate monoclonal antibodies that specifically recognize AβpE3 with affinities of 1–10 nm and inhibit AβpE3 fibril formation in vitro. In vivo application of one of these resulted in improved memory in AβpE3 oligomer-treated mice. Crystal structures of Fab-AβpE3 complexes revealed two distinct binding modes for the peptide. Juxtaposition of pyroglutamate pE3 and the F4 side chain (the “pEF head”) confers a pronounced bulky hydrophobic nature to the AβpE3 N terminus that might explain the enhanced aggregation properties of the modified peptide. The deep burial of the pEF head by two of the antibodies explains their high target specificity and low cross-reactivity, making them promising candidates for the development of clinical antibodies.
Keywords: Alzheimer disease, amyloid-beta (Aβ), crystal structure, drug development, monoclonal antibody, thermodynamics, ligand binding
Introduction
Alzheimer disease (AD) 4 remains one of the most feared consequences of aging, affecting one in nine individuals over the age of 65 (1) and with most cases of dementia linked to AD pathology (2). The amyloid cascade hypothesis is now widely accepted as a major route to Alzheimer dementia (3). According to this hypothesis, a variety of processes leads to accumulation of amyloid β (Aβ) peptides in the form of monomers, oligomers, and fibrils in the brain, resulting in plaque deposition. As a consequence, neurosynaptic functions are impaired, the number of neurons decreases, and finally the brain loses functionality (4). Multiple drug discovery efforts targeting Aβ production and accumulation are currently being explored, with passive immunotherapeutic treatment by Aβ-specific antibodies being a promising approach to cure or prevent the progress of AD (5, 6).
It is commonly accepted that the N-terminal residues of Aβ are freely accessible within amyloid fibrils and oligomeric mixtures (7–9). These epitopes are therefore a particularly attractive target for passive immunotherapy, because binding to them can initiate antibody-mediated clearance of the toxic oligomers and fibrils. A number of antibodies directed to the Aβ N terminus have been reported, with binding modes determined for the Fab PFA1 (10–12) and the antibodies gantenerumab (13), bapineuzumab (14), and aducanumab (15). The latter preferentially recognizes an N-terminal conformational epitope that is present on aggregated Aβ but absent from monomers.
The majority of clinical trials with general anti-Aβ antibodies has been disappointing so far (16, 17), possibly because of patient and trial design issues, off-target saturation by soluble Aβ forms (18), and/or interference with physiological functions of soluble Aβ (19) such as regulation of synaptic activity (20) and action as an antimicrobial peptide (21). Interest has therefore shifted toward the new N-terminal-specific monoclonal antibodies such as gantenerumab and aducanumab (2), which specifically recognize oligomeric and fibrillar Aβ species and largely ignore monomeric Aβ. Recently, aducanumab has been shown to slow cognitive decline in mild AD subjects in early clinical trials (22), providing substantial support for these therapeutic approaches.
In human AD brain deposits, more than 60% of Aβ species are N-terminally truncated (23), with Aβ peptides starting with either amino acid Phe-4 (24), Arg-5, Ser-8, Gly-9, or Glu-11 (23, 25). Of particular interest is a peptide species lacking the first two amino acids Asp-1–Ala-2, in which the neo-N terminus (formerly E3) is converted into pyroglutamic acid (pE3) by glutaminyl cyclase (26–28). The modified peptide AβpE3 is abundant in cored and diffuse Aβ deposits, as well as in vascular amyloids in AD, presenilin-linked familial AD, and brains of Down syndrome patients (29–32). AβpE3 has been reported to comprise 15–45% of total Aβ in brains of AD patients (26, 33). Importantly, AβpE3 exhibits higher aggregation propensity (34), stability against degradation by aminopeptidases (35), and an increased neurotoxicity (36) compared with full-length Aβ. It has also been suggested that AβpE3 reverses Aβ1–42 fibrillogenesis via a prion-like mechanism (36), leading to increased formation of toxic oligomers. Thus the AD-associated peptide AβpE3, potentially a critical etiological agent for AD, represents an attractive therapeutic target.
Several antibodies against AβpE3 have been evaluated within the last 5 years. Treatment with the AβpE3 oligomer-specific antibody 9D5 in an AD transgenic mouse model (5XFAD) reduced the amount of cerebral Aβ levels and plaque burden (including general Aβ, Aβ40, Aβ42, and AβpE3) after only 6 weeks of passive immunization, leading to improved performance in the elevated plus maze test (37). A pilot passive immunization study using the antibody mAb07/1 reduced general Aβ, AβpE3 and fibrillar amyloid deposits in the hippocampus and cerebellum of APPswe/PS1Δ E9 mice (38) and induced behavioral improvements (39). The monoclonal antibody mE8 successfully lowered deposited Aβ without inducing microhemorrhage in vivo (18), although it failed to show significant plaque lowering when applied in a preventive manner.
In this work, we compare the in vitro and in vivo efficacy of three monoclonal antibodies raised against AβpE3, c#6 (mAb07/1), c#24, and c#17 and determine their target binding affinity and specificity. In addition, crystal structure analyses of the Fab-peptide complexes reveal two distinct ligand binding modes, as well as a distinctive bulky hydrophobic nature of the pE3–Phe-4 N-terminal region of AβpE3. Our results provide a framework for the engineering of humanized anti-AβpE3 antibodies, a prerequisite for their potential therapeutic application in passive AD immunotherapy.
Results
AβpE3-specific antibodies inhibit AβpE3–42 but not Aβ1–42 fibrillation
Three antibodies (c#6, c#17, and c#24) against pEFRHDS (the pyroglutamate-modified N-terminal fragment of the AβpE3 peptide) were selected from hybridoma cell supernatants. Western blot analyses confirm their specificity for full-length AβpE3 and demonstrate their ability to recognize and bind oligomers of AβpE3–42 (supplemental Fig. S1). Antibodies c#6 and c#24 belong to the murine IgG1 subclass, whereas c#17 is of subclass IgG2b. In a thioflavin T fibrillation assay, all three antibodies inhibit fibril formation of the (full-length) AβpE3–42-peptide (Fig. 1, A and B). Fibril formation of full-length non-pE-modified Aβ1–42 was not inhibited by any of the AβpE3-specific antibodies (Fig. 1C), underlining their specificity for pyroglutamate-modified Aβ.
Figure 1.
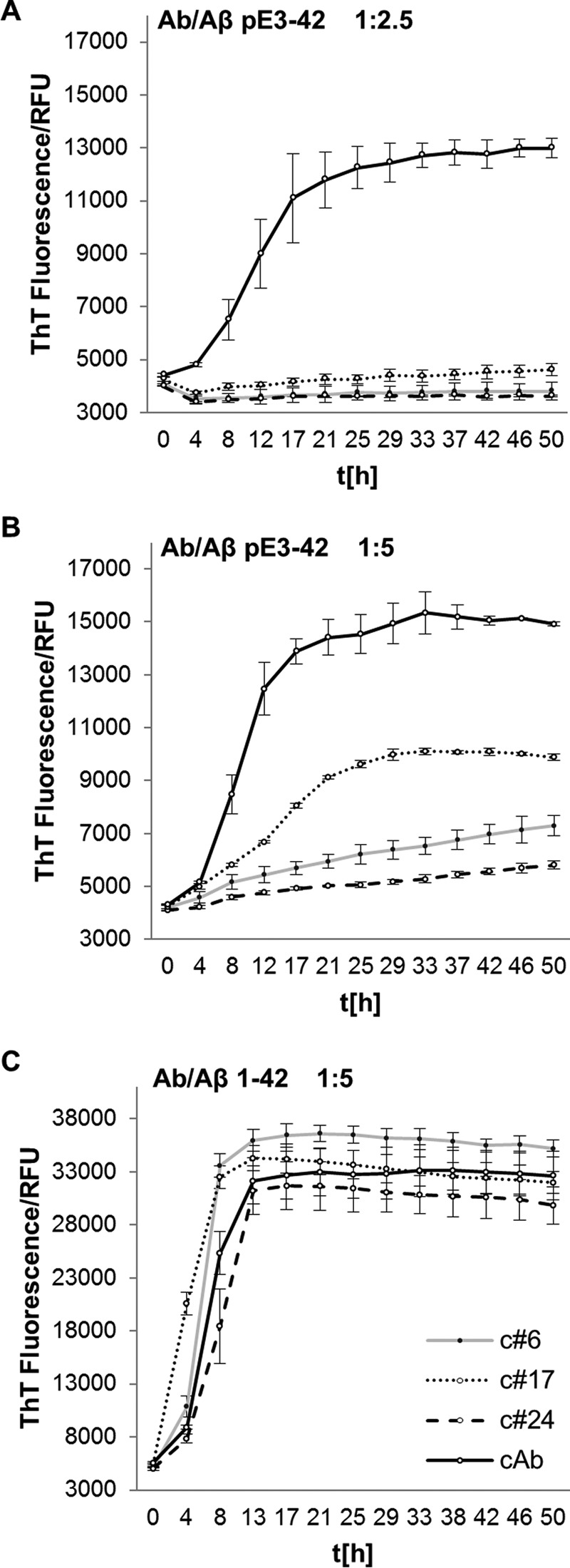
AβpE3-specific antibodies inhibit fibril formation. A and B, fibril formation, monitored by thioflavin T (ThT) fluorescence (measured in relative fluorescence units (RFU)) as a function of time [h], is inhibited by addition of AβpE3-specific antibodies (c#6, c#24, c#17) to 10 μm AβpE3–42 peptide in a molar ratio of 1:2.5 (4 μm Ab) (A) and 1:5 (2 μm Ab) (B). The MCP1-specific antibody, which does not recognize AβpE3, was used as control antibody (supplemental Fig. S3A). C, the antibodies have no influence on full-length Aβ1–42 peptide fibril formation (4 μm Ab, molar ratio of 1:5). The mean values and standard deviations were calculated based on three measurements. Because full-length (Aβx-42) peptides were used at high concentration for the inhibition studies, the enhanced aggregation rate described for AβpE3 is not apparent here (58).
Antibody c#6 inhibits AβpE3–42 oligomer-induced neuronal death and improves behavioral performance of AβpE3–42-treated animals
Exposure of rat cortical neurons to AβpE3–42 oligomers, which have been implicated to be a major toxic species in the development of Alzheimer disease (36), resulted in a significant and dose-dependent decrease in cell viability to 64.3 and 36.9% when treated with 1 and 5 μm AβpE3–42 oligomers, respectively (Fig. 2A), in agreement with previously published data (24, 40). Preincubation of AβpE3–42 oligomers with antibody c#6 increased cell viability in a dose-dependent manner, indicating that the antibody acts as an effective neuroprotectant against toxic AβpE3–42 oligomers.
Figure 2.

Antibody c#6 protects against oligomer-induced toxicity in cortical neurons and reverses AβpE3–42 oligomer induced impairment of spatial working memory. A, cortical neurons were incubated for 24 h with vehicle or AβpE3–42 oligomers at the indicated concentrations in the absence or presence of humanin (HNG, positive control) or antibody c#6 with increasing concentrations. Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (n = 3 determinations/condition). The data are represented as percentages of control. B, 4 days following ICV infusion of vehicle or AβpE3–42 oligomers, spontaneous alternation behavior (left) and the number of arm entries (right) were recorded during 5-min trials in the Y-maze test. The mice were divided in four groups: A, B, C, and D dependent on treatment with/without AβpE3–42 and/or antibody c#6 (see text for details). The data are represented as means ± standard deviation (n = 9–10) and considered statistically different at p < 0.05 as described under “Experimental procedures.”
In vivo effects on cognitive performance were investigated using the Y-maze test (Fig. 2B, left panel) (24, 40) with which we have previously shown that intracerebroventricular (ICV) injection of AβpE3–42 oligomers into mouse brains results in a dramatic impairment of spontaneous alternation behavior (40). Mice treated intraperitoneally with vehicle followed by ICV injection of AβpE3–42 oligomers (group B) demonstrated reduced alternation in the Y-maze test compared with the control group (A), reflecting a strong decrease in short term memory capacity (Fig. 2B, left panel). In contrast, mice administered intraperitoneally with antibody c#6 and challenged with AβpE3–42 oligomers (group D) exhibit alternation behavior equivalent to the control group (group C). Thus antibody c#6 prevents AβpE3–42 oligomer-induced impairment of spatial working memory. The number of arm entries did not differ statistically between experimental groups (Fig. 2B, right panel), indicating that the observed changes in alternation behavior were not due to generalized exploratory, locomotor, or motivational effects and thereby providing further validation of the Y-maze assay.
Affinity and kinetics of antibody binding to Aβ peptides
All three pyroglutamate-specific antibodies c#6, c#17, and c#24 bind to AβpE3–18 in the low nanomolar range (KD = 1–10 nm) as determined by isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR), irrespective of whether complete antibodies (two binding sites) or Fab fragments (one binding site) were analyzed (Table 1 and supplemental Figs. S2 and S3A). Binding in each case is exothermic (ΔG = ∼−11 kcal/mol) and enthalpically driven (ΔH > TΔS). Remarkably, each antibody possesses a distinct thermodynamic profile (quantified by the enthalpic and entropic contributions) for AβpE3–18 binding (supplemental Table S1). KD values derived from rate constants kon (binding) and koff (dissociation) measured using SPR are in good agreement with the binding affinities to AβpE3–18 determined by ITC (Table 1). Significant differences in the association and dissociation kinetics of the three antibodies from the ligand AβpE3–18 are observed: c#24 exhibits a 10-fold slower dissociation rate compared with c#6 and c#17 (Table 1 and supplemental Fig. S3A). The slower dissociation rate of c#24 is offset by a slower association rate (kon) (∼3-fold slower than c#6 and ∼10-fold slower than c#17), resulting in similar KD values for all three anti-AβpE3–18 antibodies. In addition, antibody c#6 exhibited high affinities for AβpE3–12 octamers (29.3 pm) and fibrils (3.75 pm) (supplemental Fig. S3, B and C), which are about 230 times (octamers) or 1800 times (fibrils) higher than that for AβpE3–12 monomer (KD = 6.7 nm) and are probably due to avidity effects.
Table 1.
Antibody/Fab binding analysis using ITC and SPR
| Clone | Aβ peptide | Kon in 1/Ms | koff in 1/s |
KD |
|
|---|---|---|---|---|---|
| SPR | ITC | ||||
| nm | nm | ||||
| 6 | Human pE3–18 pEFRHDSGYEVHHQKLV | 6.89 × 105 | 4.58 × 10−3 | 6.7 | 4.85 ± 0.35 |
| 24 | 2.41 × 105 | 0.42 × 10−3 | 1.8 | 6.85 ± 1.56 | |
| 17 | 25.0 × 105 | 3.30 × 10−3 | 1.3 | 0.99 ± 0.31 | |
| 6 | Human 1–18 DAEFRHDSGYEVHHQKLV | 184 | >5.70 × 10−3 | >30000 | |
| 24 | 69.9 | >0.88 × 10−3 | >12600 | ||
| 17 | 961 | 3.10 × 10−3 | 3120 | ||
| 6 | Human 2–18 AEFRHDSGYEVHHQKLV | — | — | — | |
| 24 | — | — | — | ||
| 17 | 0.0964 × 105 | 2.83 × 10−3 | 293 | ||
| 6 | Human 3–18 EFRHDSGYEVHHQKLV | 0.027 × 105 | 4.6 × 10−3 | 1720 | — |
| 24 | 0.012 × 105 | >0.657 × 10−3 | > 500 | — | |
| 17 | 0.074 × 105 | 2.41 × 10−3 | 326 | 16.3 × 103 | |
| 6/24/17 | Human 4–18 FRHDSGYEVHHQKLV | — | — | — | |
| 6 | mouse pE3–18 pEFGHDSGFEVRHQKLV | — | — | — | |
| 24 | — | — | — | ||
| 17 | 14.9 × 105 | 28.6 × 10−3 | 19 | ||
—, no detectable binding (maximal peptide concentration, 1000 nm; maximal 10,000 nm for Aβ4–18).
Antibody specificity for pyroglutamate in position 3 of AβpE3–18 was further probed using SPR to measure binding to different Aβ peptide species lacking pE3 (Aβ1–18, Aβ2–18, and Aβ3–18; Table 1 and supplemental Fig. S3, D–F). In accordance with results from ITC, antibodies c#6 and c#24 show no or very weak interaction with any of these peptides. In contrast, antibody c#17 possesses a measurable affinity for Aβ2–18 (KD = 293 nm) and Aβ3–18 (KD = 326 nm; 16.3 μm by ITC), albeit >200-fold weaker than for AβpE3–18 (KD = 1.3 nm). Similarly, no measurable binding of c#6 and c#24 was observed to the murine AβpE3–18 peptide, which differs by three amino acid exchanges compared with the human peptide (see Table 1 for sequence details). In contrast, c#17 binds the murine peptide with a KD of 19 nm. In summary, all three antibodies exhibit high affinity for the human AβpE3–18 peptide and little or no cross-reactivity with other Aβ species.
Crystal structures of Fab fragments in complex with AβpE3 peptides
Crystals diffracting from 1.5 to 2.2 Å were obtained for Fab fragments from all three antibodies complexed to various human and murine AβpE3 peptides (see Table 2 for data collection and refinement statistics). In each case, electron density could be assigned unequivocally to the first six N-terminal residues of the ligands (supplemental Fig. S4, A–D). Two distinct binding modes (I and II) were observed for the pE-modified Aβ peptide ligand (Fig. 3), and both differ substantially from those described previously for Aβ-specific antibodies (supplemental Fig. S5). For Fab c#6 and Fab c#24, which differ by only four amino acids in the variable domain of the light chain, the AβpE3 peptide buries a surface area of 818.4 and 932.3 Å2, respectively, with favorable surface complementarity (41). The peptide occupies a deep hydrophobic cavity between the Fab light and heavy chains lined by residues Tyr-31LC, Tyr-37LC, Leu-41LC, Val-94LC, Phe-99LC, Phe-101LC, Phe-103LC, Val-37HC, Trp-47HC, and Trp-109HC (Fig. 4, A and B) (residue numbers with the suffix LC/HC are from the light/heavy chains, respectively). The N-terminal pyroglutamate residue pE3 is buried deeply in the interface, with multiple hydrogen bonds between the peptide and residues of the Fab framework (between the backbone amides of pE3, Phe-4 and Arg-5 and the carboxylate group of Glu-99HC, between the carbonyl moieties of pE3 and Phe-4 and Asn-39LC and between the pE3 5-oxo group and the side chain of Thr-97HC (c#6) or Asn-35HC (c#24)). The guanidinium moiety of Arg-5 is held in place by hydrogen bonds to the main chain carbonyl groups of Gly-96LC and Thr-97LC. Slight differences in peptide orientation and binding are found for residues His-6, Asp-7, and Ser-8. In particular, AβpE3 peptide residue Asp-7 makes hydrogen bonds to Lys-35LC in c#24, whereas the latter residue is involved in an intramolecular salt bridge to Glu-E103HC in c#6.
Table 2.
X-ray data collection and refinement statistics
| Fab c#6/AβpE3–12PEGb | Fab c#24/AβpE3–18 | Fab c#17/AβpE3–12PEGb | Fab c#17/mAβpE3–18PEGb | |
|---|---|---|---|---|
| Data collection statistics | ||||
| Radiation source | Rotating anode | BESSY BL 14.1 | Rotating anode | BESSY BL 14.2 |
| Wavelength (Å) | 1.5418 | 0.9184 | 1.5418 | 0.9184 |
| Space group | P1 | C2 | P21 | P1 |
| Unit cell length (Å) | 53.79, 65.99, 67.77 | 115.63, 95.95, 90.38 | 43.17, 87.14, 58.03 | 40.91, 42.94, 57.98 |
| Unit cell angles (°) | 62.9, 82.9, 84.2 | 90.0, 101.2, 90.0 | 90.0, 96.06, 90.0 | 83.9, 83.3, 90.3 |
| Resolution range (Å) | 30–1.59 | 30–1.49 | 30–2.21 | 30–1.60 |
| Highest resolution shell (Å) | 1.69–1.59 | 1.58–1.49 | 2.31–2.21 | 1.70–1.60 |
| Rmerge | 3.4 (33.3) | 4.7 (64.4) | 8.1 (22.9) | 2.9 (41.3) |
| I /σI | 17.8 (2.0) | 19.0 (2.2) | 14.2/(3.4) | 16.3 (2.0) |
| Completeness (%) | 89.3 (54.3) | 98.3 (90.0) | 96.8 (78.5) | 94.5 (93.7) |
| CC (1/2) | 99.9 (79.0) | 99.9 (71.8) | 99.6 (89.6) | 99.9 (75.1) |
| Multiplicity | 2.8 (1.4) | 4.2 (4.0) | 4.6 (1.9) | 2.0 (2.0) |
| Solvent content/Fab per ASU | 41%/2 | 49%/2 | 45%/1 | 41.7% /1 |
| Model used for MR | 1ZEA | 1ZEA | 2DQT | c#17 |
| Refinement statistics | ||||
| Number of reflections (working/test set) | 99419/4972 | 154378/7719 | 40700/2042 | 48636/2433 |
| Rwork/Rfree | 0.180/0.215 | 0.181/0.213 | 0.186/0.249 | 0.173/0.210 |
| No. atoms | ||||
| Protein | 6941 | 6776 | 3321 | 3346 |
| Ligand | 86 | 246 | 54 | 47 |
| Water | 1157 | 1462 | 191 | 392 |
| B-factor (Å2) | ||||
| Protein | 18.4 | 18.7 | 23.8 | 24.6 |
| Ligand | 35.4 | 26.25 | 26.2 | 25.5 |
| Water | 29.8 | 31.5 | 26.1 | 34.3 |
| Bond length r.m.s.d. (Å) | 0.006 | 0.005 | 0.008 | 0.006 |
| Bond angles r.m.s.d. (°) | 0.86 | 0.83 | 0.99 | 0.86 |
| Ramachandran plot: (%) favored/outliers regions) | 97.7/0.1 | 98.2/0.1 | 97.5/0.0 | 98.6/0.0 |
| MolProbity clash score | 4.58 | 2.38 | 5.74 | 3.31 |
| PDB accession code | 5MYO | 5MYX | 5MY4 | 5MYK |
Figure 3.

AβpE3-specific antibodies bind the peptide in two different binding modes. A and B, N-terminal residues of the AβpE3 peptide (yellow spheres) bound in the binding pockets of Fab c#24 (binding mode I) (A) and Fab c#17 (binding mode II) (B) viewed along the direction of the antigen-binding groove. In binding mode I (observed also for Fab c#6), the N-terminal residues are deeply buried in a V-shaped cavity between the light (LC) and heavy (HC) chains, whereas in binding mode II the AβpE3-peptide is surface-located. The structures were superimposed using the Fv heavy chain Cαs.
Figure 4.

Binding pockets of the three AβpE3-specific antibodies. A–C, orientation of the AβpE3 peptide N-terminal residues (yellow sticks) in the binding pockets (surface representation) of Fab c#6 (A), Fab c#24 (B), and Fab c#17 (C). A and B, open book representation following rotation of light (LC, left panels) and heavy (HC, middle panels) chains by ±90° about a vertical axis respectively. C, top view obtained by rotation of 90° around a horizontal axis compared with Fig. 3B. Complementarity-determining region and framework residues of the LC are depicted in orange and light orange, respectively, whereas those of the HC are shown in cyan and light cyan; residues that form the interface between LC and HC are in gray. Underlined labels represent hydrophobic residues contacting the AβpE3 peptide. Right panels, 2D representations of the interactions of Fab c#6, c#24, and c#17 with AβpE3. Hydrophobic interactions ≥ 4.5 Å are separated by a gray line. Residues of all three Fabs that contact the AβpE3–12/18 peptide are summarized in supplemental Table S2.
In contrast, the Fab of c#17 binds AβpE3–18 in a more conventional manner, occupying a shallow surface groove formed by the complementarity-determining regions (Figs. 3B and 4C) and burying an accessible surface area of 616.4 Å2. The interaction is largely hydrophobic, involving only two residues from the light chain (Val-99LC and Pro-101LC), as well as heavy chain residues Trp-47HC, Phe-50HC, Tyr-59HC, and Tyr-99HC that surround the N terminus of AβpE3. A short antiparallel β-sheet is formed between pE3–Phe-4 and the Asp-106HC main chain together with a hydrogen bond between the pE3 5-oxo group and the Tyr-99HC hydroxyl. The surface location of the peptide explains the measurable yet weak affinity of c#17 for non-modified variants of the human Aβ peptide (Aβ2–18 and Aβ3–18; Table 1) and for murine AβpE3–18 (which as mentioned above shows three amino acid exchanges compared with the human peptide). The crystal structure of c#17 bound to murine AβpE3–18 reveals a very similar binding mode (supplemental Fig. S4D), with only residues pE3 to Asp-7 bound by the Fab fragment. The substitution of the human Arg-5 for G in mouse AβpE3–18 results in the loss of a single hydrogen bond between the Arg-5 guanidinium group and the backbone carbonyl oxygen of Ile-104HC. In contrast, the loss of multiple hydrogen bonds found between Arg-5 and c#6/c#24 (supplemental Fig. S4, A and B) provides an explanation for the lack of affinity of these two Fabs for murine AβpE3–18 (Table 1).
The AβpE3 N-terminal pE3–Phe-4 dipeptide forms a distinctive bulky hydrophobic “pEF head”
Although the ligand binding pockets of the three AβpE3-specific antibodies exhibit significant differences, the bound AβpE3 peptides possess striking structural similarities (Fig. 5). In each case, the γ-lactam ring of pE3 stacks parallel to the aromatic side chain of F4 to form what we term the pEF head (Fig. 5, A and B), with the hydrophilic moieties clustering at the periphery (supplemental Fig. S6). A search of the PDB for other pyroglutamate-containing proteins revealed the same pEF head structure at the N termini of a bacterial cytochrome c′ (pE1-F2, PDB code 2YLI; Fig. 5C) (42) and human α-amylase (pE1-Y2, PDB code 4GQR; Fig. 5D) (43). Interestingly, Phe-4 shows differences in NMR solution chemical shifts between non-modified AβE3Q-40 and AβpE3–40 (44). We therefore surmise that the pEF head is an inherent feature of the pE–Phe sequence and that its bulky hydrophobic nature might provide an explanation for the enhanced aggregation and fibrillation properties of AβpE3 peptides.
Figure 5.

The N-terminal dipeptide of AβpE3 exhibits a distinctive hydrophobic pEF head. Superposition of pE3–Phe-4 of the bound AβpE3 peptides from the binding pockets of Fab c#6 (yellow; see also supplemental Fig. S6) and Fab c#24 (A, magenta) and Fab c#17 (B, cyan) reveals equivalent juxtapositions of pE3 and Phe-4 to form the pEF head. This arrangement is also found at the N termini of the unrelated proteins cytochrome c (C, forest green; PDB code 2YLI) and human α-amylase (D, violet; PDB code 4X9Y).
Discussion
The use of anti-Aβ antibodies represents a promising treatment against AD. Although a number of monoclonal antibodies have been developed, initial clinical trials with anti-Aβ antibodies have by and large been disappointing (2, 16, 17). Nevertheless, recent data concerning the application of aducanumab, which preferentially binds aggregated Aβ rather than monomeric peptide, demonstrate that immunotherapy can indeed lead to reduction of brain Aβ plaques in AD patients (22). To circumvent existing pharmacological and clinical problems, we have chosen to specifically target AβpE3 for passive immunotherapy. In comparison to full-length Aβ1–42, the N-terminal pE3 modification renders AβpE3–42 more resistant to proteolytic degradation (35) and prone to aggregation (34) to yield oligomers with a toxic, transferrable structure (36). Hence, antibodies directed against AβpE3–42 harbor the potential to remove these particularly toxic species without interfering with the biological function of Aβ1–40/42 and the precursor protein (APP). Moreover, AβpE3 peptides have not been detected outside the CNS or in the cerebrospinal fluid in humans (33, 39, 45). Thus antibodies directed against AβpE3 should not accumulate with bound Aβ peptide in the bloodstream, possibly favoring central activity (18). In agreement with this, no increase of total Aβ in the blood (which has been seen for other Aβ binding antibodies) was observed upon treatment of APPswe/PS1DE9 mice with the anti-AβpE3 antibody c#6 (38). Conversely, these mice showed a decrease of total Aβ load (including both pE3 and non-pE3 Aβ peptide species) in the hippocampus and cerebellum, and prophylactic administration of c#6 in a preclinical murine model of AD resulted in cognitive improvements.
The structural investigations presented here provide valuable clues to understanding the exquisite specificity of our antibodies for the modified peptide AβpE3 without any significant cross-reactivity to Aβ1–42. Although a monomeric truncated N-terminal peptide sequence was used for co-crystallization, several lines of evidence provide support that the observed binding modes mirror their interaction with monomeric, fibrillary, and oligomeric forms of AβpE3–40/42. First, antibody binding to monomeric AβpE3–42 decreases or completely prevents fiber formation in a concentration-dependent manner in vitro. Second, SPR measurements demonstrate that the antibodies bind fibrils with KD values in the picomolar range (supplemental Fig. S3C), suggesting that the pyroglutamate modification is also accessible in the fibers, as has been shown for the free N terminus of full-length unmodified Aβ fibers (7–9). Finally, preincubation of toxic AβpE3 oligomers with antibody c#6 results in a strong suppression of oligomer-induced cortical rat neuron cell death in a concentration-dependent manner. These in vitro results were corroborated by improved cognitive behavior of c#6-pretreated mice in the Y-maze test following challenge with toxic AβpE3–42 oligomers, in line with similar in vivo studies in which the oligomeric AβpE3-specific antibody 9D5 reduced total Aβ levels and improved performances in the elevated plus maze test (37).
Although the Aβ1-x specific Fab PFA1 has also been shown to bind AβpE3–8 with low (3 μm) affinity (10), our antibodies c#6, c#24, and c#17 exhibit affinities between 1 and 10 nm for monomeric human AβpE3 peptides. Binding of c#6 to AβpE3 oligomers and fibrils is in the low picomolar range (30 and 4 pm, respectively), comparable with data published for mE8 (18). Despite similar KD values, target binding of the individual antibodies involves different enthalpic and entropic contributions, with c#24 exhibiting the most negative ΔH (−22 kcal/mol) and TΔS (−11 kcal/mol) values. Antibodies c#6 and c#24 are the most specific for AβpE3–18, characterized by a deep burial of the pEF head between light and heavy chain framework residues and little to no detectable binding to non-modified Aβ peptides. In contrast, the surface location of the peptide bound to antibody c#17 allows a low level degree of binding of unmodified peptides. In this respect, c#17 exhibits parallels to previously reported Aβ1-x specific Fabs (PFA1 and PFA2 (10), WO2 (11), 12A11, 12B4, and 10D5 (12)) and antibodies like gantenerumab and aducanumab (13, 15), which all bind their respective peptides in a surface-exposed extended conformation (shown for PFA1 in supplemental Fig. S5B). The one exception to this is the Aβ1-x-specific antibody bapineuzumab and its parent murine antibody 3D6 (14, 46), which bind the Aβ-N terminus with residues Asp-1–Phe-4 buried deeply in the interface between the light and heavy chains (supplemental Fig. S5C). The exquisite orthogonal selectivities of c#6 and c#24 for AβpE3 and bapineuzumab for unmodified Aβ1–40/42 (47) therefore provide valuable tools for dissecting the roles of these two peptide species in fibrillation and in Alzheimer disease.
The peculiar geometry observed for the pE3–Phe-4 N-terminal dipeptide (the pEF head) in all structures presented here—and found at the N termini of a bacterial cytochrome c′ (42) and human α-amylase (43)—may have relevance for the physicochemical properties of AβpE3. Loss of the N-terminal residues Asp-1 and Ala-2 and the E3 side chain leads not only to a reduction in hydrophilicity; juxtaposition of the pyroglutamate pE3 γ-lactam ring and the aromatic side chain of F4 results in a bulky hydrophobic moiety that would be difficult to accommodate in typical secondary structures. Indeed, NMR spectroscopy has demonstrated that the N-terminal pyroglutamate modification has a marked effect on Aβ secondary structure (48), with AβpE3–40 possessing a significantly decreased propensity to form helices compared with Aβ1–40. The pEF head is also likely to protect the modified peptide from degradation by aminopeptidases: the mammalian pyroglutamate peptidase type 2, a membrane anchored thyrotropin-releasing hormone-specific peptidase, is unable to process thyrotropin-releasing hormone (pGlu-His-Pro-NH2) when the naturally occurring second residue His-2 is substituted for Phe-F2 (49). Thus formation of the pEF head may lead to reduced clearance of Aβ carrying the pyroglutamate modification at position 3, as well as providing a plausible explanation for its enhanced aggregation and fibrillation. The deep burial of the pEF head by c#6 and c#24 may therefore confer particular advantages in neutralizing the adverse properties of AβpE3.
The thermodynamic, structural, and functional data presented here for the AβpE3-specific antibodies c#6, c#24, and c#17 provide a basis for humanization, with a view toward developing new biological entities with minimal immunogenicity for passive immunotherapies. In particular, antibodies c#6 (with its ability to inhibit AβpE3–42 oligomer toxicity, noting that the presence of soluble oligomeric Aβ correlates with key features of AD (50)) and c#24 (with its very-low koff value) represent promising candidates for the development of clinical antibodies for passive AD immunotherapy.
Experimental procedures
Cultivation of hybridoma cell lines, antibody expression, and purification
AβpE3-specific antibodies were generated by immunization of female BALB/c mice with the hexapeptide pEFRHDS conjugated to keyhole limpet hemocyanin (BioGenes). Hybridoma cell clones were produced by fusion of spleen cells of the immunized mice with the myeloma cell line SP2/0. Clones subtyped using the IsoStrip mouse monoclonal antibody isotyping kit (Roche) were selected using ELISA and SPR measurements. Antibodies were produced by growing and maintaining the hybridoma cell lines c#6, c#17, and c#24 in serum-free medium (Invitrogen) supplemented with 0.06 mg/ml gentamycin. Immunoglobulins were purified from culture supernatant by affinity chromatography using a 5-ml Protein G column (GE Healthcare). Bound antibodies were eluted using 2 m potassium thiocyanate, 40 mm Na2HPO4, pH 7, and dialyzed against PBS (138 mm NaCl, 8 mm Na2HPO4, 1.5 mm KH2PO4, 3 mm KCl, 2 mm EDTA, pH 7.13) overnight at 4 °C. Fab fragments were generated by digestion with papain (Pierce: 7BAEE for c#6 and c#17 and the more active 16–40 BAEE for c#24) for 24 h. Fc portions were removed chromatographically using a Protein G column (Pierce). The Fab fragments of antibodies c#6 and c#24, which bound to the column, were eluted using 100 mm glycine (pH 2.7) into a neutralizing solution of 1 m Tris (pH 9). In the case of antibody c#17, the Fab fragment was collected in the flow-through, with the Fc and undigested IgG remaining bound to protein G column. All Fab fragments were further purified by size exclusion chromatography (Superdex 75 column) to remove residual amounts of undigested antibody (supplemental Fig. S7). Concentrations of Fab fragments and antigen were determined by UV-visible spectrometry at a wavelength of 280 nm.
Aβ peptide preparation for in vitro and in vivo assays
Lyophilized Aβ1–42/AβpE3–42 peptides (Bachem H1368/H4796) were dissolved in 1,1,1,3,3,3-hexafluoro-2-isopropanol (HFIP) to dissolve preformed aggregates of the Aβ peptide. The peptide concentration was determined by absorption at 280 nm, applying an extinction coefficient of 1490 m−1 cm−1 for Aβx-40. A stock solution of 400 μm was prepared and divided into aliquots, and the HFIP was evaporated under a fume hood. Dry peptide films were stored at −80 °C. Immediately prior to analysis, peptide pellets were dissolved in 100 μl of 0.1 m NaOH and neutralized with 100 μl of 0.1 m HCl, and 1800 μl of phosphate buffer (50 mm Na2HPO4, 150 mm NaCl, pH8) added. Fibril formation of Aβ1–42 and AβpE3–42 was monitored following incubation of 10 μm monomeric peptide dissolved in phosphate buffer in the presence of 20 μm thioflavin T in 96-well microtiter plates (well volume, 150 μl). The plates were sealed with adhesive film and incubated in a plate reader at 37 °C for 2 days. The fluorescence intensity (excitation wavelength, 440 nm; emission wavelength, 490 nm) was measured every 20 min using a NOVOstar Microplate Reader spectrofluorometer (BGM Labtech). Triplicate assays of each sample were recorded within one plate. Aβ oligomers for the neuronal and in vivo assays were prepared according to established protocols, resulting in a mixture of stable trimers and tetramers of AβpE3–42, as well as minor traces of monomeric peptide.
Neuronal viability
All experiments were performed by SynAging at 6 days in vitro. Cortical neurons were prepared from Wistar rat fetuses on embryonic day 16 or 17 as described (24). The experiments were carried out in a 48-well plate in triplicate. AβpE3–42 oligomers (1.0 or 5.0 μm) or vehicle were incubated in 400 μl (total volume) of culture medium at room temperature for 10 min in the presence of the respective antibody at a concentration of 2.5 or 5 μm or vehicle (d-PBS; control). Afterward, an aliquot (120 μl) of this mixture per well of primary cells was added. After 24 h of incubation, neuronal viability was monitored with a BMG plate reader (Fluostar Galaxy) using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay as previously described (51). The data were collected and normalized to control (vehicle set to 100%). As positive control, cortical neurons were incubated with humanin peptide (final concentration, 0.1 μm) in the presence or absence of Aβ oligomers at the indicated concentration.
Analysis of working memory by the Y-maze test
All experiments were performed by SynAging. Immediate spatial working memory performance was assessed by recording spontaneous alternation behavior in a Y-maze as described previously (40). C57BL/6 J mice (14–16-week old; Janvier, Le Genest-Saint-Isle, France) were housed with free access to food and water, and kept in a constant environment (22 ± 2 °C, 50 ± 5% humidity, 12-h light cycle). Mice, which were housed individually from 1 week before the start of the experiment, received the antibody or vehicle by i.p. injection (12 mg/kg in a volume of 200 μl; 0.1 m PBS, pH 7.4) two times (9 and 2 days) prior to ICV injection of oligomers. After the second dose of antibody (or vehicle), the mice were anesthetized and received an ICV injection of AβpE3–42 oligomers (50 pmol in 1 μl) or vehicle (1 μl of 0.1 m PBS, pH 7.4) into the right ventricle, applying stereotaxic coordinates of the bregma AP −0.22, L-1.0 and D 2.5 in mm. The injection was carried out using a 10-μl Hamilton microsyringe fitted with a 26-gauge needle.
Cognitive performance was tested 4 days after ICV Aβ peptide dosing using the Y-maze test. Four experimental groups (12 mice/group) were used in this study: group A (control): twice i.p. vehicle and ICV injection of vehicle; group B (oligomers): twice i.p. vehicle and ICV injection of Aβ3(pE)-42 oligomers; group C (control item 1): twice i.p. antibody c#6 and ICV injection of vehicle; and group D (item 1 assay): twice i.p. antibody c#6 and ICV injection of AβpE3–42 oligomers. The maze (three arms positioned at equal angles) was made of opaque Perspex with each arm 40 cm long, 16 cm high, and 9 cm wide. The mice were placed in the middle of one arm and allowed to explore the maze freely during a 5-min session. The series of arm entries was recorded visually, and arm entry was considered to be complete when the hind paws of the mouse were completely placed in the arm. Alternation was defined as successive entries into the three arms on overlapping triplet sets. The degree of alternations was calculated as the percentage of actual (total alternations) to possible alternations (defined as the number of arm entries minus two). STAT VIEW computer software (SAS) was used for statistical analysis. A non-parametric analysis of variance (Kruskal–Wallis test) was carried out followed by non-parametric Mann–Whitney U tests to compare between the groups. The values with p < 0.05 were considered statistically significant. The data are presented as means ± standard deviation.
Peptide synthesis and purification for binding studies
Because full-length AβpE3–40/42 is poorly soluble, we used the more soluble C-terminally truncated human Aβ peptide sequences AβX-18. Peptides were synthesized on a 60-μmol scale by standard Fmoc solid phase peptide synthesis on Biotin-PEG-NovatagTM or Rink amide resin (Merck Millipore) using an automated Symphony Synthesizer (Rainin). Fmoc amino acids were activated with equimolar amounts of O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluroniumtetrafluoroborat/N-methylmorpholin in DMF. Fmoc deprotection was performed with 20% piperidine in DMF. Cleavage from the resin and global side chain deprotection was carried out using a mixture of TFA/1,2-ethandithiol/triisopropylsilane/water (94:2.5:2.5:1) for 4 h. The peptides, precipitated by means of cold diethylether, were collected by filtration, redissolved in acetonitrile/water, and purified by preparative HPLC (acetonitrile/water gradient; Phenomenex Luna C18 column).
AβpE3–12-octamers were prepared by coupling eight monomers of AβpE3–12-peptide to the branched lysine peptide {[(Pyr-FRHDSGYEV) 2-K]2-K]2-K}2-KKKKK (peptides and elephants GmbH, Hennigsdorf, Germany). Branched peptides were synthesized by Fmoc solid phase peptide synthesis using a standard Fmoc/tert-butyl protection scheme, with Fmoc-Lys (Fmoc)-OH used for lysine branch synthesis. Briefly, amino acids were coupled to Rink amide resin in 4-fold excess to the respective free N-terminal amino groups (two in case of the first branch, four in case of the second branch, and eight in case of the third branch), using 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium-hexafluorophosphate (0.9 equivalents to amino acid) and N-methylmorpholin (2 equivalents to amino acid) in DMF, as triple-couplings (3 × 15 min). Fmoc deprotection was done using standard protocols. AβpE3–42 fibrils were prepared as follows: AβpE3–42 (1 mm) dissolved in HFIP was incubated under a fume hood at room temperature for 5 h to evaporate the HFIP. Afterward the peptide film was dissolved in 100 mm NaOH to a final concentration of 500 μm for 10 min at room temperature and diluted further in 300 mm NaCl, 25 mm Na2HPO4, 25 mm KH2PO4, 0.01% (w/v) NaN3, pH 8.7, to 50 μm (pH was titrated to 8.7 with 100 mm HCl). After incubation of the fibrillation sample at 37 °C for 3 days, the resulting fibrils were centrifuged at 2000 × g for 5 min, and the fibril pellet was resuspended in 200 μl of running buffer. The purity and identity of the peptides were confirmed by analytical HPLC and MALDI-MS.
Isothermal titration calorimetry
Fab fragments were dialyzed against ITC buffer (150 mm NaCl, 25 mm Na2HPO4, 25 mm KH2PO4, 1 mm EDTA, pH 7.4) at 4 °C overnight. Measurements were performed at 20 °C using a VP-ITC MicroCalorimeter (MicroCal, Northampton, MA). A 10 μm solution of the lyophilized AβpE3–18 peptide dissolved in ITC buffer was injected in 15 cycles to the Fab fragment solution (1 μm) with a 5-min interval between injections. Binding enthalpies were corrected for dilution heat after titrating the peptide into ITC buffer. Analysis of the raw data and determination of association constants (KA), reaction stoichiometries (n), binding enthalpies (ΔH), and entropies (ΔS) were performed using the Origin Software of MicroCal, and the entropic contributions −TΔS at 20 °C calculated using the thermodynamic relation ΔG = −R·T·lnK = ΔH − TΔS. Similarly, whole antibodies were measured using 1 μm c#24 or c#17 antibody and 20 μm AβpE3–18 peptide, or 5 μm c#6 antibody and 100 μm AβpE3–18 peptide. Thermodynamic parameters were also determined for the interaction between the complete antibodies (4 μm) and the peptide lacking the N-terminal pyroglutamate modification AβE3–18 (80 μm).
Surface plasmon resonance (SPR)
Antibody affinity was also determined by kinetic analyses using a Biacore 3000 (GE Healthcare) equipped with a CM5 chip (GE Healthcare). An anti-mouse (capture) antibody (PA1 28555; Thermo Scientific) was coupled to the sensor surface via primary amino groups according to the manufacturer's instructions (amine coupling kit from Biacore; BR-1000-50), and the chip was rinsed for 1 h with HBS-EP buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, 0.005% (v/v) Surfactant P20), resulting in the covalent coupling of 16,300 response units of the anti-mouse antibody. The AβpE3-specific mouse antibodies (70 μl, diluted to 25 μg/ml) were injected at a flow rate of 10 μl/min until 1500 response units of mouse antibody was reached. Subsequently, the chip was washed with 100 μl/min HBS-EP until the response signal was stable. Sensograms were recorded for each peptide concentration (AβpE3–18: 0.2–50 nm; Aβx-18: 10 nm to 10 μm) for 60 min at a flow rate of 30 μl/min using HBS-EP running buffer. The association phase was monitored for 8 min (contact time) following injection of 240 μl of the peptide solution, and the dissociation followed by running HBS-EP over the chip surface for the remaining 52 min. After every cycle, Aβ peptides were flushed out to re-establish the baseline signal (see supplemental information for further experimental details). The data were evaluated using the BIA evaluation software 4.1 employing the 1:1 Langmuir binding model; no avidity effects are expected because of immobilization of the target antibody. Association and dissociation phases of the sensograms using all solute concentrations were fitted simultaneously to yield association rates, dissociation rates, and dissociation constants for each peptide.
Co-crystallization of antibody Fab fragments with AβpE3 peptides
Fabs c#6 and c#17 were mixed with a C-terminally biotinylated AβpE3 peptide (pEFRHDSGYEV-PEG-biotin) in a molar ratio of 1:1 to yield a final Fab concentration of 8 mg/ml. Crystals were grown at 13 °C by hanging drop vapor diffusion by mixing 1 μl of protein-peptide solution with an equal volume of reservoir and equilibrating against 500 μl of reservoir solution. Fab c#6-peptide crystals appeared after 6 days using 25.5% (w/v) PEG 4000, 15% glycerol, 170 mm ammonium sulfate, and Fab c#17-peptide crystals appeared after 14 days using 25% (w/v) PEG 3350, 100 mm bis-Tris, pH 5.5, 200 mm magnesium chloride containing 0.5% n-dodecyl-β-d-maltoside. Fab c#24 was mixed with the AβpE3–18 peptide (pEFRHDSGYEVHHQKLV) in a molar ratio of 1:1.2 and a final Fab concentration of 8 mg/ml. Crystals were grown at 13 °C using the sitting drop method by mixing 200 nl of protein with 200 nl of reservoir buffer and equilibrating against 70 μl of reservoir solution. Crystals appeared after 14 days in 20% w/v PEG 3000, 100 mm sodium citrate, pH 5.5.
Structure determination and refinement
Diffraction data from Fabs c#6 and c#17 in complex with AβpE3–12-PEG-biotin were collected in-house from single crystals at 100 K (20% ethylene glycol served as cryoprotectant) using a CCD detector (SATURN 944+; Rigaku Europe) mounted on a copper rotating anode source (RA Micro 007; Rigaku Europe). For Fab c#24 in complex with AβpE3–18, a data set was collected from a single crystal at 100 K on BESSY Beamline 14.1 (Helmholtz-Zentrum, Berlin, Germany) using a CCD detector (MX-225; Rayonics). Oscillation photographs were integrated, merged, and scaled using XDS (52), and the resolution limit was determined using CC(1/2) (54). The phases were determined by molecular replacement with the program PHASER (53), using PDB entries 1ZEA (Fab c#6 and Fab c#24) and 2DQT (Fab c#17) as search models. Model building and structure refinements were carried out using the programs COOT (55) and the PHENIX suite (56), respectively. The data collection and structure refinement statistics are summarized in Table 2. Structural figures were prepared using PyMOL (PyMOL Molecular Graphics System, version 1.5.0.3; Schrödinger) and 2D ligand binding representations using LIGPLOT (57). Buried accessible surface areas were calculated using the program QtPISA v1.18 (from the CCP4 suite) and shape complementarities using the program SC from the CCP4 suite (41).
Author contributions
A. P. conducted the majority of the experiments and analyzed the results. C. P. solved and analyzed the structure. M. K. participated in the collection and analysis of binding data. K. G. assisted in antibody production. T. P. performed cell-based assays and mouse experiments. The results were analyzed and discussed with I. L., H.-U. D., S. S., J.-U. R. and M. T. S. The project was conceived and designed by J.-U. R. and M. T. S., and all authors contributed to preparation of the manuscript.
Supplementary Material
Acknowledgments
We thank Hans-Henning Ludwig (Probiodrug) for peptide synthesis, Dagmar Schlenzig (Probiodrug/Fraunhofer Institute) for introductory assistance in the fibrillation assay, Thore Hettmann (Probiodrug) for useful discussions, and Uwe Müller (Free University Berlin at BESSY) for synchrotron time.
This work was supported in part by funds from the Deutsche Forschungsgemeinschaft GRK1026 Conformational Transitions in Macromolecular Interactions (to M. T. S.). M. K., H.-U. D., S. S., and J.-U. R. are consultants and former employees of Probiodrug. I. L. and H.-U. D. are shareholders and I. L. is chief development officer of Probiodrug.
The atomic coordinates and structure factors (codes 5MYO, 5MYX, 5MY4, and 5MYK) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains supplemental Tables S1 and S2 and Figs. S1–S7.
- AD
- Alzheimer disease
- pE
- pyroglutamate
- Aβ
- amyloid β
- AβpE3
- N-terminally truncated amyloid β with pyroglutamate at position 3
- LC
- light chain
- HC
- heavy chain
- ICV
- intracerebroventricular
- DMF
- dimethylformamide
- ITC
- isothermal titration calorimetry
- SPR
- surface plasmon resonance
- HFIP
- hexafluoro-2-propanol
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- Ab
- antibody.
References
- 1. Alzheimer Association (2016) 2016 Alzheimer's disease facts and figures. Alzheimer's Dement. 12, 1–80 [DOI] [PubMed] [Google Scholar]
- 2. Scheltens P., Blennow K., Breteler M. M., de Strooper B., Frisoni G. B., Salloway S., and Van der Flier W. M. (2016) Alzheimer's disease. Lancet 388, 505–517 [DOI] [PubMed] [Google Scholar]
- 3. Selkoe D. J., and Hardy J. (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8, 595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Querfurth H. W., and LaFerla F. M. (2010) Alzheimer's disease. N. Engl. J. Med. 362, 329–344 [DOI] [PubMed] [Google Scholar]
- 5. Lemere C. A., and Masliah E. (2010) Can Alzheimer disease be prevented by amyloid-β immunotherapy? Nat. Rev. Neurol. 6, 108–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lannfelt L., Relkin N. R., and Siemers E. R. (2014) Amyloid-β-directed immunotherapy for Alzheimer's disease. J. Intern. Med. 275, 284–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lu J. X., Qiang W., Yau W. M., Schwieters C. D., Meredith S. C., and Tycko R. (2013) Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell 154, 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Petkova A. T., Ishii Y., Balbach J. J., Antzutkin O. N., Leapman R. D., Delaglio F., and Tycko R. (2002) A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kheterpal I., Williams A., Murphy C., Bledsoe B., and Wetzel R. (2001) Structural features of the Aβ amyloid fibril elucidated by limited proteolysis. Biochemistry 40, 11757–11767 [DOI] [PubMed] [Google Scholar]
- 10. Gardberg A. S., Dice L. T., Ou S., Rich R. L., Helmbrecht E., Ko J., Wetzel R., Myszka D. G., Patterson P. H., and Dealwis C. (2007) Molecular basis for passive immunotherapy of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 104, 15659–15664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miles L. A., Wun K. S., Crespi G. A., Fodero-Tavoletti M. T., Galatis D., Bagley C. J., Beyreuther K., Masters C. L., Cappai R., McKinstry W. J., Barnham K. J., and Parker M. W. (2008) Amyloid-β-anti-amyloid-β complex structure reveals an extended conformation in the immunodominant B-cell epitope. J. Mol. Biol. 377, 181–192 [DOI] [PubMed] [Google Scholar]
- 12. Basi G. S., Feinberg H., Oshidari F., Anderson J., Barbour R., Baker J., Comery T. A., Diep L., Gill D., Johnson-Wood K., Goel A., Grantcharova K., Lee M., Li J., Partridge A., et al. (2010) Structural correlates of antibodies associated with acute reversal of amyloid β-related behavioral deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 285, 3417–3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bohrmann B., Baumann K., Benz J., Gerber F., Huber W., Knoflach F., Messer J., Oroszlan K., Rauchenberger R., Richter W. F., Rothe C., Urban M., Bardroff M., Winter M., Nordstedt C., et al. (2012) Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimers Dis. 28, 49–69 [DOI] [PubMed] [Google Scholar]
- 14. Miles L. A., Crespi G. A., Doughty L., and Parker M. W. (2013) Bapineuzumab captures the N-terminus of the Alzheimer's disease amyloid-β peptide in a helical conformation. Sci. Rep. 3, 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bussiere T., Weinreb P. H., Engber T., Rhodes K., Arndt J., Qian F., Dunstan R. W., Patel S., Grimm J., and Maier M. (2014) A method of reducing brain amyloid plaques using anti-Aβ antibodies. Patent WO2014089500 [Google Scholar]
- 16. Doody R. S., Thomas R. G., Farlow M., Iwatsubo T., Vellas B., Joffe S., Kieburtz K., Raman R., Sun X., Aisen P. S., Siemers E., Liu-Seifert H., Mohs R., Alzheimer's Disease Cooperative Study Steering Committee, and Solanezumab Study Group (2014) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N. Engl. J. Med. 370, 311–321 [DOI] [PubMed] [Google Scholar]
- 17. Salloway S., Sperling R., Fox N. C., Blennow K., Klunk W., Raskind M., Sabbagh M., Honig L. S., Porsteinsson A. P., Ferris S., Reichert M., Ketter N., Nejadnik B., Guenzler V., Miloslavsky M., et al. (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N. Engl. J. Med. 370, 322–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Demattos R. B., Lu J., Tang Y., Racke M. M., Delong C. A., Tzaferis J. A., Hole J. T., Forster B. M., McDonnell P. C., Liu F., Kinley R. D., Jordan W. H., and Hutton M. L. (2012) A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer's disease mice. Neuron 76, 908–920 [DOI] [PubMed] [Google Scholar]
- 19. Puzzo D., and Arancio O. (2013) Amyloid-β peptide: Dr. Jekyll or Mr. Hyde? J. Alzheimers Dis. 33, S111–S120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Puzzo D., Privitera L., Fa' M., Staniszewski A., Hashimoto G., Aziz F., Sakurai M., Ribe E. M., Troy C. M., Mercken M., Jung S. S., Palmeri A., and Arancio O. (2011) Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 69, 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumar D. K., Choi S. H., Washicosky K. J., Eimer W. A., Tucker S., Ghofrani J., Lefkowitz A., McColl G., Goldstein L. E., Tanzi R. E., and Moir R. D. (2016) Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci. Transl. Med. 8, 340ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sevigny J., Chiao P., Bussière T., Weinreb P. H., Williams L., Maier M., Dunstan R., Salloway S., Chen T., Ling Y., O'Gorman J., Qian F., Arastu M., Li M., Chollate S., et al. (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 537, 50–56 [DOI] [PubMed] [Google Scholar]
- 23. Sergeant N., Bombois S., Ghestem A., Drobecq H., Kostanjevecki V., Missiaen C., Wattez A., David J.-P., Vanmechelen E., Sergheraert C., and Delacourte A. (2003) Truncated β-amyloid peptide species in pre-clinical Alzheimer's disease as new targets for the vaccination approach. J. Neurochem. 85, 1581–1591 [DOI] [PubMed] [Google Scholar]
- 24. Bouter Y., Dietrich K., Wittnam J. L., Rezaei-Ghaleh N., Pillot T., Papot-Couturier S., Lefebvre T., Sprenger F., Wirths O., Zweckstetter M., and Bayer T. (2013) a N-truncated amyloid β (Aβ) 4–42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 126, 189–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Näslund J., Schierhorn A., Hellman U., Lannfelt L., Roses A. D., Tjernberg L. O., Silberring J., Gandy S. E., Winblad B., and Greengard P. (1994) Relative abundance of Alzheimer Aβ amyloid peptide variants in Alzheimer disease and normal aging. Proc. Natl. Acad. Sci. U.S.A. 91, 8378–8382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mori H., Takio K., Ogawara M., and Selkoe D. J. (1992) Mass spectrometry of purified amyloid β protein in Alzheimer's disease. J. Biol. Chem. 267, 17082–17086 [PubMed] [Google Scholar]
- 27. Saido T. C., Iwatsubo T., Mann D. M., Shimada H., Ihara Y., and Kawashima S. (1995) Dominant and differential deposition of distinct β-amyloid peptide species, Aβ N3(pE), in senile plaques. Neuron. 14, 457–466 [DOI] [PubMed] [Google Scholar]
- 28. Schilling S., Zeitschel U., Hoffmann T., Heiser U., Francke M., Kehlen A., Holzer M., Hutter-Paier B., Prokesch M., Windisch M., Jagla W., Schlenzig D., Lindner C., Rudolph T., Reuter G., et al. (2008) Glutaminyl cyclase inhibition attenuates pyroglutamate Aβ and Alzheimer's disease-like pathology. Nat. Med. 14, 1106–1111 [DOI] [PubMed] [Google Scholar]
- 29. Miravalle L., Calero M., Takao M., Roher A. E., Ghetti B., and Vidal R. (2005) Amino-terminally truncated Aβ peptide species are the main component of cotton wool plaques. Biochemistry 44, 10810–10821 [DOI] [PubMed] [Google Scholar]
- 30. Lemere C. A., Lopera F., Kosik K. S., Lendon C. L., Ossa J., Saido T. C., Yamaguchi H., Ruiz A., Martinez A., Madrigal L., Hincapie L., Arango J. C., Anthony D. C., Koo E. H., Goate A. M., et al. (1996) The E280A presenilin 1 Alzheimer mutation produces increased Aβ 42 deposition and severe cerebellar pathology. Nat. Med. 2, 1146–1150 [DOI] [PubMed] [Google Scholar]
- 31. Lemere C. A., Blusztajn J. K., Yamaguchi H., Wisniewski T., Saido T. C., and Selkoe D. J. (1996) Sequence of deposition of heterogeneous amyloid β-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol. Dis. 3, 16–32 [DOI] [PubMed] [Google Scholar]
- 32. Wirths O., Bethge T., Marcello A., Harmeier A., Jawhar S., Lucassen P. J., Multhaup G., Brody D. L., Esparza T., Ingelsson M., Kalimo H., Lannfelt L., and Bayer T. (2010) a Pyroglutamate Aβ pathology in APP/PS1KI mice, sporadic and familial Alzheimer's disease cases. J. Neural Transm. 117, 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu G., Miller R. A., Connolly B., Marcus J., Renger J., and Savage M. J. (2014) Pyroglutamate-modified amyloid-β protein demonstrates similar properties in an Alzheimer's disease familial mutant knock-in mouse and Alzheimer's disease brain. Neurodegener. Dis. 14, 53–66 [DOI] [PubMed] [Google Scholar]
- 34. Schilling S., Lauber T., Schaupp M., Manhart S., Scheel E., Böhm G., and Demuth H.-U. (2006) On the seeding and oligomerization of pGlu-amyloid peptides (in vitro). Biochemistry 45, 12393–12399 [DOI] [PubMed] [Google Scholar]
- 35. Kuo Y. M., Webster S., Emmerling M. R., De Lima N., and Roher A. E. (1998) Irreversible dimerization/tetramerization and post-translational modifications inhibit proteolytic degradation of Aβ peptides of Alzheimer's disease. Biochim. Biophys. Acta. 1406, 291–298 [DOI] [PubMed] [Google Scholar]
- 36. Nussbaum J. M., Schilling S., Cynis H., Silva A., Swanson E., Wangsanut T., Tayler K., Wiltgen B., Hatami A., Rönicke R., Reymann K., Hutter-Paier B., Alexandru A., Jagla W., Graubner S., et al. (2012) Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 485, 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wirths O., Erck C., Martens H., Harmeier A., Geumann C., Jawhar S., Kumar S., Multhaup G., Walter J., Ingelsson M., Degerman-Gunnarsson M., Kalimo H., Huitinga I., Lannfelt L., and Bayer T. (2010) Identification of low molecular weight pyroglutamate Aβ oligomers in Alzheimer disease: a novel tool for therapy and diagnosis. J. Biol. Chem. 285, 41517–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frost J. L., Liu B., Kleinschmidt M., Schilling S., Demuth H.-U., and Lemere C. A. (2012) Passive immunization against pyroglutamate-3 amyloid-β reduces plaque burden in Alzheimer-like transgenic mice: a pilot study. Neurodegener. Dis. 10, 265–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frost J. L., Liu B., Rahfeld J.-U., Kleinschmidt M., O'Nuallain B., Le K. X., Lues I., Caldarone B. J., Schilling S., Demuth H.-U., and Lemere C. A. (2015) An anti-pyroglutamate-3 Aβ vaccine reduces plaques and improves cognition in APPswe/PS1ΔE9 mice. Neurobiol. Aging 36, 3187–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Youssef I., Florent-Béchard S., Malaplate-Armand C., Koziel V., Bihain B., Olivier J.-L., Leininger-Muller B., Kriem B., Oster T., and Pillot T. (2008) N-truncated amyloid-β oligomers induce learning impairment and neuronal apoptosis. Neurobiol. Aging. 29, 1319–1333 [DOI] [PubMed] [Google Scholar]
- 41. Lawrence M. C., and Colman P. M. (1993) Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950 [DOI] [PubMed] [Google Scholar]
- 42. Antonyuk S. V., Rustage N., Petersen C. A., Arnst J. L., Heyes D. J., Sharma R., Berry N. G., Scrutton N. S., Eady R. R., Andrew C. R., and Hasnain S. S. (2011) Carbon monoxide poisoning is prevented by the energy costs of conformational changes in gas-binding haemproteins. Proc. Natl. Acad. Sci. U.S.A. 108, 15780–15785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Williams L. K., Li C., Withers S. G., and Brayer G. D. (2012) Order and disorder: differential structural impacts of myricetin and ethyl caffeate on human amylase, an antidiabetic target. J. Med. Chem. 55, 10177–10186 [DOI] [PubMed] [Google Scholar]
- 44. Dammers C., Gremer L., Neudecker P., Demuth H.-U., Schwarten M., and Willbold D. (2015) Purification and characterization of recombinant N-terminally pyroglutamate-modified amyloid-β variants and structural analysis by solution NMR spectroscopy. PLoS One 10, e0139710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bibl M., Gallus M., Welge V., Lehmann S., Sparbier K., Esselmann H., and Wiltfang J. (2012) Characterization of cerebrospinal fluid aminoterminally truncated and oxidized amyloid-β peptides. Proteomics Clin. Appl. 6, 163–169 [DOI] [PubMed] [Google Scholar]
- 46. Feinberg H., Saldanha J. W., Diep L., Goel A., Widom A., Veldman G. M., Weis W. I., Schenk D., and Basi G. S. (2014) Crystal structure reveals conservation of amyloid-β conformation recognized by 3D6 following humanization to bapineuzumab. Alzheimers Res. Ther. 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bouter Y., Socrates J., Lopez Noguerola J. S., Tucholla P., Crespi G. A., Parker M. W., Wiltfang J., Miles L. A., and Bayer T. A. (2015) Aβ targets of the biosimilar antibodies of bapineuzumab, crenezumab, solanezumab in comparison to an antibody against N-truncated Aβ in sporadic Alzheimer disease cases and mouse models. Acta Neuropathol. 130, 713–729 [DOI] [PubMed] [Google Scholar]
- 48. Sun N., Hartmann R., Lecher J., Stoldt M., Funke S. A., Gremer L., Ludwig H.-H., Demuth H.-U., Kleinschmidt M., and Willbold D. (2012) Structural analysis of the pyroglutamate-modified isoform of the Alzheimer's disease-related amyloid-β using NMR spectroscopy. J. Pept. Sci. 18, 691–695 [DOI] [PubMed] [Google Scholar]
- 49. Elmore M. A., Griffiths E. C., O'Connor B., and O'Cuinn G. (1990) Further characterization of the substrate specificity of a TRH hydrolysing pyroglutamate aminopeptidase from guinea-pig brain. Neuropeptides. 15, 31–36 [DOI] [PubMed] [Google Scholar]
- 50. Walsh D. M., and Selkoe D. J. (2007) Aβ oligomers: a decade of discovery. J. Neurochem. 101, 1172–1184 [DOI] [PubMed] [Google Scholar]
- 51. Garcia P., Youssef I., Utvik J. K., Florent-Béchard S., Barthélémy V., Malaplate-Armand C., Kriem B., Stenger C., Koziel V., Olivier J.-L., Escanye M.-C., Hanse M., Allouche A., Desbène C., Yen F. T., et al. (2010) Ciliary neurotrophic factor cell-based delivery prevents synaptic impairment and improves memory in mouse models of Alzheimer's disease. J. Neurosci. 30, 7516–7527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Karplus P. A., and Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wallace A. C., Laskowski R. A., and Thornton J. M. (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 [DOI] [PubMed] [Google Scholar]
- 58. Schlenzig D., Rönicke R., Cynis H., Ludwig H. H., Scheel E., Reymann K., Saido T., Hause G., Schilling S., and Demuth H. U. (2012) N-terminal pyroglutamate formation of Aβ38 and Aβ40 enforces oligomer formation and potency to disrupt hippocampal long-term potentiation. J. Neurochem. 121, 774–784 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.