

Abstract
Background
Mutations in PLA2G6, which encodes the calcium-independent phospholipase A2 group VI, cause neurodegeneration and diffuse cortical Lewy body formation by a yet undefined mechanism. We assessed whether altered protein glycosylation due to abnormal Golgi morphology might be a factor in the pathology of this disease.
Methods
Three patients presented with PLA2G6-associated neurodegeneration (PLAN); two had infantile neuroaxonal dystrophy (INAD) and one had adult-onset dystonia-parkinsonism. We analysed protein N-linked and O-linked glycosylation in cerebrospinal fluid, plasma, urine, and cultured skin fibroblasts using high performance liquid chromatography (HPLC) and matrix-assisted laser desorption ionisation - time of flight/mass spectrometry (MALDI-TOF/MS). We also assessed sialylation and Golgi morphology in cultured fibroblasts by immunofluorescence and performed rescue experiments using a lentiviral vector.
Results
The patients with INAD had PLA2G6 mutations NM_003560.2: c.[950G>T];[426–1077dup] and c.[1799G>A];[2221C>T] and the patient with dystonia-parkinsonism had PLA2G6 mutations NM_003560.2: c.[609G>A];[2222G>A]. All three patients had altered Golgi morphology and abnormalities of protein O-linked glycosylation and sialylation in cultured fibroblasts that were rescued by lentiviral overexpression of wild type PLA2G6.
Conclusions
Our findings add altered Golgi morphology, O-linked glycosylation and sialylation defects to the phenotypical spectrum of PLAN; these pathways are essential for correct processing and distribution of proteins. Lewy body and Tau pathology, two neuropathological features of PLAN, could emerge from these defects. Therefore, Golgi morphology, O-linked glycosylation and sialylation may play a role in the pathogenesis of PLAN and perhaps other neurodegenerative disorders.
INTRODUCTION
Biallelic mutations in the human calcium-independent phospholipase A2 gene, PLA2G6, cause PLA2G6-associated neurodegeneration (PLAN) disorders. One such disorder is infantile neuroaxonal dystrophy (INAD), which presents from age 6 months through 3 years, and is characterised by regression of intellectual and motor skills, hypotonia, decreased axial tone and paraplegia and, in 50% of the cases, brain iron accumulation.1,2 Cerebellar and optic nerve atrophy occur later in the disease course.2,3 A second variant, called ‘atypical’ neuroaxonal dystrophy (NAD) presents later in life and progresses more slowly. A third type of PLAN is juvenile or early adult-onset dystonia-parkinsonism. This disease is characterised by progressive speech, gait and neuropsychiatric problems and severe generalised brain atrophy without iron accumulation. It generally presents after the third decade of life and its presentation overlaps with atypical NAD.4–6
PLA2G6 mutations associated with INAD or atypical NAD result in loss of the 85/88 kDa calcium-independent phospholipase A2 lysophospholipase and phospholipase activities.7 In contrast, PLA2G6 mutations associated with dystonia-parkinsonism have increased activity towards phospholipid substrates, suggesting a possible gain-of-function disease mechanism.7 Splice variants of PLA2G6 encode a membrane bound protein, calcium-independent phospholipase A2 (iPLA2) VIA-1, and a cytosolic protein, iPLA2VIA-2.8 Various functions have been ascribed to PLA2G6, including phospholipid remodelling,9–13 maintenance of Golgi morphology and vesicular trafficking,14,15 mitochondrial homoeostasis,16,17 and cellular signalling leading to cell activation, proliferation, migration or apoptosis.18 However, the mechanism by which PLA2G6 mutations lead to PLAN has not yet been determined. As in some other neurodegenerative disorders postmortem brain tissue examination in PLAN revealed widespread α-synuclein-positive Lewy body pathology that is particularly severe in the neocortex.19,20 Additionally, there was accumulation of hyperphosphorylated Tau in threads, pretangles and tangles within the neocortex of early onset cases.19,20 These neuropathological features establish a possible link of PLAN to other degenerative disorders such as Parkinson’s disease and Alzheimer’s disease.
Since fragmentation of the Golgi is reported in neurodegeneration,21,22 we examined and found similar Golgi fragmentation in fibroblasts from our patients with PLAN. Consistent with the role of the Golgi in glycosylation, we observed altered O-linked protein glycosylation in fibroblasts from three patients with PLAN. An abnormality in the Golgi structure could lead to altered protein glycosylation, improper protein folding and protein aggregation;23,24 this could account for some or all of the clinical and pathological features of PLAN.
METHODS
Human subjects
Patients were enrolled in the National Institute of Health (NIH) Undiagnosed Diseases Program and in clinical protocol 76-HG-0238, ‘Diagnosis and Treatment of Patients with Inborn Errors of Metabolism or Other Genetic Disorders’. Patients or their parents gave written, informed consent.
Exome sequencing
Genomic DNA was extracted from whole blood using the Gentra Puregene Blood kit (Qiagen, Valencia, California, USA) according to the manufacturer’s specifications. Whole exome sequencing (WES) and analysis were performed as described.25–28 Variants were viewed, sorted and filtered using VarSifter,29 a graphical java tool to view, sort and filter the variants. The pathogenicity of variants was assessed using CDPred,30 in which a numerical score is assigned to each variant that can be aligned to a residue in the National Center for Biotechnology Information Conserved Domain database.31 Unaligned bases were assigned scores using the BLOSUM62 scoring matrix.32 Once we identified this PLA2G6 cohort, the exomes we had available were reanalysed with our updated pipeline, described in online supplementary data section 1 and supplementary tables S1–S3.
Sanger sequencing
The primer pairs used for the amplification of the regions of genomic DNA around the mutations are listed in online supplementary table S-4. PCR amplification was performed using Qiagen HotStarTaq master mix (Qiagen, Valencia, California, USA). The following conditions were used for amplification: 1 cycle of 95°C for 5 min, followed by 39 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 5 min. Unincorporated primers and nucleotides were removed using ExoSAP-IT reagent (USB, Cleveland, Ohio, USA). Sanger dideoxy sequencing of the PCR products was performed by Macrogen (Rockville, Maryland, USA). The sequences were aligned and analysed using Sequencher V.5.0.1 (Gene Codes, Ann Arbor, Michigan, USA). Mutation interpretation analysis was conducted using Alamut 2.0 (Interactive Biosoftware, San Diego, California, USA).
Multiplex ligation-dependent probe amplification
To identify deletions or duplications within PLA2G6 of patient 1, multiplex ligation-dependent probe amplification (MLPA) was performed by the Knight Diagnostic Laboratories at the Oregon Health and Science University.
Breakpoint analysis of the duplication
Total RNA was isolated from fibroblasts treated for 24 h with 100 μg/mL cycloheximide using the RNeasy mini kit (Qiagen, Valencia, California, USA) and 2 μg were reverse transcribed with Omniscript RT Kit (Qiagen, Valencia, California, USA) using random nanomer primers. Using this cDNA as template, the exon 7 to exon 4 junction was amplified by nested PCR with Platinum Taq High Fidelity DNA Polymerase (Life Technologies, Grand Island, New York, USA). The initial primer pair 5′-ACGTGAACAGCACCAGCTC-3′ and 5′-GAAGACGGTCTCTCCCTTG-3′ was used under the following conditions: 1 cycle of 94°C for 3 min, followed by 34 cycles of 94°C for 15 s, 65°C for 30 s, 68°C for 60 s, and a final extension at 68° C for 10 min. With the product from the initial PCR, the nested primer pair 5′-AACACGGCCCTGCACGTG-3′ and 5′-GTCGGTGACATCCATCTGAGTGTGG-3′ was used under the following conditions: 1 cycle of 94°C for 5 min, followed by 39 cycles of 94°C for 15 s, 62°C for 30 s and 68°C for 30 s. Sanger sequencing was done as described above.
Lentiviral transfer and expression of PLA2G6
Myc-DDK-tagged-Human phospholipase A2, group VI (PLA2G6) TrueORF Gold expression cDNA clones for transcript variants 1 (NM_003560) and 2 (NM_001004426) were obtained from Origene (Rockville, Maryland, USA). Both were amplified with the primer pair 5′-CACCATGCAGTTCTTT GGCCGCCTGGTC-3′ and 5′-TCAGGGTGAGAGCAGCA GCTGGATG-3′ and Platinum Pfx DNA Polymerase (Life Technologies, Grand Island, New York, USA) using 1 cycle of 94°C for 3 min, followed by 34 cycles of 94°C for 15 s, 65°C for 30 s, 68°C for 60 s, and a final extension at 68°C for 10 min. After gel purification (QIAquick Gel Extraction Kit, Qiagen, Valencia, California, USA) the PCR products were cloned into pENTR/D-TOPO using the pENTR directional Topo cloning kit (Invitrogen, Carlsbad, California, USA) and transfected into TOP10 chemically competent Escherichia coli. Plasmids were purified from single colony cultures (QIAprep Spin Miniprep Kit, Qiagen, Valencia, California, USA) and the PLA2G6 transcript variants were then cloned into pLenti6.3/V5-DEST using the Gateway cloning system (Invitrogen, Carlsbad, California, USA). The pLenti6.3 constructs were transfected into One Shot Stbl3 competent Escherichia coli and plasmids were purified from single colony cultures (QIAprep Spin Miniprep Kit, Qiagen, Valencia, California, USA). Virus was produced in 293FT cells using the ViraPower HiPerform Lentiviral expression system (Invitrogen, Carlsbad, California, USA). The virus-containing media was subsequently used in combination with polybrene infection/transfection reagent (EMD Millipore, Darmstadt, Germany) to infect fibroblasts of the three patients with PLAN. Selection for stable expression of the construct was achieved by culturing the cells in 5ug/mL blasticidin (Life Technologies, Grand Island, New York, USA).
Quantitative PCR
Total RNA was isolated from patient fibroblasts using the RNeasy mini kit and 3μg were reverse transcribed with Omniscript RT Kit (Qiagen, Valencia, California, USA). PLA2G6 quantitative PCR (qPCR) amplification was performed using 250 ng of cDNA, QuantiFast SYBR Green PCR Master Mix (Qiagen, Valencia, California, USA) and the primer pair 5′-TGATCAAGGCCCTCATCGTG-3′ and 5′-CCTTCCTGGTGACAAGTCTGC-3′. All data were standardised to glyceralde-hyde-3-phosphate dehydrogenase (GAPDH) expression measured using primer pair 5′-TGCACCACCAACTGCTTAGC-3′ and 5′-GGCATGGACTGTGGTCATGAG-3′. Real time qPCRs were performed with a 7500 Fast Applied Biosystems qPCR system with the following amplification conditions: 95°C for 10 min, followed by 50 cycles of 95°C for 15 s, 55°C for 30 s and 60°C for 30 s. Data were analysed using the manufacturer’s software (V.2.0.1) accompanying the 7500 Fast Applied Biosystems qPCR system.
Immunoblotting
Cells were grown to confluence in a 15-cm dish, washed twice with phosphate buffered saline (PBS), and cell lysate was collected in 600 μL radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, St Louis, Missouri, USA) containing Protease inhibitor cocktail (Roche, Manheim, Germany). Samples were sonicated for 10 min in an ultrasonic bath. 25μg of total protein, as determined by the DC Protein assay (BioRad, Hercules, Virginia, USA), was loaded on a 4% stacking—10% resolving polyacrylamide gel. Proteins were transferred to a 0.45-μm Immobilon-FL polyvinylidene fluoride (PVDF) transfer membrane (Millipore, Billerica, Massachusetts, USA) and probed with anti-iPLA2 (C-terminal region) produced in rabbit (SAB4200130, Sigma-Aldrich, St Louis, Missouri, USA) and monoclonal anti-GAPDH antibody produced in mouse (G8795, Sigma-Aldrich, St Louis, Missouri, USA). Li-cor Donkey antirabbit IRDye 680RD (Li-cor, Lincoln, Nebraska, USA) and Li-cor Donkey anti-mouse IRDye 800CW (Li-cor, Lincoln, Nebraska, USA) were used as secondary antibodies and fluorescence was measured on a 9140 Odyssey CLx infrared imaging system (Li-cor, Lincoln, Nebraska, USA). Bands were analysed using Image Studio software (Li-cor, Lincoln, Nebraska, USA).
Glycan analysis
Fibroblasts were cultured in Dulbecco’s modified Eagle medium, high glucose (Invitrogen, Carlsbad, California, USA) with 10% fetal bovine serum (Certified FBS; Invitrogen, Carlsbad, California, USA) and antibiotics-antimycotics (Invitrogen, Carlsbad, California, USA) until 80% confluence in a 15-cm culture disk. After a wash with PBS, the medium was changed to α modification of Eagle medium containing 1000 mg/L D-glucose (Corning Cellgro, Manassas, Virginia, USA), 15% FBS and antibiotics-antimycotics. Upon reaching 100% confluence the cells were washed twice with PBS and harvested using a cell scraper. The cells were then pelleted and washed with PBS by centrifugation. Fibroblast pellets were lysed in 200 μL PBS, and 200 μg protein from the cell lysate was denatured and precipitated with 2× volume of 100% propanol. N-linked and O-linked glycans were released from precipitated protein, and free oligosaccharides were purified from the supernatant.
N-linked and O-linked glycans from cerebrospinal fluid (CSF) were measured in a 200 μL sample that was concentrated to 30 μL using an Ultracel YM-10 filtration concentrator (EMD Millipore, Darmstadt, Germany).
N-linked glycans were released from 10 μL plasma samples, 30 μL CSF samples, and from the cell pellets using the PNGase F kit from New England Biolabs (Ipswich, Massachusetts, USA).33 O-linked glycans were released using β-elimination with sodium borohydride.33 Free oligosaccharides from urine samples containing 0.09 mg creatine were directly purified by solid phase extraction.34
The released N-linked glycans from plasma or cells and free oligosaccharides from urine or deprotonated total cell lysate were purified and desalted by solid phase extraction using a SepPak C18 and carbograph column. O-linked glycans from plasma or cells were purified and desalted using an AG 50W-X8 resin cation exchange column. N-linked glycans, O-linked glycans and oligosaccharides were permethylated with sodium hydroxide and iodomethane in dimethyl sulfoxide (DMSO).33,34 After permethylation, glycans were extracted with water/chloroform (2:1, vol/vol) in four steps and dried. Samples were then dissolved in 50% methanol, spotted with 11% 2,5-dihydroxylbenzoic acid matrix (1:1 vol/vol), and measured by MALDI-TOF using the positive mode on Ultraflex MALDI-TOF/TOF system (BrunkerDaltonics, Billerica, Massachusetts, USA). The quantification of O-linked glycans in fibroblast lysates was achieved by spiking glycans harvested from 150 μg of glycoproteins with 25 μM of C13-labelled T antigen (m/z 543) and C13-labelled sialylated T antigen (m/z 909). The disialylated T antigen and core 2 species were quantified using to C13-labelled sialylated T antigen.33
Lectin staining of cultured fibroblasts
For lectin staining of cultured fibroblasts, biotinylated Maackia amurensis lectin II (MAL II), fluorescein Sambucus nigra (elderberry) bark lectin (SNA), rhodamine peanut agglutinin (PNA), and fluorescein succinylated wheat germ agglutinin (s-WGA) were obtained from Vector Laboratories (Burlingame, California, USA).
Patient and control fibroblasts were seeded at 0.5×105 per 12-mm round coverslip in a 24-well dish. After an overnight incubation at 37°C, the cells were fixed with 4% paraformaldehyde in PBS and subsequently permeabilised with 0.5% NP-40 in PBS. Cells were then incubated on a rocker for 1 h at room temperature in 4% bovine serum albumin (BSA) in PBS and subsequently for 2 h with lectins for the detection of terminal sialic acid (20 μg/mL MAL II; 20 μg/mL SNA in 4% BSA in PBS), a lectin against terminal galactose (20 μg/mL PNA in 4% BSA in PBS), or with a lectin for the detection of terminal N-Acetylglucosamine (GlcNAc) (20 μg/mL s-WGA in 4% BSA in PBS). For detection of biotinylated MAL II, cells were secondarily incubated with Streptavidin Alexa Fluor 350 (Life Technologies, Grand Island, New York, USA) for 1 h. Before fixing and staining with MAL II, SNA and PNA, one set of control fibroblasts was treated for 1 h with 1 mU/mL neuraminidase (Sigma-Aldrich, St Louis, Missouri, USA) in Hank’s balanced salt solution (Life Technologies, Grand Island, New York, USA). Coverslips were mounted on slides using Prolong Gold antifade reagent (Life Technologies, Grand Island, New York, USA) and imaged using a 20× objective on a Zeiss LSM 700 confocal laser-scanning microscope (Carl Zeiss Microscopy GmbH, Jena, Germany). Ten images were obtained per coverslip and processed with LSM software ZEN Black 2012 software (Carl Zeiss Microscopy GmbH, Jena, Germany); mean pixel intensities were obtained in the regions where cells were present.
Analysis of fibroblast ER-Golgi intermediate compartment and Golgi by immunofluorescence
Fibroblasts were seeded, fixed and permeabilised as described above. Next, coverslips were incubated on a rocker for 1 h at room temperature in 4% BSA in PBS. To visualise the ER-Golgi intermediate compartment (ERGIC), coverslips were incubated for 2 h with anti-ERGIC-53/p58 antibody produced in rabbit (E1031; Sigma-Aldrich, St Louis, Missouri, USA), co-stained with anti-GM130 produced in mouse (610822; BD Biosciences, Franklin Lakes, New Jersey, USA) and Hoescht 33342 (Life Technologies, Grand Island, New York, USA) to visualise the cis-Golgi and nucleus. After three washes with PBS, the coverslips were incubated for 1 h with secondary Alexa fluor-488 goat antirabbit IgG (H+L; A11034) and Alexa fluor-555 goat antimouse IgG (H+L; A21424; Life Technologies, Grand Island, New York, USA).
For measurement of the Golgi area, coverslips were incubated for 2 h with anti-GM130 for the cis-Golgi and anti-TGN-46 produced in sheep (GTX74290; GeneTex, Irvine, California, USA) for the trans-Golgi, co-stained with Hoescht to visualise the nucleus. After three washes with PBS, the coverslips were incubated for 1 h with secondary Alexa fluor-488 goat antimouse IgG (H+L; A11029) and Alexa fluor-555 donkey antisheep IgG (H +L; A21436; Life Technologies, Grand Island, New York, USA). A second set of coverslips was incubated for 2 h with anti-golgin-97 mouse monoclonal (CDF4) antibody (A21270; Life Technologies, Grand Island, New York, USA). After three washes with PBS, the coverslips were incubated for 1 h with secondary Alexa fluor-488 goat anti-mouse IgG (H+L; A21270; Life Technologies, Grand Island, New York, USA). The coverslips were mounted on slides and imaged as described above. Ten images were obtained per coverslip and processed with LSM software ZEN Black 2012 software to measure the intensity of the ERGIC stain and the area occupied by the Golgi.
Statistical analyses
Statistical significance for immunofluorescence intensities and Golgi area between patient fibroblasts and control fibroblasts as well as between levels in fibroblasts with and without overexpression of PLA2G6 was determined using a two-tailed Student’s t test.
RESULTS
Patients
The clinical findings of patients 1 and 3 with INAD and patient 2 with adult-onset dystonia-parkinsonism were typical for these respective disorders and are listed in table 1.
Table 1.
Clinical features of three patients with biallelic mutations in PLA2G6
| Patients
|
|||
|---|---|---|---|
| Clinical features | 1 | 2 | 3 |
| Age of onset | 18 months | 29 years | 9 months |
| Gender | Female | Female | Male |
| Normal early motor development | + | + | + |
| Normal early cognitive development | + | + | + |
| Developmental regression/cognitive decline | + | + | + |
| Difficulty swallowing | + | + | + |
| Upgaze palsy | + | + | * |
| Bilateral optic atrophy | + | + | |
| Oculomotor apraxia | + | * | |
| Generalised hypotonia | + | + | |
| Cerebellar atrophy | + | + | + |
| Midbrain atrophy | + | + | |
| Brain iron accumulation | + | + | |
| Cerebral atrophy | + | + | + |
| Axial dystonia | + | ||
| Parkinsonism | + | ||
| Blepharospasm | + | ||
| Lower limb spasticity | + | ||
| Axonal sensorimotor neuropathy | + | + | |
| Family history of parkinsonism | + | + | |
For patient 3, upgaze palsy and oculomotor apraxia could not be assessed because this patient was too debilitated to produce reliable eye movements on command.
Molecular studies identify biallelic PLA2G6 mutations in each individual
Different transcripts of PLA2G6 encode the two active isoforms, membrane bound iPLA2VIA-1 and cytosolic iPLA2VIA-2 (figure 1A). Our three patients’ mutations affected both isoforms (figure 1A).
Figure 1.
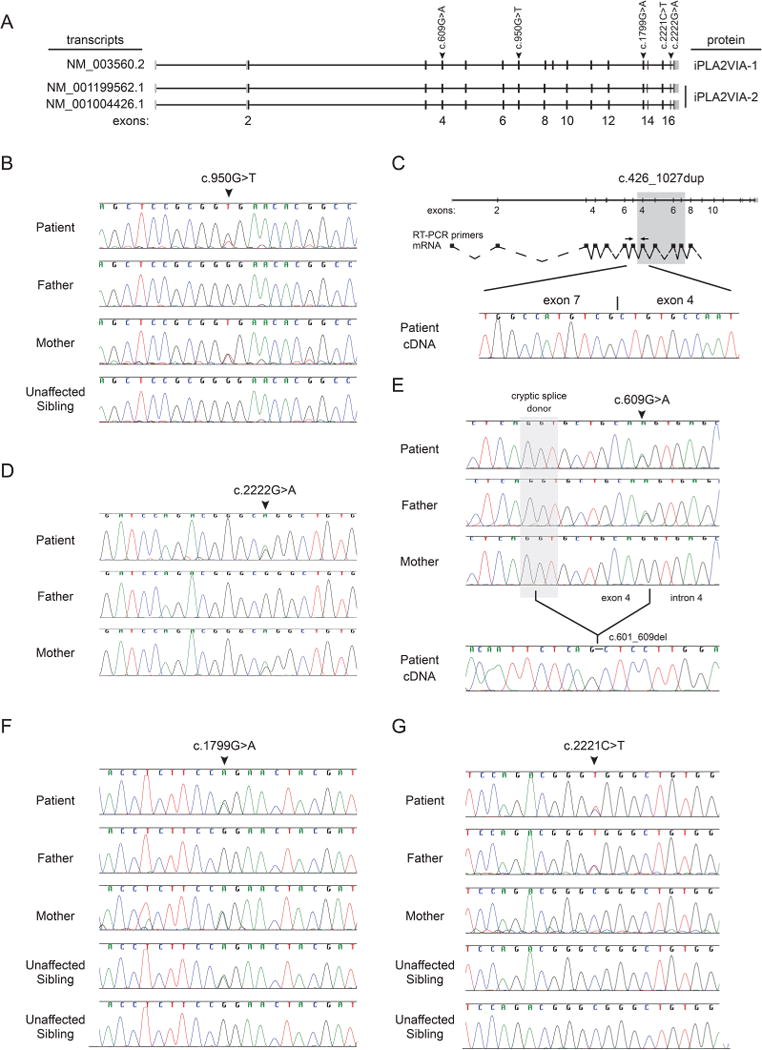
Mutation analysis of the biallelic PLA2G6 mutations in each patient. (A) Schematic overview of PLA2G6 transcripts encoding the two active isoforms, membrane bound iPLA2VIA-1 and cytosolic iPLA2VIA-2, and the mutations observed in the three patients. (B) gDNA sequence analysis of the region around the maternally inherited mutation NM_003560.2:c.950C>T in the PLA2G6 gene for each member of the family of patient 1. (C) Sequence analysis of the paternally inherited duplication of exons 4–7 (NM_003560.2:c.426–1077dup) with the resulting transcript and the location of the PCR primers on top. The cDNA amplification products, showing the exon 7–exon 4 junction in patient 1, are on the bottom. (D) gDNA sequence analysis of the region around the maternally inherited mutation NM_003560.2:c.2222G>A in the PLA2G6 gene for each member of the family of patient 2. (E) gDNA sequence analysis of the region around the paternally inherited synonymous mutation NM_003560.2: c.609G>A in the PLA2G6 gene for each member of the family of patient 2 and cDNA sequence analysis showing the splicing mutation encoding an in-frame 9 base pair deletion. (F) gDNA sequence analysis of the region around the maternally inherited mutation NM_003560.2:c.1799G>A in the PLA2G6 gene for each member of the family of patient 3. (G) gDNA sequence analysis of the region around the paternally inherited mutation NM_003560.2:c.2221C>T in the PLA2G6 gene for each member of the family of patient 3.
For patient 1, WES (see online supplementary data section 1) and targeted Sanger sequencing identified the novel maternally inherited PLA2G6 mutation NM_003560.2: c.950G>T (figure 1B). The paternally inherited duplication (NM_003560.2: c.426-1077dup) that was previously reported in an unrelated patient with PLAN,35 was ascertained using Multiplex ligation-dependent probe amplification (MLPA). Sanger sequencing confirmed the exon 7 - exon 4 junction in the cDNA that leads to a frameshift and premature termination (figure 1C).
For patient 2, WES identified the PLA2G6 mutations NM_003560.2: c.[2222G>A];[609G>A] (figure 1D, E, see online supplementary data section 1). The former was previously associated with dystonia-parkinsonism.5 The latter, a previously unreported mutation, alters the last base of exon 4 and leads to splicing from a cryptic splice donor to cause a deletion of NM_003560.2: c.601–609 (figure 1E).
For patient 3, targeted Sanger sequencing identified PLA2G6 mutations NM_003560.2: c.[1799G>A];[2221C>T] (figure 1F, G). Both mutations have previously been identified in unrelated patients with PLAN.2,7,20
PLA2G6 mutations decrease expression of iPLA2VIA-1 but not iPLA2VIA-2
We used cultured skin fibroblasts to analyse mRNA and protein levels from each affected individual. qPCR results indicated that the amount of PLA2G6 mRNA produced by each of the three patients’ fibroblasts was reduced 2.4–3.4-fold (figure 2A). Results from a qPCR specific for NM_003560.2, encoding iPLA2VIA-1, detected a decrease of 2.5–3.4-fold in all three patients (figure 2A). Despite these relatively minor reductions in NM_003560.2 mRNA levels, iPLA2VIA-1 was undetectable by immunoblotting, whereas iPLA2A-2 was present at control levels (figure 2B). This suggests that, although the mutations are present in both isoforms, they have profoundly different effects on the stability of the membrane and cytosolic forms of iPLA2VIA.
Figure 2.
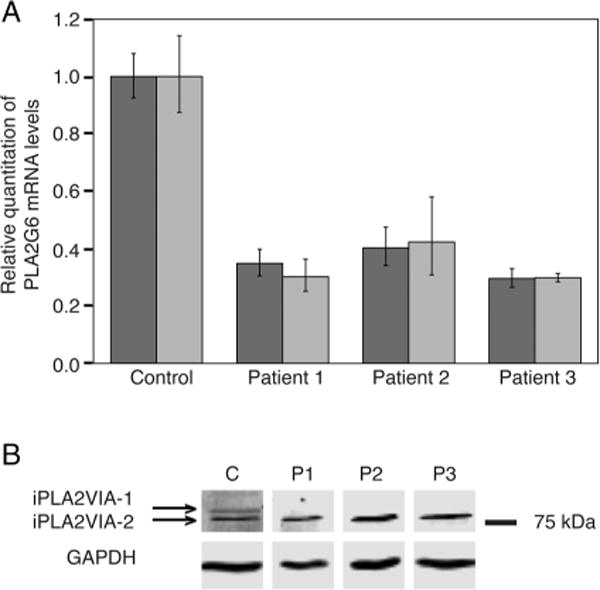
mRNA and protein expression analysis in fibroblasts. (A) Expression analysis of iPLA2GIVA-1 (dark bars) and iPLA2VIA-2 (light bars) mRNA levels in fibroblasts of each patient and a control cell line. Expression was normalised to GAPDH and plotted relative to the control. Error bars show SEM (B) Analysis of protein expression by western blot. GAPDH protein served as a loading control.
Patients with INAD and dystonia-parkinsonism have altered protein O-linked glycosylation in fibroblasts that is rescued by iPLA2VIA-1
All three patients had markedly altered protein O-linked glycosylation profiles in cultured skin fibroblasts (figure 3); the sialylated species were particularly decreased (table 2). Additionally, some mild decreases in galactosylation and sialylation were observed in the N-glycan profiles (data not shown). Lentiviral introduction of wild type iPLA2VIA-1 into patient fibroblasts normalised the O-linked glycosylation profiles (table 2, figure 3 and see online supplementary figure S-1), whereas introduction of iPLA2VIA-2 had minimal effect (data not shown). Lectin staining against α-2,3- and α-2,6-sialic acid (figure 4A, see online supplementary figure S-2 and S-3), as well as against terminal GlcNAc (see online supplementary figure S-2C and S-4), confirmed the rescue with iPLA2VIA-1 but not iPLA2VIA-2.
Figure 3.
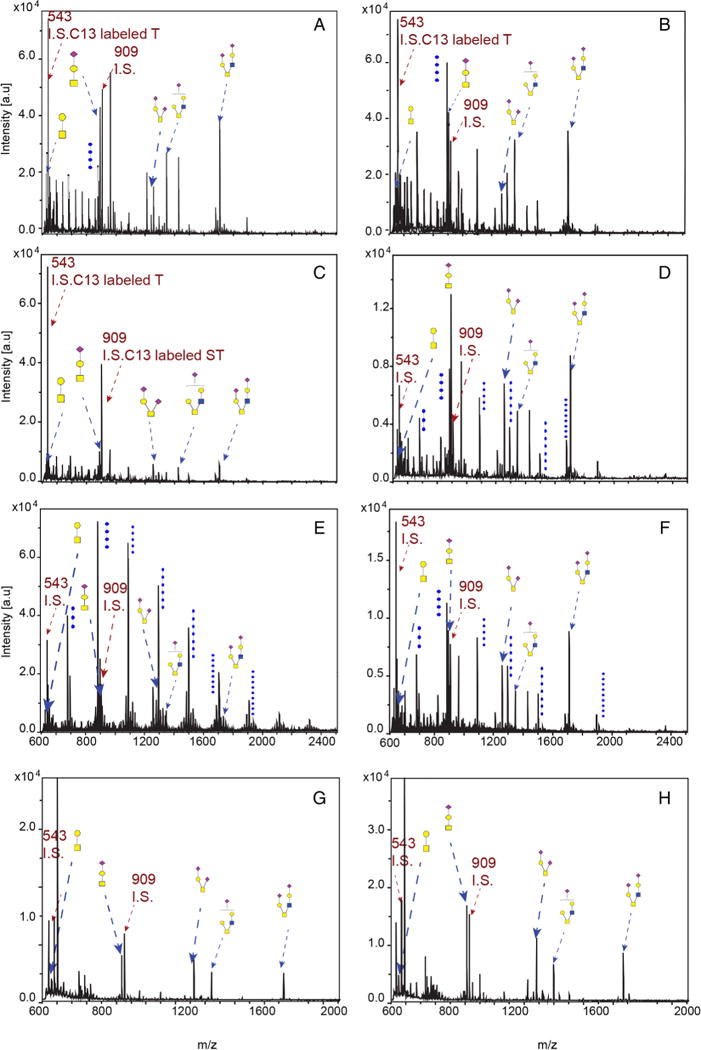
O-linked glycan profiles in cultured skin fibroblasts with and without overexpression of iPLAVIA-1. O-linked glycan profiles of control fibroblasts (A), control fibroblasts with iPLA2VIA-1 overexpression (B), fibroblasts from patient 1 (C), fibroblasts from patient 1 with iPLA2VIA-1 overexpression (D), fibroblasts from patient 2 (E), fibroblasts from patient 2 with iPLA2VIA-1 overexpression (F), fibroblasts from patient 3 (G) and fibroblasts from patient 3 with iPLA2VIA-1 overexpression (H). O-linked glycans are highlighted with dashed arrows. Samples were spiked with the internal standards (IS) C13-labelled T antigen and C13-labelled sialylated T.
Table 2.
Fibroblast O-linked glycan analysis
| O-glycan (m/z) | Control | Patient 1 | Patient 1 Rescue | Patient 2 | Patient 2 Rescue | Patient 3 | Patient 3 Rescue |
|---|---|---|---|---|---|---|---|
| Core 1 | |||||||
| m/z 534 | 1.21 | 0.14 | 0.10 | 0.00 | 0.34 | 0.43 | 0.71 |
| m/z 895* | 7.04 | 0.60 | 1.72 | 0.18 | 4.97 | 0.85 | 1.91 |
| m/z 1256† | 2.86 | 1.09 | 1.88 | 0.60 | 3.43 | 1.50 | 1.20 |
| Core 2 | |||||||
| m/z 1344* | 5.65 | 0.48 | 1.18 | 0.10 | 2.77 | 1.30 | 1.30 |
| m/z 1705† | 4.96 | 1.51 | 5.22 | 0.29 | 9.13 | 1.78 | 2.72 |
Values are in (pmole/μg protein).
Monosialylated.
Disialylated.
Rescue was established by lentiviral overexpression of wild type iPLA2VIA-1 (Also see online supplementary figure S-1).
Quantification of the core 1 T antigen (m/z=534) and sialylated T antigen (m/z=895) levels in fibroblast lysates was accomplished by obtaining their relative MALDI-TOF intensity compared with the C13-labelled standard at m/z 543. Disialylated core 1 T antigen (m/z 1256) and the core 2 species (m/z 1344 and 1705) were quantified using the C13-labelled sialylated T antigen at m/z 909 as internal standard (see figure 3).
Figure 4.
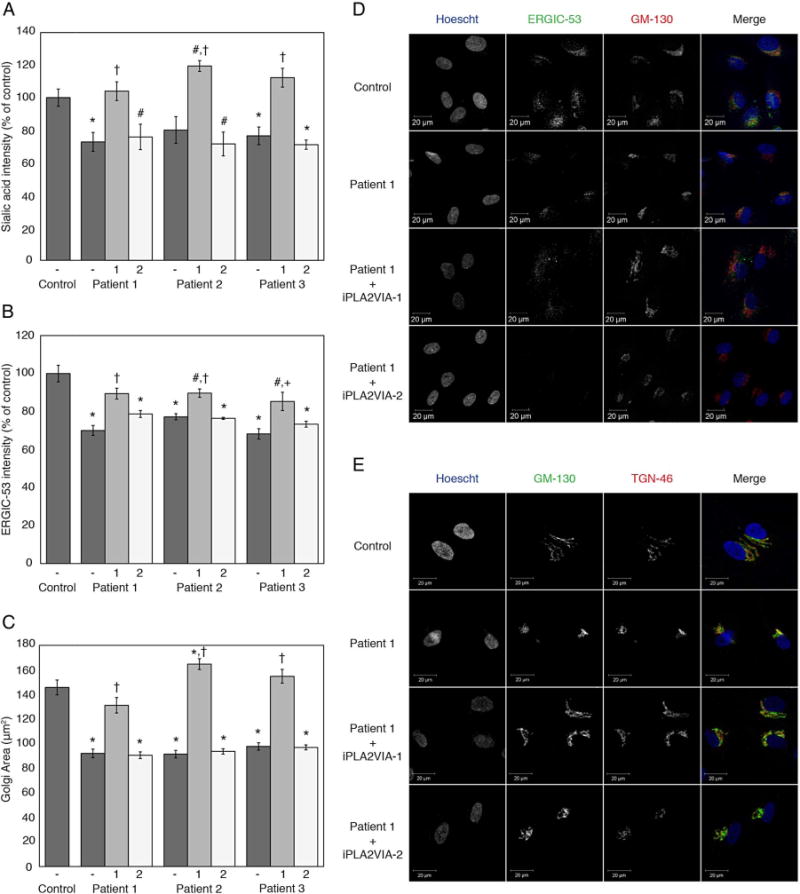
Quantification of ER-Golgi intermediate compartment (ERGIC) and Golgi area by immunofluorescence and quantification of terminal sialic acid and terminal GlcNAc by lectin staining. (A) Graph of relative fluorescent intensity for Maackia amurensis lectin II (MAL II) and Sambucus nigra (elderberry) bark lectin (SNA) staining of terminal sialic acid. Data from the patient fibroblasts without (−) or with overexpression of iPLA2VIA-1 (1) or iPLA2VIA-2 (2) are plotted relative to that of control fibroblasts. Also see online supplementary figure S-2 and S-3. (B) Graph showing relative ERGIC-53 fluorescent intensity. Data from the patient fibroblasts without (−) or with overexpression of iPLA2VIA-1 (1) or iPLA2VIA-2 (2) are plotted relative to that of control fibroblasts. (C) Graph representing 2D-Golgi area of control fibroblasts and patient fibroblasts without (−) or with overexpression of iPLA2VIA-1 (1) or iPLA2VIA-2 (2). Golgi area was assessed by immunofluorescence staining with anti-GM-130 and anti-TGN-46. (D) Representative immunofluorescence images for ERGIC staining in Control and Patient 1 fibroblasts, with and without overexpression of iPLA2VIA-1 or iPLA2VIA-2. (E) Representative immunofluorescence images for staining of cis-Golgi with GM-130 and trans-Golgi with TGN-46 in Control and Patient 1 fibroblasts, with and without overexpression of iPLA2VIA-1 or iPLA2VIA-2. #p<0.05 versus control fibroblasts, *p<0.005 versus control fibroblasts, +p<0.01 versus fibroblasts from the same patient without overexpression of PLA2G6, and †p<0.005 versus fibroblasts from the same patient without overexpression of PLA2G6. Error bars represent SEM.
Cultured skin fibroblasts from patients with INAD and dystonia-parkinsonism have reduced ERGIC and altered Golgi morphology rescued by iPLA2VIA-1
Since O-linked glycosylation predominantly occurs in the Golgi,24,36 and PLA2G6 functions in lipid remodelling in the ERGIC,37 we hypothesised that mutations in PLA2G6 affect ERGIC formation and subsequently Golgi structure. Using an antibody against ERGIC-53, we measured a significant decrease in intensity in patient fibroblasts compared with unaffected control fibroblasts (figure 4B, D). Comparison of Golgi morphology in the patient fibroblasts to that of unaffected control fibroblasts showed a significant decrease in the area occupied by the Golgi (figure 4C, E, see online supplementary figure S-5). Lentiviral introduction of wild type iPLA2VIA-1, but not of iPLA2VIA-2, rescued the decrease in ERGIC and the Golgi morphology in patient fibroblasts.
Patients with INAD and dystonia-parkinsonism have altered protein O-linked glycosylation in vivo
To determine if alterations in protein glycosylation could be detected in vivo, we screened CSF, plasma and urine. All patients had a reduction of O-linked glycans in their CSF, whereas only patients 1 and 2 had detectable anomalies in their plasma protein O-linked glycosylation profiles. The CSF of patient 2 was deficient for disialylated core 2 O-glycan (see online supplementary figure S-6). No changes were detected in plasma N-linked glycosylation and free oligosaccharides in urine (data not shown).
DISCUSSION
We describe markedly altered Golgi morphology and protein O-linked glycosylation profiles and mildly reduced galactosylation and sialylation of N-linked glycans in cultured skin fibroblasts of three individuals with PLAN. These patients also had a reduction of O-linked glycans in their CSF.
The mechanisms by which mutations in PLA2G6 cause PLAN20,35,38,39 remain undefined. Among its many functions,9–18 PLA2G6 regulates membrane tubule formation in the ERGIC, a precursor of the cis-Golgi stack.37 PLA2G6 thus indirectly regulates Golgi morphology and trafficking, and loss of functional PLA2G6 might be expected to alter Golgi morphology in a manner consistent with the fragmentation of the Golgi in cultured fibroblasts from each of our patients. Furthermore, given that O-linked glycosylation and terminal sialylation of nearly all glycans occur in the Golgi,24,36 and that disruption of Golgi morphology is associated with altered glycosylation in other diseases,40–45 we hypothesise that the global protein O-linked glycosylation and mild N-linked glycosylation changes observed in our patients’ fibroblasts, are secondary to the changes in Golgi morphology.
Interestingly, only introduction of the wild type iPLA2VIA-1 isoform into cultured fibroblasts from our patients with PLAN rescued the abnormal Golgi morphology and O-linked protein glycosylation. This suggests that iPLA2VIA-1 and iPLA2VIA-2 have different effects on disruption of membrane homoeostasis and membrane tubule formation in the ERGIC. So far, insufficient information is available on these two isoforms to determine the mechanism by which these differences arise and further study is required to define and validate this.
Disruption of Golgi structure has been reported in Alzheimer’s disease, Parkinson’s disease and other neurodegenerative disorders.46,47 Such changes in Golgi morphology are thought to occur early in Alzheimer’s disease, before amyloid-β accumulation or α-synuclein pathology is detected.47 Consequently, Golgi disruption may be a common factor in several neurological disorders including PLAN.46,47 This mechanism posits that disruption of Golgi morphology causes the abnormal glycosylation observed in PLAN and the other diseases,40–45 and that these changes in protein glycosylation contribute to protein aggregation, cellular stress, and ultimately, apoptosis and neurodegeneration. For example, decreases in terminal sialic acid on cellular glycoproteins, such as that observed for our patients with PLAN, has been associated with amyloid β precursor protein (APP) plaques in Alzheimer’s disease and with other neurodegenerative processes.48–50 In the case of PLAN, we conclude that the decrease in sialic acid is due to a global reduction in O-linked glycans because the PNA staining did not increase, as it would for a sialylation-specific decrease. Furthermore, the reductions in MALII and SNA lectin staining were global and not limited to the plasma membrane; this excludes altered cell surface expression as the sole cause of the observed decrease.
Underscoring the importance of glycosylation in neuronal health, nuclear O-GlcNAcylation is proposed to protect the brain from aggregation and downstream toxicity of Tau, amyloid β precursor protein (APP) and α-synuclein.51–55 While we cannot currently link the altered Golgi morphology observed in cultured cells from patients with PLAN to aggregation and downstream toxicity of Tau, APP and α-synuclein, we suggest this as a hypothesis warranting future investigation.
In conclusion, our study describes for the first time morphological changes in the Golgi and O-linked glycosylation of fibroblasts from individuals with PLA2G6 mutations. We propose that structural changes of the Golgi, also seen in other neurodegenerative diseases, cause aberrations in global protein glycosylation that possibly contribute to Tau, APP and α-synuclein misfolding and aggregation, eliciting cellular stress leading to apoptosis and neurodegeneration.
Supplementary Material
Acknowledgments
The authors thank Drs John Hanover and Paul Lee for critical review of the manuscript.
Funding: This work was supported by the Intramural Research Program of the National Human Genome Research Institute and the Common Fund of the NIH Office of the Director of the National Institutes of Health, Bethesda, Maryland, USA.
Footnotes
Contributors: Study concept and design: MD, MSK and CFB. Recruitment of patients and collection of clinical information: LAW, WAG and CT. Acquisition, analysis and interpretation of whole exome sequencing data: MD, KRC, WPB and CT. Acquisition of cell imaging and mutation analysis data: MD and MSK. Acquisition of glycome profiles by mass spectrometry: XL, MAR and MH. Analysis and interpretation of data: MD, MSK, MH and CFB. Drafting of the manuscript: MD and CFB. Critical review of the manuscript: MD, MSK, MH, LAW, XL, MAR, KRC, WPB, CFB, WAG and CT. Study supervision: CFB and WAG.
Competing interests: CFB currently works at University of British Columbia, Vancouver, Canada.
Ethics approval: Institutional Review Board of the National Human Genome Research, Institute National Institutes of Health, Bethesda, Maryland, USA.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: Data will be shared through PUBMED upon publication.
References
- 1.Carrilho I, Santos M, Guimaraes A, Teixeira J, Chorao R, Martins M, Dias C, Gregory A, Westaway S, Nguyen T, Hayflick S, Barbot C. Infantile neuroaxonal dystrophy: what’s most important for the diagnosis? Eur J Paediatr Neurol. 2008;12:491–500. doi: 10.1016/j.ejpn.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 2.Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, Coryell JC, Nguyen TM, Nardocci N, Zorzi G, Rodriguez D, Desguerre I, Bertini E, Simonati A, Levinson B, Dias C, Barbot C, Carrilho I, Santos M, Malik I, Gitschier J, Hayflick SJ, Guimaraes A, Teixeira J, Chorao R, Martins M, Westaway S, Nguyen T, Hayflick S. Neurodegeneration associated with genetic defects in phospholipase A(2) Neurology. 2008;71:1402–9. doi: 10.1212/01.wnl.0000327094.67726.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurian MA, Morgan NV, MacPherson L, Foster K, Peake D, Gupta R, Philip SG, Hendriksz C, Morton JE, Kingston HM, Rosser EM, Wassmer E, Gissen P, Maher ER, McNeill A, Lin JP, Hayflick SJ. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN) Neurology. 2008;70:1623–9. doi: 10.1212/01.wnl.0000310986.48286.8e. [DOI] [PubMed] [Google Scholar]
- 4.Kurian MA, Hayflick SJ. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int Rev Neurobiol. 2013;110:49–71. doi: 10.1016/B978-0-12-410502-7.00003-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, Hardy J, Houlden H, Singleton A, Schneider SA. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65:19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M, Hashimoto T, Takashima S, Hattori N. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology. 2010;75:1356–61. doi: 10.1212/WNL.0b013e3181f73649. [DOI] [PubMed] [Google Scholar]
- 7.Engel LA, Jing Z, O’Brien DE, Sun M, Kotzbauer PT. Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One. 2010;5:e12897. doi: 10.1371/journal.pone.0012897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larsson PK, Claesson HE, Kennedy BP. Multiple splice variants of the human calcium-independent phospholipase A2 and their effect on enzyme activity. J Biol Chem. 1998;273:207–14. doi: 10.1074/jbc.273.1.207. [DOI] [PubMed] [Google Scholar]
- 9.Balsinde J, Bianco ID, Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc Natl Acad Sci USA. 1995;92:8527–31. doi: 10.1073/pnas.92.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balsinde J, Dennis EA, Balboa MA, Saez Y, Fuentes L, Perez R, Nieto ML, Melero R, Shirai Y. Function of calcium-independent phospholipase A2 in arachidonic acid metabolism in P388D1 macrophages. Adv Exp Med Biol. 1997;407(Pt 3):99–103. doi: 10.1007/978-1-4899-1813-0_15. [DOI] [PubMed] [Google Scholar]
- 11.Atsumi G, Murakami M, Kojima K, Hadano A, Tajima M, Kudo I. Distinct roles of two intracellular phospholipase A2s in fatty acid release in the cell death pathway. Proteolytic fragment of type IVA cytosolic phospholipase A2alpha inhibits stimulus-induced arachidonate release, whereas that of type VI Ca2+-independent phospholipase A2 augments spontaneous fatty acid release. J Biol Chem. 2000;275:18248–58. doi: 10.1074/jbc.M000271200. [DOI] [PubMed] [Google Scholar]
- 12.Murakami M, Shimbara S, Kambe T, Kuwata H, Winstead MV, Tischfield JA, Kudo I. The functions of five distinct mammalian phospholipase A2S in regulating arachidonic acid release. Type IIa and type V secretory phospholipase A2S are functionally redundant and act in concert with cytosolic phospholipase A2. J Biol Chem. 1998;273:14411–23. doi: 10.1074/jbc.273.23.14411. [DOI] [PubMed] [Google Scholar]
- 13.Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A(2) research: from cells to animals to humans. Prog Lipid Res. 2011;50:152–92. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 14.Ben-Tekaya H, Kahn RA, Hauri HP. ADP ribosylation factors 1 and 4 and group VIA phospholipase A(2) regulate morphology and intraorganellar traffic in the endoplasmic reticulum-Golgi intermediate compartment. Mol Biol Cell. 2010;21:4130–40. doi: 10.1091/mbc.E10-01-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt JA, Kalkofen DN, Donovan KW, Brown WJ. A role for phospholipase A2 activity in membrane tubule formation and TGN trafficking. Traffic. 2010;11:1530–6. doi: 10.1111/j.1600-0854.2010.01115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malhotra A, Edelman-Novemsky I, Xu Y, Plesken H, Ma J, Schlame M, Ren M. Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc Natl Acad Sci USA. 2009;106:2337–41. doi: 10.1073/pnas.0811224106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J Biol Chem. 2006;281:22275–88. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lei X, Zhang S, Bohrer A, Barbour SE, Ramanadham S. Role of calcium-independent phospholipase A(2)beta in human pancreatic islet beta-cell apoptosis. Am J Physiol Endocrinol Metab. 2012;303:E1386–95. doi: 10.1152/ajpendo.00234.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. 2009;46:73–80. doi: 10.1136/jmg.2008.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paisan-Ruiz C, Li A, Schneider SA, Holton JL, Johnson R, Kidd D, Chataway J, Bhatia KP, Lees AJ, Hardy J, Revesz T, Houlden H. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging. 2012;33:814–23. doi: 10.1016/j.neurobiolaging.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rendon WO, Martinez-Alonso E, Tomas M, Martinez-Martinez N, Martinez-Menarguez JA. Golgi fragmentation is Rab and SNARE dependent in cellular models of Parkinson’s disease. Histochem Cell Biol. 2013;139:671–84. doi: 10.1007/s00418-012-1059-4. [DOI] [PubMed] [Google Scholar]
- 22.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–8. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lis H, Sharon N. Protein glycosylation. Structural and functional aspects. Eur J Biochem. 1993;218:1–27. doi: 10.1111/j.1432-1033.1993.tb18347.x. [DOI] [PubMed] [Google Scholar]
- 24.Freeze HH. Genetic defects in the human glycome. Nat Rev Genet. 2006;7:537–51. doi: 10.1038/nrg1894. [DOI] [PubMed] [Google Scholar]
- 25.Gahl WA, Markello TC, Toro C, Fajardo KF, Sincan M, Gill F, Carlson-Donohoe H, Gropman A, Pierson TM, Golas G, Wolfe L, Groden C, Godfrey R, Nehrebecky M, Wahl C, Landis DM, Yang S, Madeo A, Mullikin JC, Boerkoel CF, Tifft CJ, Adams D. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med. 2012;14:51–9. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S, Jaffe DB, Lander ES, Nusbaum C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–9. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teer JK, Bonnycastle LL, Chines PS, Hansen NF, Aoyama N, Swift AJ, Abaan HO, Albert TJ, Program NCS, Margulies EH, Green ED, Collins FS, Mullikin JC, Biesecker LG. Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 2010;20:1420–31. doi: 10.1101/gr.106716.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara ECM, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo S, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning Z, Ling Ng B, Novo SM, O’Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang J, Worsley GJ, Yan J, Yau L, Zuerlein M, Rogers J, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012;28:599–600. doi: 10.1093/bioinformatics/btr711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnston JJ, Teer JK, Cherukuri PF, Hansen NF, Loftus SK, NIH Intramural Sequencing Center (NISC) Chong K, Mullikin JC, Biesecker LG. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am J Hum Genet. 2010;86:743–8. doi: 10.1016/j.ajhg.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–9. doi: 10.1093/nar/gkq1189. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA. 1992;89:10915–19. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia B, Zhang W, Li X, Jiang R, Harper T, Liu R, Cummings RD, He M. Serum N-glycan and O-glycan analysis by mass spectrometry for diagnosis of congenital disorders of glycosylation. Anal Biochem. 2013;442:178–85. doi: 10.1016/j.ab.2013.07.037. [DOI] [PubMed] [Google Scholar]
- 34.Xia B, Asif G, Arthur L, Pervaiz MA, Li X, Liu R, Cummings RD, He M. Oligosaccharide analysis in urine by maldi-tof mass spectrometry for the diagnosis of lysosomal storage diseases. Clin Chem. 2013;59:1357–68. doi: 10.1373/clinchem.2012.201053. [DOI] [PubMed] [Google Scholar]
- 35.Crompton D, Rehal PK, MacPherson L, Foster K, Lunt P, Hughes I, Brady AF, Pike MG, De Gressi S, Morgan NV, Hardy C, Smith M, MacDonald F, Maher ER, Kurian MA. Multiplex ligation-dependent probe amplification (MLPA) analysis is an effective tool for the detection of novel intragenic PLA2G6 mutations: implications for molecular diagnosis. Mol Genet Metab. 2010;100:207–12. doi: 10.1016/j.ymgme.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 36.Roth J. Cytochemical localization of terminal N-acetyl-D-galactosamine residues in cellular compartments of intestinal goblet cells: implications for the topology of O-glycosylation. J Cell Biol. 1984;98:399–406. doi: 10.1083/jcb.98.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ha KD, Clarke BA, Brown WJ. Regulation of the Golgi complex by phospholipid remodeling enzymes. Biochim Biophys Acta. 2012;1821:1078–88. doi: 10.1016/j.bbalip.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, Zorzi G, Pasha S, Rodriguez D, Desguerre I, Mubaidin A, Bertini E, Trembath RC, Simonati A, Schanen C, Johnson CA, Levinson B, Woods CG, Wilmot B, Kramer P, Gitschier J, Maher ER, Hayflick SJ. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet. 2006;38:752–4. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gregory A, Hayflick SJ. Genetics of neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Rep. 2011;11:254–61. doi: 10.1007/s11910-011-0181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bexiga MG, Simpson JC. Human diseases associated with form and function of the Golgi complex. Int J Mol Sci. 2013;14:18670–81. doi: 10.3390/ijms140918670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Condon KH, Ho J, Robinson CG, Hanus C, Ehlers MD. The Angelman syndrome protein Ube3a/E6AP is required for Golgi acidification and surface protein sialylation. J Neurosci. 2013;33:3799–814. doi: 10.1523/JNEUROSCI.1930-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foulquier F, Amyere M, Jaeken J, Zeevaert R, Schollen E, Race V, Bammens R, Morelle W, Rosnoblet C, Legrand D, Demaegd D, Buist N, Cheillan D, Guffon N, Morsomme P, Annaert W, Freeze HH, Van Schaftingen E, Vikkula M, Matthijs G. TMEM165 deficiency causes a congenital disorder of glycosylation. Am J Hum Genet. 2012;91:15–26. doi: 10.1016/j.ajhg.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hucthagowder V, Morava E, Kornak U, Lefeber DJ, Fischer B, Dimopoulou A, Aldinger A, Choi J, Davis EC, Abuelo DN, Adamowicz M, Al-Aama J, Basel-Vanagaite L, Fernandez B, Greally MT, Gillessen-Kaesbach G, Kayserili H, Lemyre E, Tekin M, Turkmen S, Tuysuz B, Yuksel-Konuk B, Mundlos S, Van Maldergem L, Wevers RA, Urban Z. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum Mol Genet. 2009;18:2149–65. doi: 10.1093/hmg/ddp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller VJ, Ungar D. Re’COG’nition at the Golgi. Traffic. 2012;13:891–7. doi: 10.1111/j.1600-0854.2012.01338.x. [DOI] [PubMed] [Google Scholar]
- 45.Pokrovskaya ID, Willett R, Smith RD, Morelle W, Kudlyk T, Lupashin VV. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology. 2011;21:1554–69. doi: 10.1093/glycob/cwr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonatas NK, Stieber A, Gonatas JO. Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J Neurol Sci. 2006;246:21–30. doi: 10.1016/j.jns.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 47.Tillement JP, Papadopoulos V. Subcellular injuries in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2014;13:593–605. doi: 10.2174/18715273113126660197. [DOI] [PubMed] [Google Scholar]
- 48.Wielgat P, Braszko JJ. Significance of the cell adhesion molecules and sialic acid in neurodegeneration. Adv Med Sci. 2012;57:23–30. doi: 10.2478/v10039-012-0011-0. [DOI] [PubMed] [Google Scholar]
- 49.McFarlane I, Georgopoulou N, Coughlan CM, Gillian AM, Breen KC. The role of the protein glycosylation state in the control of cellular transport of the amyloid beta precursor protein. Neuroscience. 1999;90:15–25. doi: 10.1016/s0306-4522(98)00361-3. [DOI] [PubMed] [Google Scholar]
- 50.Schedin-Weiss S, Winblad B, Tjernberg LO. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014;281:46–62. doi: 10.1111/febs.12590. [DOI] [PubMed] [Google Scholar]
- 51.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–58. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marotta NP, Cherwien CA, Abeywardana T, Pratt MR. O-GlcNAc modification prevents peptide-dependent acceleration of alpha-synuclein aggregation. Chembiochem. 2012;13:2665–70. doi: 10.1002/cbic.201200478. [DOI] [PubMed] [Google Scholar]
- 53.Yuzwa SA, Cheung AH, Okon M, McIntosh LP, Vocadlo DJ. O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J Mol Biol. 2014;426:1736–52. doi: 10.1016/j.jmb.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 54.Lazarus BD, Love DC, Hanover JA. O-GlcNAc cycling: implications for neurodegenerative disorders. Int J Biochem Cell Biol. 2009;41:2134–46. doi: 10.1016/j.biocel.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu Y, Shan X, Yuzwa SA, Vocadlo DJ. The emerging link between O-GlcNAc and Alzheimer disease. J Biol Chem. 2014;289:34472–81. doi: 10.1074/jbc.R114.601351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.