

Abstract
Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are the most common genetic causes of Parkinson’s disease (PD), and also one of the strongest genetic risk factors in sporadic PD. The LRRK2 protein contains a GTPase domain and a kinase domain, and several protein-protein interaction domains. Both in vitro and in vivo assays in different model systems have provided tremendous insights into the molecular mechanisms underlying LRRK2 induced dopaminergic neurodegeneration. Among all the model systems, animal models are crucial tools to study the pathogenesis of human disease. How do the animal models recapitulate LRRK2 induced dopaminergic neuronal loss in human PD? To answer this question, this review focuses on the discussion of the animal models of LRRK2 associated PD including genetic and viral-based models.
Keywords: LRRK2, Animal models, Parkinson’s disease
Introduction
Parkinson’s disease (PD) is recognized as the most common movement disorder, affecting up to 1% of the population above the age of 60 and 4–5% above the age of 85 [1]. Clinical symptoms in PD patients include akinesia, resting tremor, muscle rigidity, and postural imbalance [1]. The cardinal symptoms are caused by the progressive degeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) [2]. Although the majority of PD cases appear to be sporadic, in the past couple of decades several genes have been identified to be responsible for this progressive neurodegenerative disease [3]. To date, genes encoding LRRK2 (leucine-rich repeat kinase 2), α-synuclein, parkin, DJ-1, PINK1 (phosphatase and tensin homologue deleted on chromosome 10-induced putative kinase 1), VPS35 (vacuolar protein sorting 35), DNAJC13, GBA (Glucocerebrosidase), and EIF4G1 (eukaryotic initiation factor 4G1), among others are associated with genetic forms of PD [3–8]. Mutations in the LRRK2 gene (PARK8, dardarin, OMIM 609007) cause late-onset, autosomal dominant PD and is the most frequent genetic cause of PD, accounting for 4% of familial PD and 1% of sporadic PD across all populations. Importantly, LRRK2 mediated PD is clinically and pathologically indistinguishable from sporadic PD [9, 10], suggesting that understanding LRRK2 associated PD may lead to an understanding of sporadic PD.
The LRRK2 protein contains two enzymatic domains, a GTPase and a kinase domain, and multiple protein-protein interaction domains including a leucine-rich repeat (LRR), a WD40 repeat, and a LRRK2 specific repeat domain (Figure 1) [11, 12]. LRRK2 interaction domains are thought to serve as protein binding modules where LRRK2 acts as a signaling scaffold. LRRK2 GTPase and kinase enzyme activity are important in regulating LRRK2 dependent cellular signaling pathways and may reciprocally regulate each other to direct LRRK2’s ultimate function [13]. Pathogenic mutations of LRRK2 are centered on LRRK2 enzymatic domains (Figure 1). Thus, LRRK2 enzymatic activity is important in PD. The most prevalent LRRK2 mutation, G2019S is within the kinase domain. It accounts for 5 – 6% of autosomal-dominant PD patients and ~1% of sporadic late-onset PD. Patients with the G2019S mutation exhibit Lewy bodies in most cases [1]. However, mutations in the GTPase domain and COR domain, such as R1441 C/G and Y1669C often vary on Lewy body pathology [10, 14]. This raises the possibility that these mutations cause disease via distinct pathogenic mechanisms.
Figure 1. Schematic showing the domain structure of LRRK2 protein and the position of pathogenic mutations.
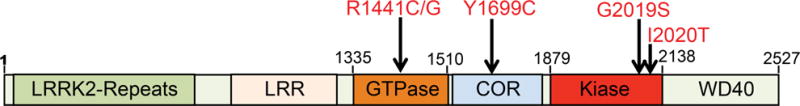
Residues 1-660 encode LRRK2 specific repeat sequences, 984-1278 encode the leucine-rich repeat (LRR), 1335-1510 encode the Roc GTPase domain, 1519-1795 encode the C-terminal of Ras (COR) domain and 1879-2138 encode the kinase domain. Five confirmed LRRK2 pathogenic mutations: R1441C/G, Y1699C, G2019S, I2020T.
Tremendous work in both in vitro and in vivo systems suggests that LRRK2 is involved in diverse pathways and cellular signaling including regulation of protein translation, vesicle trafficking, neurite outgrowth, autophagy, and cytoskeletal dynamics [15–17]. Several model systems have been developed to study LRRK2 function from yeast to invertebrates such as Drosophila and C. elegans, rodents, and patient-derived induced pluripotent stem cells (iPScs) [18]. Yeast, a eukaryotic single-cell organism, has been widely used to uncover the fundamental pathobiology of proteins associated with neurodegenerative diseases including PD. The first LRRK2 yeast model revealed LRRK2 GTPase function plays a key role in LRRK2 pathobiology [19, 20]. The toxicity is closely associated with GTPase activity and defects in endocytic vesicular trafficking and autophagy [19]. More importantly, using this yeast model, the first GTPase activating protein (GAP) for LRRK2, ArfGAP1, was identified and characterized [20]. Patient-derived iPScs provide highly relevant models for PD studies as the well-developed capacity to generate iPSc-derived DA neurons. Several LRRK2 iPSc models have been developed. DA neurons derived from LRRK2 iPScs display reduced neurite length, accumulation of α-synuclein and tau, increased vulnerability to cellular stress, and impaired autophagy and mitochondrial function [18]. IPSc models allow us to study LRRK2 pathobiology directly in human context. However, both yeast and iPSc models cannot recapitulate the physiological cell diversity in the intact mammalian brain and the complexity of brain circuits.
While the eukaryotic yeast and iPS cell models provide an important yet complementary insights to animal models on understanding disease mechanisms, this review focuses on the animal models of LRRK2 associated PD and discusses the advantages and disadvantages of each model and how each of these different models have contributed to understanding the role of LRRK2 in PD pathogenesis.
1. Genetic animal models of LRRK2
1.1 LRRK2 Drosophila models
Animal models are crucial tools for LRRK2 research. Invertebrate animals, especially Drosophila, have proven to play an important role in studying LRRK2 pathogenic mechanisms and developing therapeutics. Drosophila have well-defined nerve systems, which share similar neuronal transmitters with mammals. Importantly, Drosophila have DA neuronal clusters and can perform complicated behavioral tests, which mimic some DA dependent behaviors in human. Several steps have been taken to establish and utilize LRRK2 Drosophila models: 1) generation of Drosophila strains carrying mutations in genes linked to disease, 2) determination of the Drosophila models to see if they recapitulate the pathogenesis of the disease and in turn are good models of the disease, 3) investigation of the detailed molecular mechanisms underlying the phenotypes, 4) identification of genetic modifiers to dissect the signaling pathways involved in pathogenesis, 5) drug candidate screening. Several LRRK2 Drosophila models have been generated and are listed in Table 1.
Table 1.
Drosophila models for LRRK2-associated PD.
| LRRK2 variants | Genetic manipulations | Motor deficits/life span | DA neuronal loss | nigrostriatal dysfunction | Sensitivity to oxidative stress | Other phenotypes | References |
|---|---|---|---|---|---|---|---|
| dLRRK | Loss of function | Locomotor activity ↓ | No changes | TH staining ↓ DA neurons shrunken | ND | ND | Lee, et al., 2007 |
| dLRRK | Loss of function | Life span ↓ | No changes | No changes | Sensitive to hydrogen peroxide, not to paraqua, rotenone and β-mercaptoethanol | ND | Wang, et al., 2008 |
| dLRRK | Loss of function | Life span ↓ Fertility ↓ Malformed abdomen |
No changes | DA content ↑ | Hydrogen peroxide ↓ Paraqua ↓ |
ND | Imai et al., 2008 |
| dLRRK | Loss of function | Locomotor activity ↓ | ND | ND | ND | ND | Tain, et al., 2009 |
| dLRRK | O/E | No changes | No changes | No changes | ND | ND | Lee, et al., 2007 |
| hLRRK2 | O/E | Locomotor activity ↓ Life span ↓ Response to L-Dopa |
TH neurons ↓ No response to L-Dopa |
ND | ND | Retinal degeneration | Liu, et al., 2008 |
| hG2019S | Locomotor activity ↓ Life span ↓ Response to L-Dopa |
TH neurons ↓↓ No response to L-Dopa |
ND | ND | Retinal degeneration | ||
| dLRRK | O/E | ND | No changes | No changes | No changes | ND | Imai et al., 2008 |
| dY1383C dI1915T |
ND | No changes | TH staining ↓ DA content ↓ |
Hydrogen peroxide ↑ Paraqua ↑ |
ND | ||
| hLRRK2 | O/E at 29°C | Locomotor activity: 10 days↓ 20days↑ Life span ↑ Fertility↑ |
TH neurons ↓ | ND | Rotenone↑ | Retinal degeneration | Venderova et al., 2009 |
| hI1122V hY1699C hI2020T |
Locomotor activity: 10 days↓ 20days↑ Life span ↑ in hY1699C, hI2020T Fertility ↑in hI1122V, hI2020T |
TH neurons ↓ the most with I 2020T | ND | Rotenone↑ | Retinal degeneration | ||
| hLRRK2 | O/E | No changes | No changes | ND | No changes | No changes | Ng et al., 2009 |
| hG2019S hY1699C hG2385R |
Locomotor activity ↓ Life span ↓ |
TH neurons ↓ | ND | hG2019S, hG2385R↑ hY1699C no change |
No changes | ||
| hLRRK2 | O/E | Locomotor activity: no changes Life span ↓ |
ND | Dendritic ends↓ | ND | ND | Lin et al., 2010 |
| hG2019S | Locomotor activity ↓↓ Life span ↓↓ |
TH neurons ↓ | Dendritic ends↓↓ Axon degeneration↑ |
ND | ND | ||
| hR1441C hG2385R |
Locomotor activity ↓ Life span ↓ |
ND | Dendritic ends↓ | ND | ND | ||
| hLRRK2 | O/E | ND | ND | ND | ND | Visual function: no changes | Hindle, et al., 2013 |
| hG2019S | ND | ND | ND | ND | Visual function ↓ | ||
| hI1122V hR1441C hY1383C hI1915T hI2020T hG2385R hG2019/K 1906M |
ND | ND | ND | ND | Visual function: no changes | ||
| hLRRK2 hG2019S |
O/E | Locomotor activity: no changes | ND | Axon transport: no changes | ND | ND | Godena et al., 2014 |
| hR1441C hY1699C |
Locomotor activity ↓ | ND | Axon transport ↓ | ND | ND | ||
| dR1069C dY1383C |
Locomotor activity ↓ | ND | Axon transport ↓ | ND | ND |
ND: not determined; O/E, overexpression; ↑ increased; ↓decreased.
1.1.1 LRRK2 knockout Drosophila models
Drosophila has one human LRRK2 homolog dLRRK and residues affected by PD-causing mutations in LRRK2 are conserved in Drosophila LRRK2. To study the function of endogenous wild type (WT) LRRK2, Drosophila LRRK2 knockouts have been generated [21–24]. Several groups reported that the homozygous mutant fly develops normally with a normal life span as well as unchanged number and pattern of DA neurons [21, 24, 23], although one group reported LRRK loss-of-function mutants exhibited severely impaired locomotive activity and a severe reduction in tyrosine hydroxylase immunostaining and shrunken morphology of DA neurons in LRRK mutants [22]. In addition, Wang et al. showed that mutant flies containing C-terminal kinase domain truncated dLRRK are selectively sensitive to H2O2, but not to paraquat, rotenone or β-mercaptoethanol [23]. By contrast, Imai et al. showed that dLRRK null flies are relatively resistant to general oxidative stress, such as paraquat and H2O2 treatment, compared to WT flies [21]. Thus, the exact role of dLRRK in oxidative stress remains unclear. The different phenotypes is possibly due to the different genomic locus of insertion for gene disruption and the different genetic backgrounds. As the majority of the reports support that dLRRK is dispensable for survival of DA neurons in flies and this is consistent with the phenotypes in LRRK2 knockout mice, the general consensus is that LRRK2 toxicity is from a gain-of-function and not a loss-of-function mechanism.
1.1.2 LRRK2 transgenic Drosophila models
In contrast to dLRRK knockout Drosophila, overexpression of both human LRRK2 and dLRRK pathogenic mutations in Drosophila leads to age-dependent DA-responsive reductions in locomotor activity and loss of DA neurons (Table 1) [21, 25–28]. Interestingly, in addition to the DA neurodegeneration, different LRRK2 mutations cause different phenotypes related to the degeneration. One recent study showed that LRRK2 G2019S induced extensive neurodegeneration throughout the visual system [29]. This degeneration is LRRK2 G2019S mutation specific and occurs in a kinase dependent manner. Dopaminergic expression of LRRK2 G2019S led to non-autonomous cell death reminiscent of that seen in PD [29]. Another report showed that LRRK2 R1441C or Y1699C mutations in the GTPase-COR domain preferentially associates with de-acetylated microtubules, and inhibits axonal transport in Drosophila, causing locomotor deficits in vivo. These features are not seen with the LRRK2 G2019S mutation, suggesting these defects are GTPase activity dependent [30]. A previous study suggested that reduced axonal transport rates caused by alpha-synuclein mutants might contribute to accumulation of alpha-synuclein and hence Lewy body formation and neuritic abnormalities in PD brain [31]. Taken together, reduced axonal transport rates may contribute to the formation of Lewy bodies or lewy neurites in some PD cases carrying R1441C or Y1669C mutation. These studies suggest that different LRRK2 pathogenic mutations act at distinct pathways and cause varied neuropathology in the DA neurodegeneration.
1.1.3 Using LRRK2 Drosophila models to study molecular mechanisms underlying LRRK2 associated PD
Do LRRK2 Drosophila models reveal the pathogenic mechanisms underlying LRRK2 induced DA neurodegeneration? To address this, Drosophila offers a wide variety of genetic tools including genetic screens, which allow genome-wide analyses of genetic interactions based on the modification of a given phenotype, and candidate gene approaches, in which only those suspected genes are analyzed for modifications of the phenotype. Both strategies allow identification of components of signaling pathways involved in PD pathogenesis. Using LRRK2 Drosophila models, several in vivo LRRK2 interactors have been identified and characterized in different signaling pathways.
LRRK2 function in protein synthesis/translation
Drosophila dLRRK was shown to regulate protein translational pathways. Imai et al. first provided evidence that both dLRRK and human LRRK2 can phosphorylate eukaryotic initiation factor 4E-binding protein (4E-BP), a negative regulator of eukaryotic initiation factor 4E-mediated protein translation and a key mediator of various stress responses [21]. A link between dLRRK and protein synthesis was further strengthened by the observation from the same group that LRRK2 interacts with the microRNA pathway to regulate protein synthesis [32]. However, these Drosophila studies have yet to be extended to mammalian systems. A recent study, using a combination of a LRRK2 Drosophila model and human dopamine neurons, demonstrated that LRRK2 phosphorylates ribosome protein s15 to enhance protein translation and mediate LRRK2 induced neurodegeneration [33]. Taken together, there is strong convergent evidence that LRRK2 regulates protein translation machinery in diverse species and tissues.
LRRK2 function in vesicular trafficking
Studies using LRRK2 Drosophila models have revealed potential roles for LRRK2 in multiple aspects of vesicle trafficking including endolysosomal pathways, synaptic vesicle (SV) endocytosis, ER-Golgi and retromer trafficking. First, dLRRK was reported to localize to the membranes of late endosomes and lysosomes, physically interacts with the crucial mediator of late endosomal transport Rab7 and negatively regulates Rab7-dependent perinuclear localization of lysosomes [34]. LRRK2 has been further shown to localize at endosomes and interacts with clathrin-light chains (CLCs) to limit Rac1 activation. These data identify a new pathway in which CLCs function with LRRK2 to control Rac1 activation on endosomes [35]. The function of LRRK2 in endolysosomal pathways is further strengthened by a study on novel ethyl methanesulfonate (EMS)-induced nonsense alleles in dLRRK, which cause striking defects in the endolysosomal and autophagy pathways [36]. Second, a study in Drosophila shows that LRRK2 functions on SV endocytosis at the neuromuscular junctions by phosphorylating endophilin A (EndoA) at S75 and mediating EndoA-dependent membrane tubulation and membrane association [37]. In addition, dLRRK has been demonstrated to regulate Golgi outpost (GOP) dynamics in dendrites through the golgin Lava lamp [38]. Moreover, genetic interactions between VPS35, Rab7L1, ArfGAP1 and LRRK2 in Drosophila highlight LRRK2’s role in retromer and ER-Golgi trafficking [39, 40, 20]. All data taken together strongly support that LRRK2 plays a crucial role in vesicular trafficking pathway, which may provide potential mechanisms for accumulation of α-synuclein in LRRK2 associated PD.
LRRK2 function in dendritic degeneration and synaptic morphology
Expression of LRRK2 G2019S in Drosophila dendritic arborization neurons induces mislocalization of the axonal protein tau in dendrites and causes dendrite degeneration. This may act through a mechanism LRRK2 G2019S promotes tau phosphorylation by the glycogen synthase kinase 3β (GSK3β) [28]. In addition, LRRK2 regulates synaptic morphology through interacting with 4E-BP at the postsynaptic site and phosphorylating Futsch at the presynaptic compartments of the Drosophila neuromuscular junctions [41]. These studies point out a possible role for LRRK2 in dendrite degeneration and synaptic dysfunction.
LRRK2 genetic interaction with other PD genes
As the number of genetic alterations linked to PD pathogenesis increases, establishing functional pathways and whether these genes or risk factors interact with each other will be crucial. Drosophila as a classical genetic model provides powerful tools to study genetic interactions between different genes. Using LRRK2 Drosophila models, genetic dissection revealed that LRRK2 interacts with other PD genes or risk factors such as VPS35, RAB7L, Parkin, DJ-1, and PINK1 [39, 40, 27] and implicates several potential LRRK2 functions. Genetic interaction between LRRK2 and VPS35 and Rab7L implicate LRRK2 function in retromer and lysosomal pathways that contribute to PD [39, 40]. Coexpression of human parkin in LRRK2 G2019S-expressing flies provides significant protection against DA neurodegeneration that occurs with age or in response to rotenone, suggesting a potential link between LRRK2, Parkin and mitochondria in the pathogenesis of LRRK2 related parkinsonism [27]. Genetic interaction between LRRK2 and Parkin, DJ-1 and PINK1 also suggest that dominant PD genes may act via common pathways with the recessive PD genes.
1.1.4 Using LRRK2 Drosophila models to identify potentially therapeutic compounds
The genetic LRRK2 Drosophila model represents a promising platform for inhibitor identification and validation. Studies have shown that GW5074, curcumin, or sorafenib significantly suppressed LRRK2 PD-like phenotypes in Drosophila [42, 43]. Although candidate compounds have been used in these studies, they open the possibility of performing compound screens, which may be useful for finding new drugs for treatment of LRRK2 associated PD.
1.2 LRRK2 C. elegans models
The nematode Caenorhabditis elegans has a well-defined and genetically tractable nervous system that offers an effective model to explore basic mechanistic pathways that might underpin complex human neurological diseases. C. elegans contains only one lrk-1 gene encoding a LRRK-like protein. Lrk-1 is localized in the Golgi apparatus and is required for polarized localization of SV proteins. Loss of lrk-1 causes SV protein mislocalization to both presynaptic and dendritic endings in neurons, which are dependent on the AP-1 clathrin adaptor UNC-101 [44]. The results raise the possibility that the LRK-1 functions on the trans-Golgi network (TGN) to exclude SV proteins from the dendrite-specific transport mechanisms mediated by the AP-1 clathrin adaptor complex. This study suggest that LRRK2 might function in the Golgi network. Recent identification of ArfGAP1, a Golgi protein that reciprocally regulates LRRK2 induced toxicity both in vitro and in vivo, might provide a new insight into LRRK2 function in ER to Golgi trafficking [20]. Other loss of function studies in C. elegans revealed that LRRK2 acts to protect C. elegans DA neurons from the toxicity of 6-hydroxydopamine and/or human α-synuclein, possibly through the p38 pathway, by supporting upregulation of GRP78 [45]. Loss of lrk-1 renders animals hypersensitive to the endoplasmic reticulum stressor tunicamycin, which is rescued by PINK1 [46]. These studies suggest a functional link between LRRK2 and ER stress [46, 45].
While loss of the LRRK2 homolog in C. elegans provided information of the biological function of LRRK2, overexpression of human LRRK2 in C. elegans established a model that recapitulates key features of PD. Overexpression of human LRRK2 WT, R1441C or G2019S in DA neurons in C. elegans causes age-dependent DA neurodegeneration, behavioral deficits, and locomotor dysfunction that are accompanied by a reduction of dopamine levels in vivo [47, 48]. Several studies suggested that these phenotypes could be caused by mitochondrial dysfunction, autophagy inhibition and ER stress. Expressing human LRRK2 WT increased nematode survival by protecting against mitochondrial stress, but mutant forms of LRRK2 (G2019S or R1441C) enhanced vulnerability to mitochondrial dysfunction and inhibition of autophagy [47, 49]. Although LRRK2 G2019S consistently inhibits autophagy in multiple studies, the effects of LRRK2 WT appear to vary between studies even from the same group [47, 49]. The explanation for this variation appears to be depend on whether or not α-synuclein is present [50]. Coexpressing LRRK2 WT with α-synuclein produces a modest age-dependent inhibition of autophagy [50]. Since C. elegans, like Drosophila, does not express endogenous α-synuclein, caution needs to be taken in interpreting studies using C. elegans models.
The observations from LRRK2 C. elegans models support a role for LRRK2 kinase and GTPase activity as a critical mediators of neurotoxicity induced by mutant LRRK2. Overexpression of the GTP binding defective mutant, K1347A prevents the LRRK2 induced neurodegeneration and behavioral abnormalities [48]. LRRK2 kinase inhibitors TTT-3002 and LRRK2-IN1 protect against LRRK2 R1441C- or LRRK2 G2019S-induced neurodegeneration [48, 42]. These studies suggested that both LRRK2 GTPase and kinase activity play crucial roles in LRRK2 induced neurodegeneration in C. elegans.
1.3 LRRK2 zebrafish models
Although zebrafish has been established as an excellent vertebrate model for study of human disease, zebrafish LRRK2 (zLRRK2) models are not well developed. There is one human LRRK2 homolog in zebrafish, zLRRK2, which has a high degree conservation of amino acid sequences with human LRRK2 (hLRRK2) proteins and the highest conservation within the kinase domain. Two groups reported the generation of loss of function zLRRK2 models, but with conflicting results. Sheng et al. first reported that the deletion of the WD40 domain of zLRRK2 by morpholino targeted splicing caused Parkinsonism-like phenotypes, including loss of DA neurons in the diencephalon and locomotion defects [51]. These neurodegenerative and locomotion defects could be rescued by over-expressing zLRRK2 or hLRRK2 mRNA. The administration of L-dopa could also rescue the locomotion defects, but not the neurodegeneration [51]. However, a later study reported by Ren et al. demonstrated that the deletion of the WD40 domain of zLRRK2 using the same methods does not cause the loss of DA neurons [52]. Given the opposite results from two similar studies, the loss of function zLRRK2 models need further evaluation. Transient co-overexpress of human WT or GS LRRK2 with GFP tagged ubiquitin in WT zebrafish embryos causes impaired clearance of transiently expressed ubiquitin, suggesting of ubiquitin proteasome system disruption [53]. The characterization on DA system was not performed [53]. Taken together, LRRK2 zebrafish models are underdeveloped and need more evaluation and characterization.
1.4 LRRK2 mouse models
Whereas all the models are important and can be used in a variety of research directions, generally more effort is placed on developing mouse models to study human genetic disorders because mice possess similar neuronal networks and basal ganglia circuitry with high conservation of homologs with the human disease causing genes. Then, what are the criteria for the effective modeling of human diseases in mice? A good model should recapitulate the genetic and pathological features of the disease in human patients while avoiding spurious phenotypes that are not involved in human diseases [54–56]. For PD, mouse models that faithfully recapitulate the characteristic neurodegeneration and motor deficits as well as other hallmarks of PD such as alpha-synuclein aggregation are necessary. They would provide in vivo platforms to validate pathogenic molecular pathways and therapeutic strategies in more controlled physiological systems [55].
1.4.1 LRRK2 knockout mouse models
A question frequently raised is: whether LRRK2 pathology could be the result of a loss of function? To address this question, several groups generated and analyzed LRRK2 knockout mice. Consistent among the knockouts is that observation that there is no DA neurodegeneration although some abnormalities are observed outside the nervous system (Table 2) [57–63]. Andres-Mateo et al. reported the first LRRK2 knockout mouse model showing an intact nigrostriatal DA pathway up to 2 years of age and no altered sensitivity to MPTP-induced neurotoxicity [57]. Tong et al. demonstrated LRRK2 knockouts develop striking kidney pathology and impaired autophagy lysosomal function [62, 63]. The kidney phenotype was observed in two other LRRK2 knockouts, although the defects in autophagy changes were not observed [58, 59]. A recent study using LRRK2 knockouts suggests that LRRK2 influences neurogenesis and particularly neuronal morphogenesis [61].
Table 2.
Mouse models for LRRK2-associated PD.
| LRRK2 variants | Genetic manipulations | Motor deficits | DA neuronal loss | Nigrostriatal dysfunction | Other phenotypes | References |
|---|---|---|---|---|---|---|
| LRRK2−/− | Knockout | ND | No changes | No changes | Lack of hypersensitivity to MPTP | Andres- Mateos, et al., 2009 |
| LRRK2−/− | Knockout | No changes | No changes | ND | ND | Lin, et al., 2009 |
| LRRK2−/− | Knockout | ND | No changes | No changes | Accumulation of alpha-synuclein and ubiquitinated proteins; impaired autophagy-lysosomal pathway; Increased apoptotic cell death, inflammatory responses, and oxidative damage | Tong et al., 2010 |
| LRRK2 −/− | Knockout | No changes | No changes | No changes | An early-onset increase in number and size of secondary lysosomes in kidney; hypertension blood pressure | Herzig, et al., 2011 |
| LRRK2 −/− | Knockout | Abnormal exploratory behavior at 7 &16 months | No changes | No changes | Degeneration in the kidney, increased autophagic activity | Hinkle, et al., 2012 |
| WT | BAC transgenic | No changes | No changes | No changes | ND | Li, et al, 2009 |
| R1441G | Decline in rearing starting from 3–6 months; Response to L-Dopa | No changes | Axonal and tau pathology; Impaired dopamine release | ND | ||
| WT | BAC transgenic | Hyperactive at 6 months | No changes | No changes | No changes | Li, et al, 2010 |
| G2019S | No changes at 12 months | No changes | Decreased DA content and DA release and uptake | Enhanced kinase activity and phospho-tau | ||
| WT | BAC transgenic | No changes | No changes | Reduction of extracellular dopamine levels | No changes | Melrose et al., 2011 |
| G2019S | No changes | No changes | Reduction of extracellular dopamine levels | Increased phospho-tau | ||
| WT | Tet-off/CamKII-tTA inducible transgenic | No changes | ND | ND | ND | Lin et al., 2009 |
| G2019S | Increased ambulatory activities at 12 months | ND | ND | ND | ||
| WT | Tet-off/Pitx3- tTA inducible transgenic | No changes | No changes | No changes | Decreased expression of DA genes | Liu et al., 2015 |
| G2019S | No changes | No changes | Impaired dopamine homeostasis and release | Decreased expression of DA genes | ||
| G2019S | CMVE-PDGFβ drived transgenic | No changes | TH neurodegeneration at 20 months | No changes | Reduced neurite complexity and autophagic abnormalities | Ramonet, et al., 2011 |
| R1441C | Decreased at 15 months | No changes | No changes | ND | ||
| WT | CMVE-PDGFβ drived transgenic | No changes | No changes | No changes | No changes | Chen et al., 2012 |
| G2019S | Impaired motor activity, Response to L-Dopa | TH neurodegeneration starting from 12 months | Decreased dopamine transporters or TH staining | Activated MKK4-JNK pathway | ||
| R1441C | ROSA26 drived transgenic | No changes | No changes | No changes | Nuclear abnormalities | Tsika, et al., 2014 |
| WT | Thy1.2 drived transgenic | ND | No changes | No changes | No changes on neurite outgrowth | Garcia- Miralles, et al., 2015 |
| G2019S | ND | No changes | No changes | No changes on neurite outgrowth | ||
| R1441C | Knock-in | Reduced response to AMPH in locomotor activity | No changes | Reduced catecholamine release in cultured mutant chromaffin cells | ND | Tong, et al., 2009 |
| G2019S | Knock-in | No changes | No changes | No changes | No changes in autophagy | Herzig, et al., 2011 |
| G2019S | Knock-in | Homo-G2019Smice travel longer distances at 12 months | No changes | Impaired dopamine release, altered DA metabolism | Mitochondrial abnormalities; Elevated glutamate release; increased phosphor-tau | Beccano-Kelly, et al., 2014; Yue et al., 2015 |
ND: not determined
Since the majority of LRRK2 PD patients exhibit α-synuclein deposition, the role of LRRK2 in α-synuclein pathology has been explored. Lin et al. showed that knockout of LRRK2 rescued A53T α-synuclein overexpression induced Golgi fragmentation, α-synuclein accumulation and aggregation, microglial activation, and forebrain neuronal degeneration [60]. On the other hand, Tong et al. demonstrated that LRRK2 knockout mice develop striking accumulation and aggregation of α-synuclein and Daher et al. showed that deletion of LRRK2 had no influence on the lethal neurodegenerative phenotype of the A53T α-synuclein transgenic mice [64, 63]. The difference findings between these studies could be due to the different level of α-synuclein expression and or technical concerns. Whether inhibition of LRRK2 could be employed as a therapeutic strategy to attenuate α-synuclein-mediated neuronal damage relevant to PD needs further investigation.
All the observations from the LRRK2 knockout mice suggest that LRRK2 plays little if any role in the development and survival of DA neurons under physiologic conditions. Thus, PD caused by LRRK2 mutations are likely not due to a loss of LRRK2 function.
1.4.2 LRRK2 transgenic mouse models
Many groups have generated LRRK2 related PD mouse models expressing LRRK2 WT or PD-associated mutant LRRK2 G2019S or R1441C/G (Table 2) [65–67, 60, 68–73, 58, 74, 75]. Several transgenic techniques for LRRK2 related PD modeling in mice have been utilized, including conventional [65, 73, 69, 70], BAC transgenic [66–68], tet-inducible transgenic [60, 71] and mutant LRRK2 knock-in techniques [72, 58, 74, 75]. However, to date only two of the LRRK2 models exhibit age-dependent SNpc DA neurodegeneration [65, 69]. Most LRRK2 transgenic animals manifest deficits in DA transmission and DA-responsive behavior. Between the two studies with SNpc DA neurodegeneration, both used conventional transgenic techniques utilizing the PDGF-β promoter to generate LRRK2 mutant G2019S mouse lines. Ramonet et al. show that LRRK2 G2019S mice developed about 20% SNpc DA neurodegeneration at 20 months of age [69] while Chen et al. demonstrated more robust degeneration in the SNpc starting from 12 months of age with about 50% degeneration at 16 months of age without a phenotype in LRRK2 WT transgenic mice [65]. The different degree of the degeneration may be due to the different overexpression level of the transgenes.
Why don’t most LRRK2 transgenic models exhibit SNpc DA degeneration? One potential explanation could be a lack of robust transgene overexpression in SNpc DA neurons. The BAC and knock-in models express mutant LRRK2 during development and thus there may be compensatory mechanisms in the mouse that prevent loss of DA neurons. Thus, conditional and selective expression of LRRK2 in SNpc DA neurons may overcome this problem. A recent study reported a LRRK2 G2019S conditional transgenic mouse model using the tet-off system and a PitX3-tTA driver line to drive transgene expression in DA neurons. However, no SNpc DA degeneration was observed in this model [71]. The reason is unclear but may be related to not aging the mice to old enough such as 24 months, or perhaps expression of LRRK2 only in DA neurons is not enough for DA degeneration to occur given that the endogenous LRRK2 expression levels are comparatively low in SNpc DA neurons and LRRK2 is also expressed in other neurons. Thus, overexpresion of LRRK2 in other neurons at the same time as in DA neurons or other genetic and/or environmental factors may be required for degeneration of DA neurons.
1.5 LRRK2 rat models
For the last several decades, investigators have chosen to use mouse models because of the technologies that were available. Now the same technologies are available in the rat. As a model of human disease, the rat offers many advantages over the mouse and other organisms. Physiology is easier to monitor in the rat. Moreover, in many cases, the physiology is more like the corresponding human condition. The rat is more intelligent than the mouse and is capable of learning a wider variety of tasks that are important in mimicking human behavioral symptoms. Recently, both LRRK2 knockout and transgenic rat models have been generated and characterized.
1.5.1 LRRK2 knockout rat models
Like other LRRK2 animal models, LRRK2 knockout rats have no significant loss of SNpc neurons. Similar to LRRK2 knockout mice, loss of LRRK2 in rats leads to abnormal phenotypes in peripheral organs. Two studies have observed abnormal kidneys [76, 77]. Besides the kidney phenotype, Ness et al. observed significant weight gain in the LRRK2 knockout rats accompanied by significant increases in insulin and insulin-like growth factors [77]. They also found significant alterations in the cellular composition of the spleen in LRRK2 knockout animals, which Baptista et al. did not observe [76, 77]. Instead, they found LRRK2 knockout rats displayed an abnormal lung and liver phenotype. Using LRRK2 knockout rats, the West group demonstrated resistance to DA neurodegeneration elicited by intracranial administration of LPS and protection from α-synuclein-induced DA neurodegeneration and rhabdomyolysis-induced kidney injury [78, 79]. The abnormal peripheral phenotype of the LRRK2 knockout rat is suggestive of a complex LRRK2 biology influencing metabolism, immune function and kidney homeostasis. The phenotype of LRRK2 knockout rat is consistent with LRRK2 knock out in other organisms such as Drosophila, C. elegans and mouse, supporting the concept that LRRK2 plays little role in the development and survival of DA neurons under physiologic conditions.
1.5.2 LRRK2 transgenic rat models
The first LRRK2 transgenic rat model was developed by Zhou et al. using an inducible system [80]. Temporal expression of human LRRK2 G2019S in rats did not lead to DA neurodegeneration, but enhanced locomotor activity with age accompanied with impaired dopamine reuptake by the dopamine transporter (DAT) was observed. As a result of compromised DAT activity, amphetamine-evoked dopamine release and amphetamine-elicited locomotor activity were reduced in LRRK2 G2019S transgenic rats [80]. Since only two copies of LRRK2 transgene was expressed in this model, there may have been insufficient protein to produce DA neurodegeneration. Human BAC-LRRK2 G2019S or R1441G rats were developed and mutant LRRK2 expression was approximately 5~8 times higher than endogenous rat LRRK2. However, both BAC-LRRK2 R1441G and G2019S transgenic rats do not show signs of neurodegeneration and do not develop significant motor or cognitive deficits with age [81–83]. Instead, LRRK2 G2019S induced oxidative stress in the striatum and substantia nigra, increased inducible nitric oxide synthase expression in SNpc DA neurons, and abnormal morphology of SNpc DA neurons [81, 83]. Although this model does not reproduce the key features of end-stage PD, it may be useful in studying gene-environment interactions. However, a recent study indicates that BAC-LRRK2 R1441G transgenic rats did not show increased vulnerability to sub-toxic doses of paraquat [82]. Since these studies lacked a wild type human LRRK2 transgenic rat as a control, it is not possible to conclude that the phenotype induced by mutant LRRK2 is due to the LRRK2 PD mutation or overexpression of the LRRK2 protein. All results from different LRRK2 transgenic rats suggest that rats compensate and accommodate LRRK2’s toxic effects.
2. Viral-mediated animal models of LRRK2
While the genetic LRRK2 models shed light on LRRK2 cellular functions and pathogenic pathways, development of recombinant viral vectors for in vivo delivery of transgenes has opened up a new possibility to model diseases in the CNS. The viral mediated gene transfer approach in adulthood bypasses the development of compensatory effects. This approach also allows researchers to target specific neuronal populations, such as SNpc DA neurons. Another advantage of the viral mediated gene transfer approach is that it allows researchers to control transgene dosage by modulating copy number of the transgene. While viral models allow us to recapitulate some of the neurodegeneration processes observed in PD patients that has so far been difficult to show in other models, there are caveats of non-physiological doses of transgenes and potential alterations in RNA translation.
Due to large size of LRRK2 gene and the limited packaging capacity of different viral vectors, so far only two LRRK2 viral models have been developed and characterized.
2.1. Herpes simplex virus (HSV)-LRRK2 viral model
The first LRRK2 viral model was developed by Lee et al. by carrying LRRK2 into HSV amplicons expressing a CMV-driven GFP reporter [84]. One advantage of HSV is that it is injected in the striatum and retrogradely transported into SNpc DA neurons, which avoids non-specific inflammatory damage to the substantia nigra. In this model, after 3 weeks injection, the HSV-WT-LRRK2 induced modest SNpc DA neurodegeneration of about 10–20%, whereas the HSV-LRRK2 G2019S induced up to 50% neuronal loss in SNpc DA neurons. Interestingly, the kinase-dead LRRK2 does not induce neuronal loss, which strongly suggested that kinase activity of LRRK2 mediates LRRK2 induced DA neurodegeneration. This notion is further supported by the protective effects of pharmacological inhibition of LRRK2 kinase activity in this HSV model [84].
2.2. Adenoviruses (rAd)-LRRK2 viral model
Second-generation E1, E3, E2a-deleted recombinant human serotype 5 adenoviruses (rAd) carrying LRRK2 WT and G2019S were generated by Dusonchet, et al. [85]. Similar to HSV, adenoviral particles can be efficiently retrogradely transported to DA neurons within the SNpc following intrastriatal injections. Injection of rAd-LRRK2 G2019S into rat striatum causes a progressive loss of TH-positive DA neurons in the SNpc, reaching about 21% at 42 d post injection but no cell loss is detected in the rAd-GFP- or rAd-LRRK2 WT injected groups. Abnormal transient hyperphosphorylation of tau in dystrophic SNpc neuritic processes was observed upon LRRK2 overexpression at 10 days [85]. Tsika et al. further characterized the striatal pathology in this model [86]. Expression of LRRK2 G2019S selectively induces the accumulation of neuronal ubiquitin-positive inclusions accompanied by neurite degeneration and the altered distribution of axonal phosphorylated neurofilaments in striatum. The pathological phenotype is dependent on LRRK2 kinase activity as a kinase-inactive mutation (LRRK2 G2019S/D1994N) completely ameliorates the pathological effects of LRRK2 G2019S [86].
Another LRRK2 viral model has been briefly mentioned in another study. The authors delivered lentiviral vectors carrying enhanced green fluorescent protein (eGFP)-tagged LRRK2 G2019S in adult mouse striatum and observed LRRK2 function in TGN turnover [87]. However, there was no characterization of this model in terms of pathology in the nigrostriatal pathways.
Concluding remarks
Modeling of LRRK2 associated PD in various animal models has provided unprecedented insights into the potential mechanisms of LRRK2 mediated neurodegeneration such as regulation of protein translation, vesicle trafficking, neurite outgrowth, autophagy and cytoskeletal dynamics. However, none of the current LRRK2 animal models fulfills all the key features of PD. Different LRRK2 animal models recapitulate different clinical and neuropathological features of LRRK2 -associated PD, including the degeneration of nigrostriatal DA neurons, neuropathology, α-synuclein accumulation, abnormal striatal DA neurotransmission, and behavioral deficits.
Why are the animal models ‘imperfect’ for modeling LRRK2 associated PD? First, for the simple animal models such as Drosophila and C. elegans, they do not have α-synuclein homolog and a true human LRRK2 homolog. PD patients harboring LRRK2 mutations frequently exhibit α-synuclein neuropathology in the form of Lewy bodies. A question about whether α-synuclein is required for LRRK2 pathology or vice visa has been raised. The challenge remains to validate the mechanisms identified in these model systems in human PD. Second, for LRRK2 rodent models, perhaps rodent DA neurons are particularly resistant to LRRK2 toxicity. In addition, there may be compensatory mechanisms in the rodents that prevent loss of DA neuron. Third, the fact that LRRK2 mutations in humans are partially penetrant, implicating that there may be additional factors such as genetic and/or environmental stressors that are required for degeneration of DA neurons. In deed, in both LRRK2 Drosophila and C. elegans models, treatments with mitochondrial function inhibitors exacerbate neurodegeneration. Fourth, the HSV or adenovirus-mediated LRRK2 rodent models induce robust DA neurodegeneration, supporting the notion that both non-cell-autonomous and cell-autonomous processes contribute to the degeneration of DA neurons. The transgene can be virally expressed in both neurons and glia to activate the inflammatory pathway in glial cells and elicit neurodegeneration in DA neurons, which is largely absent in the genetic LRRK2 models. Therefore, non-cell autonomous effects may provide a promising mechanism for LRRK2 induced PD in humans. All these possibilities need to be taken into consideration in developing future LRRK2 animal models.
Acknowledgments
This work was supported by grants from the NIH/NINDS NS38377 (VLD and TMD), the JPB Foundation (TMD), NIH/NIA K01-AG046366 (YX) and The William N. & Bernice E. Bumpus Foundation Innovation Awards (YX). TMD is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation’s Parkinson’s Disease Programs, M-1, M-2.
References
- 1.Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet. 2009;373(9680):2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 2.Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116(7):1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson’s disease. Annual review of genomics and human genetics. 2011;12:301–325. doi: 10.1146/annurev-genom-082410-101440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, Reinthaler E, Harutyunyan A, Kralovics R, Peters A, Zimprich F, Brucke T, Poewe W, Auff E, Trenkwalder C, Rost B, Ransmayr G, Winkelmann J, Meitinger T, Strom TM. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89(1):168–175. doi: 10.1016/j.ajhg.2011.06.008. doi:S0002-9297(11)00261-8 [pii] 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chartier-Harlin MC, Dachsel JC, Vilarino-Guell C, Lincoln SJ, Lepretre F, Hulihan MM, Kachergus J, Milnerwood AJ, Tapia L, Song MS, Le Rhun E, Mutez E, Larvor L, Duflot A, Vanbesien-Mailliot C, Kreisler A, Ross OA, Nishioka K, Soto-Ortolaza AI, Cobb SA, Melrose HL, Behrouz B, Keeling BH, Bacon JA, Hentati E, Williams L, Yanagiya A, Sonenberg N, Lockhart PJ, Zubair AC, Uitti RJ, Aasly JO, Krygowska-Wajs A, Opala G, Wszolek ZK, Frigerio R, Maraganore DM, Gosal D, Lynch T, Hutchinson M, Bentivoglio AR, Valente EM, Nichols WC, Pankratz N, Foroud T, Gibson RA, Hentati F, Dickson DW, Destee A, Farrer MJ. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet. 2011;89(3):398–406. doi: 10.1016/j.ajhg.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, Ghika J, Hentati F, Krygowska-Wajs A, Lynch T, Melamed E, Rajput A, Rajput AH, Solida A, Wu RM, Uitti RJ, Wszolek ZK, Vingerhoets F, Farrer MJ. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89(1):162–167. doi: 10.1016/j.ajhg.2011.06.001. doi:S0002-9297(11)00242-4 [pii] 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vilarino-Guell C, Rajput A, Milnerwood AJ, Shah B, Szu-Tu C, Trinh J, Yu I, Encarnacion M, Munsie LN, Tapia L, Gustavsson EK, Chou P, Tatarnikov I, Evans DM, Pishotta FT, Volta M, Beccano-Kelly D, Thompson C, Lin MK, Sherman HE, Han HJ, Guenther BL, Wasserman WW, Bernard V, Ross CJ, Appel-Cresswell S, Stoessl AJ, Robinson CA, Dickson DW, Ross OA, Wszolek ZK, Aasly JO, Wu RM, Hentati F, Gibson RA, McPherson PS, Girard M, Rajput M, Rajput AH, Farrer MJ. DNAJC13 mutations in Parkinson disease. Hum Mol Genet. 2014;23(7):1794–1801. doi: 10.1093/hmg/ddt570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scholz SW, Jeon BS. GBA mutations and Parkinson disease: when genotype meets phenotype. Neurology. 2015;84(9):866–867. doi: 10.1212/WNL.0000000000001321. [DOI] [PubMed] [Google Scholar]
- 9.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600. doi: 10.1016/j.neuron.2004.10.023. doi:S0896627304006890 [pii] 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 10.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci. 2006;29(5):286–293. doi: 10.1016/j.tins.2006.03.006. doi:S0166-2236(06)00069-5 [pii] 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci. 2010;11(12):791–797. doi: 10.1038/nrn2935. doi:nrn2935 [pii] 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berwick DC, Harvey K. LRRK2 signaling pathways: the key to unlocking neurodegeneration? Trends Cell Biol. 2011 doi: 10.1016/j.tcb.2011.01.001. doi:S0962-8924(11)00002-X [pii] 10.1016/j.tcb.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Xiong Y, Dawson VL, Dawson TM. LRRK2 GTPase dysfunction in the pathogenesis of Parkinson’s disease. Biochem Soc Trans. 2012;40(5):1074–1079. doi: 10.1042/BST20120093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cookson MR. LRRK2 Pathways Leading to Neurodegeneration. Curr Neurol Neurosci Rep. 2015;15(7):42. doi: 10.1007/s11910-015-0564-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin I, Kim JW, Dawson VL, Dawson TM. LRRK2 pathobiology in Parkinson’s disease. J Neurochem. 2014;131(5):554–565. doi: 10.1111/jnc.12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nikonova EV, Xiong Y, Tanis KQ, Dawson VL, Vogel RL, Finney EM, Stone DJ, Reynolds IJ, Kern JT, Dawson TM. Transcriptional responses to loss or gain of function of the leucine-rich repeat kinase 2 (LRRK2) gene uncover biological processes modulated by LRRK2 activity. Hum Mol Genet. 2012;21(1):163–174. doi: 10.1093/hmg/ddr451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniel G, Moore DJ. Modeling LRRK2 Pathobiology in Parkinson’s Disease: From Yeast to Rodents. Curr Top Behav Neurosci. 2015;22:331–368. doi: 10.1007/7854_2014_311. [DOI] [PubMed] [Google Scholar]
- 19.Xiong Y, Coombes CE, Kilaru A, Li X, Gitler AD, Bowers WJ, Dawson VL, Dawson TM, Moore DJ. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet. 2010;6(4):e1000902. doi: 10.1371/journal.pgen.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiong Y, Yuan C, Chen R, Dawson TM, Dawson VL. ArfGAP1 is a GTPase activating protein for LRRK2: reciprocal regulation of ArfGAP1 by LRRK2. J Neurosci. 2012;32(11):3877–3886. doi: 10.1523/JNEUROSCI.4566-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27(18):2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun. 2007;358(2):534–539. doi: 10.1016/j.bbrc.2007.04.156. [DOI] [PubMed] [Google Scholar]
- 23.Wang D, Tang B, Zhao G, Pan Q, Xia K, Bodmer R, Zhang Z. Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol Neurodegener. 2008;3(3) doi: 10.1186/1750-1326-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci. 2009;12(9):1129–1135. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, Ross CA, Montell C, Smith WW. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A. 2008;105(7):2693–2698. doi: 10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng CH, Mok SZ, Koh C, Ouyang X, Fivaz ML, Tan EK, Dawson VL, Dawson TM, Yu F, Lim KL. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci. 2009;29(36):11257–11262. doi: 10.1523/JNEUROSCI.2375-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venderova K, Kabbach G, Abdel-Messih E, Zhang Y, Parks RJ, Imai Y, Gehrke S, Ngsee J, Lavoie MJ, Slack RS, Rao Y, Zhang Z, Lu B, Haque ME, Park DS. Leucine-Rich Repeat Kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson’s disease. Hum Mol Genet. 2009;18(22):4390–4404. doi: 10.1093/hmg/ddp394. [DOI] [PubMed] [Google Scholar]
- 28.Lin CH, Tsai PI, Wu RM, Chien CT. LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ss. J Neurosci. 2010;30(39):13138–13149. doi: 10.1523/JNEUROSCI.1737-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hindle S, Afsari F, Stark M, Middleton CA, Evans GJ, Sweeney ST, Elliott CJ. Dopaminergic expression of the Parkinsonian gene LRRK2-G2019S leads to non-autonomous visual neurodegeneration, accelerated by increased neural demands for energy. Hum Mol Genet. 2013;22(11):2129–2140. doi: 10.1093/hmg/ddt061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Godena VK, Brookes-Hocking N, Moller A, Shaw G, Oswald M, Sancho RM, Miller CC, Whitworth AJ, De Vos KJ. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat Commun. 2014;5:5245. doi: 10.1038/ncomms6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saha AR, Hill J, Utton MA, Asuni AA, Ackerley S, Grierson AJ, Miller CC, Davies AM, Buchman VL, Anderton BH, Hanger DP. Parkinson’s disease alpha-synuclein mutations exhibit defective axonal transport in cultured neurons. J Cell Sci. 2004;117(Pt 7):1017–1024. doi: 10.1242/jcs.00967. [DOI] [PubMed] [Google Scholar]
- 32.Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466(7306):637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin I, Kim JW, Lee BD, Kang HC, Xu JC, Jia H, Stankowski J, Kim MS, Zhong J, Kumar M, Andrabi SA, Xiong Y, Dickson DW, Wszolek ZK, Pandey A, Dawson TM, Dawson VL. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell. 2014;157(2):472–485. doi: 10.1016/j.cell.2014.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dodson MW, Zhang T, Jiang C, Chen S, Guo M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet. 2012;21(6):1350–1363. doi: 10.1093/hmg/ddr573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schreij AM, Chaineau M, Ruan W, Lin S, Barker PA, Fon EA, McPherson PS. LRRK2 localizes to endosomes and interacts with clathrin-light chains to limit Rac1 activation. EMBO Rep. 2015;16(1):79–86. doi: 10.15252/embr.201438714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dodson MW, Leung LK, Lone M, Lizzio MA, Guo M. Novel ethyl methanesulfonate (EMS)-induced null alleles of the Drosophila homolog of LRRK2 reveal a crucial role in endolysosomal functions and autophagy in vivo. Dis Model Mech. 2014;7(12):1351–1363. doi: 10.1242/dmm.017020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock PJ, Morais VA, Vilain S, Haddad D, Delbroek L, Swerts J, Chavez-Gutierrez L, Esposito G, Daneels G, Karran E, Holt M, Gevaert K, Moechars DW, De Strooper B, Verstreken P. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75(6):1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 38.Lin CH, Li H, Lee YN, Cheng YJ, Wu RM, Chien CT. Lrrk regulates the dynamic profile of dendritic Golgi outposts through the golgin Lava lamp. J Cell Biol. 2015 doi: 10.1083/jcb.201411033. pii: jcb.201411033 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linhart R, Wong SA, Cao J, Tran M, Huynh A, Ardrey C, Park JM, Hsu C, Taha S, Peterson R, Shea S, Kurian J, Venderova K. Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson’s disease mutant of Leucine-Rich Repeat Kinase 2 (LRRK2) Mol Neurodegener. 2014;9:23. doi: 10.1186/1750-1326-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77(3):425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee S, Liu HP, Lin WY, Guo H, Lu B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J Neurosci. 2010;30(50):16959–16969. doi: 10.1523/JNEUROSCI.1807-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Hamamichi S, Lee BD, Yang D, Ray A, Caldwell GA, Caldwell KA, Dawson TM, Smith WW, Dawson VL. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum Mol Genet. 2011;20(20):3933–3942. doi: 10.1093/hmg/ddr312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang D, Li T, Liu Z, Arbez N, Yan J, Moran TH, Ross CA, Smith WW. LRRK2 kinase activity mediates toxic interactions between genetic mutation and oxidative stress in a Drosophila model: suppression by curcumin. Neurobiol Dis. 2012;47(3):385–392. doi: 10.1016/j.nbd.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 44.Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol. 2007;17(7):592–598. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- 45.Yuan Y, Cao P, Smith MA, Kramp K, Huang Y, Hisamoto N, Matsumoto K, Hatzoglou M, Jin H, Feng Z. Dysregulated LRRK2 signaling in response to endoplasmic reticulum stress leads to dopaminergic neuron degeneration in C. elegans. PLoS One. 2011;6(8):e22354. doi: 10.1371/journal.pone.0022354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Samann J, Hegermann J, von Gromoff E, Eimer S, Baumeister R, Schmidt E. Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J Biol Chem. 2009;284(24):16482–16491. doi: 10.1074/jbc.M808255200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, Ghosh J, Hsu CH, Segal L, Raghavan K, Matsumoto K, Hisamoto N, Kuwahara T, Iwatsubo T, Moore L, Goldstein L, Cookson M, Wolozin B. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J Neurosci. 2009;29(29):9210–9218. doi: 10.1523/JNEUROSCI.2281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yao C, El Khoury R, Wang W, Byrd TA, Pehek EA, Thacker C, Zhu X, Smith MA, Wilson-Delfosse AL, Chen SG. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol Dis. 2010;40(1):73–81. doi: 10.1016/j.nbd.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saha S, Liu-Yesucevitz L, Wolozin B. Regulation of autophagy by LRRK2 in Caenorhabditis elegans. Neurodegener Dis. 2014;13(2–3):110–113. doi: 10.1159/000355654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saha S, Ash PE, Gowda V, Liu L, Shirihai O, Wolozin B. Mutations in LRRK2 potentiate age-related impairment of autophagic flux. Mol Neurodegener. 2015;10(1):26. doi: 10.1186/s13024-015-0022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheng D, Qu D, Kwok KH, Ng SS, Lim AY, Aw SS, Lee CW, Sung WK, Tan EK, Lufkin T, Jesuthasan S, Sinnakaruppan M, Liu J. Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS Genet. 2010;6(4):e1000914. doi: 10.1371/journal.pgen.1000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ren G, Xin S, Li S, Zhong H, Lin S. Disruption of LRRK2 does not cause specific loss of dopaminergic neurons in zebrafish. PLoS One. 2011;6(6):e20630. doi: 10.1371/journal.pone.0020630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lichtenberg M, Mansilla A, Zecchini VR, Fleming A, Rubinsztein DC. The Parkinson’s disease protein LRRK2 impairs proteasome substrate clearance without affecting proteasome catalytic activity. Cell Death Dis. 2011;2:e196. doi: 10.1038/cddis.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu Q, Shenoy S, Li C. Mouse models for LRRK2 Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S186–189. doi: 10.1016/S1353-8020(11)70058-X. [DOI] [PubMed] [Google Scholar]
- 55.Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron. 2010;66(5):646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee Y, Dawson VL, Dawson TM. Animal models of Parkinson’s disease: vertebrate genetics. Cold Spring Harb Perspect Med. 2012;2(10) doi: 10.1101/cshperspect.a009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andres-Mateos E, Mejias R, Sasaki M, Li X, Lin BM, Biskup S, Zhang L, Banerjee R, Thomas B, Yang L, Liu G, Beal MF, Huso DL, Dawson TM, Dawson VL. Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) J Neurosci. 2009;29(50):15846–15850. doi: 10.1523/JNEUROSCI.4357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S, Schnell CR, Mueller M, Kinzel B, Grevot A, Bolognani F, Stirn M, Kuhn RR, Kaupmann K, van der Putten PH, Rovelli G, Shimshek DR. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. 2011;20(21):4209–4223. doi: 10.1093/hmg/ddr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hinkle KM, Yue M, Behrouz B, Dachsel JC, Lincoln SJ, Bowles EE, Beevers JE, Dugger B, Winner B, Prots I, Kent CB, Nishioka K, Lin WL, Dickson DW, Janus CJ, Farrer MJ, Melrose HL. LRRK2 knockout mice have an intact dopaminergic system but display alterations in exploratory and motor co-ordination behaviors. Mol Neurodegener. 2012;7(25) doi: 10.1186/1750-1326-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron. 2009;64(6):807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paus M, Kohl Z, Ben Abdallah NM, Galter D, Gillardon F, Winkler J. Enhanced dendritogenesis and axogenesis in hippocampal neuroblasts of LRRK2 knockout mice. Brain Res. 2013;1497:85–100. doi: 10.1016/j.brainres.2012.12.024. [DOI] [PubMed] [Google Scholar]
- 62.Tong Y, Giaime E, Yamaguchi H, Ichimura T, Liu Y, Si H, Cai H, Bonventre JV, Shen J. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol Neurodegener. 2012;7(2) doi: 10.1186/1750-1326-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ, 3rd, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010;107(21):9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Daher JP, Pletnikova O, Biskup S, Musso A, Gellhaar S, Galter D, Troncoso JC, Lee MK, Dawson TM, Dawson VL, Moore DJ. Neurodegenerative phenotypes in an A53T alpha-synuclein transgenic mouse model are independent of LRRK2. Hum Mol Genet. 2012;21(11):2420–2431. doi: 10.1093/hmg/dds057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen CY, Weng YH, Chien KY, Lin KJ, Yeh TH, Cheng YP, Lu CS, Wang HL. (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 2012;19(10):1623–1633. doi: 10.1038/cdd.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J Neurosci. 2010;30(5):1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, Beal MF, Burke RE, Li C. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat Neurosci. 2009;12(7):826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Melrose HL, Dachsel JC, Behrouz B, Lincoln SJ, Yue M, Hinkle KM, Kent CB, Korvatska E, Taylor JP, Witten L, Liang YQ, Beevers JE, Boules M, Dugger BN, Serna VA, Gaukhman A, Yu X, Castanedes-Casey M, Braithwaite AT, Ogholikhan S, Yu N, Bass D, Tyndall G, Schellenberg GD, Dickson DW, Janus C, Farrer MJ. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol Dis. 2010;40(3):503–517. doi: 10.1016/j.nbd.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland NG, Galter D, Thomas B, Lee MK, Dawson TM, Dawson VL, Moore DJ. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One. 2011;6(4):e18568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsika E, Kannan M, Foo CS, Dikeman D, Glauser L, Gellhaar S, Galter D, Knott GW, Dawson TM, Dawson VL, Moore DJ. Conditional expression of Parkinson’s disease-related R1441C LRRK2 in midbrain dopaminergic neurons of mice causes nuclear abnormalities without neurodegeneration. Neurobiol Dis. 2014;71:345–358. doi: 10.1016/j.nbd.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu G, Sgobio C, Gu X, Sun L, Lin X, Yu J, Parisiadou L, Xie C, Sastry N, Ding J, Lohr KM, Miller GW, Mateo Y, Lovinger DM, Cai H. Selective expression of Parkinson’s disease-related Leucine-rich repeat kinase 2 G2019S missense mutation in midbrain dopaminergic neurons impairs dopamine release and dopaminergic gene expression. Hum Mol Genet. 2015 doi: 10.1093/hmg/ddv249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beccano-Kelly DA, Volta M, Munsie LN, Paschall SA, Tatarnikov I, Co K, Chou P, Cao LP, Bergeron S, Mitchell E, Han H, Melrose HL, Tapia L, Raymond LA, Farrer MJ, Milnerwood AJ. LRRK2 overexpression alters glutamatergic presynaptic plasticity, striatal dopamine tone, postsynaptic signal transduction, motor activity and memory. Hum Mol Genet. 2015;24(5):1336–1349. doi: 10.1093/hmg/ddu543. [DOI] [PubMed] [Google Scholar]
- 73.Garcia-Miralles M, Coomaraswamy J, Habig K, Herzig MC, Funk N, Gillardon F, Maisel M, Jucker M, Gasser T, Galter D, Biskup S. No dopamine cell loss or changes in cytoskeleton function in transgenic mice expressing physiological levels of wild type or G2019S mutant LRRK2 and in human fibroblasts. PLoS One. 2015;10(4):e0118947. doi: 10.1371/journal.pone.0118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, Shen J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci U S A. 2009;106(34):14622–14627. doi: 10.1073/pnas.0906334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yue M, Hinkle KM, Davies P, Trushina E, Fiesel FC, Christenson TA, Schroeder AS, Zhang L, Bowles E, Behrouz B, Lincoln SJ, Beevers JE, Milnerwood AJ, Kurti A, McLean PJ, Fryer JD, Springer W, Dickson DW, Farrer MJ, Melrose HL. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol Dis. 2015;78:172–195. doi: 10.1016/j.nbd.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baptista MA, Dave KD, Frasier MA, Sherer TB, Greeley M, Beck MJ, Varsho JS, Parker GA, Moore C, Churchill MJ, Meshul CK, Fiske BK. Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS One. 2013;8(11):e80705. doi: 10.1371/journal.pone.0080705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ness D, Ren Z, Gardai S, Sharpnack D, Johnson VJ, Brennan RJ, Brigham EF, Olaharski AJ. Leucine-rich repeat kinase 2 (LRRK2)-deficient rats exhibit renal tubule injury and perturbations in metabolic and immunological homeostasis. PLoS One. 2013;8(6):e66164. doi: 10.1371/journal.pone.0066164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boddu R, Hull TD, Bolisetty S, Hu X, Moehle MS, Daher JP, Kamal AI, Joseph R, George JF, Agarwal A, Curtis LM, West AB. Leucine-rich repeat kinase 2 deficiency is protective in rhabdomyolysis-induced kidney injury. Hum Mol Genet. 2015;24(14):4078–4093. doi: 10.1093/hmg/ddv147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Daher JP, Volpicelli-Daley LA, Blackburn JP, Moehle MS, West AB. Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc Natl Acad Sci U S A. 2014;111(25):9289–9294. doi: 10.1073/pnas.1403215111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou H, Huang C, Tong J, Hong WC, Liu YJ, Xia XG. Temporal expression of mutant LRRK2 in adult rats impairs dopamine reuptake. Int J Biol Sci. 2011;7(6):753–761. doi: 10.7150/ijbs.7.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee JW, Tapias V, Di Maio R, Greenamyre JT, Cannon JR. Behavioral, neurochemical, and pathologic alterations in bacterial artificial chromosome transgenic G2019S leucine-rich repeated kinase 2 rats. Neurobiol Aging. 2015;36(1):505–518. doi: 10.1016/j.neurobiolaging.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shaikh KT, Yang A, Youshin E, Schmid S. Transgenic LRRK2 (R1441G) rats-a model for Parkinson disease? PeerJ. 2015;3:e945. doi: 10.7717/peerj.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Walker MD, Volta M, Cataldi S, Dinelle K, Beccano-Kelly D, Munsie L, Kornelsen R, Mah C, Chou P, Co K, Khinda J, Mroczek M, Bergeron S, Yu K, Cao LP, Funk N, Ott T, Galter D, Riess O, Biskup S, Milnerwood AJ, Stoessl AJ, Farrer MJ, Sossi V. Behavioral deficits and striatal DA signaling in LRRK2 p.G2019S transgenic rats: a multimodal investigation including PET neuroimaging. J Parkinsons Dis. 2014;4(3):483–498. doi: 10.3233/JPD-140344. [DOI] [PubMed] [Google Scholar]
- 84.Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, Lee YI, Maguire-Zeiss KA, Bowers WJ, Federoff HJ, Dawson VL, Dawson TM. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med. 2010;16(9):998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dusonchet J, Kochubey O, Stafa K, Young SM, Jr, Zufferey R, Moore DJ, Schneider BL, Aebischer P. A rat model of progressive nigral neurodegeneration induced by the Parkinson’s disease-associated G2019S mutation in LRRK2. J Neurosci. 2011;31(3):907–912. doi: 10.1523/JNEUROSCI.5092-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsika E, Nguyen AP, Dusonchet J, Colin P, Schneider BL, Moore DJ. Adenoviral-mediated expression of G2019S LRRK2 induces striatal pathology in a kinase-dependent manner in a rat model of Parkinson’s disease. Neurobiol Dis. 2015;77:49–61. doi: 10.1016/j.nbd.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 87.Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe K, Ding J, Nalls MA, C. International Parkinson’s Disease Genomics, C. North American Brain Expression. Olszewski M, Hauser DN, Kumaran R, Lozano AM, Baekelandt V, Greene LE, Taymans JM, Greggio E, Cookson MR. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A. 2014;111(7):2626–2631. doi: 10.1073/pnas.1318306111. [DOI] [PMC free article] [PubMed] [Google Scholar]