

Summary
Mcm2-7 helicase is loaded onto double stranded origin DNA as an inactive double hexamer (DH) in G1 phase. The mechanisms of Mcm2-7 remodeling to trigger helicase activation in S phase remain unknown. Here, we develop an approach to detect and purify the endogenous DHs directly. Through cellular fractionation, we provide in vivo evidence that DHs are assembled on chromatin in G1 phase and separated during S phase. Interestingly, Mcm10, a robust MCM interactor, co-purifies exclusively with the DHs in the context of chromatin. Deletion of the main interaction domain, Mcm10 C-terminus, causes growth and S phase defects, which can be suppressed through Mcm10-MCM fusions. By monitoring the dynamics of MCM DHs, we show a significant delay in DH dissolution during S phase in the Mcm10-MCM interaction deficient mutants. Therefore, we propose an essential role for Mcm10 in Mcm2-7 remodeling through formation of a cell cycle regulated supercomplex with DHs.
Keywords: DNA replication, cell cycle, helicase activation, Mcm2-7, Mcm10
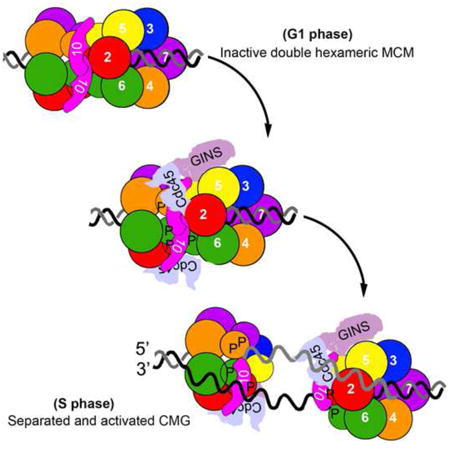
Graphical abstract
Introduction
The assembly of the DNA replication machinery and initiation of synthesis are controlled in a tightly orchestrated manner according to different stages of the cell cycle (Costa et al., 2013; Heller et al., 2011; Labib, 2010; Siddiqui et al., 2013). The origin licensing step involving recruitment and assembly of the replicative helicase, mini-chromosome maintenance (MCM), into the pre-replication complexes (pre-RC) has been reconstituted in vitro through purified yeast proteins (Evrin et al., 2009; Remus et al., 2009). Notably, two Mcm2–7 hexameric rings are sequentially loaded onto double-stranded (ds) DNA as an inactive head-to-head double hexamer (DH) (Evrin et al., 2009; Gambus et al., 2011; Remus et al., 2009; Ticau et al., 2015). These findings raise an intriguing question: How is the double hexameric MCM activated to initiate bidirectional DNA replication in eukaryotes (Boos et al., 2012; Li and Araki, 2013; Tognetti et al., 2014)?
Mcm2-7 in solution exhibits primarily a single hexameric structure stabilized upon ATP binding (Bochman and Schwacha, 2009; Coster et al., 2014). The pre-RC intermediates are very sensitive to salt wash, while the MCM DHs remain very stable on chromatin in the presence of high salt (Gambus et al., 2011; Remus et al., 2009). It is of particular importance to ensure that MCM hexamers be poised on chromatin before S phase ready for activation given the fact that helicase reloading is blocked during S phase (Bell and Dutta, 2002; Masai et al., 2010). The DH state may be maintained in the initial holo-helicase Cdc45–Mcm2–7–GINS (CMG) complex (Costa et al., 2014). However, the two helicase rings need to be separated and remodeled to encircle the leading strands to initiate bidirectional replication (Fu et al., 2011; Yardimci et al., 2010). The two rings are dimerized through an interface composed of the N-termini of Mcm2-7 subunits (Evrin et al., 2009; Fletcher et al., 2003; Remus et al., 2009), which bear multiple critical target sites for protein kinases, such as Dbf4-dependent kinase Cdc7 (DDK) and CDK (Hoang et al., 2007; Sheu and Stillman, 2010; Sheu et al., 2014). Phosphorylation is thought to be required, but not sufficient to activate the helicase (On et al., 2014; Yeeles et al., 2015).
Mcm10 is among the recently published minimal set of the essential firing factors for reconstituted DNA synthesis in vitro (Yeeles et al., 2015), and has been inferred to be important in Mcm2-7 helicase activation post CMG formation, as indicated by Mcm10 depletion in yeast (Kanke et al., 2012; van Deursen et al., 2012; Watase et al., 2012) and Xenopus (Pacek et al., 2006). However, the mechanistic details of Mcm10 function have yet to be defined (Thu and Bielinsky, 2013; Thu and Bielinsky, 2014).
In this study we developed an approach to purify the endogenous MCM complexes from yeast cells which allows us to monitor the formation and separation of MCM DHs in vivo. Using this assay we were able to show that Mcm10 defines an essential role in splitting DHs. Interestingly Mcm10 does not associate with MCM complexes until being loaded onto chromatin as the DHs. Though Mcm10-DH association occurs in G1, it is enhanced in S phase. Their direct interaction is mainly mediated by a previously uncharacterized C-terminus of Mcm10. Loss of Mcm10 C-terminus causes the S phase defects, which can be suppressed by artificially fusing Mcm10 and MCM. Furthermore, we showed that mcm1OΔC displays a significant delay in separating the double hexameric CMG complexes. We propose that MCM10 C-terminus mediated specific interaction with the DHs plays critical role in MCM DH splitting.
Results
Isolation of the endogenous MCM DH species
To uncover the mechanism of Mcm2-7 helicase activation, first we developed an approach to detect the DH form of Mcm2-7 in vivo, which has been extensively studied in an in vitro pre-RC reconstitution system with purified yeast proteins (Evrin et al., 2009; Remus et al., 2009; Ticau et al., 2015). To this end, we introduced a second copy of Mcm4 with a 3HA tag while the endogenous Mcm4 was tagged with 5FLAG. The tagged strains showed nearly the same growth as wild-type (WT) (Figure S1A). The MCM complexes containing both Mcm4-FLAG and Mcm4-HA should result from the formation of MCM DH because a single heterohexameric ring contains only one copy of each Mcm2-7 subunit (Figure 1A) (Costa et al., 2014; Sun et al., 2014). This enables us to isolate the putative double hexameric MCM species specifically via sucrose gradient centrifugation or sequential immunoprecipitations (IP) coupled with peptide elution. As illustrated in Figure 1A, to enrich for the chromatin-loaded MCM, we modified a protocol developed previously to prepare a large scale native chromatin fraction (SN2) from yeast cells (Sheu and Stillman, 2006; van Deursen et al., 2012). The non-chromatin-bound (SN1) and SN2 fractions prepared from G1 cells were first subjected to FLAG-IP. To examine the oligomeric states of MCM in SN1 and SN2, we applied the FLAG peptide eluted samples (FLAG elution) to a 5-30% sucrose gradient. After centrifugation, fractions were collected and subjected to SDS-PAGE (polyacrylamide gel electrophoresis) and immunoblots. The Mcm4-FLAG containing complexes from SN1 did not contain any Mcm4-HA and appeared to be about 669 kDa (Figure 1B, upper panel). In SN2, the major portion of the Mcm4-FLAG complexes displayed a molecular size larger than 669 kDa. More interestingly, Mcm4-HA was present and co-sedimented with Mcm4-FLAG complexes exclusively in the more rapidly sedimenting fractions from SN2, verifying formation of the MCM DHs (Figure 1B, fractions 4-7, lower panel). These results indicated that Mcm2-7 exists as a single hexamer before being loaded onto chromatin (Gambus et al., 2011) and is assembled on chromatin in vivo into the DHs whose high resolution structure has been reported recently (Li et al., 2015). Next, FLAG elutes were precipitated using HA antibodies. The bound or HA peptide eluted fraction was analyzed either by PAGE and silver staining (Figure 1C) or immunoblotting (IB) (Figure 1D). The double hexameric MCM complexes were purified to near homogeneity through this sequential affinity purification from SN2 (Figure 1C). Each band corresponding to Mcm2-7 subunits was verified by mass spectrometry. Mcm4-FLAG and Mcm4-HA associated with each other in SN2 (Figure 1D, lanes 10-12 and 13-15), but barely in SN1 (lanes 7-9). These data verify that this procedure purifies the DH MCM species from yeast cells.
Figure 1. Purification of the endogenous DH Mcm2-7 species.
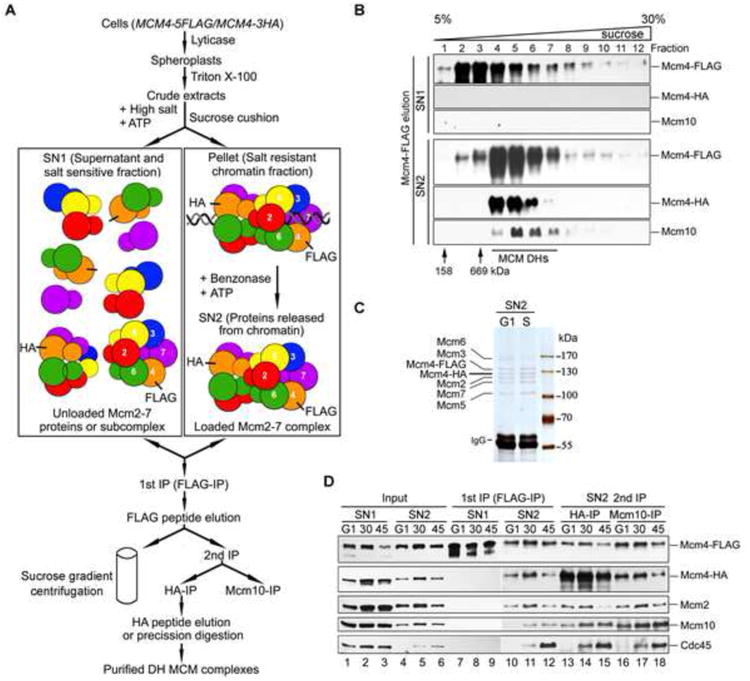
(A) A diagram of procedures to purify the native Mcm2-7 DHs from yeast cells. The MCM4-5FLAG/MCM4-3HA strain was constructed (Table S2, QY793; Table S3). Cells were synchronized in G1 or S phase by α–factor and subsequent release. A chromatin fraction (SN2) was prepared as described in Experimental Procedures. FLAG-IP and peptide elution was followed by sucrose gradient centrifugation (B) or by HA-IP (C,D). See also Figure S1 and Table S1.
(B) Mcm2-7 exists mainly as single hexamers and DHs in SN1 and SN2, respectively. Mcm4-FLAG elutes from G1 cells were applied to a 5-30% sucrose gradient. After centrifugation, the fractions were separated by SDS-PAGE and analyzed by western blotting with indicated antibodies.
(C, D) The final HA precipitates or elutes from G1 or S cells were subjected to SDS-PAGE and silver staining (C) or IB with indicated antibodies (D).
Mcm10 preferentially associates with the MCM DHs
Interestingly, Mcm10 was repeatedly found to co-purify with MCM complexes (Figure 1B, fractions 4-8; Figure 1D, lanes 10-12). Mcm10 showed positive interactions with multiple Mcm2-7 subunits including Mcm2, 4, 6 and 7 in the yeast two hybrid assay (Table S1, Figure S1B) as reported previously (Christensen and Tye, 2003; Homesley et al., 2000; Izumi et al., 2000). More important, Mcm10 was assembled into a supercomplex with both Mcm4-FLAG and Mcm4-HA, i.e., double hexameric MCM, exclusively in SN2 (Figure 1B; Figure 1D, compare lanes 10-12 to 7-9). These results indicate that Mcm10 interacts with the MCM DHs on chromatin in vivo, in agreement with previous reports that Mcm10 preferentially associates with the loaded MCM (Pacek et al., 2006; van Deursen et al., 2012). It is worth noting that although Mcm10 can be initially loaded onto the DHs weakly in G1, the Mcm10-DH association is enhanced coincidently with the accumulating recruited Cdc45 during S phase (Figure 1D, compare lanes 4-6). Moreover, the loaded Mcm10 and Cdc45 molecules were associated with the MCM DHs (lanes 13 to 15), suggesting that MCM may persist in the DH state in the context of CMG complexes (Costa et al., 2014) and Mcm10 is assembled into dimeric CMG supercomplexes in S phase. More interestingly, if we conducted an Mcm10-IP subsequent to the FLAG elution step, Mcm4-FLAG, Mcm4-HA and Cdc45 were stably recovered in association with Mcm10 (Figure 1D, lanes 16 to 18). Taken together, these data allow us to conclude that Mcm10 is assembled into a cell cycle regulated supercomplex with MCM DHs in vivo.
Separation of the MCM DHs during S phase requires Mcm10
To further quantify the possible change of MCM DHs during cell cycle progression in vivo, as illustrated in Figure 2A, we modified the assay by adding a GFP tag to the second copy of Mcm2 while the endogenous Mcm2 copy was kept intact (Figure S2A). The Mcm2 and Mcm2-GFP proteins can be separated on a gel and detected simultaneously on anti-Mcm2 immunoblots. When we trapped Mcm2-GFP by GBP (GFP binding protein) beads, the co-trapped untagged version of Mcm2 should represent the DH species of MCM prior to separation (Figure 2A). The relative amount of DHs in each sample can be estimated by the ratio of untagged Mcm2 to Mcm2-GFP in the precipitates. Consistent with observations in Figure 1D, untagged Mcm2 co-precipitated in the Mcm2-GFP trap exclusively in the 500 mM salt resistant SN2 but not in the SN1 (Figure 2B, compare lane 3 to 4), further validating the in vivo DH assay.
Figure 2. Mcm10 is required for MCM DH separation in vivo.
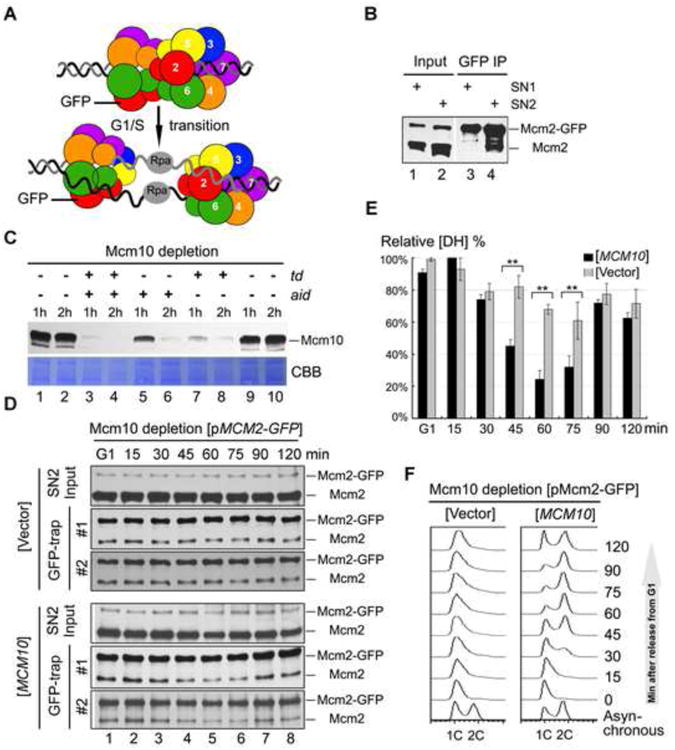
(A) An in vivo DH separation assay modified from the strategy illustrated in Figure 1A. The MCM2-GFP/MCM2 strain was constructed (Table S2, QY713; Table S3). Two versions of Mcm2 were separated on a gel and detected by anti-Mcm2 immunoblots. When we trapped Mcm2-GFP by GBP beads, the co-trapped untagged version of Mcm2 should reflect the relative amount of MCM DH prior to separation into two single hexamers during S-phase. See also Figure S2.
(B) Asynchronous cells were fractionated, incubated with GBP beads, then subject to IB with indicated antibodies. Note that MCM DHs are only detected in SN2, but not in SN1.
(C) Efficient depletion of endogenous Mcm10 protein through a two-degron strategy. Temperature-inducible (td) and auxin-inducible (aid) degrons were added to the N- and C-terminus of Mcm10ΔC (Table S2, QY394). The corresponding two ubiquitin ligases (E3), UBR1 and OsTIR1, were integrated into the genomic UBR1 locus under control of the galactose inducible Gal1 promoter.QY394 strain was first grown at 25°C in rich medium supplemented with 2% raffinose before transfer to 2% galactose to induce the expression of two E3. Two degrons were turned on by adding 500 μM indole-3-acetic acid (IAA) (aid) or switching to 37°C (td) for 1h or 2h, as indicated above each lane. The effect of depletion by each degron system or their combination was detected by IB with anti-Mcm10.
(D) In vivo DH separation assay in Mcm10-depleted background harboring an empty vector (upperpanel) or a WT MCM10 construct (lower panel). Cells were synchronized in G1 by α-factor and released for the indicated time at 37°C. Mcm2-GFP in the SN2 fraction was trapped by GBP beads and probed with anti-Mcm2 antibodies. “#1” and “#2” denote two independent samples.
(E) The amounts of Mcm2 and Mcm2-GFP in precipitates were quantified. The ratio of Mcm2/Mcm2-GFP in the IP fraction is calculated to indicate the relative amount of MCM DH in the samples. The maximum amount of DH MCM is normalized to 100%. The average and standard eviation are calculated from the results of at least three independent experiments. P-value <0.01 and0.05 are donated as “**” and “*”, respectively. To ensure the signals are within the linear range, immunoblots with different exposure were quantified by Quantity One (Biorad). See Figure S2C for the raw data and quantification.
(F) Representative cell cycle profiles of the samples used for in vivo DH separation assays. Cells were collected at the indicated time after released from G1 arrest. Cell cycle profiles were analyzed by flow cytometry.
Mcm10, as the most enigmatic essential firing factor (Yeeles et al., 2015), has been proposed to participate in a novel step during CMG activation and origin unwinding through Mcm10 depletion in yeast and Xenopus (Kanke et al., 2012; Pacek et al., 2006; van Deursen et al., 2012; Watase et al., 2012). To directly address whether Mcm10 functions in MCM DH splitting prior to or during origin unwinding, we monitored the relative DH levels during S phase progression in a conditional Mcm10 depletion background. We combined both temperature induced (td) and auxin induced (aid) degrons to deplete endogenous Mcm10 proteins (van Deursen et al., 2012; Watase et al., 2012). The td and aid degrons were turned on by switching to growth at 37°C and adding indole-3-acetic acid (IAA), respectively. The Ubr1 and Tir1 ubiquitin ligases were induced by galactose. The cellular Mcm10 protein was barely detectable after incubation in galactose for 1h and shifting to 37°C in the presence of auxin for another 2h (Figure 2C, lane 4), leading to cell death in the absence of WT Mcm10 (Figure S2B). Strikingly, under such an efficient Mcm10 depletion condition, the ratio of untagged Mcm2 to Mcm2-GFP in the GFP-trap remained almost constant after being released from α–factor into S phase for 120 min, indicating a failure in separation of MCM DHs (Figure 2D, upper panel and 2E, Figure S2C). Furthermore, no DNA synthesis was detected by flow cytometry (Figure 2F). In contrast, if WT Mcm10 was expressed from a plasmid in the same strain, the untagged Mcm2 in precipitates decreased quickly upon S phase entry (Figure 2D, lower panel and 2E, Figure S2C), indicating efficient DH separation. There is little untagged Mcm2 left in the precipitates at 60 min, which correlates well with the time of completion of DNA replication, as shown in the flow cytometry profiles (Figure 2F). Taken together, these data suggest an essential role of Mcm10 in MCM DH splitting.
Mcm10 directly binds the Mcm2, Mcm4 and Mcm6 N-termini, which form the DH interface
To investigate whether the DH splitting function of Mcm10 can be attributed to its interaction with MCM DHs, we first mapped the interaction domain(s). In the yeast two hybrid assays, the N-terminus (a.a. 1-390) of Mcm2 recapitulated the interaction between full length Mcm2 and Mcm10 (compare Figure S3A to S3B). We then constructed a set of truncations of Mcm2-7 subunits and affinity purified them for pull-down assays. Direct binding to Mcm10 was observed for fragments of Mcm2 (a.a. 1-299) (Figure 3A, lane 2), Mcm4 (a.a. 1-471) (lane 6) and Mcm6 (a.a. 1-439) (lane 5). These results indicate that Mcm10 shows robust association with the N-termini of Mcm2, 4 and 6, all of which are near the head-to-head interface in the MCM DH (Evrin et al., 2009; Li et al., 2015; Remus et al., 2009). Indeed, direct associations between Mcm2 and Mcm4 or Mcm6 N-terminal fragments themselves were identified as well (Figure 3A, lanes 3 and 4). Taken together with Mcm10 preferential association of MCM DH in vivo, it seems likely that Mcm10 binds near the interface of MCM DH.
Figure 3. Mcm10 C-terminus is mainly responsible for association with Mcm2-7.
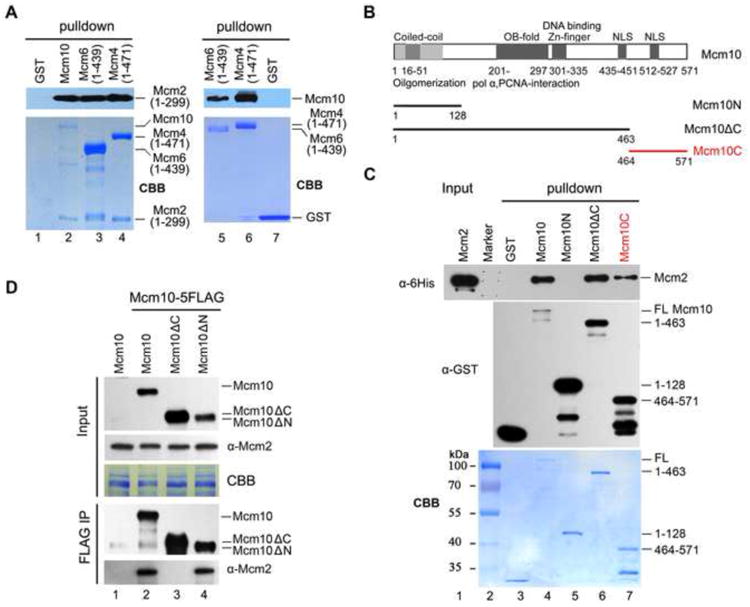
(A) Mcm10 binds directly to the N-terminal fragments of Mcm2, Mcm4 and Mcm6. The GST pull-down assay was conducted using affinity purified GST-Mcm10, 6His-Mcm2 (1-299), GST-Mcm4 (1-471) or GST-Mcm6 (1-439) for the left panel. For the right panel, the GST tag was removed from Mcm10 protein by precision protease before binding to glutathione Sepharose. The exact length of each fragment is indicated in parentheses.
(B) A diagram of Mcm10 and its truncations used in GST pull-down assays. Conserved regions of Mcm10 are indicated as dark and light grey bars according to their high and moderate sequence similarity among eukaryotic orthologs, respectively. OB-fold: oligonucleotide binding-fold; NLS: nuclear localization sequence.
(C) Multiple sites of Mcm10 bind directly to Mcm2 in vitro. Purified recombinant GST-Mcm10 or its truncations and 6His-Mcm2 were incubated with glutathione Sepharose in the binding buffer containing 1 μg/μl BSA. The Mcm2 and Mcm10 bands were revealed by Coomassie blue staining (CBB), or via IB against anti-His and anti-GST antibodies, respectively. See also Figure S3.
(D) The mcm10ΔC mutant shows compromised interaction with Mcm2 in vivo. Mcm10-5FLAG was immunoprecipitated by M2 affinity beads and subjected to IB with indicated antibodies.
Mcm10 C-terminus is mainly responsible for association with MCM
Next, we mapped the interaction domain(s) within Mcm10 via GST pull-down assays. Previous studies have focused on the highly conserved internal domain, which is composed of an OB-fold which mediates interaction with pol α and PCNA, and a zinc finger responsible for DNA binding (Figure 3B) (Du et al., 2012; Thu and Bielinsky, 2014). Through truncation of Mcm10 protein, we first deduce that the Mcm10 internal domain also binds Mcm2 (Figure 3C, compare lanes 5 and 6). To our surprise, a previously uncharacterized region, the Mcm10 C-terminus (a.a.464-571, Mcm10C) alone is sufficient to bind to Mcm2 directly (lane 7). Overexposure also shows a very weak binding of the Mcm10 N-terminus (a.a.1-128, Mcm10N) with Mcm2 (data not shown). We conclude that the Mcm10-MCM interactions are mediated by multiple sites in Mcm10. When we compare their ability to co-precipitate Mcm2, mcm10 truncations showed compromised association with Mcm2 compared to WT (Figure 3D). Notably, deletion of the C-terminus resulted in barely detectable Mcm10-Mcm2 interaction in vivo (lane 3). These data, consistent with the in vitro pull-down results, suggest that the Mcm10 C-terminus is mainly responsible for mediating the Mcm10-MCM interaction.
A cell cycle regulated Mcm10-MCM DH supercomplex
Next, we monitored Mcm10-MCM interaction throughout the cell cycle in both WT and mcm10ΔC. A strain carrying Cdc45-3HA and Mcm4-5FLAG at their genomic loci was grown and synchronized at G1 by α–factor. Cells were collected at different time points after being released into S phase. First, we carried out Mcm10-IP using whole cell extracts (WCE) (Figure 4A). During G1, small amounts of Mcm2 and Mcm4 co-precipitate with Mcm10 (Figure 4B, lanes 1 and 2), indicating formation of a Mcm10-MCM complex. This result is in agreement with previous findings that Mcm10 could associate with the chromatin-loaded MCM independent of S-CDK activity (van Deursen et al., 2012). Notably, the G1-associated Mcm4 is the fast-migrating form, indicating that the formation of the Mcm10-MCM complex occurs prior to Mcm4 phosphorylation. Although the levels of Mcm10 and Mcm2-7 subunits did not fluctuate significantly within the cell cycle (Figure 4A), the amounts of Mcm2 and Mcm4 bound with Mcm10 gradually increased and peaked during S phase (Figure 4B, lanes 2-4). Meanwhile, Cdc45 protein gradually accumulated (Figure 4A) and became associated with Mcm10 (Figure 4B, lanes 2-4), suggesting higher affinity or stability of Mcm10 as assembled into a Mcm10-CMG supercomplex during S phase. However, Mcm2-7 and Cdc45 were barely detectable in precipitates of Mcm10ΔC (Figure 4B, lanes 5-8). These data suggest a cell cycle regulated Mcm10-DH supercomplex, whose formation is dependent on the C-terminus of Mcm10.
Figure 4. Dynamics of the Mcm10-DH supercomplexes during the cell cycle.
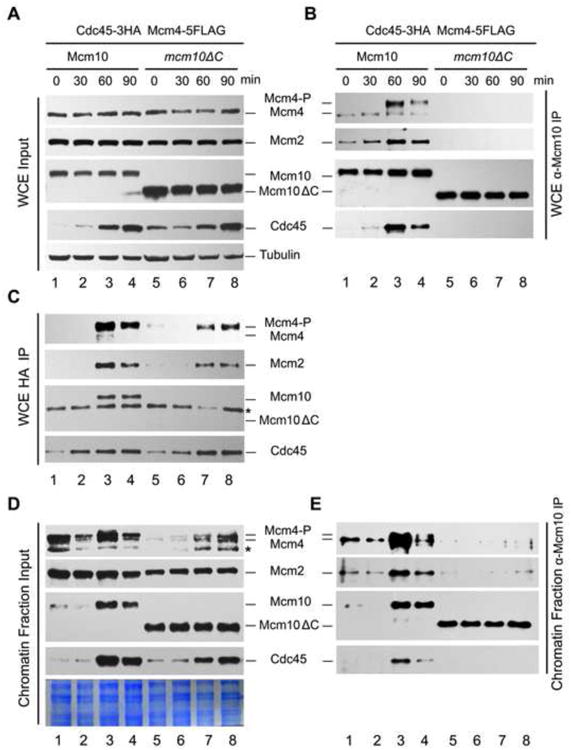
All samples were released from G1 and collected at the indicated time points. Cells were fractionated as described in Figure 1. IP assays were conducted with WCE (A, B, C) or SN2 fractions (D, E), then subjected to IB with indicated antibodies. The flow cytometry profiles of the samples used in this experiment are presented in Figure S4. Tubulin was loaded as a control in (A).
Mcm10-MCM interaction is not essential for CMG assembly and Mcm4 phosphorylation
To more precisely determine the time of association of Mcm10 with Cdc45, Cdc45-3HA immunoprecipitates were prepared (Figure 4C). Neither Mcm10 nor Mcm2-7 co-precipitated together with Cdc45 in G1 (lanes 1 and 2), but both associated in early S phase (lanes 3 and 4; Figure S4). Although Mcm10ΔC protein barely associated with Cdc45, the assembly of Cdc45-MCM complex was not significantly affected in the mcm10ΔC background (lanes 7 and 8). Meanwhile, only phosphorylated Mcm4 (Mcm4-P) associated with Cdc45 but not the non-phosphorylated form, which was present in vast excess over Mcm4-P in WCE, suggesting Mcm4 phosphorylation may occur prior to CMG assembly (Figure 4C, upper panel)(Masai et al., 2006). More interestingly, the profile of Mcm4 phosphorylation, like CMG assembly, was not significantly affected in the interaction defective mutant mcm10ΔC (Figure 4C, upper panel, lanes 7 and 8). Taken together, these data indicate that Mcm10-MCM interaction is unlikely to be required for Mcm4 phosphorylation or CMG assembly.
An enhanced Mcm10-CMG association during early S phase
Since the assembly of Mcm10-MCM occurs exclusively in the MCM DH context as demonstrated in Figure 1, we next analyzed Mcm10-associated complexes using the chromatin bound (SN2) fraction instead of WCE (Figure 4D). Intriguingly, IP using the SN2 recapitulated the results from that of WCE, verifying that Mcm10-MCM supercomplexes are assembled in the chromatin context, wherein MCM is loaded as the DHs (Figure 4E). We designated this complex as MCM-Mcm10-MCM. In the Mcm10-IP, a low level of MCM-Mcm10-MCM complex was detectable in G1 phase (Figure 4E, lanes 1-2). Upon S phase entry, Mcm10 recruitment was enhanced (Figure 4D, lanes 3-4) and assembled into the Mcm10-CMG supercomplex (Figure 4E, lanes 3-4, bottom panel). These results indicate a cell cycle regulated Mcm10 recruitment to the loaded MCM complexes on chromatin, which is largely dependent on Mcm10 C-terminus.
Mcm10 C-terminus mediated interaction with MCM plays important role in chromosome replication
Given the importance of Mcm10 C-terminus in association with the MCM DHs, we next asked whether the interaction defective mcm10 mutants affect cell growth. Since MCM10 is essential for cell viability, the mcm10 mutant strains were constructed via plasmid shuffling. WT MCM10 was cloned and expressed on a pRS316/URA3 single-copy vector to allow growth of mcm10Δ. The mcm10 allele was constructed in a second vector, pRS313/HIS3 and introduced into the same strain. The pRS316-MCM10 plasmid can be eliminated on 5-FOA plates due to its expression of URA3, which converts 5-FOA to a toxin. Thus, growth on 5-FOA plates reflects the physiological function of the copy of the mcm10 mutant expressed on pRS313. Five-fold serial dilution of log phase cells were spotted on SC-His plates in the presence or absence of 5-FOA. Correlating with the relatively greater contribution of Mcm10C than Mcm10N to interact with MCM, the mcm10ΔC allele showed much weaker growth than mcm10ΔN (Figure 5 A).
Figure 5. Mcm10 C-terminus mediated interaction is required for normal DNA replication and growth.
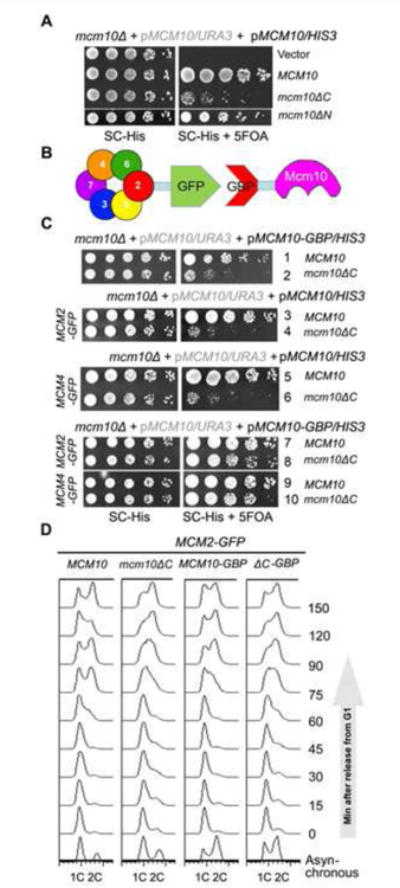
(A) The mcm10ΔC allele shows significant slow growth. 5-fold serial dilution of log phase cells were spotted on the indicated plates and incubated for 2 days at 30°C before being photographed.
(B) An in vivo GFP trap strategy to enforce the Mcm10-MCM interaction. Mcm2 or Mcm4 was tagged with GFP, while Mcm10 or Mcm10ΔC was fused with GBP. Interaction between any of two proteins might be restored artificially via the GFP-GBP pair through in vivo GFP trap experiments.
(C) In vivo GFP trap of Mcm10 with either Mcm2 or Mcm4 suppresses the growth defect of mcm10ΔC.
(D) The S phase defects of mcm10ΔC can be rescued by Mcm10-Mcm2 fusion. Cells were released from α-factor synchronization and analyzed for DNA content by flow cytometry.
If the sickness of mcm10ΔC is specifically caused by compromised Mcm10-MCM interaction, it should be suppressed by enforcing an interaction between Mcm10 and MCM. We adopted an in vivo GFP trap strategy to achieve this (Figure 5B). If we add a GFP tag to one protein and a GBP (GFP binding protein) tag to another protein, these two proteins can be tethered to each other through strong affinity between the GFP and GBP pair. In Figure 5C, we introduced a pRS313/HIS3 plasmid expressing each mcm10 allele with or without a GBP tag at the C-terminus by plasmid shuffling. Control experiments showed that Mcm10 and Mcm2 carrying a GBP or GFP tag, respectively, supported normal cell growth (lines 1 and 3). Expression of both WT Mcm10-GBP and WT Mcm2-GFP also showed growth comparable to untagged or single-tagged WT strains (Figure 5C, compare lines 1, 3 and 7). This control suggests that dissociation of Mcm10 and Mcm2 is not important for normal growth, validating the fusion approach. We then tested the various alleles. In Mcm2 untagged background, mcm10ΔC showed slow growth (line 2). The slow growth of mcm10ΔC-GBP, but not of untagged mcm10ΔC, was significantly overcome by addition of Mcm2-GFP (compare line 8 to 4). Since multiple Mcm2-7 subunits are partners of Mcm10, we next asked if trapping interaction defective Mcm10 through other partners has a similar effect as Mcm2-GFP As shown side by side in Figure 5C, mcm10ΔC was restored to near WT growth by fusion of Mcm4-Mcm10, providing direct evidence that the defects of mcm10ΔC allele are solely attributable to compromised interaction with MCM. Moreover, flow cytometry profiles indicated that mcm10ΔC was defective in S phase, which can be suppressed by Mcm10-Mcm2 fusion (Figure 5D). These data provide genetic evidence that Mcm10 C-terminus mediated interaction with MCMdefines an unanticipated crucial role in replication initiation.
Mcm10-DH interaction is important for the MCM DHs remodeling into single hexamers
Next, we examined whether Mcm10-DH interaction is required for origin firing. First, we examined single-stranded DNA (ssDNA) production. To do this, we measured S phase checkpoint activation which is known to be dependent on the accumulation of ssDNA. Synchronized cells were released from G1 into medium supplemented with 100 mM hydroxyurea, a potent inhibitor of ribonucleotide reductase. In WT, the checkpoint kinase Rad53 became hyperphosphorylated in about 60 min (Figure 6A). However, hyperphosphorylation of Rad53 was dramatically impaired in mcm10ΔC. In previous studies, depletion of Mcm10 mutants showed a similar checkpoint defect, which was interpreted as a defect in unwinding (Kanke et al., 2012; van Deursen et al., 2012; Watase et al., 2012). This result implies that Mcm10-DH association may be required for origin unwinding.
Figure 6. Mcm10-DH interaction is important for its function in MCM DH separation.
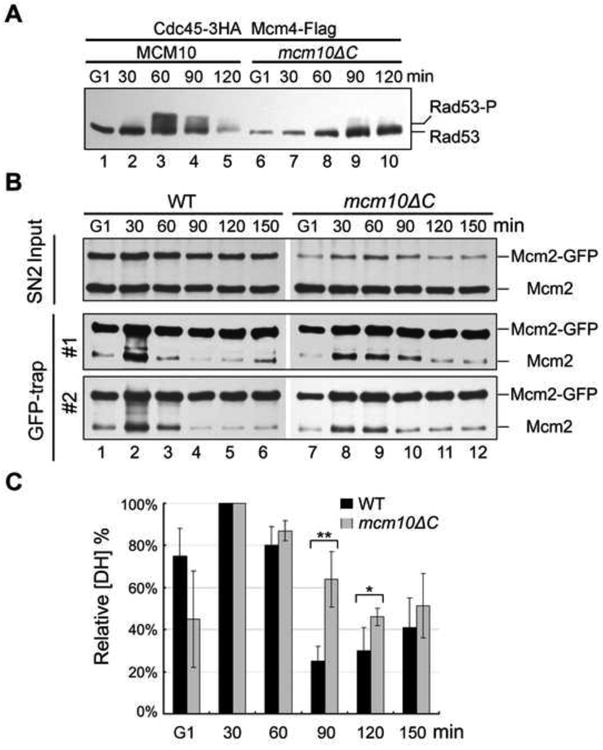
(A) Defects in S-phase checkpoint activation in mcm10ΔC mutant. The cells were synchronized and released into fresh medium supplemented with 100 mM hydroxyurea. To detect Rad53 phosphorylation, cell lysates from the indicated time points were analyzed by IB with anti-Rad53 antibodies.
(B) DH separation is largely delayed in the interaction-defective mcm10ΔC mutant. In vivo DH separation assay in WT (left panel) or mcm10ΔC mutant (right panel) was carried out as described in Figure 1F. Cells were synchronized in G1 and released for the indicated time at 25°C. The SN2 fraction was prepared. Mcm2-GFP in the SN2 fraction was precipitated by GBP beads and probed with anti-Mcm2 antibodies. Results from independent experiments are presented as “#1”, “#2” and “#3”. The representative flow cytometry profiles are shown in Figure S5A.
(C) The relative amounts of DH MCM during cell cycle progression. Quantification was carried out as described in Figure 2F. The average and standard deviation are calculated from the results of at least three independent experiments. P-value <0.01 and 0.05 are donated as “**” and “*”, respectively. See Figure S5B for the raw data and detailed quantification.
Lastly, we examined whether the essential role of Mcm10-DH interaction lies in DH separation function of Mcm10 by using the in vivo DH splitting assay described in Figure 2. In WT cells, the untagged Mcm2 in precipitates decreased gradually after being released into S phase, indicating the separation of MCM DHs (Figure 6B, left panel; 6C and Figure S5B). There are few DHs left at 90 min, correlating with the completed DNA replication (Figure S5A). However, in the interaction defective mcm10ΔC mutant, the separation of MCM DHs was significantly compromised (Figure 6B, right panel; 6C and Figure S5B), consistent with the relative slow S phase progression (Figure S5A). Taken together, these data provide direct evidence that the crucial role for Mcm10 in DH splitting and activation suggested by Figure 2 is due to the interaction between Mcm 10 and MCM DH.
Discussion
Although the close link between Mcm10 and Mcm2-7 has been inferred from a series of genetic and physical interactions (Christensen and Tye, 2003; Homesley et al., 2000; Lee et al., 2010; Merchant et al., 1997; van Deursen et al., 2012), the exact role of Mcm10 in replication initiation still remains controversial (Thu and Bielinsky, 2013). Here, we show that Mcm10 directly associates with the MCM DHs on chromatin in a cell cycle regulated manner. More importantly, their interaction is required for remodeling the MCM DHs into single hexamers that leads to activation of the helicase complex and replication initiation.
MCM double hexameric structure has been extensively studied in yeast in recent years. However, these studies are mainly based on the in vitro pre-RC reconstitution system by purified proteins. Here, we provide in vivo evidence to support that MCMs form the DHs on the yeast chromatin. Through a sequential IP and peptide elution procedure, we are able to obtain the native MCM DHs with high purity. Based on a similar rationale, we also developed an in vivo DH splitting assay, which enables us to monitor the dynamic changes of DH, i.e. formation and separation, during cell cycle progression. We showed that Mcm10 depletion delays MCM DH separation and that this can be accounted for by the direct interaction between Mcm10 and the MCM DHs that we characterize extensively. It is worth pointing out that a caveat in interpreting our DH splitting assay is that disappearance of DHs could be due to helicase activation in the initiation step or passive replication during the fork progression stage. Therefore, the rate of DH disappearance by itself cannot distinguish a role of Mcm10 in helicase activation from one in fork progression. Mcm10 has been proposed to be involved in both replication initiation and progression, although they are not mutually exclusive. In our cell cycle profiling experiments, both depletion of Mcm10 and mutation weakening the MCM interaction, mcm10ΔC, cause defects in very early S phase, i.e., the initiation step. Furthermore, a series of evidence from other groups consistently suggest that Mcm10 participates in a step downstream of CMG assembly and upstream of origin unwinding (Kanke et al., 2012; van Deursen et al., 2012; Watase et al., 2012; Yeeles et al., 2015). Therefore, we favor the idea that the in vivo DH splitting deficiency observed in mcm10 mutants should reflect its role in the DH splitting step during helicase activation rather than in replication progression.
We demonstrate that Mcm10 C-terminus mediated interaction is required for efficient DH splitting. The in vitro and in vivo physical interaction profiles presented here provide evidence to support a specific Mcm10-DH interaction, consistent with previous reports that Mcm10 preferentially associates with the loaded MCM (Pacek et al., 2006; van Deursen et al., 2012). The interaction interface is mediated mainly by the Mcm10 C-terminus, whereas it is the internal domain that is involved in association with ssDNA, dsDNA, pol α and other proteins documented previously (Eisenberg et al., 2009; Ricke and Bielinsky, 2006; Robertson et al., 2008; Zhu et al., 2007). Direct evidence for the important role of Mcm10-MCM interaction came from the significant rescue effect by artificially restoring the interaction in mcm10ΔC. The ability of the fusions to do this is somewhat remarkable, considering the numerous other interactions reported for the scaffold protein Mcm10 (Thu and Bielinsky, 2014). Under the conditions we used, Mcm10-GBP was expressed from its native promoter. The cellular amount of Mcm10 is estimated to be lower than that of each Mcm2-7 subunit. Due to robust and irreversible binding between GFP and GBP, stable heterodimerization between Mcm10 and Mcm2 or Mcm4 might have been expected to interfere with Mcm10 interaction with other proteins (e.g. pol α, Orc2/5, Dpb11, Cdc45 and PCNA) (Du et al., 2012; Thu and Bielinsky, 2014). However, stable fusion between Mcm10 and Mcm2 or Mcm4 does not pose a detectable threat to growth in WT cells. It is possible that the stable fusion of Mcm10 with MCM does not significantly impede Mcm10 from chaperoning other proteins, or there might be some Mcm10-GBP molecules accessible to other partners under our fusion conditions. Nevertheless, we also tried to fuse Mcm10 with other partners such as pol α, and found that fusions exacerbate the defect of mcm10 alleles we tested instead of alleviating them (data not shown). These data argue along with the rest of our data that Mcm10-MCM interaction contributes to the essential function of Mcm10 in origin firing, which was recently demonstrated by in vitro reconstitution experiments (Yeeles et al., 2015).
Moreover, Mcm10 recruitment and association with MCM is temporally and spatially regulated, though the detailed mechanism needs to be addressed in the future. Given the conserved interactions between Mcm10 and Mcm2-7 from yeast to human, we speculate that the critical role of the direct Mcm10-DH interaction in helicase remodeling and activation will likely take place in higher eukaryotes as well.
Experimental Procedures
Isolation of the native double hexameric Mcm2-7 species
A plasmid expressing a second copy of Mcm4 with a 3HA tag was introduced into the Mcm4-5FLAG background strain (Table S2 and S3, strain QY793). FLAG-IP/peptide elution and HA-IP/peptide elution were sequentially conducted to purify the putative MCM complexes containing both Mcm4-FLAG and Mcm4-HA, which result from dimerization of two heterohexameric Mcm2-7 rings. The MCM DH species was enriched by taking advantage of its resistance to high salt (Donovan et al., 1997; Gambus et al., 2011; Remus et al., 2009). A high salt resistant chromatin fraction (SN2) was prepared as previously (Sheu and Stillman, 2006) with some modifications described in Supplemental Experimental Procedures.
The EBX-2 buffer used here is {50 mM HEPES/KOH pH7.5, 150 mM KGlu, 2.5 mM MgOAc, 0.1 mM ZnOAc, 2 mM NaF, 0.5 mM spermidine, 20 mM β-Glycerophosphate, 3 mM ATP, 1 mM DTT, 1 mM PMSF, Protease inhibitor tablets (EDTA free, Roche)}. 500 μl of SN2 was first mixed with M2 beads for 3h with rotation at 4°C, and washed three times with 1 ml EBX-2 buffer. The bound fraction was eluted by 1 μg/μl FLAG peptide. The FLAG elutes were incubated with anti-HA and protein G beads. After the same wash, the bound or HA peptide eluted fraction was resolved by 10% SDS-PAGE and analyzed by silver staining, mass spectrometry or western blots.
In vivo double hexamer splitting assay
A second copy of Mcm2 with GFP tag at its C-terminus was cloned (pRS317-MCM2-GFP) and expressed under control of its native promoter. Plasmid was transformed into conditional Mcm10 depletion background (Table S2 and S3, strain QY394), WT (strain QY6129) or mcm10ΔC (strain QY6131) mutant cells. The high salt (500 mM NaCl) resistant SN2 was prepared basically as described above. 500 μl of SN2 was mixed with GBP beads for 3h with rotation at 4°C, and washed three times with 1 ml EBX-2 buffer. The bound fraction was dissolved by 6% SDS-PAGE and IB by anti-Mcm2. The detection of untagged endogenous Mcm2 reflects the DH portion of MCM complex. The ratio of untagged/tagged Mcm2 represents the relative level of MCM DH comprised Mcm2 and Mcm2-GFP in vivo. The change in the level of MCM DH is used to estimate the dynamics (e.g., assembly or splitting) of the dimeric rings.
Supplementary Material
Acknowledgments
We thank Drs. Bruce Stillman, Karim Labib, Li-Lin Du, Quanwen, Jin, Wei Xiao, and Junbiao Dai for reagents; Drs. John Diffley, Karim Labib, Philippe Pasero, Qun He, Li-Lin Du, Jing Li and members of the Lou lab for helpful discussion and comments on the manuscript.
This work was supported by National Natural Science Foundation of China 31271331 and 31071095 to HL, 31200052 to QC; Program for New Century Excellent Talents in University of China NCET-09-0733 to HL; NIH grant GM080338 to XSC; Research Fund for the Doctoral Program of Higher Education of China 20120008110017; Project (2014SKLAB1-7) and Program for Extramural Scientists (2015SKLAB6-26) of the State Key Laboratory of Agrobiotechnology; Chinese Universities Scientific Fund 2015TC039 and 2014JD075.
Footnotes
Author Contributions: Author Contributions: H.L., Y.Q. Y.X. and J.L.C. conceived and designed research; Y.X., YQ. purified the native MCM complexes; Y.Q. Y.X., L.L., J.C. and Q.C. performed other experiments; Y.Q. and Z.L. did mass spectrometry analysis; H.L., J.L.C, X.S.C, Y.Q., and Y.X. analyzed the data and wrote the paper.
No conflicts of interest declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bell SP, Dutta A. DNA replication in eukaryotic cells. Annual Review of Biochemistry. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Bochman ML, Schwacha A. The Mcm Complex: Unwinding the Mechanism of a Replicative Helicase. Microbiology and Molecular Biology Reviews. 2009;73:652–683. doi: 10.1128/MMBR.00019-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos D, Frigola J, Diffley JFX. Activation of the replicative DNA helicase: breaking up is hard to do. Current Opinion in Cell Biology. 2012;24:423–430. doi: 10.1016/j.ceb.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Christensen TW, Tye BK. Drosophila Mcm10 Interacts with Members of the Prereplication Complex and Is Required for Proper Chromosome Condensation. Molecular Biology of the Cell. 2003;14:2206–2215. doi: 10.1091/mbc.E02-11-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Hood IV, Berger JM. Mechanisms for initiating cellular DNA replication. Annual Review of Biochemistry. 2013;82:25–54. doi: 10.1146/annurev-biochem-052610-094414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Renault L, Swuec P, Petojevic T, Pesavento JJ, Ilves I, MacLellan-Gibson K, Fleck RA, Botchan MR, Berger JM. DNA binding polarity, dimerization, and ATPase ring remodeling in the CMG helicase of the eukaryotic replisome. eLife. 2014;3:e03273. doi: 10.7554/eLife.03273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coster G, Frigola J, Beuron F, Morris Edward P, Diffley John F. Origin Licensing Requires ATP Binding and Hydrolysis by the MCM Replicative Helicase. Molecular Cell. 2014;55:666–677. doi: 10.1016/j.molcel.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan S, Harwood J, Drury LS, Diffley JFX. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proceedings of the National Academy of Sciences. 1997;94:5611–5616. doi: 10.1073/pnas.94.11.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Stauffer ME, Eichman BF. Structural biology of replication initiation factor mcm10. Subcellular Biochemistry. 2012;62:197–216. doi: 10.1007/978-94-007-4572-8_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg S, Korza G, Carson J, Liachko I, Tye BK. Novel DNA binding properties of the Mcm10 protein from Saccharomyces cerevisiae. Journal of Biological Chemistry. 2009;284:25412–25420. doi: 10.1074/jbc.M109.033175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proceedings of the National Academy of Sciences. 2009;106:20240–20245. doi: 10.1073/pnas.0911500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher RJ, Bishop BE, Leon RP, Sclafani RA, Ogata CM, Chen XS. The structure and function of MCM from archaeal M. Thermoautotrophicum. Nature structural & molecular biology. 2003;10:160–167. doi: 10.1038/nsb893. [DOI] [PubMed] [Google Scholar]
- Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Schärer OD, Walter JC. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell. 2011;146:931–941. doi: 10.1016/j.cell.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambus A, Khoudoli GA, Jones RC, Blow JJ. MCM2-7 Form Double Hexamers at Licensed Origins in Xenopus Egg Extract. Journal of Biological Chemistry. 2011;286:11855–11864. doi: 10.1074/jbc.M110.199521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RC, Kang S, Lam WM, Chen S, Chan CS, Bell SP. Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell. 2011;146:80–91. doi: 10.1016/j.cell.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang ML, Leon RP, Pessoa-Brandao L, Hunt S, Raghuraman MK, Fangman WL, Brewer BJ, Sclafani RA. Structural Changes in Mcm5 Protein Bypass Cdc7-Dbf4 Function and Reduce Replication Origin Efficiency in Saccharomyces cerevisiae. Molecular and Cellular Biology. 2007;27:7594–7602. doi: 10.1128/MCB.00997-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homesley L, Lei M, Kawasaki Y, Sawyer S, Christensen T, Tye BK. Mcm10 and the MCM2-7 complex interact to initiate DNA synthesis and to release replication factors from origins. Genes & Development. 2000;14:913–926. [PMC free article] [PubMed] [Google Scholar]
- Izumi M, Yanagi K, Mizuno T, Yokoi M, Kawasaki Y, Moon KY, Hurwitz J, Yatagai F, Hanaoka F. The human homolog of Saccharomyces cerevisiae Mcm10 interacts with replication factors and dissociates from nuclease-resistant nuclear structures in G(2) phase. Nucleic acids research. 2000;28:4769–4777. doi: 10.1093/nar/28.23.4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanke M, Kodama Y, Takahashi TS, Nakagawa T, Masukata H. Mcm10 plays an essential role in origin DNA unwinding after loading of the CMG components. EMBO Journal. 2012;31:2182–2194. doi: 10.1038/emboj.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib K. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes & Development. 2010;24:1208–1219. doi: 10.1101/gad.1933010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Liachko I, Bouten R, Kelman Z, Tye BK. Alternative mechanisms for coordinating polymerase alpha and MCM helicase. Mol Cell Biol. 2010;30:423–435. doi: 10.1128/MCB.01240-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zhai Y, Zhang Y, Li W, Yang M, Lei J, Tye BK, Gao N. Structure of the eukaryotic MCM complex at 3.8 A. Nature. 2015;524:186–191. doi: 10.1038/nature14685. [DOI] [PubMed] [Google Scholar]
- Li Y, Araki H. Loading and activation of DNA replicative helicases: the key step of initiation of DNA replication. Genes to Cells. 2013;18:266–277. doi: 10.1111/gtc.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M. Eukaryotic chromosome DNA replication: where, when, and how? Annual Review of Biochemistry. 2010;79:89–130. doi: 10.1146/annurev.biochem.052308.103205. [DOI] [PubMed] [Google Scholar]
- Masai H, Taniyama C, Ogino K, Matsui E, Kakusho N, Matsumoto S, Kim JM, Ishii A, Tanaka T, Kobayashi T, et al. Phosphorylation of MCM4 by Cdc7 Kinase Facilitates Its Interaction with Cdc45 on the Chromatin. Journal of Biological Chemistry. 2006;281:39249–39261. doi: 10.1074/jbc.M608935200. [DOI] [PubMed] [Google Scholar]
- Merchant AM, Kawasaki Y, Chen Y, Lei M, Tye BK. A lesion in the DNA replication initiation factor Mcm10 induces pausing of elongation forks through chromosomal replication origins in Saccharomyces cerevisiae. Molecular and Cellular Biology. 1997;17:3261–3271. doi: 10.1128/mcb.17.6.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- On KF, Beuron F, Frith D, Snijders AP, Morris EP, Diffley JF. Prereplicative complexes assembled in vitro support origin-dependent and independent DNA replication. EMBO Journal. 2014;33:605–620. doi: 10.1002/embj.201387369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacek M, Tutter AV, Kubota Y, Takisawa H, Walter JC. Localization of MCM2-7, Cdc45, and GINS to the Site of DNA Unwinding during Eukaryotic DNA Replication. Molecular Cell. 2006;21:581–587. doi: 10.1016/j.molcel.2006.01.030. [DOI] [PubMed] [Google Scholar]
- Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JFX. Concerted Loading of Mcm2-7 Double Hexamers Around DNA during DNA Replication Origin Licensing. Cell. 2009;139:719–730. doi: 10.1016/j.cell.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke RM, Bielinsky AK. A conserved Hsp10-like domain in Mcm10 is required to stabilize the catalytic subunit of DNA polymerase-alpha in budding yeast. Journal of Biological Chemistry. 2006;281:18414–18425. doi: 10.1074/jbc.M513551200. [DOI] [PubMed] [Google Scholar]
- Robertson PD, Warren EM, Zhang H, Friedman DB, Lary JW, Cole JL, Tutter AV, Walter JC, Fanning E, Eichman BF. Domain architecture and biochemical characterization of vertebrate Mcm10. Journal of Biological Chemistry. 2008;283:3338–3348. doi: 10.1074/jbc.M706267200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu YJ, Stillman B. Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Molecular cell. 2006;24:101–113. doi: 10.1016/j.molcel.2006.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu YJ, Stillman B. The Dbf4–Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature. 2010;463:113–117. doi: 10.1038/nature08647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu YJ, Kinney JB, Lengronne A, Pasero P, Stillman B. Domain within the helicase subunit Mcm4 integrates multiple kinase signals to control DNA replication initiation and fork progression. Proceedings of the National Academy of Sciences. 2014;111:E1899–E1908. doi: 10.1073/pnas.1404063111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui K, On KF, Diffley JFX. Regulating DNA Replication in Eukarya. Cold Spring Harbor Perspectives in Biology. 2013;5:a012930. doi: 10.1101/cshperspect.a012930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Fernandez-Cid A, Riera A, Tognetti S, Yuan Z, Stillman B, Speck C, Li H. Structural and mechanistic insights into Mcm2–7 double-hexamer assembly and function. Genes & Development. 2014;28:2291–2303. doi: 10.1101/gad.242313.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thu YM, Bielinsky AK. Enigmatic roles of Mcm10 in DNA replication. Trends in Biochemical Sciences. 2013;38:184–194. doi: 10.1016/j.tibs.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thu YM, Bielinsky AK. MCM10: One tool for all-Integrity, maintenance and damage control. Seminars in cell & developmental biology. 2014;30:121–130. doi: 10.1016/j.semcdb.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticau S, Friedman Larry J, Ivica Nikola A, Gelles J, Bell Stephen P. Single-Molecule Studies of Origin Licensing Reveal Mechanisms Ensuring Bidirectional Helicase Loading. Cell. 2015;161:513–525. doi: 10.1016/j.cell.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognetti S, Riera A, Speck C. Switch on the engine: how the eukaryotic replicative helicase MCM2–7 becomes activated. Chromosoma. 2014:1–14. doi: 10.1007/s00412-014-0489-2. [DOI] [PubMed] [Google Scholar]
- van Deursen F, Sengupta S, De Piccoli G, Sanchez-Diaz A, Labib K. Mcm10 associates with the loaded DNA helicase at replication origins and defines a novel step in its activation. EMBO Journal. 2012;31:2195–2206. doi: 10.1038/emboj.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase G, Takisawa H, Kanemaki Masato T. Mcm10 Plays a Role in Functioning of the Eukaryotic Replicative DNA Helicase, Cdc45-Mcm-GINS. Current Biology. 2012;22:343–349. doi: 10.1016/j.cub.2012.01.023. [DOI] [PubMed] [Google Scholar]
- Yardimci H, Loveland AB, Habuchi S, van Oijen AM, Walter JC. Uncoupling of sister replisomes during eukaryotic DNA replication. Molecular Cell. 2010;40:834–840. doi: 10.1016/j.molcel.2010.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JTP, Deegan TD, Janska A, Early A, Diffley JFX. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature. 2015;519:431–435. doi: 10.1038/nature14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Ukomadu C, Jha S, Senga T, Dhar SK, Wohlschlegel JA, Nutt LK, Kornbluth S, Dutta A. Mcm10 and And-1/CTF4 recruit DNA polymerase α to chromatin for initiation of DNA replication. Genes & Development. 2007;21:2288–2299. doi: 10.1101/gad.1585607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.