

Abstract
Fluorescence anisotropy is used to follow the binding of RecA to short single-stranded DNA (ssDNA) sequences (39 bases) at low DNA and RecA concentration where the initial phase of polymerization occurs. We observe that RecA condensation is extremely sensitive to minute changes in DNA sequences. RecA binds strongly to sequences that are rich in pyrimidines and that lack significant secondary structure and base stacking. We find a correlation between the DNA folding free energy and the onset concentration for RecA binding. These results suggest that the folding of ssDNA and base stacking represent a barrier for RecA binding. The link between secondary structure and binding affinity is further analyzed with two examples: discrimination between two naturally occurring polymorphisms differing by one base and RecA binding on a molecular beacon. A self-assembly model is introduced to explain these observations. We propose that RecA may be used to sense ssDNA sequence and structure.
Keywords: RecA–DNA interaction, DNA secondary structure, fluorescence anisotropy, single nucleotide polymorphism, molecular beacon
Collective interaction of proteins with DNA is fundamental to biological regulation. In some cases, such as gene activation, the interaction is carried out by a handful of different proteins that nucleate on a specific and relatively short DNA sequence to direct synthesis of mRNA (1). In other cases, such as DNA recombination, multiple copies of the same protein interact over hundreds of DNA bases and polymerize in a nonspecific manner. So far, it is not clear whether this second type of interaction is specific and if so, what are the mechanisms involved. This question is addressed in this work by using RecA, the protein responsible for recombination in bacteria. A sensitive detection system, fluorescence anisotropy, is used to investigate the initial stages of RecA interaction with its substrate, single-stranded DNA (ssDNA).
A comprehensive review of RecA, its role in recombination, and its DNA binding properties are given in ref. 2. To summarize, on binding ATP or a nonhydrolyzable ATP analog ([γ-thio]ATP, ATPγS), RecA polymerizes on DNA with a site size of three bases per protein (3, 4) and forms a right-handed helix with a 6-RecA pitch, which stretches and rigidifies the DNA (5–7). RecA is also a DNA-dependent ATPase (8, 9); it hydrolyzes ATP when bound to DNA and switches between a high-affinity state, RecA-DNA-ATP, and a low-affinity state, RecA-DNA-ADP (10). Substrates for RecA are both ssDNA and double-stranded DNA (dsDNA). A nucleation barrier characterizes polymerization on dsDNA, whereas binding to ssDNA occurs readily at a high polymerization rate (11, 12). It has generally been considered that RecA binding is not specific, with the exception of a few homopolymers, such as poly(T), to which RecA has enhanced affinity (13, 14). However, recent in vitro selection experiments demonstrated that RecA has a preferential affinity for DNA sequences rich in GT composition, some of which are similar to the recombination hotspot χ (15, 16). Differences in affinity are difficult to detect with classical biochemical techniques because the onset of collective binding occurs at low concentration, and, in fact, it was recently shown, by using surface plasmon resonance, that RecA polymerizes efficiently on certain repeat sequences (17).
Recently, we introduced an approach to study with high resolution RecA polymerization on ssDNA in solution, by using fluorescence anisotropy. This was made possible by using a microdispensing pump, which accurately controlled the addition of small amounts of RecA into the DNA solution over a wide range of concentration (unpublished data). Binding curves as a function of RecA concentration were measured with fluorescent end-labeled ssDNA in the form of poly(T), a polymer that has no secondary structure or base stacking. At low DNA concentration we observed a condensation of RecA on ssDNA. This study showed that the fluorescence anisotropy signal is mostly sensitive to RecA binding in close proximity to the tagged end of the DNA. We examined the effect of ssDNA length on polymerization, and introduced a model of protein assembly that computes the equilibrium distribution of bound RecA domains with good agreement to the data.
In this paper we extend the approach to probe the recognition properties of RecA–ssDNA interaction, simplifying the reactions by using ATPγS, thus avoiding ATP hydrolysis. To identify high- and low-affinity binding sites, we used a variety of triplet repeats (the size of the binding unit) as substrates, as well as nonrepeat sequences that probe the ability to distinguish small differences. The results obtained suggest simple rules for substrate discrimination. Both secondary structure of self-hybridized sequences and stacked purine-rich sequences represent obstacles for RecA polymerization. When the free energy of ssDNA secondary structure can be computed, we find that RecA concentration at the onset of polymerization depends exponentially on the folding free energy. These elastic and entropic barriers provide a mechanism to discriminate a single base substitution in 63 bases, when such small differences affect local secondary structure or rigidity. Finally, we use a molecular beacon, as a sensitive probe of ssDNA structure, to show that RecA binding in the presence of a well-defined folded state reduces to a two-state system.
Materials and Methods
ssDNA and RecA Protein.
In all cases ssDNA oligomers, typically 39 bases long with 5′-fluorescein tags, were used (Midland Certified Reagents, Midland, TX), and purified with reverse-phase HPLC. Triplet repeats are denoted in the text as (NNN)13. Other sequences used are defined as follows: hemoglobin-β (HBB) = ATG GTG CAC CTG ACT CCT GXG GAG AAG TCT GCC GTT ACT, with single point mutation replacing X = A (HBB-N) with X = T (HBB-S), resulting in sickle cell anemia (http://ncbi.nlm.nih.gov, GenBank GI = 183944). Single nucleotide polymorphism in p53 (GenBank GI = 4732146) (p53) = GAA GCT CCC AGA ATG CCA GAG GCT GCT CCC CXC GTG GCC CCT GCA CCA GCG ACT CCT ACA CCG, where X = C (p53–1) is replaced by X = G (p53–2). A sequence with minimal secondary structure and equal base composition (MIX)38 = CTA ATGC CGTA ATGC CGTA ATGC CGTA ATGC CGTA ATC. The oligonucleotides p53–1 and p53–2 were purchased with 3′-fluorescein and 5′-trityl to enhance HPLC purification of the correct length. Molecular beacon (BCN)T15 = (fluorescein-dT)GCGAGT15CTCGC-Dabcyl. DNA oligomer concentrations were measured at both 492 and 260 nm. RecA protein (New England Biolabs) binding assays were done in 25 mM Tris⋅HCl (pH 7.5)/150 mM NaCl/1 mM MgCl2/1 mM DTT/100 μM ATPγS. This buffer is similar to the one used in Biet et al. (17).
Anisotropy of RecA-Bound ssDNA.
The theoretical principles and experimental considerations of fluorescence anisotropy are described in Lakowicz (18). On vertical polarized excitation, light is emitted with polarization primarily in the vertical direction Iv but also in the perpendicular direction Ip. The anisotropy is defined by (18):
 |
The fluorescent tag has an intrinsic anisotropy, A0, which is reduced due to thermal rotational motion according to:
 |
where θ is the rotational correlation time and τ is the fluorescent lifetime. Large molecules have a longer rotational time than small molecules (18, 19) and, hence, have a higher fluorescence anisotropy value. Our recent work showed that the fluorescence anisotropy of poly(T) is small, and its value increases when RecA binds close to the labeled end. At high RecA coverage, the rotation of the bound complex slows down so that the signal saturates at the intrinsic anisotropy A0 = 0.31 (unpublished data; see Fig. 1). For the curves presented below, each naked ssDNA has a different fluorescence anisotropy, ADNA, and we define the onset of binding Ronset as the concentration of RecA at which (A − ADNA)/(A0 − ADNA) = 0.1.
Figure 1.

(Upper) Purines versus pyrimidines. RecA binding curves on ssDNA triplet repeats, 39 bases long, as designated. The anisotropy of the naked ssDNA, ADNA, is subtracted from the signal. Concentration of ssDNA is 1 nM oligomer (13 nM binding sites). (Lower) Effect of ssDNA base pairing. RecA binding curves on ssDNA triplet repeats with variable G⋅C base pairing.
Measurement Setup.
Fluorescence was excited by the 488-nm line of an air-cooled argon laser (532 Omnichrome, Melles Griot, Irvine, CA), which was polarized vertically (Glan–Thompson, Newport, Fountain Valley, CA) and directed to the center of a quartz cuvette. Light was collected in a “T” format (90° to the incident beam), projected onto two analyzers oriented parallel and perpendicular to the polarization vector, and detected by two sensitive photomultiplier tubes (Thorn EMI Electron Tubes, Rockaway, NJ). The solution was continually mixed by a stirring magnet. A simple protein-dispensing pump was constructed by using a computer-controlled dc motor (860, Newport), a linear stage, and a Hamilton syringe, dispensing 40–200 μl of RecA solution into 3 ml of binding buffer. Light intensities were measured for each of the two polarization directions by using a photon counter (Hewlett–Packard). The temperature was stabilized by using a water circulation bath at 21.0°C. Binding assays were obtained by using a computer program (LABVIEW, National Instruments, Austin, TX) to control the pumping and acquisition. The protein was dispensed in small aliquots and stirred for 2 min before taking measurements.
Results and Discussion
Base-Stacked Purine-Rich Sequences Hinder RecA Polymerization.
We show that stacked purine-rich sequences (adenine and guanine) hinder RecA polymerization, whereas pyrimidine-rich sequences (cytosine and thymine) form high-affinity substrates. Fig. 1 Upper presents binding curves of RecA to (i) singlet repeats: poly(T), poly(C), poly(A), and (ii) two triplet repeats, one with pyrimidines, (TTC)13, and one with purines, (GAA)13. Poly(T) and poly(C) are flexible homopolymers of pyrimidines and, hence, pose no elastic resistance to RecA binding. For both sequences, the onset of polymerization is at low RecA concentration, ≈30 nM, with reduced sharpness for poly(C), indicating less cooperativity (20). In contrast, poly(A) and poly(G) are rigid purine homopolymers. As a result, the polymerization onset for poly(A) is shifted to high concentration, 0.6 μM, reaching no saturation even at 3 μM RecA. Unfortunately, pure poly(G) cannot be synthesized. The low-affinity substrate is kept when purines are mixed, as in (GAA)13, with a slight enhancement with respect to poly(A). Mixing pyrimidines, (TTC)13, leaves RecA affinity high, but reduces the sharpness of the condensation. This may be related to a reduced diffusion, and hence cooperativity (20), of RecA monomers along a (TTC)13 chain, whereas on poly(T) they diffuse freely. Finally, to verify that stacking is indeed important we tested a repeat with 50% purines, (AC)21, for which stacking of the purines is broken (not shown). Essentially we find that (AC)21 behaves like poly(C).
Secondary Structure of ssDNA Hinders RecA Polymerization.
Secondary structures caused by self-hybridization of ssDNA are a major limitation to RecA polymerization. Fig. 1 Lower shows binding curves for a selected number of triplet repeats, which have mixed base composition and various degrees of secondary structure: (TAC)13, (CCG)13, (CAG)13, (CGG)13, and poly(T) — as a reference. They are presented in a decreasing order of affinity to RecA. (TAC)13 is a flexible polymer with  pyrimidines (TC) with very weak secondary structure, and thus forms a high-affinity substrate. (CCG)13 is also flexible, with an equivalent pyrimidine content (CC), but has secondary structure because of G⋅C base pairing. As a result, RecA binding is weaker and less cooperative. (CAG)13 is purine rich (AG), with more base stacking and tighter secondary structure than (CCG)13 and, indeed, the affinity is further reduced. Last, (CGG)13 is purine rich with a tight secondary structure, and is, hence, the lowest-affinity substrate. Finally, we embedded one “defect”—a weak binding site (AAA)—in the middle of a poly(T) sequence. The defect induces base pairing of TTT-AAA to form a weakly folded loop. This additional secondary structure decreases the overall affinity of RecA for this sequence, as shown in Fig. 2. These triplet repeats are only a fraction of all of the possible repeats, but they clearly show that secondary structures resulting from base pairing present a barrier for RecA binding.
pyrimidines (TC) with very weak secondary structure, and thus forms a high-affinity substrate. (CCG)13 is also flexible, with an equivalent pyrimidine content (CC), but has secondary structure because of G⋅C base pairing. As a result, RecA binding is weaker and less cooperative. (CAG)13 is purine rich (AG), with more base stacking and tighter secondary structure than (CCG)13 and, indeed, the affinity is further reduced. Last, (CGG)13 is purine rich with a tight secondary structure, and is, hence, the lowest-affinity substrate. Finally, we embedded one “defect”—a weak binding site (AAA)—in the middle of a poly(T) sequence. The defect induces base pairing of TTT-AAA to form a weakly folded loop. This additional secondary structure decreases the overall affinity of RecA for this sequence, as shown in Fig. 2. These triplet repeats are only a fraction of all of the possible repeats, but they clearly show that secondary structures resulting from base pairing present a barrier for RecA binding.
Figure 2.

Effect of “lattice defect.” Introducing a weak binding site AAA in the middle of poly(T) (TTT-d = T18A3T18), forms a weak base pairing hairpin structure, shifting the onset of binding.
Folding of ssDNA as a Nucleation Barrier and Implication for Base Stacking.
The previous sections presented a trend by which secondary structures induced by base pairing and base stacking hinder RecA polymerization. The fluorescence anisotropy of ssDNA reflects its rigidity, as shown in Fig. 3 (Upper). When ssDNA is folded into a rigid state, its anisotropy is no longer that of a flexible polymer, and this anisotropy increases with rigidity, as expected for globular molecules (18). Can we condense the observed sequence-dependent binding into a more quantitative unified picture? If we consider that RecA polymerizes on ssDNA by unzipping self-hybridization and by unfolding or stretching stacked bases, we can attempt to relate RecA polymerization onset to the secondary structure free energy of ssDNA, whenever the latter is computable. The folding secondary structure and free energy of a ssDNA sequence that has some appreciable self-hybridization can be readily computed, by using, for example, the program mfold (ref. 21; http://mfold2.wustl.edu/mfold). This program does not compute the free energy of a stacked sequence, such as poly(A), because stacking interactions are not well understood, as we discuss below. In Fig. 3 Lower we plot RecA onset concentration (Ronset; see Materials and Methods) as a function of the computed ssDNA folding free energy ΔG for all our 36- to 39-base-long sequences, excluding those that do not self-fold into stable structures (see below). From this plot we obtain a clear correlation: the tighter the folded structure, the harder it is for RecA to bind. In fact, the data fit well to an exponential function: Ronset ≈ exp(0.09ΔG). This result suggests that the binding onset of RecA is activated with a nucleation barrier that is directly related to the folded state of ssDNA. In the plot we set ΔG = 0 for poly(T) and poly(C) because they do not fold and stack only weakly. Likewise, (TAC)13 and (TCA)13 do not fold into stable structures, and we set ΔG = 0.
Figure 3.

(Upper) Anisotropy of naked DNA. Anisotropy of ssDNA as a function of computed folding free energy for various sequences: triplet repeats, as designated; HBB-N(S), as described in the text; alternating sequences Fig. 5: (+−), (TTC-CGG)6; (−+), (CGG-TTC)6; (+−+), (TTC)3(CGG)6(TTC)3; (−+−), (CGG)3(TTC)6(CGG)3. TTT-d = T18A3T18. Poly(C), poly(T), (TAC)13, and (TCA)13 do not fold into stable structures, and we set ΔG = 0. (Lower) RecA binding to folded ssDNA. Measured onset of RecA binding versus computed folding free energy (●). Solid line, exponential fit: Ronset = 0.03exp(0.09ΔG). Free energy values for poly(A) and (GAA)13 were included in the graph by extrapolating the fit (○).
The correlation between RecA binding onset and ssDNA structure may have implications for base stacking. Although it is generally known that stacking interactions of adjacent bases order according to purine–purine > purine–pyrimidine > pyrimidine–pyrimidine, there are significant variations in reported base-stacking parameters. For example, values for AA stacking free energy vary between 1 and 6 kcal/mol (1 kcal = 4.18 kJ) (22). Alternatively, some sources provide stacking interaction parameters only in the context of dsDNA (23). We give an order of magnitude estimate and compare with RecA onset: 1 kcal/mol per AA pair implies 38 kcal/mol for a 39-mer poly(A) and, if we extrapolate our fit in Fig. 3 Lower to include the onset of RecA binding to poly(A), we deduce a value of 30 kcal/mol, which is in reasonable agreement. We therefore propose to use the correlation between self-hybridization folding free energy and RecA onset to estimate the free energy of sequences for which we cannot compute the stacking free energy by extrapolating the fit to read off the free energy at a measured RecA onset. We have included the extrapolated free energy values for poly(A) and (GAA)13 in Fig. 3 Lower (with different symbols) as examples.
Single Base Substitution.
If the onset of binding is exponentially sensitive to secondary structure, can RecA polymerization discriminate a single base substitution? Here we show two examples in which a single base substitution can be detected, one where the base substitution changes the folding and one where it affects local rigidity. We chose known naturally occurring substitutions for the purpose of illustrating the sensitivity, and both examples are not related to RecA function in vivo. The first example is a naturally occurring polymorphism of p53 (codon 72 Arg/Pro), and we designed a 63-mer with the single base change in the middle (p53–1, p53–2, see Methods and Materials). This base substitution changes the secondary structure and its folding free energy (21) from 5.7 kcal/mol (p53–1) to 7.5 kcal/mol (p53–2). This change is reflected in the binding; RecA polymerizes better on p53–1, as shown in Fig. 4, with a discrimination between the two sequences that is within the error bars.
Figure 4.

Detecting single base substitution. RecA binding to hemoglobin-[β] HBB-N and HBB-S sequences, which differ by 1 base in 39, and to p53–1 and p53–2, which differ by 1 base in 63 (see text).
The second case is a single substitution that does not induce a computable change in secondary structure. We selected the single base mutation occurring in hemoglobin-[β] subunit and designed two sequences, HBB-N and HBB-S (see Methods and Materials). Fig. 4 clearly shows that RecA binding to the normal sequence (HBB-N) is weaker than to the mutated one (HBB-S). Interestingly, the computation of folded structure does not include stacking and hence the folding free energy of the two sequences is identical (Fig. 3 Lower). However, on close inspection of the sequences, we can rationalize the discrimination. The mutation occurs within a rigid segment of 9 purine bases. Replacement of a base with a pyrimidine reduces this rigidity, and we expect enhanced RecA binding, as observed.
Alternating Sequences: Design of Recognition Elements.
One can construct sequences that distribute rigidity and secondary structure. Cooperative monomer–monomer interaction on the DNA determines the average length of the RecA-binding unit. This average length corresponds to an effective Hill coefficient, which, in turn, determines the slope of the binding curve, or the degree of nonlinearity. By using simple triplet repeats, we cannot impose any particular form of cooperativity. However, the classification of high and low affinity binding sites, as well as the role of secondary structure, provides a basis for rational design of ssDNA recognition elements, in which the binding affinity and degree of cooperativity in RecA binding can be tuned. An example of such a design is presented in Fig. 5. With TTC and CGG as good (+) and bad (−) sites, these linear sequences are “bipolar.” In the (TTC)13 repeat, each bound monomer enhances the binding of its nearest neighbor, whereas in an alternating TTC-CGG sequence [denoted (+−) and (−+)], every bound monomer antagonizes its nearest neighbor. The sequences (+−+) and (−+−) have “domain walls” between uniform regions of good and bad sites. Each of these sequences folds into a particular structure and, hence, this design relates induced cooperativity to secondary structure. The change in onset and in cooperativity follows the general trend of Fig. 3 (Lower).
Figure 5.

Alternating sequences. RecA binding to mixed sequences, which distribute secondary structure and rigidity, as designated.
A Self-Assembly Model.
What is the origin of the activation barrier (Fig. 3 Lower)? Consider the binding of RecA to ssDNA that has a local self-hybridized folded state with a free energy ΔG. Recently, we introduced a simple adsorption model in which monomers in solution self-assemble on DNA (unpublished work). This model, which is similar to micelle formation, explained the condensation of RecA on poly(T), its length dependence, for both 5′ and 3′ labeling. Here we modify the model to include a folded state of ssDNA. Because RecA polymerizes in the direction 5′ to 3′ in equilibrium, there will be more domains close to the 3′-end. For short DNA substrates, as in our case, we consider only a single RecA domain per DNA bound to the 3′-end, and compute the distribution of domain lengths. Each DNA polymer of length L is uniquely defined by the length of the bound RecA domain, l. The system is at multiple chemical equilibria among the species: l-domains, free RecA, and free DNA, at concentrations Dl, R0, and D0, respectively. The free energy of the system is the sum of the adsorption energy (first term) and the ideal gas entropies of each species (second and third terms), given in units of kBT, by
 |
1 |
Here, ɛ is a sequence-averaged monomer adsorption energy and δl is the “end-cap” energy, so that the energy of an adsorbed RecA at the end of a domain is ɛ − δl. Why do we subtract δl? In the absence of any ssDNA folding, the end-cap energy is constant independent of RecA domain length. However, if the DNA sequence has local folding, RecA binding will be penalized by the energy needed to overcome this fold. This implies that above some effective domain length, leff, the end-cap energy increases by an amount ΔG and, hence, we write
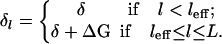 |
2 |
Conservation of total RecA and DNA molecules, RT and DT, implies R0 + Σl Dll = RT and D0 + Σl Dl = DT. Minimizing the free energy and using these conservation laws, we obtain an exponential equilibrium distribution of domain lengths:
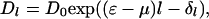 |
3 |
were μ = −log(R0) is the chemical potential of the free RecA. The mean anisotropy measured is then an ensemble average over the RecA occupancies
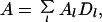 |
4 |
where Al is the anisotropy signal of a DNA molecule on which l RecA monomers are bound. The fluorescence anisotropy signal increases significantly only when RecA binds close to the labeled end, implying that for 5′-labeling, the onset is determined primarily by the long RecA domains (unpublished data). Therefore, to estimate the onset concentration we consider only one term in Eq. 4, l = leff and set Dleff ∼ α, with α being a small fractional occupancy ≈0.1. From Eqs. 3 and 2, and because the RecA concentration is not depleted, R0 ≈ RT, we obtain:
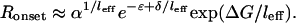 |
We thus recover the exponential shift of the onset with the DNA folded free energy. Comparing the factor in our exponential fit (Fig. 3 Lower) we obtain leff ≈ 7 monomers.
RecA Polymerization on a Molecular Beacon: A Two-State System.
Lastly, we use a molecular beacon, a sensitive ssDNA hairpin probe that emits light according to its conformation, to obtain further insight into the mechanism of RecA binding to folded DNA. The beacon comprises a loop and a stem with a fluorophore and its quencher at the opposite ends of the stem (24). When the beacon is closed, in the hairpin conformation, fluorescence is quenched because of the proximity of the fluorophore and the quencher.
Fig. 6 shows that RecA binding unfolds the hairpin structure of a beacon, (BCN)T15 (see Materials and Methods), resulting in a 28-fold increase of total fluorescence as well as an increase in fluorescence anisotropy. Note that at low RecA concentration the anisotropy signal is larger than the intensity one, a surprising result that is explained below. The combined signal of the beacon provides more information, as compared with end-labeled ssDNA without a quencher. This will suggest that the constraint set by the closed stem on the self-assembly of RecA effectively reduces the beacon to two states: (i) closed with few RecA monomers bound to the loop, and (ii) open and almost fully covered with RecA. We will also obtain an estimate for the DNA folding energy.
Figure 6.

Molecular beacon. (Upper) Fluorescence anisotropy and total intensity of RecA binding to a beacon with a 15-T loop, (BCN)T15. The total intensity of the beacon increases by 28-fold on binding. (Inset) Normalized beacon anisotropy as a function of its intensity (circles) and a two-state model (solid line). (Lower Left) of RecA Polymerization (ellipses) opens the beacon. F, fluorophore; Q, quencher. (Lower Right) sketch of RecA domain length distribution in the vicinity of the half-intensity point, where 50% of the beacons are open.
Closed beacons may have RecA domains no longer than a length L★, above which the stem must unzip due to the rigidification of ssDNA by the RecA domain. L is the maximal number of monomers. As explained above, Eq. 3, the distribution of RecA domains bound to ssDNA is exponential in domain length l, Dl ∼ exp((ɛ − μ)l − δl), where μ, ɛ, and δl are the RecA chemical potential, adsorption, and end-cap energies. For the short domains l < L★, the end-cap energy is a constant, δl = δ, whereas for l > L★, it must include the stem folding energy, δl = δ + ΔGf. This creates a gap in the length distribution, breaking it into two piece-wise exponentials that separate open and closed beacons, as sketched in the bottom of Fig. 6 for the region where half of the beacons are open.
If closed beacons emit less light than open ones by a factor η ≫ 1, it is straightforward to show that the total intensity and the mean anisotropy are a weighted ensemble average over the intrinsic anisotropies Al (18):
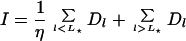 |
5 |
and,
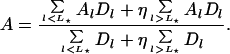 |
6 |
The distribution Dl is normalized so that Σl Dl = 1, and for η = 1 we get I = 1, A = Σl AlDl, as the simple ensemble average (Eq. 4). Close to the half-intensity point most of the closed beacons have L★ bound monomers and most of the open ones have L monomers, effectively reducing the system to two states, DL★ and DL, with DL★ + DL ≈ 1. We therefore reduce (Eqs. 5 and 6) to two states, and define the normalized total intensity and anisotropy of the beacon:
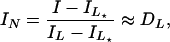 |
7 |
and
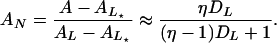 |
8 |
Combining Eqs. 7 and 8, we obtain:
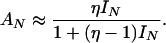 |
9 |
The Inset of Fig. 6 shows that, indeed, the beacon fits well to this two-state model with the measured signal-to-noise ratio, η = 28, and no adjustable parameters. Note that Eq. 9 correctly predicts that the anisotropy precedes the intensity, AN > IN.
Finally, we estimate the beacon folding free energy. From the intensity and anisotropy signals we find the RecA half-point concentrations (IN = 1/2, AN = 1/2) RI and RA, where DL★ = DL and DL★ = ηDL, respectively. When our exponential distribution Dl is used, these two relations yield an expression for the beacon folding free energy ΔGf:
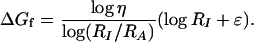 |
With ɛ = 15.5kBT measured for poly(T) (unpublished data) we obtain ΔGf = 4.3kBT, which is smaller by a factor 2 than the folding computation.
Summary and Conclusions
This paper addresses the issue of collective binding of RecA protein to DNA and the physical mechanisms that modulate the specificity of this interaction. We presented experiments of RecA polymerization on ssDNA oligomers using fluorescence anisotropy with improved resolution due to a microdispensing system. This sensitive system allowed us to probe the onset of binding at a low concentration regime in which RecA condenses on ssDNA. We have previously shown that the onset is very sensitive to various parameters, such as sequence length and salt (unpublished data). Here we focused on RecA sequence-recognition properties. First, RecA binding sites of low and high affinity were identified by using triplet repeats in the presence of ATPγS, a nonhydrolyzable analog of ATP. We find that RecA binds well to sequences rich in pyrimidines and that ssDNA rigidity of stacked purine sequences and secondary structure caused by self-hybridization all hinder polymerization. We find a correlation between the onset concentration for polymerization and the free energy of ssDNA folding: the onset of polymerization on all sequences depends exponentially on the folded state free energy. This result is consistent with a simple nucleation model in which RecA domains must unfold DNA secondary structure, which poses an activation barrier for polymerization. A similar idea has recently been proposed in single-molecule experiments that have shown that RecA binds well to stretched DNA, thus implying an important role for thermal fluctuations in the binding (26). The correlation between folding free energy and polymerization onset does not exclude sequence-dependent specificity, especially to purine-rich stacked sequences.
The observation that the onset of binding is exponentially sensitive to ssDNA structure and sequence led us to consider the limit of a single base substitution and how it might affect the binding. Two known, naturally occurring substitutions were studied as examples, a single point mutation (1 in 39 bases: sickle cell anemia) and a single nucleotide polymorphism (1 in 63 bases: p53). In both cases, RecA binding curves on the normal and the altered sequences were distinguishable and followed the general trend that rigidity and secondary structure pose resistance to binding. In the first case, the base change affects the local rigidity, whereas in the second case it directly alters the ssDNA folding. We also studied the binding of RecA to a molecular beacon, as an example of a well-defined secondary structure. The barrier for binding imposed by the hairpin reduces the problem to a two-state system.
These measurements lead us to conclude that RecA polymerization, at its onset, is a sensitive detector of DNA sequence and structure. This conclusion may have importance both for the basic understanding of RecA–ssDNA interaction and for the purpose of molecular computation, in which recognition elements of DNA may be useful. The significance of the present study to RecA–DNA recognition in vivo requires further work under various conditions, in particular in the presence of ATP. Nevertheless, the identification of simple rules for DNA sequence discrimination and the activation picture presented may provide a basis for identification of possible recombination hotspots. It is instructive to compare our results with experiments of in vitro molecular selection that showed that RecA has a preferential affinity for DNA sequences rich in G and T composition (15), some of which are similar to the χ sequence (16). We have looked at the selected sequences (15) and, in fact, the best RecA substrates have minimal secondary structure and lack significant base stacking, which follows the same trends that we have found independently. Specifically, one of best selected sequences, CLONE1 (GCG TGT GTG GTG GTG TGC) (15), folds into an unstable large loop held by the G⋅C base pairs of the ends and any possible stacking of purines is broken by intervening pyrimidines.
Acknowledgments
We thank G. Bonnet, M. Calame, B. Dubertret, N. Goddard, D. Lubensky, M. Morpurgo, D. Nelson, B. Shraiman, and D. Thaler for helpful discussions and suggestions. We are especially grateful to K. Adzuma for his support and to T. Tlusty for his help with the theoretical model. This work was supported by the Mather Foundation. R.B.-Z. acknowledges a Burroughs Wellcome Fellowship.
Abbreviations
- ssDNA
single-stranded DNA
- dsDNA
double-stranded DNA
- ATPγS
[γ-thio]ATP
References
- 1.Ptashne M, Gann A. Curr Biol. 1998;8:R812–R822. doi: 10.1016/s0960-9822(07)00508-8. [DOI] [PubMed] [Google Scholar]
- 2.Roca R, Cox M. Prog Nucleic Acid Res Mol Biol. 1997;56:129–223. doi: 10.1016/s0079-6603(08)61005-3. [DOI] [PubMed] [Google Scholar]
- 3.Egelman M H, Stasiak A. J Mol Biol. 1986;191:677–697. doi: 10.1016/0022-2836(86)90453-5. [DOI] [PubMed] [Google Scholar]
- 4.Takahashi M, Kubitsa M, Norden B. J Mol Biol. 1989;205:137–147. doi: 10.1016/0022-2836(89)90371-9. [DOI] [PubMed] [Google Scholar]
- 5.Stasiak A, DiCapua E, Koller T. J Mol Biol. 1981;151:557–564. doi: 10.1016/0022-2836(81)90010-3. [DOI] [PubMed] [Google Scholar]
- 6.Leahy M C, Radding C M. J Biol Chem. 1986;261:6954–6960. [PubMed] [Google Scholar]
- 7.Egelman E H, Stasiak A. J Mol Biol. 1988;200:329–349. doi: 10.1016/0022-2836(88)90245-8. [DOI] [PubMed] [Google Scholar]
- 8.Ogawa T, Wabiko H, Tsurimoto T, Horii T, Masukata H, Ogawa H. Cold Spring Harbor Symp Quant Biol. 1979;43:909–915. doi: 10.1101/sqb.1979.043.01.099. [DOI] [PubMed] [Google Scholar]
- 9.Roberts J W, Roberts C W, Craig N L, Phizicky E M. Cold Spring Harbor Symp Quant Biol. 1979;43:917–920. doi: 10.1101/sqb.1979.043.01.100. [DOI] [PubMed] [Google Scholar]
- 10.Menetski J P, Kowalczykowski S C. J Mol Biol. 1985;181:281–295. doi: 10.1016/0022-2836(85)90092-0. [DOI] [PubMed] [Google Scholar]
- 11.Kowalczykowski S C, Clow J, Krupp R A. Proc Natl Acad Sci USA. 1987;84:3127–3131. doi: 10.1073/pnas.84.10.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pugh B F, Cox M M. J Mol Biol. 1988;203:479–493. doi: 10.1016/0022-2836(88)90014-9. [DOI] [PubMed] [Google Scholar]
- 13.McEntee K, Weinstock G M, Lehman I R. J Biol Chem. 1981;256:8835–8844. [PubMed] [Google Scholar]
- 14.Cazenave C, Chabbert M, Toulme J-J, Helene C. Biochim Biophys Acta. 1984;781:7–13. doi: 10.1016/0167-4781(84)90117-9. [DOI] [PubMed] [Google Scholar]
- 15.Tracy R B, Kowalczykowski S C. Genes Dev. 1996;10:1890–1903. doi: 10.1101/gad.10.15.1890. [DOI] [PubMed] [Google Scholar]
- 16.Anderson D G, Kowalczykowski S C. Cell. 1997;90:77–86. doi: 10.1016/s0092-8674(00)80315-3. [DOI] [PubMed] [Google Scholar]
- 17.Biet E, Sun J S, Dutreix M. Nucleic Acids Res. 1999;27:596–600. doi: 10.1093/nar/27.2.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lakowicz J R. Principles of Fluorescence Spectroscopy. 2nd Ed. Boston: Kluwer; 1999. [Google Scholar]
- 19.Berg H C. Random Walks in Biology. Princeton: Princeton Univ. Press; 1993. , Expanded Ed. [Google Scholar]
- 20.McGhee J D, von Hippel P H. J Mol Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 21.SantaLucia J., Jr Proc Natl Acad Sci USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Firedman R A, Honig B. Biophys J. 1995;69:1528–1535. doi: 10.1016/S0006-3495(95)80023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cantor C R, Smith C L. Genomics. New York: Wiley; 1993. p. 73. [Google Scholar]
- 24.Tyagi S, Kramer F R. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 25.Bonnet G, Krichevsky O, Libchaber A. Proc Natl Acad Sci USA. 1998;95:8602–8606. doi: 10.1073/pnas.95.15.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leger J F, Robert J, Bourdieu L, Chatenay D, Marko J F. Proc Natl Acad Sci USA. 1998;95:12295–12299. doi: 10.1073/pnas.95.21.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]