

Abstract
Retrotransposons are “copy-and-paste” insertional mutagens that substantially contribute to mammalian genome content. Retrotransposons often carry long terminal repeats (LTRs) for retrovirus-like reverse transcription and integration into the genome. We report an extraordinary impact of a group of LTRs from the mammalian endogenous retrovirus-related ERVL retrotransposon class on gene expression in the germline and beyond. In mouse, we identified more than 800 LTRs from ORR1, MT, MT2, and MLT families, which resemble mobile gene-remodeling platforms that supply promoters and first exons. The LTR-mediated gene remodeling also extends to hamster, human, and bovine oocytes. The LTRs function in a stage-specific manner during the oocyte-to-embryo transition by activating transcription, altering protein-coding sequences, producing noncoding RNAs, and even supporting evolution of new protein-coding genes. These functions result, for example, in recycling processed pseudogenes into mRNAs or lncRNAs with regulatory roles. The functional potential of the studied LTRs is even higher, because we show that dormant LTR promoter activity can rescue loss of an essential upstream promoter. We also report a novel protein-coding gene evolution—D6Ertd527e—in which an MT LTR provided a promoter and the 5′ exon with a functional start codon while the bulk of the protein-coding sequence evolved through a CAG repeat expansion. Altogether, ERVL LTRs provide molecular mechanisms for stochastically scanning, rewiring, and recycling genetic information on an extraordinary scale. ERVL LTRs thus offer means for a comprehensive survey of the genome's expression potential, tightly intertwining with gene expression and evolution in the germline.
Repetitive mobile sequences are a common genome component intertwining with genome stability and evolution of new traits. A particular type is retrotransposons, interspersed repetitive elements that amplify by a copy-and-paste mechanism entailing integration of reverse-transcribed DNA into the genome. Retrotransposon amplification threatens genome integrity through insertional mutations and chromosomal aberrations; consequently, defensive mechanisms evolved that suppress retrotransposons (for review, see Crichton et al. 2014). Retrotransposons, however, can also provide functional gene parts, such as promoters, enhancers, exons, terminators, or splice junctions (for review, see de Souza et al. 2013; Gerdes et al. 2016; Göke and Ng 2016; Thompson et al. 2016). Retrotransposons thus explore the space where their mutagenic potential coexists with other occasional contributions to gene expression.
Retrotransposons are broadly divided by the presence of long terminal repeats (LTRs) and retrotransposition autonomy (Craig et al. 2015). Murine LTR retrotransposons can be further classified into three classes (Mager and Stoye 2015). Here, we focus on a selected set of LTR sequences from class III whose more than 400,000 copies comprise ∼5.5% of the mouse genome (Mouse Genome Sequencing Consortium 2002; McCarthy and McDonald 2004). Class III is an assorted group of endogenous retrovirus-related elements termed ERVL (Supplemental Table S1), which includes autonomous endogenous retroviruses, e.g., Mouse Endogenous Retrovirus type-L (MuERV-L) (Bénit et al. 1997) and nonautonomous Mammalian apparent LTR Retrotransposons (MaLRs) (Smit 1993) that are sometimes recognized as a separate ERVL-MaLR group (Crichton et al. 2014). Here, we investigate related MuERV-L and MaLR LTRs that make remarkable contributions to maternal and zygotic transcriptomes.
MuERV-L is a recent endogenous retrovirus that no longer retrotransposes although it produces virus-like particles (Bénit et al. 1997; Costas 2003; Ribet et al. 2008). MuERV-L LTRs are annotated as MT2_Mm LTRs and share sequence similarity with members of the MT2 family (MT2A-C) and MaLR LTRs, suggesting a common ancestry (McCarthy and McDonald 2004; Hubley et al. 2016). In MaLR elements (∼85% of ERVL insertions [Mouse Genome Sequencing Consortium 2002]), LTRs flank a noncoding 1- to 2-kb internal fragment. Rodent MaLRs include the ancestral mammalian MLT family and rodent-specific ORR1 and MT families; ORR1A and MTA subfamilies are the youngest (Smit 1993; Hubley et al. 2016). About a quarter of murine MaLR insertions are polymorphic, whereas the rest are fixed (Nellaker et al. 2012).
MaLR and MT-2 sequences were found in transcripts from oocytes and early embryos, including 5′ exon fusions with protein-coding transcripts (Peaston et al. 2004; Veselovska et al. 2015; Karlic et al. 2017). Furthermore, MuERV-L expression was associated with zygotic genome activation (ZGA) and embryonic stem cell (ESC) potency (Kigami et al. 2003; Svoboda et al. 2004; Macfarlan et al. 2011, 2012; Schoorlemmer et al. 2014). MaLR and MT2 LTRs thus offer a unique opportunity for adaptation and separation of maternal/zygotic expression programs. Here, we provide a systematic analysis of the contribution of MaLR and MT2 LTRs to gene evolution and expression during the oocyte-to-embryo transition (OET) in rodents and other mammals, exploring their role as “plug-and-play” promoter platforms that insert into existing transcriptional units or create new ones.
Results
Elementary features of ERVL LTRs
MLT, MT, ORR1, and MT2 LTRs (collectively referred to as “ERVL LTRs” hereafter) provide a unique model of LTR evolution and co-option during the last 80 million years. These LTRs were organized into 19 subgroups (Supplemental Fig. S1A), in which MLT1 and MLT2 represent the ancestral mammalian LTR subgroups, and the MT, ORR1, and MT2 subgroups radiated during rodent evolution (Fig. 1A–C; Supplemental Fig. S1B). Sequence analysis also showed that, except for MT2, ERVL LTRs are depleted of CpG dinucleotides, suggesting that MT2 LTRs evolved differently (Supplemental Fig. S1C). Depletion of CpGs in MT and ORR1 LTRs appears progressive, resulting in a minimal CpG frequency in the most recent subfamilies (Supplemental Fig. S1C).
Figure 1.

Sequence properties of selected ERVL LTRs. (A) Organization of ORR1, MT, and MuERV-L retrotransposons. Internal sequences of ORR1 and MT elements do not encode any protein. (B) Abundance of selected ERVL LTRs in mammalian genomes. The brown areas indicate misannotated ORR1F, ORR1G, and MTC LTRs in genomes of other rodents. (C) Nucleotide substitution rate for the closest pairs among 200 random inserts in each LTR subfamily. (D) Three types of LTR retrotransposon inserts and their frequencies among the selected youngest ERVL subfamilies. (E) A schematic depiction of an MT LTR gene-remodeling platform. (F) A combined SD sequence logo of MT, ORR1, and MT2 LTR families. (G) Conserved position of the splice consensus sequence at the 3′ end of selected LTRs. Gray rectangles depict consensus lengths of LTRs aligned by the 3′ end to the top. Red or black points represent positions of TGTAAGY consensus motif or AATAAA polyadenylation signal, respectively, in 200 randomly chosen LTRs in each subfamily.
MaLR LTRs have a relatively uniform chromosome-wide distribution and exhibit known biases of LTR retrotransposon distribution (van de Lagemaat et al. 2006), such as reduced incidence of intronic inserts oriented sense to gene expression (Supplemental Fig. S2). The majority of ERVL genomic inserts are solo LTRs, which account for ∼65%–70% of the annotated inserts in MTA and ORR1A0 subfamilies (Fig. 1D). Solo MT2 LTRs are also common although less than MaLRs (Fig. 1D).
At the time of insertion, an LTR carries a functional promoter, a transcription start site (TSS), and a polyadenylation site [poly(A)]. In addition, ERVL LTRs may carry a splice donor (SD) and even a functional AUG codon (Peaston et al. 2004; Flemr et al. 2013), but no splice acceptor (SA) (Fig. 1E). Sequence analysis revealed similar but distinct SDs in MaLR LTRs (Fig. 1F; Supplemental Fig. S1D) that might have a common ancestry because they are in approximately the same position at the 3′ end of the LTRs (Fig. 1G). At the same time, SD presence in different subfamilies negatively correlates with evolutionary age suggesting a gradual loss of SDs in inserted LTRs.
Genome-wide gene remodeling by ERVL LTR co-option across mammals
Co-option refers to a contribution of an LTR sequence to gene transcription without necessarily being an exaptation, i.e., a co-option that provides a novel host function. To annotate co-option events from retrotransposon reservoirs that could influence OET in mammals, we annotated overlaps of exons and retrotransposons in mouse, golden hamster (Mesocricetus auratus), human, and bovine genomes, and selected co-options supported by next-generation sequencing (NGS) data. We used four co-option categories according to a retrotransposon's contribution to gene structure (Fig. 2A): (I) 5′ exon (retrotransposon-derived promoter, TSS, and/or SD); (II) internal exon (retrotransposon-derived SD and/or SA); (III) 3′ exon [retrotransposon-derived SA and/or poly(A)]; and (IV) intraexonic, in which a retrotransposon sequence does not contribute to mRNA processing. Categories I–III were further divided into full or partial contributions depending whether an exon came fully or partially from the retrotransposon. For example, a full 5′ exon co-option means that TSS (the promoter presumably as well) and SD come from the same retrotransposon (Fig. 2A), whereas a partial contribution means that either TSS or SD was provided.
Figure 2.

Gene remodeling by LTRs. (A) Four categories of LTR co-option according to the co-opted exon boundaries. LTR co-options may affect gene expression but not the encoded protein, remodel a gene and change its protein product, or create a new transcriptional unit, such as an lncRNA gene. (B) Whole-genome analyses of impacts of LTR, LINE, and SINE elements on gene structure according to the classification depicted in A. Repeatmasker (Smit et al. 2013–2015) was used for Class I–III LTR annotation. The y-scale depicts the ratio of observed co-option events and annotated insertions, which are listed in Supplemental Tables S2 (mouse), S5 (hamster), S6 (human), and S7 (cow). (C) Impact of MaLR and MT2 LTRs on gene structure according to the classification depicted in A in four mammals. The y-scale depicts the number of co-opted insertion events. B and C display both full and partial contributions.
We identified more than 75,000 gene-affecting events involving all classes of retrotransposons in mouse (∼half were intraexonic SINE insertions). Class III (ERVL) LTR retrotransposons have the highest frequency of 5′ exon contribution in mouse and hamster genomes (Fig. 2B). In humans and cows, Class III LTRs also substantially contribute to co-option events but their 5′ exon contribution does not stand out as in rodents (Fig. 2B). The difference seems mainly due to co-option of the MT family insertions (Fig. 2C). Importantly, MaLR 5′ exon co-option exists beyond rodents; 252 and 125 5′ exons derived from MLT LTRs were identified in human and bovine genomes, respectively (Fig. 2C). Because annotations of the human and mouse genomes are more exhaustive, the extent of co-option in bovine and hamster genomes is likely underestimated.
Co-option of ERVL LTR promoter platforms
We investigated usage of ERVL LTRs as “plug-and-play” gene-remodeling platforms in mouse oocytes and early embryos. Notably, contributions of LTR subfamilies to 5′ exon co-option anti-correlated with their age: the youngest ORR1, MT, and MT2 subfamilies showed a remarkably high proportion of 5′ exon co-option compared to the older subfamilies (Fig. 3A). Most of the identified 5′ exon co-option events concerned MTA LTRs, whereas the oldest MTE LTRs made a minimal contribution. The MTA and MT2 LTRs had the highest frequency of 5′ exon co-option (>3%), MTA having the highest absolute number of co-option events (Fig. 3B).
Figure 3.

Evolution of exon co-option in mice. (A) Frequency of co-options in selected LTR subfamilies (full and partial contribution). (B) Numbers of full LTR 5′ exon co-options in protein-coding genes and lncRNAs expressed in oocytes and early embryos. (C) MT LTR family phylogeny and bursts of gene rewiring events. The left tree shows a phylogenetic tree of 5000 randomly selected MT LTRs combined with 596 LTRs co-opted as complete 5′ exons. The right tree highlights in red the co-opted LTRs.
In total, we found 1574 ERVL LTRs contributing to promoters and first exons of long noncoding RNA (lncRNA) and protein-coding genes expressed in oocytes and early embryos (Supplemental Table S2); 509 LTRs in protein-coding genes and 333 LTRs in lncRNA genes made full 5′ exon contributions (Fig. 3B). These 842 loci thus represent cases of the complete “plug-and-play” gene remodeling by ERVL LTRs in the reference mouse strain C57Bl/6. A phylogenetic analysis of MT LTRs that underwent 5′ exon co-option suggested that the bulk of murine 5′ exon co-options is a consequence of several bursts of retrotransposition of parental elements (Fig. 3C).
ERVL LTR-mediated control of gene expression during OET
Vertical expansion of retrotransposons requires expression and retrotransposition in the germline. We thus analyzed abundance of ERVL LTR-derived RNAs in poly(A) RNA NGS data from the germline cycle (Fig. 4A). Although combining such data sets has limited significance, it nonetheless suggests that LTRs of the most recent ERVL subfamilies make highly pronounced contributions to poly(A) transcriptomes of specific stages of the germline cycle, particularly during oogenesis (MTA) and ZGA (ORR1A and MT2). Such patterns also emerge in independent NGS data (Supplemental Fig. S3A; Abe et al. 2015) and are consistent with the literature (Kigami et al. 2003; Peaston et al. 2004; Svoboda et al. 2004; Flemr et al. 2013). A survey of individual co-opted ERVL LTR promoters reveals two main expression patterns during OET (Fig. 4B; Supplemental Fig. S3B). MT LTRs are expressed maternally and their RNA is extensively degraded before the major ZGA phase (two-cell stage), whereas MT2 expression transiently associates with ZGA. Expression of ORR1 elements is not uniform; we find maternal or zygotic expression among ORR1A and ORR1B inserts, suggesting evolving transcriptional control within MaLR subfamilies.
Figure 4.

Transcriptional control by co-option of MaLR and MT2 LTRs. (A) LTR RNA abundance in transcriptomes of germline cycle stages presented as log10 RPM of selected LTR sequences in poly(A) NGS data sets (Supplemental Table S3). Included are profiles of LINE1 and IAP, the presently active mouse autonomous retrotransposons (Maksakova et al. 2006; Sookdeo et al. 2013). (B) Maternal and zygotic expression of solo LTRs during oocyte-to-embryo transition. UCSC Genome Browser (Kent et al. 2002) snapshots exemplify expression patterns of co-opted 5′ exons. For each LTR, all stages were set for the maximum CPM values indicated on the top of each column. Most LTR subfamilies have distinct maternal or zygotic expression patterns corresponding to the specific patterns shown here (Supplemental Fig. S3). At the same time, some variability within an LTR subfamily is occasionally observed as shown for two different ORR1B LTR insertions. Developmental stages: (GV) full-grown GV oocyte; (MII) metaphase II oocyte; (1C) one-cell (fertilized egg); (2C) two-cell; (4C) four-cell; (Mo) morula; (Bl) blastocyst. MT2xORR1 is the 3′ MT2 LTR of MuERV-L that is preceded by an 87-bp fragment of ORR1A3 internal sequence. (C) Expression of MaLR and MT2 LTR-derived 5′ exons from lncRNAs and protein-coding genes ordered by the maternal/ZGA expression ratio (GV+MII)/(2C+4C). The heatmap shows log2 FPKM values of the annotated LTR 5′ exons (full contribution) with FPKM >0.1 in at least one sample. The colored bar indicates the LTR family. (D) Expression of genes containing LTR-derived 5′ exons in mouse and hamster oocytes. Points represent log2 FPKM values of genes in mouse and hamster oocytes (GSE86470). Point colors indicate whether the 5′ LTR-derived exon is present in the mouse (black) or hamster (red) genome or in genomes of both species (blue), and gray points depict remaining genes. (E) Comparison of oocyte expression of genes that have an LTR-derived 5′ exon in mice or hamsters with expression of other genes. The x-axis represents gene expression (log2 FPKM), whereas the y-axis is fraction of genes.
Co-option of ERVL LTR promoters and 5′ exons contributed to phased gene expression during OET with the majority of co-opted LTRs supporting maternal expression (Fig. 4C). The ratio LTR [FPKM]/gene [FPKM] suggests that LTRs provide the main promoter for lncRNAs during OET, whereas protein-coding genes often have other promoters supporting higher expression than that controlled by the LTR: 3/333 lncRNA and 56/509 protein-coding genes had gene FPKM at least 4× higher than the LTR-derived 5′ exon FPKM value (6/333 and 163/509 for a twofold difference, respectively).
To estimate the impact of LTR co-option on gene expression and evolution, we compared transcriptomes of mouse and golden hamster oocytes. Both rodents have similar parameters of ovulation cycle, gestation period, and litter size. Their maternal transcriptomes retain sufficient similarity (Pearson and Spearman correlations 0.66 and 0.67, respectively) to analyze expression of protein-coding genes that co-opted LTR promoters and 5′ exons. Most of the genes with differentially co-opted LTRs are expressed in oocytes of both species, although a small group (6%) is expressed in species in which LTR co-option occurred but not in species lacking the co-option (Fig. 4D). Furthermore, genes with co-opted LTRs have higher expression in oocytes of both species relative to the whole transcriptome (Fig. 4E). However, a minority of co-opted LTRs yields high expression. Using Dicer1 as a benchmark for MT-driven expression, 20 mouse genes have higher MT-driven expression. These results imply that ERVL LTR co-option typically enhances stage-specific gene expression, but it less often yields a strong LTR-driven expression or adds a new gene into existing gene expression.
Variability and plasticity of MT LTR promoter activity in control of gene expression
The impact of an LTR on the local transcriptional landscape depends on many factors. In the Dicer1 model case, we identified plasticity and latency of LTR-controlled expression that demonstrates how transcriptional activity of a specific LTR can be influenced by the genomic context. We reported earlier that an MTC solo LTR functions as an oocyte-specific promoter for a truncated Dicer1 isoform (denoted Dicer1O) that is responsible for highly active RNAi in mouse oocytes (Flemr et al. 2013). The MTC insertion is present in genomes of golden and Chinese hamsters (annotated MTD there) but not in genomes of mole rat Nannospalax galili, jerboa Jaculus jaculus, or Guinea pig (Fig. 5A), placing the insertion event ∼30–40 million years ago (MYA) (Supplemental Fig. S4A).
Figure 5.
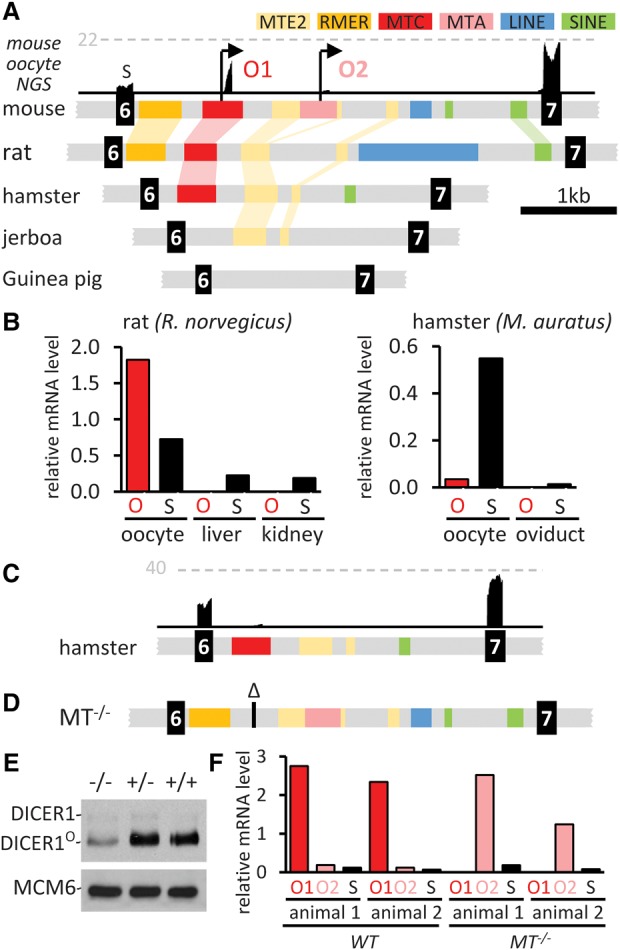
Dicer1 rewiring and remodeling by MT LTRs. (A) Retrotransposon content changes during evolution of Dicer1 intron 6 in rodents. Above the mouse sequence is a snapshot of a UCSC Genome Browser track with mouse oocyte NGS data. The gray dashed line indicates CPM. O1, O2—two oocyte-specific promoters. (B) qPCR analysis of Dicer1 isoform mRNA expression in rat and hamster oocytes. Dicer1O (O) and full-length somatic Dicer1 isoform (S) expression are shown relative to Hprt. (C) NGS data support minimal Dicer1O expression in hamster oocytes. Shown is a UCSC Genome Browser snapshot. The horizontal dashed line represents the number of reads. (D) A schematic view of the intron 6 in Dicer1MT−/− mice with MTC (O1 promoter) deletion. (E) Oocytes lacking the MTC LTR (O1 promoter) still produce a detectable amount of DICER1O. Shown is an immunoblot from C57Bl/6NCrl oocytes. A low amount of the full-length DICER1 is visible above the DICER1O isoform. Each lane represents roughly 500 oocytes. (F) qPCR analysis of Dicer1 transcripts driven by MTC (O1) and MTA (O2) LTRs. Dicer1 expression is shown relative to Hprt.
Dicer1O mRNA was detected in rat and hamster oocytes but not in somatic cells (Fig. 5B). Despite the common design of primers, the Dicer1 isoform qPCR in hamster oocytes yielded a much stronger signal for the somatic transcript than for the Dicer1O transcript (Fig. 5B), a result consistent with NGS analysis of hamster oocytes where the Dicer1O transcripts had a much lower NGS signature than full-length Dicer1 transcripts (Fig. 5C). Thus, unlike in mice, Dicer1O mRNA comprises a minority of Dicer1 transcripts in hamster oocytes.
The MTC LTR in Dicer1 is essential for fertility, and its loss correlates with up-regulation of transcripts targeted by endogenous RNAi (Flemr et al. 2013). Interestingly, after crossing mice lacking the MTC element (Dicer1MT−/−) (Fig. 5D) onto a CD1 strain background, only 25% of the Dicer1MT−/− females exhibited sterility, and the rest produced viable progeny, suggesting that the sterile phenotype is not fully penetrant in this mixed genetic background. Surprisingly, residual amounts of DICER1O protein were found in oocytes of Dicer1MT−/− mice (Fig. 5E) and brought into focus an MTA LTR insertion localized ∼600 bp downstream from the MTC (Fig. 5A; Supplemental Fig. S4B). We discovered that the MTA LTR can function as an alternative promoter and produce a second Dicer1O-like transcript. Typical expression from the MTA LTR is minimal (<10% considering NGS data) (Smallwood et al. 2011; Abe et al. 2015; Veselovska et al. 2015; Karlic et al. 2017), possibly because the upstream MTC LTR promoter reduces transcription from the MTA LTR. Upon the MTC LTR deletion, the MTA LTR promoter activity in the mixed genetic background increases (Fig. 5F), yielding DICER1O protein that can rescue the sterile phenotype.
Scanning of genetic information downstream from ERVL LTRs
The concept of a mobile remodeling “plug-and-play” platform entails an LTR insertion that remodels the local transcriptional landscape via transcription extending downstream from the insertion. Mouse ZGA offers a unique model for examining such transcription because the maternal transcriptome has a reduced intronic and intergenic NGS signal (Abe et al. 2015), thereby facilitating detection of nascent transcripts in the zygote. Furthermore, promoter activity of several LTRs, including MT2 and ORR1A, appears during ZGA (Fig. 4A,B). Indeed, low levels of transcripts far downstream from MuERV-L are apparent during ZGA, especially in two-cell embryos treated with aphidicolin (Fig. 6A; Supplemental Fig. S5A), which prevents replication-dependent formation of transcriptionally repressive chromatin (for review, see Svoboda et al. 2015). Because the downstream transcript levels appeared minimal at individual loci, we assessed downstream transcription by analyzing cumulative expression 150 kb around the hundred most expressed MuERV-L inserts using two independent NGS data sets (Park et al. 2013; Abe et al. 2015). We found significantly higher transcript levels in two-cell embryos as far as 40 kb (Supplemental Fig. S5B; Park et al. 2013) and 120 kb (Fig. 6B; Abe et al. 2015) downstream when compared with corresponding upstream regions. Whether the transcription also extends through active genes could not be determined from the data. A lower but apparent transcript accumulation was also found downstream from ORR1A and MT2 solo LTRs that are less expressed during ZGA (Supplemental Fig. S5C,D).
Figure 6.

Genome scanning by LTRs and emergence of new genes. (A) Transcription downstream from MuERV-L is apparent during ZGA, especially in two-cell embryos treated with aphidicolin (2Ca). Shown is a representative UCSC Genome Browser snapshot of an MuERV-L insertion expressed during ZGA. The gray horizontal lines represent five CPM. Stages: (GV) full-grown GV oocyte; (1C) one-cell (fertilized egg); (2C) two-cell; (4C) four-cell. (B) Cumulative display of transcription in 150-kb genomic flanks around the hundred MuERV-L elements most expressed during ZGA. (C–F) UCSC Genome Browser snapshots of selected genomic loci with mapped NGS data (Abe et al. 2015; Karlic et al. 2017). Gray dashed lines indicate CPMs. Positions of repetitive sequences (D–F) are indicated by gray rectangles in rows from the top: SINE, LINE, LTR, and DNA transposon elements. The conservation tracks (D,F) display homology with rat (top), rabbit, human, dog, and cow genomes. (C) A lncRNA gene with MuERV-L-derived 5′ exons and downstream exons from the genomic flank. (D) A new lncRNA gene formed by MaLR LTR insertions. The promoter and exon 1 come from an MTA solo LTR, exon 2 through exonization of an ORR1F solo LTR. (E) Examples of antisense pseudogene sequence rewiring yielding a lncRNA substrate for endosiRNAs where an MTB solo LTR was inserted into a locus already containing a pseudogene (Nme3) or a pseudogene (Dlgap5) was inserted into a locus already containing an MTA. (F) An example of a sense pseudogene (Speer4E pseudogene) rewiring yielding a CPAT positive transcript.
Novel multiexon lncRNA genes may emerge by deploying an ERVL LTR into a new genomic locus. Of the 333 lncRNA loci co-opting ERVL LTR promoter platforms, 87 apparently co-opted them as sole promoters after separation of mice from rats (exemplified in Fig. 6C,D). Such co-options are thus key candidate events for lncRNA genesis. “Scanning” of the downstream genomic flank with transcription, however, does not universally produce defined spliced transcripts such as in Figure 6C. Of the 100 most expressed MuERV-L loci, only 16 contained downstream exons recognized during lncRNA transcript model assembly (Karlic et al. 2017).
Pseudogene recycling by ERVL LTRs
ERVL LTRs bridge lncRNA and protein-coding gene evolution by providing promoters for retrotransposed mRNAs. Among 842 LTRs that made the full 5′ exon contribution, 78 (9.3%) produced transcript models that included processed pseudogene sequences (Supplemental Table S8). Pseudogene recycling by ERVL LTRs occurs in two ways; an LTR insertion recycles an already integrated pseudogene, or a pseudogene integrates into a locus that already carries an LTR (Fig. 6E). The latter case reiterates that LTR insertions may potentially affect gene expression, which may manifest upon a change in the genomic context, and that they can retain this capacity for millions of years.
“Pseudogene recycling” may have distinct functional outcomes. An antisense-transcribed pseudogene can generate a lncRNA base-pairing with the original mRNA. In mouse oocytes, such double-stranded RNA can give rise to small interfering RNAs (Fig. 6E) and subject the parental gene to post-transcriptional regulation by RNAi. If the sense strand of a processed pseudogene retains protein-coding capacity, it can be recycled into a protein-coding homolog of the parental gene (Fig. 6F); should the protein-coding capacity be lost, a new lncRNA will form.
MaLR LTR contribution to de novo evolution of a protein-coding gene
In one remarkable case, a co-opted MTD solo LTR contributed to de novo genesis of a protein-coding gene (Fig. 7). The gene, first annotated as an anonymous expressed DNA segment D6Ertd527e (Piao et al. 2001), formed between glutamine fructose-6-phosphate transaminase 1 (Gfpt1) and anthrax toxin receptor 1 (Antxr1) genes (Supplemental Fig. S6A). Its evolution apparently started from a lncRNA gene in the common ancestor of mice and hamsters where it acquired four promoters—A, B, C (MTD-derived), and D—controlling expression of four first exons spliced to a common 3′ terminal exon (Fig. 7A); the 3′ end of the terminal exon sequence is conserved across mammals. Remarkably, the syntenic human locus carries an MLT1-derived 5′ exon of an oocyte-specific unannotated lncRNA (Supplemental Fig. S6B). The human and rodent lncRNAs are unrelated; the conserved D6Ertd527e 3′ exon region resides in the first intron of the human lncRNA. Thus, this is a case of evolution of two novel mammalian genes involving two independent co-options of MaLR LTR promoters in one locus.
Figure 7.

A solo MTD LTR contribution to de novo evolution of a protein-coding gene. (A) Genomic organization of the D6Ertd527e locus in Mus musculus and Mesocricetus auratus. Shown are UCSC Genome Browser snapshots of D6Ertd527e loci with mapped oocyte RNA NGS. The gray dashed lines indicate CPMs. Below the conservation track is the RepeatMasker track with MT LTR insertions in red, SINE insertions in green, and a large LINE-1 insert in blue. The conserved 3′ UTR region is framed. (B) CAG trinucleotide density in MTD-driven transcripts in mice, rat, and hamster. Each CAG is represented by a vertical line. The widening depicts the coding sequence; the initiation codon is in the MTD exon. (C) Virtual translation of MTD-driven D6Ertd527e transcripts from rodent species (black) and mouse strains (blue). The phylogenetic tree was adopted from Nellaker et al. (2012). (D) D6ERTD527E protein expression in NIH3T3 cells. Transiently transfected cells expressing C-terminally HA-tagged D6ERTD527E or N-terminally HA-tagged PACT (control) were analyzed 48 h post-transfection by immunoblotting. (E) Ectopically expressed C-terminally HA-tagged D6ERTD527E protein (red) has cytoplasmic localization in mouse NIH3T3 cells. DNA (blue) was stained with DAPI. Untransfected cells lacking the HA signal demonstrate staining specificity. Scale bar, 10 μm.
D6Ertd527e is expressed maternally (Supplemental Fig. S6C). In mouse oocytes, the MTD LTR promoter yields high expression, whereas the remaining promoters are essentially not used. In hamster oocytes, expression is distributed among the promoters (Fig. 7A). D6Ertd527e shows varying protein coding capacity intertwined with an expanding CAG trinucleotide repeat ([CAG]n) at the beginning of the terminal exon (Fig. 7A). Analysis of mouse and hamster D6Ertd527e transcript variants (Supplemental Material) with the Coding Potential Assessing Tool (CPAT) suggested that the locus initially produced lncRNAs that began to acquire coding capacity. In hamster, promoters B, C, and D were predicted to produce lncRNAs (CPAT scores 0.02, 0.02, and 0.23, respectively). Promoter A was predicted to produce a protein-coding transcript (CPAT score 0.87), but the coding sequence (CDS) producing the high score is preceded by AUG codons for two short open reading frames. In mice, transcripts from all promoters were predicted to be protein-coding (CPAT scores for A–D: 0.9998, 0.9087, 0.9998, and 0.9998). However, only the transcript from the MTD LTR (promoter C) has the first available AUG associated with the longest open reading frame. In contrast, none of the putative D6Ertd527e rat transcripts has any predicted protein-coding capacity (CPAT values 0.15, 0.15, 0.16, and 0.16). The most striking difference between mouse and rat D6Ertd527e loci is a complex [CAG]n expansion in mice that gave rise to a large CDS and, consequently, the CPAT positive scoring of the MTD LTR-driven transcript (Fig. 7B). In the rat and hamster, the initiation codon from the MTD element is not in frame with the expanded repeat.
Unlike rat and hamster, all examined mouse species and strains have the polyS frame of [CAG]n expanded across the entire CDS (Fig. 7C). CDS variability could be attributed to mutations in [CAG]n and small sequence duplications (Fig. 7C). Mus pahari (Coelomys), which separated from Mus musculus ancestors about 7 MYA (Veyrunes et al. 2005), has the shortest CDS. Mus spretus, which separated about 2 MYA (Veyrunes et al. 2005), has an extended CDS because it underwent a small internal duplication shared with Mus musculus strains. There is considerable variability of D6Ertd527e alleles among laboratory strains, whose ancestors radiated in less than 1 MYA. Notably, the C57BL/6NJ strain has the longest CDS, whereas the nearest C57B/6J strain (mouse genome reference sequence) has a D6Ertd527e CDS variant like the more distant BALB/cJ or 129S1/SvlmJ strains (Fig. 7C). These data demonstrate that the CDS is dynamically evolving and, despite the origin from a trinucleotide expansion, the coding sequence diverged from a perfectly homopolymeric amino acid sequence.
We ectopically expressed D6Ertd527e fused to a C-terminal hemagglutinin (HA) tag in cultured mammalian cells (mouse NIH3T3 and human U2OS and HeLa cell lines) and detected a protein of the expected size (Fig. 7D; Supplemental Fig. S6D). The protein diffusely localized to the cytoplasm, with no noticeable effect on the expressing cells (Fig. 7E). Ectopic expression of D6ERTD527E–HA demonstrates that the MTD LTR provides a functional 5′ UTR and AUG codon, whereas the [CAG]n-derived CDS is translated into a detectable nonaggregating protein. Furthermore, D6ERTD527E peptides were identified in mouse oocyte proteomes (Wang et al. 2010, 2016; Pfeiffer et al. 2011). Thus, D6Ertd527e encodes an expressed protein implying that [CAG]n expansion can furnish the genetic material to generate a novel protein-coding gene.
Discussion
Although retrotransposons can be harmful genomic parasites bringing disease-causing mutations (Hancks and Kazazian 2012) they also provide distinct paths for genome remodeling. A rapidly growing body of literature shows in different model systems diverse contributions of LTR retrotransposons to gene function by providing promoters, enhancers, splice sites, or polyadenylation sites (for review, see de Souza et al. 2013; Gerdes et al. 2016; Göke and Ng 2016; Thompson et al. 2016). Co-option of LTR sequences may have clear biological implications, as shown for human innate immunity (Chuong et al. 2016), mammalian development (Chuong et al. 2013; Lynch et al. 2015; Nishihara et al. 2016), evolution of mammalian gene regulatory networks (Xie et al. 2010; Sundaram et al. 2014), or RNA interference in mice (Flemr et al. 2013). Our study represents a remarkable case of ERVL LTRs that function as gene-remodeling platforms that stochastically sculpt gene expression and function in oocytes and zygotes. In comparison to enhancer evolution, LTR co-option presented here is more complex (involving simultaneous co-option of TSSs and SDs) and occurring at a large scale. Although fusion of ERVL LTRs with mRNAs was reported more than a decade ago (Peaston et al. 2004) and MT2 association with gene expression has been explored in considerable detail in ESCs (Macfarlan et al. 2011, 2012; Maksakova et al. 2013), we extend these data by showing that ERVL LTRs provide a widespread system for evolving rodent maternal/zygotic programs in terms of both temporal control of gene expression and modification of gene-encoded information.
MaLR elements achieved remarkable expansion considering their nonautonomous nature. Their survival and expansion during >75 million years of rodent evolution required endogenous retroviruses, which typically invade a host, burst in copy number, and eventually become fossils (Katzourakis et al. 2005; Maksakova et al. 2006; Mager and Stoye 2015). The number of MaLR insertions, which exceeds by an order of magnitude the number of protein-coding genes, may be regarded as much a success of a parasite as a benefactor to the host. “Domesticated” MaLRs producing mainly solo LTRs could be beneficial in restraining bursts of autonomous ERVLs by competing for retrotransposition factors and using them for less damaging MaLR retrotransposition. MaLRs thus combine features of genome parasites, gene remodelers, and genome maintenance factors.
MaLR LTRs exhibit several features that could contribute to their evolutionary success and affect the germline gene expression. First, we observed reduced frequency of CpG dinucleotides, possibly from purifying selection of CpGs through methylation and subsequent deamination (Sved and Bird 1990). Human promoters with low CpG density show no significant correlation between DNA methylation and promoter activity (Weber et al. 2007). If the same applies to mice, recent MaLR LTR promoters would have reduced sensitivity to repressive DNA methylation. Second, a distinct ERVL LTR feature is the conserved SD at the 3′ end and its more frequent presence in younger ERVL LTR subfamilies. The consensus sequence AASTgtaag (S = G/C) was found in human and mouse MaLRs (Peaston et al. 2004). Thus, sequence data suggest that the functional SD is an ancestral feature retained during ERVL expansion in rodents, implying that it confers benefit for ERVL elements. NGS data show that splicing has no apparent use for internal ERVL transcripts because partnering SAs are downstream from the LTR insertion (Veselovska et al. 2015; Karlic et al. 2017). We speculate that mimicking the exon–intron gene structure and recruiting the splicing machinery to MaLRs has a positive effect on MaLR expression. This proposal is consistent with experience from transgenic mice, in which adding exon–intron boundaries improves transgene expression, whereas transgenes lacking introns (i.e., intronless insertions in the genome) are prone to silencing (van de Sluis and Voncken 2011). In any case, this SD is the key feature for the gene-remodeling platform because it defines the intrinsic 5′ ERVL LTR-derived exon that is poised to splice with exons downstream from the insertion site.
Importantly, each new LTR insertion is by definition a copy of an active LTR promoter with the potential to initiate germline transcription extending tens of kilobases downstream from the insertion site. This scenario implies that during the last 75 million years, essentially each sequence in the lineage leading to mice might have been transcribed in the oocyte or early embryo from a MaLR or MT2 LTR promoter and probed as a potential downstream exon. Oocytes or early embryos would thus be a major testing ground of phenotypic manifestations of LTR-mediated gene remodeling, which is somewhat counterintuitive given assumptions about the critical need to protect the germline.
The outcome of this impressive genome-recycling machine underscores the low probability at which new traits emerge. More than 99% of ERVL insertions show no sign of co-option; the number of full 5′ exon co-options appears relatively modest: 333 LTRs in lncRNA and 509 LTRs in protein-coding genes. These results, however, must be considered from an evolutionary perspective, in which novel beneficial traits emerge with a low probability. In fact, the number of MaLR and MT2 5′ exon co-options reported here is the highest observed number among vertebrates of a single group of repetitive sequence contributions to gene structure and expression.
Interestingly, the co-option frequency of MTA and MT2, the most recent ERVL subfamilies, is more than an order of magnitude higher than the average 5′ exon co-option frequency for LTRs of the entire ERVL class, suggesting that 5′ exon co-options are transient and mostly lost during evolution. In other words, we observe an evolutionary gradient of 5′ co-option events, ranging from the highest occurrence in the youngest LTR subfamilies and lowest in the more ancestral ones. The lower co-option rates of ORR1A LTRs than those of MTA, MTB, and MT2 LTRs suggest that MT and MT2 LTRs could have some selective advantage for co-opting 5′ exons. Co-option of MTA LTRs as promoters and full 5′ exons occurring with a 3.6% frequency (two-thirds are protein-coding genes) contrasts with B2 SINE elements that were reported to create regulated Pol II transcription at novel genomic sites (Ferrigno et al. 2001). We find that 0.1% (142) of B2s contribute to promoters and first exons of genes expressed during OET, but none made a full 5′ exon contribution.
It is unknown how many LTR insertions acquired function in reproduction, although some of the co-option events suggest such a role. For example, an MTD insertion in mice and hamsters yields abundant oocyte-specific mRNA encoding a truncated estrogen receptor beta. MTB LTRs function as the main promoters of Spin1, a maternal gene essential for OET (Chew et al. 2013). We showed a bona fide exaptation in Dicer1, which is essential for normal meiotic progression (Flemr et al. 2013). In any case, the more than 800 LTR promoters engaged during OET in mouse represent a strong regulatory potential, in which ERVL LTRs support evolution of maternal/zygotic gene expression programs. This concept extends previous studies on MuERV-L in ESCs (Macfarlan et al. 2012; Schoorlemmer et al. 2014) into the physiological regulation of maternal/zygotic gene expression and expands the scale of effects on gene expression by an order of magnitude relative to earlier observations (Peaston et al. 2004).
Several studies provided insights into transcriptional control of ERVL LTRs, which could explain the distinct maternal/zygotic expression patterns seen in Figure 4. These patterns are defined by transcriptional activation mediated by maternal or zygotic transcription factors and transcriptional repression. Multiple mechanisms can mediate retrotransposon recognition and silencing (for review, see Crichton et al. 2014; Friedli and Trono 2015; Wolf et al. 2015; Thompson et al. 2016). Transcriptional silencing involves distinct histone modification patterns, such as the loss of “active” (e.g., acetylation or H3K4me3) and emergence of “repressive” histone marks (e.g., H3K9me2/3 or H3K27me3). It remains unknown whether silencing of ERVL elements also employs Krüppel-associated box zinc-fingers proteins (KRAB-ZFPs), which function as sensors guiding silencing of Class II elements (Rowe et al. 2010, 2013; Maksakova et al. 2013; Ecco et al. 2016). Although LTRs exhibiting the maternal expression pattern (e.g., MT family) seem to be targeted by maternal silencing mechanisms associated with nuage and small RNAs (Lim et al. 2013, 2016), they overcome this repression and remain expressed during the growth phase. These LTRs utilize not yet identified maternal transcription factors, which disappear before ZGA, resulting in lack of expression in the zygote. LTRs with zygotic expression, exemplified by MT2, are expressed during ZGA and then become silenced (Kigami et al. 2003; Svoboda et al. 2004). The silencing involves the H3K4 demethylase KDM1A, transcriptional corepressor TRIM28 (also known as KAP1), and H3K9 methyltransferases EHMT2 (also known as G9a) and EHMT1 (also known as GLP) (Macfarlan et al. 2011; Maksakova et al. 2013).
Dicer1 gene remodeling and pseudogene sequence recycling integrate LTR co-option with endogenous RNAi more than previously thought (Tam et al. 2008; Watanabe et al. 2008; Flemr et al. 2013; Karlic et al. 2017). We also show that Dicer1 has been twice remodeled and rewired by LTR insertions into intron 6 during rodent evolution. Unknown selective pressures yielded low Dicer1O expression in hamster and high in mice from an MTC solo LTR insertion in their common ancestor. Although the additional recent MTA LTR insertion contributes little to Dicer1O expression in mice, LTR insertions nevertheless may retain gene rewiring and remodeling potential for millions of years that become manifest upon a genetic (or possibly epigenetic) change. Furthermore, the high expression observed at a small number of specific loci such as Dicer1 in mice likely represents a later adaptation rather than the original state. It is conceivable that lower transcriptional output of a newly inserted LTR would be better tolerated while allowing for accumulating additional mutations that would tune or eliminate the LTR promoter activity.
Finally, ERVL LTRs contribute to evolution of new genes. Given the properties of these LTRs, emergence of new lncRNAs and recycled pseudogenes is predictable. Nevertheless, we discovered a case of de novo emergence of a protein-coding gene. There are several vertebrate de novo protein-coding genes described in the literature that are of a comparable age or even younger than D6Ertd527e (for review, see Long et al. 2003; McLysaght and Hurst 2016). D6Ertd527e is unique, however, in how its protein-coding potential emerged from two common mutations: an LTR insertion provided transcriptional control and functional translation start, which combined with CDS formed through [CAG]n expansion. Notably, [CAG]n provides an effective substrate for de novo evolution of a diversified CDS. Reading frames in [CAG]n encode three possible amino acid chains: polyQ, polyS, or polyA. The murine D6Ertd527e CDS variants always use the polyS reading frame (Fig. 7C). The unused polyQ frame accumulated stop codons (Supplemental Fig. S6E), presumably because a single point mutation in [CAG]n can form a stop codon only in this frame. Natural selection might also contribute to the demise of the polyQ frame because polyglutamines from expanding [CAG]n are associated with protein aggregation, toxicity, and pathologies (Blum et al. 2013). Importantly, point mutations in other frames of [CAG]n are either silent or cause amino acid changes, thus leading to diversification of the originally homopolymeric amino acid chain. The expansion of a low-complexity sequence is reminiscent of convergent evolution of antifreeze proteins in fish, where a tripeptide expansion was found (Chen et al. 1997a,b). In contrast to D6Ertd527e, antifreeze proteins were derived from bona fide protein-coding genes, whereas D6Ertd527e evolved completely de novo. In any case, a further systematic analysis of simple repeats in different genomes should clarify how common or isolated is this seemingly simple mechanism of a protein-coding gene evolution.
Methods
Oocyte and embryo collection and gene expression analysis
Animal experiments were approved by the Institutional Animal Use and Care Committees and were carried out in accordance with the law. Oocytes and early embryos were isolated and cultured as described previously (Nagy 2003; Flemr et al. 2013). Hamster and rat full-grown GV oocytes were collected as mouse oocytes without superovulation. Gene expression in oocytes and early embryos was analyzed in triplicates by qPCR as described previously (Flemr et al. 2013). Data were normalized to Hprt by the ΔΔCt approach using in-house software. The primers and PCR conditions are shown in the Supplemental Table S4.
DNA cloning: D6Ertd527e expression vector
Mouse D6Ertd527e (NM_001167937.1) cDNA was obtained from Nugen (clone: B020023E12). CDS was amplified using primers introducing NheI and NotI restriction sites and the C-terminal HA-tag (5′-GAGCTAGCGGAGCAAGCCTGTAACAAGTTC and 5′-GTGCGGCCGCAGAACCTTATCTAGAAGCGTAGTCTGGGACGTCGTATGGGTAGGCTTCCCATGGTGTGCGACTGTG). The PCR product was cloned via pCR4.1 plasmid (Invitrogen) into pcDNA3.1 expression vector using NheI and NotI sites.
Cell culture and transfection
Mouse fibroblasts NIH3T3, human sarcoma U2OS, and adenocarcinoma HeLa cells were cultured at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (Sigma) and penicillin/streptomycin (100 units/mL; Invitrogen). Lipofectamine 3000 Reagent (Invitrogen) was used for cell transfection using manufacturer's instructions.
Immunoblotting
Whole-cell extracts 48 h post-transfection were prepared by lysing cells in cold RIPA buffer with protease inhibitors (Protease Inhibitor Cocktail; Calbiochem). After centrifugation (16,000g, 15 min, 4°C), supernatants were collected and protein concentration was measured using Bio-Rad Protein Assay kit. Equal amounts of total protein were resolved in a 7.5% polyacrylamide gel and transferred to PVDF membrane (Millipore). Primary anti-HA (Roche) and secondary HPR-conjugated anti-rat antibodies and SuperSignal WestFemto Chemiluminescent reagents (Pierce) were used for immunodetection.
Next-generation sequencing
Total RNA was extracted from 25 oocytes using PicoPure RNA Isolation Kit with on-column genomic DNA digestion according to the manufacturer's instruction (Thermo Fisher Scientific). Each sample was spiked in with 0.2 pg synthesized Renilla luciferase mRNA before extraction as a normalization control. RNA-seq libraries were constructed using the Ovation RNA-seq system V2 (NuGEN) followed by Ovation Ultralow Library system (DR Multiplex System, NuGEN). RNA-seq libraries were pooled and sequenced by 125-bp paired-end reads using Illumina HiSeq.
Bioinformatics analyses
All bioinformatics analyses are described in detail in Supplemental Methods. Relevant R scripts are provided in the file archive Supplemental_File_S1.rar. We used mouse genome reference version mm10/NCBI38. Data for analysis of LTR insertions were obtained from RepeatMasker Viz. mm10 track in the UCSC Genome Browser (http://genome.ucsc.edu/) (Kent et al. 2002). NGS data were mapped and analyzed as described earlier (Abe et al. 2015; Karlic et al. 2017).
For the nucleotide substitution rate analysis, 200 randomly selected sequences were aligned for each LTR group, and a distance matrix of substitution rates (defined as a fraction of mismatches with indels omitted) was extracted from the multiple alignment and subjected to hierarchical clustering by mean distance. The distribution of the normalized shortest distances (i.e., normalized substitution rates between closest neighbors) within each LTR group were represented in the final box plot (Fig. 1C).
The MT LTR phylogenetic tree (Fig. 3C) was built from 5000 randomly selected LTRs and 773 5′ exon LTRs aligned using Clustal Omega (version 1.2.3, default parameters) (Sievers et al. 2011) and FastTree (version 2.1.9, parameters: -gamma -nt -gtr).
For the annotation of retrotransposon co-option, we collected transcript annotations from the UCSC mm10 database and combined them with lncRNA transcript models generated from oocyte and early embryo NGS data (Supplemental Table S3). LTR sequence co-option was classified into four categories according to the LTR sequence (entire sequences for non-LTR retrotransposons) overlap with annotated exons:
-
I.
5′ Exon contribution (retrotransposon-derived promoter, TSS, and/or SD)
-
II.
Internal exon contribution (retrotransposon-derived SD and/or SA),
-
III.
3′ Exon contribution (retrotransposon-derived SA and/or poly(A))
-
IV.
Transcript tagging, in which a transcript contains a retrotransposon sequence that does not contribute to mRNA formation.
For categories I–III, a full contribution designates that the retrotransposon overlaps both exon borders; a partial contribution, one of the two. Classification criteria included NGS support by 10 or more mapped reads from the overlapping exon in at least one of the NGS samples. For classes I–III, at least two spliced reads in at least two samples were required to support splicing.
For the analyses of cumulative RNA expression, we used 100 most-expressed MuERV-L elements and estimated FPKM values 150 kb upstream and downstream. To reduce the transcriptional signal from other promoters while retaining the signal downstream from inserts, we masked regions with FPKM >1. Because our NGS experiments were performed on total RNA (Abe et al. 2015), this cutoff is an equivalent of FPKM ∼4–5 of rRNA-depleted samples. One-base resolution coverage for flanking regions was summed up and binned into 10-kb bins; combined FPKM values were calculated for each bin.
Data access
The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE86470.
Supplementary Material
Acknowledgments
We thank the Mediterranean Institute for Life Sciences in Split for hosting data mining and Jiri Hejnar for comments. This work was funded from the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 647403, D-FENS). The P.S. laboratory was also supported by the Ministry of Education, Youth, and Sports project NPU1 LO1419. S.G. was supported by the Marie Curie Initial Training Network (project #607720, RNATRAIN). R.K., V.F., F.H., M.K., and K.V. were supported by the European Commission Seventh Framework Program (Integra-Life; grant 315997 to K.V.), and Croatian Science Foundation (grant IP-2014-09-6400 to K.V.). F.A. was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (#20062002, #25252054). R.M.S. and J.M. were supported by the grant from the National Institutes of Health HD022681. IMG institutional support was provided by RVO: 68378050. Computational resources for P.S. were provided by the CESNET LM2015042 and the CERIT Scientific Cloud LM2015085 under the programme “Projects of Large Research, Development, and Innovations Infrastructures.”
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.216150.116.
Freely available online through the Genome Research Open Access option.
References
- Abe K, Yamamoto R, Franke V, Cao M, Suzuki Y, Suzuki MG, Vlahovicek K, Svoboda P, Schultz RM, Aoki F. 2015. The first murine zygotic transcription is promiscuous and uncoupled from splicing and 3′ processing. EMBO J 34: 1523–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénit L, De Parseval N, Casella JF, Callebaut I, Cordonnier A, Heidmann T. 1997. Cloning of a new murine endogenous retrovirus, MuERV-L, with strong similarity to the human HERV-L element and with a gag coding sequence closely related to the Fv1 restriction gene. J Virol 71: 5652–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum ES, Schwendeman AR, Shaham S. 2013. PolyQ disease: misfiring of a developmental cell death program? Trends Cell Biol 23: 168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, DeVries AL, Cheng CH. 1997a. Convergent evolution of antifreeze glycoproteins in Antarctic notothenioid fish and Arctic cod. Proc Natl Acad Sci 94: 3817–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, DeVries AL, Cheng CH. 1997b. Evolution of antifreeze glycoprotein gene from a trypsinogen gene in Antarctic notothenioid fish. Proc Natl Acad Sci 94: 3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew TG, Peaston A, Lim AK, Lorthongpanich C, Knowles BB, Solter D. 2013. A tudor domain protein SPINDLIN1 interacts with the mRNA-binding protein SERBP1 and is involved in mouse oocyte meiotic resumption. PLoS One 8: e69764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Rumi MA, Soares MJ, Baker JC. 2013. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat Genet 45: 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, Feschotte C. 2016. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 351: 1083–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costas J. 2003. Molecular characterization of the recent intragenomic spread of the murine endogenous retrovirus MuERV-L. J Mol Evol 56: 181–186. [DOI] [PubMed] [Google Scholar]
- Craig NL, Chandler M, Gellert M, Lambowitz AM, Rice PA, Sandmeyer SB. 2015. Mobile DNA III. AMS Press, Washington, DC. [Google Scholar]
- Crichton JH, Dunican DS, Maclennan M, Meehan RR, Adams IR. 2014. Defending the genome from the enemy within: mechanisms of retrotransposon suppression in the mouse germline. Cell Mol Life Sci 71: 1581–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza FS, Franchini LF, Rubinstein M. 2013. Exaptation of transposable elements into novel cis-regulatory elements: is the evidence always strong? Mol Biol Evol 30: 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecco G, Cassano M, Kauzlaric A, Duc J, Coluccio A, Offner S, Imbeault M, Rowe HM, Turelli P, Trono D. 2016. Transposable elements and their KRAB-ZFP controllers regulate gene expression in adult tissues. Dev Cell 36: 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrigno O, Virolle T, Djabari Z, Ortonne JP, White RJ, Aberdam D. 2001. Transposable B2 SINE elements can provide mobile RNA polymerase II promoters. Nat Genet 28: 77–81. [DOI] [PubMed] [Google Scholar]
- Flemr M, Malik R, Franke V, Nejepinska J, Sedlacek R, Vlahovicek K, Svoboda P. 2013. A retrotransposon-driven dicer isoform directs endogenous small interfering RNA production in mouse oocytes. Cell 155: 807–816. [DOI] [PubMed] [Google Scholar]
- Friedli M, Trono D. 2015. The developmental control of transposable elements and the evolution of higher species. Annu Rev Cell Dev Biol 31: 429–451. [DOI] [PubMed] [Google Scholar]
- Gerdes P, Richardson SR, Mager DL, Faulkner GJ. 2016. Transposable elements in the mammalian embryo: pioneers surviving through stealth and service. Genome Biol 17: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göke J, Ng HH. 2016. CTRL+INSERT: retrotransposons and their contribution to regulation and innovation of the transcriptome. EMBO Rep 17: 1131–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, Kazazian HH Jr. 2012. Active human retrotransposons: variation and disease. Curr Opin Genet Dev 22: 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubley R, Finn RD, Clements J, Eddy SR, Jones TA, Bao W, Smit AF, Wheeler TJ. 2016. The Dfam database of repetitive DNA families. Nucleic Acids Res 44: D81–D89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlic R, Ganesh S, Franke V, Svobodova E, Urbanova J, Suzuki Y, Aoki F, Vlahovicek K, Svoboda P. 2017. Long non-coding RNA exchange during oocyte-to-embryo transition in mice. DNA Res 10.1093/dnares/dsw058. [DOI] [Google Scholar]
- Katzourakis A, Rambaut A, Pybus OG. 2005. The evolutionary dynamics of endogenous retroviruses. Trends Microbiol 13: 463–468. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigami D, Minami N, Takayama H, Imai H. 2003. MuERV-L is one of the earliest transcribed genes in mouse one-cell embryos. Biol Reprod 68: 651–654. [DOI] [PubMed] [Google Scholar]
- Lim AK, Lorthongpanich C, Chew TG, Tan CW, Shue YT, Balu S, Gounko N, Kuramochi-Miyagawa S, Matzuk MM, Chuma S, et al. 2013. The nuage mediates retrotransposon silencing in mouse primordial ovarian follicles. Development 140: 3819–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CY, Knowles BB, Solter D, Messerschmidt DM. 2016. Epigenetic control of early mouse development. Curr Topics Dev Biol 120: 311–360. [DOI] [PubMed] [Google Scholar]
- Long M, Betran E, Thornton K, Wang W. 2003. The origin of new genes: glimpses from the young and old. Nat Rev Genet 4: 865–875. [DOI] [PubMed] [Google Scholar]
- Lynch VJ, Nnamani MC, Kapusta A, Brayer K, Plaza SL, Mazur EC, Emera D, Sheikh SZ, Grutzner F, Bauersachs S, et al. 2015. Ancient transposable elements transformed the uterine regulatory landscape and transcriptome during the evolution of mammalian pregnancy. Cell Rep 10: 551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan TS, Gifford WD, Agarwal S, Driscoll S, Lettieri K, Wang J, Andrews SE, Franco L, Rosenfeld MG, Ren B, et al. 2011. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev 25: 594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, Firth A, Singer O, Trono D, Pfaff SL. 2012. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager DL, Stoye JP. 2015. Mammalian endogenous retroviruses. Microbiol Spectr 3: MDNA3-0009-2014. [DOI] [PubMed] [Google Scholar]
- Maksakova IA, Romanish MT, Gagnier L, Dunn CA, van de Lagemaat LN, Mager DL. 2006. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet 2: e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksakova IA, Thompson PJ, Goyal P, Jones SJ, Singh PB, Karimi MM, Lorincz MC. 2013. Distinct roles of KAP1, HP1 and G9a/GLP in silencing of the two-cell-specific retrotransposon MERVL in mouse ES cells. Epigenet Chromatin 6: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy EM, McDonald JF. 2004. Long terminal repeat retrotransposons of Mus musculus. Genome Biol 5: R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLysaght A, Hurst LD. 2016. Open questions in the study of de novo genes: what, how and why. Nat Rev Genet 17: 567–578. [DOI] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]
- Nagy A. 2003. Manipulating the mouse embryo: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Nellaker C, Keane TM, Yalcin B, Wong K, Agam A, Belgard TG, Flint J, Adams DJ, Frankel WN, Ponting CP. 2012. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol 13: R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara H, Kobayashi N, Kimura-Yoshida C, Yan K, Bormuth O, Ding Q, Nakanishi A, Sasaki T, Hirakawa M, Sumiyama K, et al. 2016. Coordinately co-opted multiple transposable elements constitute an enhancer for wnt5a expression in the mammalian secondary palate. PLoS Genet 12: e1006380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Komata M, Inoue F, Yamada K, Nakai K, Ohsugi M, Shirahige K. 2013. Inferring the choreography of parental genomes during fertilization from ultralarge-scale whole-transcriptome analysis. Genes Dev 27: 2736–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peaston AE, Evsikov AV, Graber JH, de Vries WN, Holbrook AE, Solter D, Knowles BB. 2004. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev Cell 7: 597–606. [DOI] [PubMed] [Google Scholar]
- Pfeiffer MJ, Siatkowski M, Paudel Y, Balbach ST, Baeumer N, Crosetto N, Drexler HC, Fuellen G, Boiani M. 2011. Proteomic analysis of mouse oocytes reveals 28 candidate factors of the “reprogrammome”. J Proteome Res 10: 2140–2153. [DOI] [PubMed] [Google Scholar]
- Piao Y, Ko NT, Lim MK, Ko MS. 2001. Construction of long-transcript enriched cDNA libraries from submicrogram amounts of total RNAs by a universal PCR amplification method. Genome Res 11: 1553–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribet D, Louvet-Vallée S, Harper F, de Parseval N, Dewannieux M, Heidmann O, Pierron G, Maro B, Heidmann T. 2008. Murine endogenous retrovirus MuERV-L is the progenitor of the “orphan” ε viruslike particles of the early mouse embryo. J Virol 82: 1622–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, et al. 2010. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463: 237–240. [DOI] [PubMed] [Google Scholar]
- Rowe HM, Kapopoulou A, Corsinotti A, Fasching L, Macfarlan TS, Tarabay Y, Viville S, Jakobsson J, Pfaff SL, Trono D. 2013. TRIM28 repression of retrotransposon-based enhancers is necessary to preserve transcriptional dynamics in embryonic stem cells. Genome Res 23: 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoorlemmer J, Pérez-Palacios R, Climent M, Guallar D, Muniesa P. 2014. Regulation of mouse retroelement MuERV-L/MERVL expression by REX1 and epigenetic control of stem cell potency. Front Oncol 4: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G. 2011. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet 43: 811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AF. 1993. Identification of a new, abundant superfamily of mammalian LTR-transposons. Nucleic Acids Res 21: 1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2013–2015. RepeatMasker Open-4.0. http://www.repeatmasker.org/. [Google Scholar]
- Sookdeo A, Hepp CM, McClure MA, Boissinot S. 2013. Revisiting the evolution of mouse LINE-1 in the genomic era. Mobile DNA 4: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram V, Cheng Y, Ma Z, Li D, Xing X, Edge P, Snyder MP, Wang T. 2014. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res 24: 1963–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sved J, Bird A. 1990. The expected equilibrium of the CpG dinucleotide in vertebrate genomes under a mutation model. Proc Natl Acad Sci 87: 4692–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda P, Stein P, Anger M, Bernstein E, Hannon GJ, Schultz RM. 2004. RNAi and expression of retrotransposons MuERV-L and IAP in preimplantation mouse embryos. Dev Biol 269: 276–285. [DOI] [PubMed] [Google Scholar]
- Svoboda P, Franke V, Schultz RM. 2015. Sculpting the transcriptome during the oocyte-to-embryo transition in mouse. Curr Topics Dev Biol 113: 305–349. [DOI] [PubMed] [Google Scholar]
- Tam OH, Aravin AA, Stein P, Girard A, Murchison EP, Cheloufi S, Hodges E, Anger M, Sachidanandam R, Schultz RM, et al. 2008. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 453: 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PJ, Macfarlan TS, Lorincz MC. 2016. Long terminal repeats: from parasitic elements to building blocks of the transcriptional regulatory repertoire. Mol Cell 62: 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Lagemaat LN, Medstrand P, Mager DL. 2006. Multiple effects govern endogenous retrovirus survival patterns in human gene introns. Genome Biol 7: R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Sluis B, Voncken JW. 2011. Transgene design. Methods Mol Biol 693: 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veselovska L, Smallwood SA, Saadeh H, Stewart KR, Krueger F, Maupetit-Mehouas S, Arnaud P, Tomizawa S, Andrews S, Kelsey G. 2015. Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol 16: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyrunes F, Britton-Davidian J, Robinson TJ, Calvet E, Denys C, Chevret P. 2005. Molecular phylogeny of the African pygmy mice, subgenus Nannomys (Rodentia, Murinae, Mus): implications for chromosomal evolution. Mol Phylogenet Evol 36: 358–369. [DOI] [PubMed] [Google Scholar]
- Wang S, Kou Z, Jing Z, Zhang Y, Guo X, Dong M, Wilmut I, Gao S. 2010. Proteome of mouse oocytes at different developmental stages. Proc Natl Acad Sci 107: 17639–17644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Pfeiffer MJ, Drexler HC, Fuellen G, Boiani M. 2016. Proteomic analysis of mouse oocytes identifies PRMT7 as a reprogramming factor that replaces SOX2 in the induction of pluripotent stem cells. J Proteome Res 10.1021/acs.jproteome.5b01083. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, et al. 2008. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 453: 539–543. [DOI] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. 2007. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39: 457–466. [DOI] [PubMed] [Google Scholar]
- Wolf G, Greenberg D, Macfarlan TS. 2015. Spotting the enemy within: Targeted silencing of foreign DNA in mammalian genomes by the Krüppel-associated box zinc finger protein family. Mobile DNA 6: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie D, Chen CC, Ptaszek LM, Xiao S, Cao X, Fang F, Ng HH, Lewin HA, Cowan C, Zhong S. 2010. Rewirable gene regulatory networks in the preimplantation embryonic development of three mammalian species. Genome Res 20: 804–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.